Index
Index of abbreviations
3
Index of figures
4
Introduction
6
1 Iron Sulfur cluster (Isc)
8
1.1 Structure and function
9
1.2 Iron-‐sulfur cluster biosynthesis
12
2 Isc System
14
2.1 IscR
14
2.2 IscS
15
2.3 IscU
17
2.4 IscA
18
2.5 HscA and HscB
19
2.6 Ferredoxin (FdX)
22
2.7 YfhJ
23
2.8 Mechanism of Fe-‐S cluster assembly mediated by the Isc system
24
3 Friedreich ataxia
28
4 Frataxin
31
4.1 Structure
31
4.2 Iron-‐binding properties
34
4.3 Frataxin function
37
4.4 Aconitases: examples of frataxin regulation
40
5 Reconstitution experiments
44
5.1 Results
46
5.1.1 Optimization of IscU, HscA, HscB and ATP concentration 47 5.1.2 Role of CyaY anf YfhJ in Fe-‐S cluster biosynthesis 505.1.3 Dependence of CyaY activity from iron 52 5.1.4 Aconitase2 reconstitution 54
5.2 Discussion
55
5.3 Methods
57
5.3.1 Protein Purifications 57 5.3.2 Reconstitution experiments 585.3.3 Aconitase2 reconstitution experiments 59
6 Circular Dichroism of proteins
60
6.1 Circular Dichroism spectrometers
61
6.2 CD of polypeptides and proteins
62
6.2.1 Proteins UV spectroscopy 636.2.2 Protein CD spectra 64
7 Yeast frataxin stability
69
7.1 Results
72
7.2 Discussion
77
7.3 Methods
78
7.3.1 Far-‐UV CD measurements. 78 7.3.2 Thermal unfolding curves measurements 79Final Conclusions
80
Bibliography
82
Index of abbreviations
CD Circular DichroismDTT Dithiothreitol
EDTA Ethylenediaminetetraacetic acid FdX Ferredoxin
FRDA Friedreich ataxia
GST Glutathione S-‐transferase
IPTG Isopropyl β-‐D-‐1-‐thiogalactopyranoside IRP Iron-‐responsive element-‐binding protein Isc Iron-‐sulfur cluster
nif Nitrogen fixation operon Ni-‐NTA Ni-‐nitrilotriacetic
NMR Nuclear magnetic resonance OD Optical Density
PCR Polymerase Chain Reaction PLP Pyridoxal-‐phosphate
TCEP Tris(2-‐carboxyethyl)phosphine TEV Tobacco Etch Virus
Index of figures
Introduction
1 Iron Sulfur cluster (Isc)
1.1 Various [Fe–S] clusters found in iron–sulfur proteins 10
2 Isc System
2.1 Network of protein-‐protein interactions involving IscS 16 2.2 Proposed mechanism of NIFS desulfuration reaction 17 2.3 Kinetic scheme of the Hsc66 ATPase reaction cycle 20 2.4 A model for the IscU binding cycle of HscA 21 2.5 Proposed mechanistic scheme for chaperone-‐catalyzed Fe-‐S cluster transfer 27
3 Friedreich ataxia
4 Frataxin
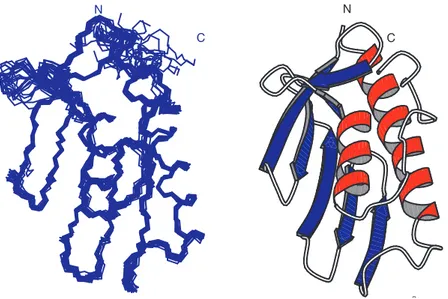
4.1 Tertiary structure of S_Fra 32 4.2 Alignment of frataxin and cyaY sequences 33
4.3 Mapping the residues involved in iron binding onto the structures of CyaY and hfra 36
4.4 The three dimensional structure of porcine mitochondrial aconitase 41
5 Reconstitution experiments
5.1 Bacterial growth curve 45 5.2 Scan of IscU concentration 47 5.3 Scan of HscA concentration 48 5.4 Scan of HscB concentration 48 5.5 Scan of ATP concentration 49 5.6 Scan of CyaY concentration 50 5.7 Scan of YfhJ concentration 51
5.8 Scan of YfhJ concentration in presence of CyaY 51 5.9 Scan of Fe2+ concentration 52 5.10 Scan of CyaY concentration 53 5.11 Aconitase reconstitution 54 5.12 Kinetic curve of Aconitase2 reconstitution 55 5.13 Glove Box 59
6 Circular Dichroism of proteins
6.1 Schematic representation of circularly polarized light 60 6.2 Schematic representation of a circular dichroism spectrometer 61 6.3 Circular dichroism spectra of pure secondary structures 65
Table 6.1 Characteristic CD bands for α-‐helix, β-‐sheets, β-‐turns and random coil
67
7 Yeast frataxin stability
7.1 representations of the structures of CyaY, Yfh1 and hfra 70 7.2 Thermal denaturation curves of Yfh1 73 7.3 Thermal denaturation curves of Yfh1 74 7.4 CD spectra of Yfh1 75 7.5 CD spectra of Yfh1 76 7.6 Comparison of the thermal denaturation curves 77
Final Conclusions
Introduction
Iron is an essential and ubiquitous element in biology. The survival of both prokaryotic and eukaryotic organisms depends on it; this ion takes part in the prosthetic group as a hemoprotein cofactor and in Fe-‐S clusters[1]. Determining its homeostasis and how its cellular forms are regulated in amount and allocation is thus of particular importance for understanding how these mechanisms can be at time so compromised to cause pathological conditions, such as metabolic and neurodegenerative diseases[2].
