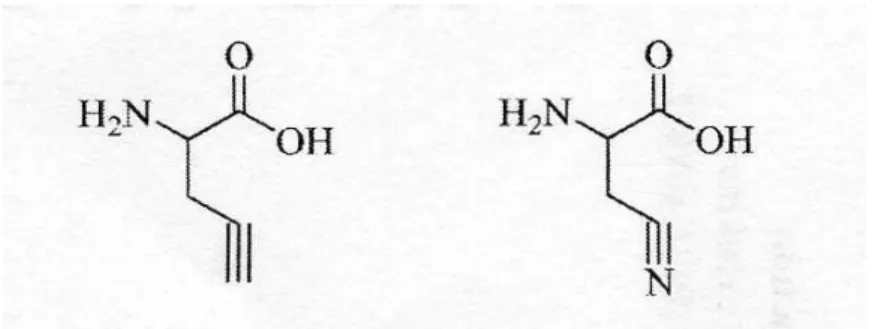UNIVERSITA’ DEGLI STUDI DI PISA
FACOLTA’ DI FARMACIA
TESI DI LAUREA
Anno Accademico 2011-2012
RUOLI ED EFFETTI DEL NUOVO
MEDIATORE ACIDO SOLFIDRICO
NELLA PATOFISIOLOGIA
VASCOLARE E NEL SISTEMA RENALE
Candidato
Barbara Oligeri
Relatore
INDICE
Indice 2 1. Introduzione 4 2. Funzioni 5 3. Biosintesi H2S 6 4. Catabolismo 145. Meccanismi delle azioni biologiche dell’H2S 16
6. Principali effetti e ruoli regolatori dell’H2S 20
6.1 Sistema Nervoso Centrale 20
6.2 Sistema Gastrointestinale 22
6.3 Sistema endocrino 25
6.4 H2S e infiammazione 26
7. Sistema cardiovascolare 28
7.1 Effetti vascolari dell’H2S nella circolazione sistemica 28
7.2 Effetti vascolari dell’H2S nella circolazione polmonare 37
7.3 H2S come sensore vascolare di ossigeno 39
8. Ruolo dell’H2S nella patofisiologia vascolare 43
8.1 H2S e lesioni ischemiche del cuore 43
8.2 H2S ed ipertensione sistemica 46
8.3 H2S e ipertensione polmonare 47
9. Ruolo di H2S nel danno renale indotto da iniezione di adriamicina 49
9.1 Discussione 50
10. Effetti dell’inibizione dell’H2S in disturbi renali strutturali e funzionali indotti da
gentamicina 53 10.1 Discussione 54
11. Approcci farmacologici possibili 57 11.1 Donatori di solfuro di idrogeno (H2S) attivati da cisteina 57
11.2 Farmaci anti-infiammatori non steroidei che rilasciano H2S 67
11.2.1 Effetti anti-infiammatori di H2S endogeno 69
11.2.2 Effetti anti-infiammatori di donatori di H2S 70
11.2.3 Derivati di farmaci anti-infiammatori che rilasciano H2S 73
11.3 H2S e latanoprost 77
11.4 H2S-sildenafil 78
11.5 H2S sartani 80
12. Conclusioni 82 13. Bibliografia 84
1 INTRODUZIONE
Il mediatore endogeno ossido nitrico (NO) è stato uno dei capisaldi che ha influenzato lo sviluppo della farmacologia cardiovascolare negli ultimi vent’anni.
Recenti ricerche hanno rivelato che nella famiglia dei gas-trasmettitori si è ormai aggiunto all’ossido nitrico (NO) e al monossido di carbonio (CO) un gas endogeno generato da vari tessuti: il solfuro di idrogeno (H2S) (Szabò, 2007).
I trasmettitori gassosi sono una famiglia di molecole regolatrici coinvolte nella regolazione a più livelli di funzioni fisiologiche e patologiche nei tessuti di mammiferi (Wang, 2002). Questo composto è noto per la sua tossicità e l’orribile odore ma sta rivelando importanti peculiarità di modulatore endogeno riportando quasi tutti gli aspetti benefici dell’ossido nitrico sul sistema cardiovascolare, senza produrre metaboliti tossici.