The concentration of iron in the cell must however be tightly regulated: while iron deficiency is lethal for the organisms, iron accumulation is highly toxic. Free iron, present in the cytosol as Fe2+ can participate together with H2O2 to Fenton reactions producing free radicals and cause damage to lipid membranes, proteins and nucleic acids[2].
In eukaryotes the majority of iron is present in mitochondria where it takes part to the heme and Fe-‐S cluster biosynthesis. The latter process is a sophisticated machinery that involves several proteins conserved in organisms. In bacteria iron is under the control of the operon isc. In E. coli, for instance, this operon encoded eight proteins: IscR, IscS, IscU, IscA, HscB, HscA, Fdx and Yfhj[3].
An intriguing issue is that of better understanding the mechanism of Fe-‐S cluster biosynthesis and the role of each component of the Isc system. This information is important not only for the interest of our basic knowledge of iron metabolism but also because working on the simpler bacterial model system may help us to understand also how iron is correlated to human diseases. Abnormal Fe-‐S protein biogenesis and mitochondrial iron accumulation in hearth and neurones are for instance part of the typical phenotype of a genetic neurodegenerative disease,
Friedreich’s Ataxia[4, 5]. This pathology is caused by the deficiency of a mitochondrial protein, frataxin, extremely conserved throughout species[6]. Although the frataxin function is still unclear, it was proved that this protein is able to bind iron in vitro and
in vivo so that it was suggested a role as iron storage or as iron chaperone in the Fe-‐S
cluster biosynthesis[7-‐13]. Despite its involvement in iron metabolism, frataxin does not show any feature commune with other iron binding protein[14].
To determine frataxin structural features and differences between species, it is fundamental to completely characterise the system and its modification through evolution. By comparing the human frataxin, the yeast Yfh1 and bacterial CyaY, it was shown that the three proteins have the same fold but different thermal stabilities[15] and iron-‐binding properties[16].
The final aim of this thesis has been to provide evidence to clarify frataxin’s role in the machinery of Fe-‐S cluster biosynthesis and to point out some frataxin’s structural properties. For these reasons:
1. I have been studying the kinetic of cluster assembly in the presence not only of the desulphurase IscS and scaffold IscU, but also the chaperons HscA and HscB focusing especially on the effect of bacterial frataxin CyaY and of the little known YfhJ.
2. I used CD spectroscopy to detect the behaviour of the less stable Yfh1 in the presence of different environmental conditions. My purpose has been to determine if the ionic composition has a strong effect on Yfh1 stability. These studies will hopefully provide important pieces of information useful to understand frataxin features and its involvement in Isc machinery.
Iron Sulfur cluster (Isc)
1
Iron is ubiquitous in the environment and in biology. Eukaryotic cells (and most prokaryotic organisms) require iron for survival and proliferation, as a constituent of hemoproteins, iron-‐sulfur (Fe-‐S) proteins and other proteins that use iron in functional groups to carry out essential housekeeping functions for cellular metabolism. Cellular iron deficiency arrests cell growth and leads to cell death[2].The biological importance of iron is largely attributable to its chemical properties: it is involved in one-‐electron oxidation-‐reduction reactions with transition between its ferric and ferrous states. However, the same chemical property explains why an excess of free and reactive iron is toxic. In the cytoplasm, a significant fraction of iron is reduced and can participate in “Fenton-‐type” redox chemistry: ferrous iron reacts with hydrogen peroxide (H2O2) or lipid peroxides to generate ferric iron, OH-‐, and the highly reactive hydroxyl radical (OH•) or lipid radicals such as LO• and LOO•. These radicals damage lipid membranes, proteins, and nucleic acids. Since both cellular iron overload and iron deficiency cause cell death, the levels of reactive iron must be carefully controlled and limited[2].
Biological iron–sulfur clusters have been identified about 40 years ago as acid-‐ labile prosthetic groups contained within a class of electron carrier proteins called ferredoxins[17]. They are widely distributed in nature and can be found in anaerobic, aerobic and photosynthetic bacteria, fungi, plants and mammals[18]. Most known Fe– S proteins of a eukaryotic cell are located in the mitochondria, but some also exist outside them. Finally, at least one Fe–S protein, the endonuclease Ntg2p, has been localized to the cell nucleus[19].
Since their discovery, [Fe-‐S] clusters have been considered ideal agents of electron transfer because of their versatile electronic properties[20]. Many different proteins that contain these clusters have been described with different functionality[21].
[Fe–S] clusters are now known to have roles in controlling protein structure and in enzyme active sites (see for instance aconitase), to act as environmental sensors (FNR), to serve as modulators of gene regulation (IRP and soxR), and to participate in radical generation[20]. Such functional diversity almost reflects the chemical versatility of iron and sulfur, leading to the suggestion that prebiotic iron–sulfur complexes could have played an important role in the emergence of life on earth. The importance of [Fe–S] clusters is furthermore underlined by their involvement in three major processes required to sustain life on earth: nitrogen fixation, photosynthesis and respiration[17].
1.1 Structure and function
The most common [Fe–S] clusters found in nature are [2Fe–2S] and [4Fe–4S] clusters and these are usually coordinated to proteins by cysteine ligands. However, biological [Fe–S] clusters of higher nuclearity have been discovered, and not all are attached to their protein partners by cysteine ligands. Despite the apparent diversity in the overall structure, reactivity, electronic properties and polypeptide environments of [Fe–S] clusters, polynuclear [Fe–S] clusters are all constructed from [2Fe–2S] rhombs[17]. For example two [2Fe-‐2S] units may be converted to the cubane [4Fe-‐ 4S]. Even if cuboidal cluster [3Fe-‐4S] is considerably less stable and it could be converted in linear [2Fe-‐2S] or cubane [4Fe-‐4S], it has been assembled from [4Fe-‐4S] via loss of one iron atom[20].
In addition, not all [Fe–S] proteins contain clusters with Fe as the only metal: the nitrogenase MoFe protein for example, contains a cluster which has a [7Fe–9S–Mo] core[17].
Figure 1.1 Various [Fe–S] clusters found in iron–sulfur proteins. The most common (a) [2Fe–2S] and (b) [4Fe–4S] clusters are coordinated to their protein partners by cysteine ligands. Iron–sulfur clusters of higher nuclearity such as (c) the [8Fe–7S] P cluster and (d) the [Mo–7Fe–9S] FeMo-‐cofactor from nitrogenase represent just some of the intricate possibilities of more complex biological iron– sulfur clusters. (Iron is represented in green, sulfur in yellow and molybdenum in magenta)[17].
Because of their structural and chemical versatility, Fe-‐S clusters present an extreme functional flexibility. The electronic structures of oligonuclear Fe-‐S clusters have been studied by a variety of spectroscopic techniques[22] showing that they are ideal agents for electron storage and transfer, signaling and regulation, via their tunable sensitivity to various oxidants or reductants. Furthermore Fe-‐S clusters are well suited to supply single electrons, as required in homolytic reactions[23].
These are also the properties that can make them sensors of molecules in their immediate environment. In fact Fe-‐S clusters are sensitive to cellular oxidants and in
favourable conditions and appropriate organization of the necessary ligands, certain biologically relevant [Fe–S] clusters have the capacity to form spontaneously. Never-theless, spontaneous, intracellular assembly of [Fe–S] clusters using free Fe2þ/3þand S2"is not an attractive physiological prospect because these elements are meta-bolic poisons, especially in the concentrations and under the conditions required to achieve chemical [Fe–S] cluster assembly. A more reasonable expectation was that [Fe–S] clusters are formed biologically by protein-directed activation and specific delivery of Fe and S to the assembly site, and this possibility turned out to be correct. Here we discuss the origins of the identification of proteins involved in [Fe–S] cluster assembly in bac-teria and our current understanding of the functions of those proteins.
Origins of the concept of molecular scaffolds for complex metallocluster assembly Observations leading to the discovery of proteins involved in [Fe–S] cluster assembly did not initially come from an analysis of the formation of simple [2Fe–2S] or [4Fe–4S] clusters, but rather emerged from attempts to understand the assembly of one of nature’s most complex [Fe–S] clusters, the FeMo-cofactor of the nitrogenase MoFe protein. FeMo-cofactor (Figure 1d) has attracted the attention of the bioinorganic community for many years because it provides the site for biological nitrogen reduction. Genetic and biochemical studies established that a consortium of proteins is required to assemble FeMo-cofactor. Two general pathways for this process were initially considered. Namely, FeMo-cofactor could either be assembled directly on the MoFe protein or it Figure 1 Cys Cys Cys Cys Cys Cys Cys Cys Cys Cys Cys Cys His Cys Homocitrate (a) (b) (c) (d)
Current Opinion in Chemical Biology
Various [Fe–S] clusters found in iron–sulfur proteins. The most common(a) [2Fe–2S] and (b) [4Fe–4S] clusters are coordinated to their protein partners by cysteine ligands. Iron–sulfur clusters of higher nuclearity such as(c) the [8Fe–7S] P cluster and (d) the [Mo–7Fe–9S] FeMo-cofactor from nitrogenase represent just some of the intricate possibilities of more complex biological iron–sulfur clusters. (Iron is represented in green, sulfur in yellow and molybdenum in magenta.)