Grazie agli studi attuali sulle funzioni biologiche dell’H2S endogeno questo mediatore sta
diventando un argomento di grande interesse scientifico tant’è che è stato chiamato da diversi autori il “nuovo NO”.
È prodotto in quantità importanti da diversi tessuti di mammiferi probabilmente ad una velocità lenta e costante in cui esercita effetti in diverse aree, in particolare sul sistema cardiovascolare.
2 FUNZIONI
L’H2S ha un ruolo in: proliferazione cellulare e apoptosi, metabolismo di glucosio,
neurotrasmissione (Abe e Kimura, 1996), ipertensione (Zhong et al, 2003), infiammazione (Li et al, 2006), percezione del dolore (Kawabata et al, 2007), integrità della mucosa gastrica (Fiorucci et al, 2006) e sistema cardiovascolare, dove sembra fornire un importante contributo alla regolazione omeostatica (Szabò, 2007).
Come altri solfuri inorganici ed organici è un efficace “spazzino”di specie reattive dell’ossigeno grazie alla sua azione antiossidante.
Induce il rilassamento della muscolatura liscia vascolare tramite i canali del potassio ATP-sensibili (KATP) e grazie a questo svolge importanti effetti cardioprotettivi per ciò che concerne la riperfusione miocardica e l’ischemia; inoltre, una ridotta produzione di H2S endogeno può avere un ruolo nelle
patologie ischemiche e nell’ipertensione.
Nelle arterie polmonari e sistemiche determina un rilassamento e/o contrazione a seconda della sua concentrazione, del tipo di vaso e della specie.
Influenza anche le attività del sistema nervoso centrale (SNC) e dei sistemi gastroenterico ed endocrino; l’H2S rilassa le cellule della muscolatura liscia
(SMC) mediante un effetto diretto sui canali KATP causando iperpolarizzazione e chiusura dei canali Ca 2+ voltaggio-dipendenti seguiti da una riduzione del calcio intracellulare.
Questo nuovo gas endogeno rilassa le SMC anche mediante il rilascio dall’endotelio del fattore iperpolarizzante endotelio-derivato (EDHF) e ossido nitrico (NO); di contro contrae le SMC mediante una riduzione della disponibilità
di ossido nitrico reagendo con lo stesso a formare un composto nitrosotiolo e mediante un effetto inibitorio sulla ossido sintetasi endoteliale (eNOS), nonchè in forma di riduzione della concentrazione di AMP ciclico nelle SMC.
Vi è infine una probabile interrelazione tra carenza di attività di H2S e il
progredire della disfunzione endoteliale.
3 BIOSINTESI
Nella cellula animale l’H2S viene sintetizzato in vari tessuti propri del sistema nervoso e
cardiovascolare e può essere generato attraverso vie diverse, sia enzimatiche che non enzimatiche.
Gli studi sul sistema enzimatico coinvolto nel processo di sintesi dell’H2S sono ancora in una
fase molto iniziale, tuttavia quantità importanti di H2S sono biosintetizzate ad opera di due
enzimi piridossal-5-fosfato dipendenti: cistationina beta-sintetasi(CBS) e cistationina gamma-liasi(CSE), responsabili della maggior produzione endogena di H2S in tessuti di mammiferi e
sono stati individuati in cellule umane e di altri mammiferi (Levonen et al, 2000).
Recentemente sono stati scoperti altri enzimi che partecipano alla biosintesi: cisteina aminotransferasi(CAT) e 3-mercaptopiruvato solfotransferasi(3MST).