Formation of iron–sulfur clusters in bacteria Frazzon and Dean 167
vitro, they may also respond to strong reducing agents or ligands for iron that are
stronger than their sulfur ligands. Oxidation of Fe-‐S clusters may cause cluster rearrangement or total disassembly. An example is FNR, an E. Coli regulator controlling the synthesis of proteins required for anaerobic respiration with nitrate, fumarate, trimethylamine oxide and similar electron acceptors replacing oxygen. FNR is functional only in its dimeric form, stabilized by [4Fe-‐4S] cluster, binding DNA with high affinity thus controlling gene expression. The [4Fe-‐4S] cluster is very sensitive to oxygen, which rapidly converts it to the more air-‐stable [2Fe-‐2S] form making FNR inactive and monomeric[21, 23].
Cytoplasmic aconitase is converted to IRP1 through loss of its Fe-‐S cluster. Binding to iron-‐responsive elements (IREs), IRPs regulate translation of ferritin mRNA (ferritin is an iron storage protein) and transferrin receptor mRNA (transferrin receptor transfers iron into the cell). The Fe-‐S clusters of the aconitases are also readily attacked by oxidants and converted to [3Fe-‐4S] clusters. As a result they can be considered as sensors for oxygen, which destroy the cluster, and also as sensors for iron, as iron is nedeed for resynthesis of the cluster[23].
SoxR contains [2Fe-‐2S] cluster and is activated on exposure of cells specifically to O2 and NO, but not to H2O2 or OH. Active SoxR activates transcription of only a single gene, soxs, leading to the formation of the SoxS protein. SoxS regulates expression of a number of genes whose products function in the defense against 02 and NO[23].
Fe-‐S clusters can serve as active sites of enzymes as, for instance, in mitochondrial aconitase involved in the tricarboxylic acid cycle. Thus, destruction or (re)synthesis of this cluster will determine the activity of aconitase and hence control the citric acid cycle[23]: the iron of a [4Fe-‐4S] cluster, which has no Cys ligand, serves as a Lewis acid in catalyzing the transformation of the substrate[24].
1.2 Iron-sulfur cluster biosynthesis
In proteins, Fe-‐S clusters can be spontaneously assembled from the required components under the proper conditions, just as we can do it in vitro from purified apoproteins. However, in both prokaryotes and in eukaryotes, enzymes have been identified that use pyridoxal phosphate for desulfurase activity, transforming cysteine in alanine and elemental sulfur. Similarly, specific carrier-‐proteins for Fe are likely to be used. Thus, the formation of clusters is driven by the availability of iron and three cysteines: one to provide sulfur for the synthesis and three others to stabilize the Fe-‐S cluster created. When only two Cys are available per peptide, the peptide will dimerize to make a [4Fe-‐4S] cluster[24].
Investigation on Fe-‐S cluster maturation in bacteria has led to the identification of two operons termed nif (nitrogen fixation) and isc (iron–sulfur cluster assembly) that function in Fe–S-‐cluster biosynthesis. The nif operon encodes proteins that execute specific functions in the assembly of nitrogenase, a complex metalloenzyme that catalyses the fixation of nitrogen. The isc operon encodes proteins necessary for the maturation of bacterial Fe–S proteins. Many of the gene products encoded by the nif and isc operons have sequence similarity to the eukaryotic components of Fe–S-‐
protein maturation[19].
The molybdenum-‐dependent nitrogenase is a complex metalloenzyme composed of two component proteins called the Fe protein and the MoFe protein. The Fe protein acts as a specific, ATP-‐binding, one-‐electron reductant of the MoFe protein, which contains the active site for substrate binding and reduction. A single [4Fe-‐4S] cluster is believed to be symmetrically bridged between the Fe protein subunits. In A.
gene products are required for the full activation or the catalytic stability of the nitrogenase Fe protein. Deletion of the nifV gene resulted in lower MoFe protein activity, probably resulting from the accumulation of an altered FeMo-‐cofactor[25].
In E. coli are present two systems for the Fe-‐S cluster biosynthesis. Although Isc and Suf are systems relied on the same activities (presented by cysteine desulfurase, scaffold, ATPase and carrier) genetic and biochemical characterizations revealed differences in the ability of the Isc and Suf systems to function under stress conditions. In fact they are considered the house-‐keeping and stress-‐dedicated systems, respectively. First of all it was pointed that the Isc system is inactivated by ROS since a solvent-‐exposed Fe–S cluster, built on the scaffold IscU, can be easily oxidized by ROS. On the other hand, Suf might have evolved to be efficient when Fe and S levels are in present in limited amount. As a result of that Isc is the primary system used in exponentially growing cells, whereas Suf is used transiently under oxidative stress and iron starvation conditions[26].
In Eukaryotes a Isu system has been discovered to be able to mediate the Fe-‐S cluster assembly and to transfer it to apo-‐acceptor. Isu and Isc proteins are very similar in sequence and function[18].
Isc system
2
Understanding the functions of Isc proteins in prokaryotes is important to provide insights into Fe-‐S cluster assembly in mitochondria, with potential relevance in iron-‐ storage diseases and the control of cellular iron uptake.The isc operon in the bacterium E. Coli encodes eight proteins: IscR, IscS, IscU, IscA, HscB, HscA, Fdx and YfhJ. The role of YfhJ is not clear yet. Furthermore mutants with inactivated iscR or orf3 showed no differences from wild-‐type cells while inactivation of the iscS gene elicited the most drastic alteration. Strains with mutations in the iscU,
hscB, hscA, and fdx genes exhibited conspicuous phenotypical consequences almost
identical to one another. The effect of the inactivation of iscA was small but appreciable on Fe-‐S enzymes[27].
2.1 IscR
IscR shares amino acids similarity with MarA (multiple antibiotic resistance), suggesting a function as a regulator. Schwartz et al. discovered that iscR is co-‐ transcribed with iscSUA and that transcription of iscRSUA originates from a single promoter upstream of iscR that is repressed by IscR itself[28].
IscR contains a [2Fe-‐2S] cluster bound with an atypical ligation scheme of three cysteines and one histidine[29]. Its activity is decreased in mutants lacking the Fe-‐S cluster assembly proteins leading to consider that there may be a link between the levels of [2Fe-‐2S]IscR and the rate of Fe-‐S cluster formation. In fact when Fe-‐S cluster assembly becomes rate limiting, levels of [2Fe-‐2S]IscR would decrease as a result of a diminution in its rate of synthesis, and repression of iscRSUA would be relieved. The resulting increase in the Isc assembly proteins would subsequently lead to an
thus reset repression of the isc operon[28].
IscR acquires an Fe-‐S cluster via the Isc proteins once the cellular demand for Fe-‐S cluster biogenesis is satisfied, suggesting that the Isc proteins might be able to distinguish between IscR and other apo-‐protein targets. The proposed (Cys)3(His)1 ligation scheme of IscR could differentiate it from other apo-‐proteins. Perhaps by making IscR a poor substrate for the Isc proteins via one atypical amino acid ligand, IscR is able to sense the cellular demand for Fe-‐S cluster biogenesis indirectly by the availability of the Isc machinery to associate with IscR[29].
Giel et al. reported that the isc operon was more repressed under anaerobic conditions rather than under aerobic conditions. Some Fe–S clusters are sensitive to O2 and/or reactive oxygen species[30] so it is likely that Fe–S clusters are continually being damaged or destroyed during aerobic growth. As a result of that, levels of substrate proteins that need Fe–S biogenesis or repair increases. The higher rate of cluster turnover in aerobic condition leads to decrease isc repression. It suggests that there may be more competition between IscR and substrate proteins for the Isc machinery when O2 is present. In contrast, under anaerobic conditions, the Isc machinery appears to satisfy the Fe–S demand more efficiently due to decreased general cluster turnover, and thus less competition among substrate proteins. The increased repression of isc under anaerobic conditions is due to increased of [2Fe– 2S]IscR[31].
These mechanisms would provide an additional level of global regulation to ensure that Fe-‐S clusters are synthesized when there is an increased demand.
2.2 IscS
is a pyridoxal phosphate (PLP) binding enzyme that catalyzes the desulfurization of L-‐cysteine to yield L-‐alanine and sulfur S0 or sulphide in presence of a reducing agent[32].
Increasing evidence has revealed an important role of IscS in the biosyntheses of Fe-‐S clusters, thiamine, thionucleosides in tRNA, biotin, lipoic acid, molybdopterin, and NAD. The enzymes are also proposed to be involved in cellular iron homeostasis and in the biosynthesis of selenoproteins[33].
Figure 2.1 Network of protein-protein interactions involving IscS. IscS initiates intracellular sulfur trafficking, delivering the sulfur to several sulfur-‐accepting proteins such as IscU, ThiI, TusA, and MoaD/MoeB that commit the sulfur to different metabolic pathways. IscU is the primary scaffold for assembly of Fe-‐S clusters. Frataxin/CyaY has been postulated as an Fe chaperone, an Fe donor for Fe-‐S cluster assembly, or a regulator of Fe-‐S cluster formation. In the schematic, sulfur delivering is indicated by red arrows and IscS-‐interacting proteins are framed by ovals (red, in sulfur accepting proteins)[34].
Similarly to what was suggested for NIFS (the A. vinelandii homologue) mechanism, IscS catalyzes the formation of an external aldimine Shiff base between the amino group of the substrate and PLP. The cysteinyl thiolate anion of Cys328 generated in the active site makes a nucleophilic attack on the sulfur of the cysteine-‐
Fe-S cluster formation [14]. The network of known IscS protein interactions is shown in Figure 1.
Thiolated nucleotides are found in several tRNAs. In E. coli and Salmonella enterica serovar Typhimurium, these are s4U8, s2C32, ms2i(o)6A37, and (c)mnm5s2U34, which, with the exception of s4U8,
are located within the anticodon loop and are crucial for proper mRNA decoding [21]. The base thiolations are mediated by several acceptor proteins, falling into two distinct pathways [21]. In the iron-sulfur cluster independent pathway, direct transfer of sulfur from IscS to the acceptor ThiI leads to the s4U8 modification [22], while transfer to TusA results in the (c)mnm5s2U34 modification [23]. ThiI also participates in thiamine biosynthesis [24]. The second pathway proceeds through the formation of an iron-sulfur cluster and is dependent on the IscU acceptor protein. The enzymes TtcA and MiaB accept sulfur from IscU [3] and are responsible for the s2C32 [25] and ms2i(o)6A37 modification [26], respectively. The unique tRNA thiolation pattern associated with sulfur transfer from IscS to TusA, IscU or ThiI provides a convenient readout system to assess the in vivo effects of IscS mutations on its interaction with these proteins.
The proteins involved in sulfur utilization have been extensively studied both functionally and structurally. Structures of IscS [27], the sulfur acceptor proteins TusA [28], ThiI [29], IscU [30,31], rhodanese [32], and the modulators human frataxin [33,34] and its bacterial homologue CyaY [35,36], as well as IscX [16,37] have been determined by X-ray crystallography or NMR. All of these proteins adopt different folds and the acceptor proteins receive sulfur from IscS by molecular mechanisms that are not fully understood.
Despite this wealth of structural information, the question of how IscS is able to communicate with such a broad spectrum of proteins and deliver sulfur to a wide range of structurally divergent partners is unresolved as no structural information on its complex(es) with binding partner(s) is presently known. To begin addressing this question, we have determined the crystal structure of the IscS-TusA and the IscS-IscU complexes, which reveal different modes of binding of these proteins and provide a framework for understanding sulfur transfer from IscS. Further, we performed extensive mutagenesis of the IscS surface followed by in vitro (pull-down) and in vivo (tRNA complementation assay)
Figure 1. Network of protein-protein interactions involving IscS. IscS initiates intracellular sulfur trafficking, delivering the sulfur to several sulfur-accepting proteins such as IscU, ThiI, TusA, and MoaD/MoeB that commit the sulfur to different metabolic pathways. IscU is the primary scaffold for assembly of Fe-S clusters. Frataxin/CyaY has been postulated as an Fe chaperone, an Fe donor for Fe-S cluster assembly, or a regulator of Fe-S cluster formation. In the schematic, sulfur delivering is indicated by red arrows and IscS-interacting proteins are framed by ovals (red, in sulfur accepting proteins).
doi:10.1371/journal.pbio.1000354.g001
Author Summary
Sulfur is incorporated into the backbone of almost all proteins in the form of the amino acids cysteine and methionine. In some proteins, sulfur is also present as iron–sulfur clusters, sulfur-containing vitamins, and cofac-tors. What’s more, sulfur is important in the structure of tRNAs, which are crucial for translation of the genetic code from messenger RNA for protein synthesis. The biosyn-thetic pathways for assembly of these sulfur-containing molecules are generally well known, but the molecular details of how sulfur is delivered from protein to protein are less well understood. In bacteria, one of three pathways for sulfur delivery is the isc (iron-sulfur clusters) system. First, an enzyme called IscS extracts sulfur atoms from cysteine. This versatile enzyme can then interact with several proteins to deliver sulfur to various pathways that make iron–sulfur clusters or transfer sulfur to cofactors and tRNAs. This study describes in atomic detail precisely how IscS binds in a specific and yet distinct way to two different proteins: IscU (a scaffold protein for iron–sulfur cluster formation) and TusA (which delivers sulfur for tRNA modification). Furthermore, by introducing mutations into IscS, we have identified the region on the surface of this protein that is involved in binding its target proteins. These findings provide a molecular view of the protein– protein interactions involved in sulfur transfer and advance our understanding of how sulfur is delivered from one protein to another during biosynthesis of iron–sulfur clusters.
IscS Interactions with Partner Proteins
17 PLP adduct and finally the cysteine persulfide and an enamine derivative of alanine are formed[35, 36].
Figure 2.2 Proposed mechanism of NIFS desulfuration reaction[35].
2.3 IscU
IscU has been widely conserved throughout evolution and is considered to be one of the most conserved protein sequences in nature[37]. Since it contains three conserved cysteine residues (Cys37, Cys63, and Cys106)[38], IscU has the ability to accommodate both [2Fe-‐2S]2+ and [4Fe-‐4S]2+ clusters. This supports the proposal that this ubiquitous protein provides a scaffold for the transient assembly of clusters mediated by IscS and subsequently used for maturation of apo Fe-‐S proteins[39]. IscU interacts with IscS to accept a sulphur atom and, since E. coli IscU does not interact directly with iron ions[40], it is suggested that CyaY, the bacterial orthologue of
Mechanism of L-Cysteine Desulfurase
Scheme 3: Proposed Mechanism of NIFS Desulfuration Reaction
"'7,
H H-S-vCOi N F S + H , S y C O i N Y ' L-Cy*&WBiochemistry, Vol. 33, No. I S , 1994 4119 deuterated. Thus, during the formation of the alanine product, the a- and @-hydrogens are all readily exchanged. The observed complete exchange of the @-hydrogens can be explained by the rapid equilibration of the enamine form of the product (compound I, Scheme 3) with the ketimine form
of the product (compound 11, Scheme 3) before rearrangement and separation of thealanine from theenzyme. It wasobserved that the a-hydrogen of all the cysteine remaining in the reaction mixture had been exchanged as well. Thus, it appears that the slow step in the reaction involves displacement of the sulfur from the bound cysteine.
It was also found that the a-hydrogen and all three 8-hydrogens of a small fraction of L-alanine becamedeuterated when NIFS was incubated with L-alanine in 2H20. Thus, exchange occurred at all four positions or not at all. This result indicates that L-alanine reacts slowly with the enzyme but once the alanine-PLP adduct is formed there is rapid exchange of all four hydrogens. The mechanism for the rapid exchange of the 8-hydrogens is most likely explained as discussed above for the rapid equilbration of compounds I
and I1 shown in Scheme 3. The exchange of the a-hydrogen
would occur during the rearrangement of compound I1 to alanine. These results are similar to those of Babu and Johnston, who reported the complete exchange of the a- and 8-hydrogens of alanine catalyzed by glutamic-pyruvic tran- saminase and glutamic-oxaloacetic transaminase (Babu &
Johnston, 1976). Our results and the observation that millimolar amounts of L-alanine do not significantly inhibit NIFS-catalyzed cysteine desulfurase activity thus reflect a rather slow binding/dissociation of L-alanine with NIFS relative to formation of the enamine intermediate.
DISCUSSION
We have previously shown that the product of the n i p gene is an L-cysteine desulfurase which catalyzes the removal of cysteine sulfur to form L-alanine and elemental sulfur (Zheng et al., 1993). Our current hypothesis is that the reaction catalyzed by NIFS represents a step in the formation of the Fe-S cores contained within the nitrogenase component proteins. In particular, we have suggested that NIFS catalyzes formation of an enzyme-bound persulfide which is the active species for providing the inorganic sulfide necessary for Fe-S cluster biosynthesis. In Scheme 3 a model that describes the proposed mechanism for the formation of an enzyme-bound persulfide using PLP chemistry and cysteine substrate is shown. The salient and novel feature of the model is nucleophilic attack by an active site cysteinyl thiolate anion on the sulfur of a cysteine-PLP adduct. This nucleophilic attack results in formation of a cysteinyl persulfide and an enamine derivative of alanine. In the present study, three basic features predicted by such a mechanism were experimentally confirmed: An essential active site cysteinyl thiolate ( C Y S ~ ~ ~ ) was identified, formation of an enzyme-bound persulfide was demonstrated, and indirect evidence for formation of an enamine intermediate during L-alanine formation was obtained. Furthermore, derivatization of the active site cysteinyl residue by incubation of NIFS with the mechanism-based inhibitors, allylglycine or vinylglycine, to form an enzyme-bound y-methylcystathionyl or cystathionyl residue, respectively, clearly demonstrates that a cysteinyl thiolate is poised for nucleophilic attack at the appropriate substrate position (see Schemes 1-3). The observation that treatment of NIFS with L-allylglycine or L-vinylglycine specifically blocks alkylation of the Cys32s residue and the results of site-directed mutagenesis experi- ments, which show that Cys32s is required for cysteine
BH' H
\\
BH+ H B:YK=Tx\\
H H w (I)+-*
-
N Y ' L - M h Henzyme-bound analog adducts to form either a y-methyl- cystathionyl or a cystathionyl residue.
Persul'de Formation Is an Intermediate in NIFS-
Catalyzed Desulfuration of L-Cysteine. In Scheme 3 a mechanism for NIFS is proposed in which L-cysteine des- ulfurization occurs by nucleophilic attack of the active site Cys325 thiolate on the substrate cysteine PLP adduct. If correct, this mechanism predicts the formation of an enzyme- bound persulfide as an intermediate in the reaction. We therefore tested for the formation of such a persulfide during NIFS catalysis by reacting the substrate-treated enzyme with thealkylating reagent 1 ,5-I-AEDANS and asking if a disulfide linkage was formed from an enzyme-bound persulfide and the alkylating reagent. In a separate control experiment the untreated enzyme was also reacted with 1S-I-AEDANS to form the stable thioether derivative. The rationale and results of these experiments are presented in Figure 1. The results show that reaction of 1,5-I-AEDANS with the substrate- treated form of NIFS results in formation of a DTT-reducible disulfide bond in more than 80% of the enzyme. The fluorescent species released from the substrate-treated enzyme by reducing this disulfide bond was shown to be N-(thioacetyl)-
N'-(5-sulfo-l-naphthyl)ethylenediamine. In contrast, the
fluorescent NIFS derivative obtained by reacting the untreated enzyme with 1,5-I-AEDANS could not be released by treatment with DTT.
Evidence for the Reversible Formation of an Enamine Intermediate during L-Cysteine Desulfurization Catalyzed by NIFS. To further characterize the mechanism by which NIFS catalyzes the desulfurization of L-cysteine, the reaction was carried out in the presence of 2H20 and the deuterium
incorporated into the reactants and products was determined by GC-MS analysis of their n-butyl trifluoroacetyl derivatives. Under these reaction conditions it was found that alanine
frataxin, might supply iron ions[10].
Whereas IscU homologues generally have been described as dimers, e.g. the IscU from T. maritima was shown to form a homodimer[41], monomeric form were identified for the human protein[42] and for Haemophilus influenzae and E. coli IscUs[40].
Controversial results were found from biophysical studies on IscU. Mansy et al. reported the characterization of IscU from Thermatoga maritima, an evolutionarily ancient hyperthermophilic bacterium, stabilized by a D40A mutation. T. maritima IscU is a thermally stable protein with a thermally unstable cluster. It possesses a high degree of secondary structure represented by 36.7% α-‐helix, 13.1% antiparallel β-‐sheet, 11.3% parallel β-‐sheet, 20.2% β-‐turn, and 19.1% other at 20 °C. Furthermore cluster coordination and temperature change have no effect on the secondary structure of the protein[43].
Even if the dispersion of signals in 1H-‐15N heteronuclear single quantum correlation NMR spectra of T. maritima IscU supports the presence of significant tertiary structure for the apo-‐protein and shows that the cofactor coordination is not necessary for proper protein folding, consistently with a scaffolding role [43], Bertini at al. stated that its tertiary structure could not be determined because the protein behaves as a flexible molten globule-‐like state[41]. In reverse, Adinolfi et al. showed that E. coli IscU is well folded with a high melting temperature reversibility of the thermal unfolding curve. This view is confirmed by the excellent dispersion of cross peaks in the 1H-‐15N NMR correlation spectrum[40].
2.4 IscA
role is yet unknown. Deletion of iscA gene causes slight but appreciable decreases in these Fe-‐S cluster assembly thus IscA protein does not contribute to a crucial step[27].
The comparison of the IscA sequence of different species shows the presence of three invariant cysteine residues (C35, C99, C101) in IscA implicated in Fe-‐S cluster binding[44].
Bilder et al. had recently solved and refined the 2.3 Å resolution crystal structure of E. coli IscA, a polipeptide of 107 residue with a novel fold in which mixed β-‐sheets form a compact α-‐β sandwich domain. The great majority of the amino acids that are conserved in IscA homologues are located in elements that constitute a well-‐ordered fold. However the C-‐terminal decapeptide that contains two of the three invariant cysteines (C99 and C101), is not visible in the electron density map. The lack of well-‐ defined electron density for this region is consistent with either a dynamic disorder or multiple ordered conformations of the region. In addition, the crystal packing reveals a helical assembly that is based on domain swapping of two possible tetramericoligomers of IscA.[44].
Probabily IscA is involved in the assistance of Fe-‐S cluster transfer to apo-‐ protein[3], infact holo-‐IscA and eukaryotic homologues of IscA have been shown to complex and transfer [2Fe-‐2S] clusters to the apo-‐form of E. coli ferredoxin, also synthesized from the isc operon [45].
2.5 HscA and HscB
The hscA and hscB genes of Escherichia coli encode novel chaperone and co-‐ chaperone proteins, designated Hsc66 and Hsc20, respectively. Purified HscA exhibits a low intrinsic ATPase activity (0.6 min-‐1 at 37°C and pH 7.5), and HscB was found to
![Figure
1.1
Various
[Fe–S]
clusters
found
in
iron–sulfur
proteins.
The
most
common
(a)
[2Fe–2S]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7648494.119000/10.892.310.640.264.634/figure-various-fe-clusters-iron-sulfur-proteins-common.webp)

![Figure
2.2
Proposed
mechanism
of
NIFS
desulfuration
reaction[35].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7648494.119000/17.892.324.674.220.686/figure-proposed-mechanism-nifs-desulfuration-reaction.webp)
![Figure
2.3
Kinetic
scheme
of
the
Hsc66
ATPase
reaction
cycle[47].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7648494.119000/20.892.270.648.819.951/figure-kinetic-scheme-hsc-atpase-reaction-cycle.webp)
![Figure
2.4
A
model
for
the
IscU
binding
cycle
of
HscA.
Kinetic
constants
shown
correspond
to
rates
in
the
presence
of
IscU
and
HscB[50].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7648494.119000/21.892.319.671.693.1035/figure-model-iscu-binding-kinetic-constants-correspond-presence.webp)
![Figure
2.5
Proposed
mechanistic
scheme
for
chaperone-catalyzed
Fe-S
cluster
transfer
from
IscU 2 [2Fe2S]
to
apoacceptor
proteins[70].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7648494.119000/27.892.266.772.117.449/figure-proposed-mechanistic-chaperone-catalyzed-transfer-apoacceptor-proteins.webp)