In particolare CSE è considerato come la fonte principale di H2S nel sistema cardiovascolare:
aorta, arteria mesenterica, vena porta ed altri tessuti vascolari (Zhao et al, 2001), dove si stima che il livello di produzione di H2S sia nell’ordine di 3-6 nanomoli/min/g di tessuto (Zhao et
Ciò si può comprendere dal fatto che quando lo specifico inibitore di CSE, DL-propargil-glicina (PAG), è stato aggiunto a omogenati di tessuti di arterie di differenti ratti, la produzione endogena di H2S da cisteina esogena è stata completamente abolita.(Zhao et al,
2001).
La distribuzione di questi enzimi non è omogenea; pertanto la CSE viene espressa in particolare nelle cellule della muscolatura liscia vascolare, non in cellule endoteliali (Wang, 2003); al contrario si pensa che la CBS sia espressa in prevalenza nel sistema nervoso centrale: ippocampo, cervelletto, corteccia cerebrale e tronco cerebrale e assente nei tessuti vascolari dove può essere indotta solo in particolari condizioni.
La CSE è una proteina di 45 aminoacidi ed è un tetramero formato da due omodimeri che contribuiscono entrambi alla tasca del sito attivo; dall’altra parte, la CBS è un enzima che forma cistationina anche se, in generale, catalizza reazioni di beta-sostituzione tra serina, L-cisteina, cisteina tioetere o altri alfa-L- aminoacidi beta sostituiti e una varietà di mercaptani (Braunstein et al, 1971).
I domini CBS sono piccoli gruppi proteici, di solito associati in tandem, che sono coinvolti nel legame con gruppi adenosil (Kery et al, 1994).
La CBS umana è un omotetramero che consiste di 63 sottounità KDa che legano due cofattori, piridossal 5,-fosfato (PLP) ed eme.
Ciascuna sottounità CBS di 551 residui aminoacidici lega due substrati (omocisteina e serina) e viene ulteriormente regolata da S-adenosil-L-metionina (AdoMet) (Kery et al, 1994).
Mentre il ruolo di eme nell’attività CBS è sconosciuto, l’attività catalitica della CBS può essere spiegata esclusivamente tramite la partecipazione di PLP nel meccanismo di reazione (Kery et al, 1999).
Negli esseri umani diverse malattie genetiche sono state associate a mutazioni nei domini di CBS e possono essere considerate come obiettivi promettenti per la progettazione razionale di nuovi farmaci.
In alcuni tessuti, CSE e CBS sono entrambi richiesti per la sintesi di H2S mentre in altri solo
uno di essi è necessario (Wang, 2002).
Il sistema enzimatico CAT/3MST è localizzato nell’endotelio (Shibuya et al, 2009).
Nei tessuti dei mammiferi l’H2S può essere generato attraverso almeno quattro diverse vie
biosintetiche sia enzimatiche sia non enzimatiche:
Il primo processo di sintesi è catalizzato dall’enzima CBS che idrolizza la L-cisteina con formazione di uguali quantità di L-serina e H2S.
CBS
L-cisteina L-SERINA + H
2S
VIT B6
La seconda via biosintetica comporta la dimerizzazione di due molecole di L-cisteina per formare L-cistina; quest’ultima, a sua volta, viene scissa ad opera dell’enzima CSE per formare tiocisteina, piruvato e NH3.
A questo punto la tiocisteina può essere soggetta a due diverse vie, una non enzimatica e una enzimatica: nel primo caso si arriva alla formazione di L-cisteina e H2S (Cavallini et al,
1962), nel secondo, la tiocisteina reagisce con un composto tiolico R-SH come cisteina o glutatione per dare H2S e CysSR tramite l’enzima CSE (Stipanuk e Beck, 1982).
dimerizzazione
2 L-CISTEINA L-CISTINA
VIT B6
CSE
TIOCISTEINA + PIRUVATO + NH
3+
non enzimatica
R-SH
VIT B6 CSE
L-CISTEINA + H
2S CysSR + H
2S
La terza via di sintesi comporta la reazione tra L-cisteina e a-chetoglutarato con formazione di 3-mercaptopiruvato e L-glutammato,; l’enzima catalizzante il processo è la cisteina aminotransferasi(CAT).
Dopodiché il 3-mercaptopiruvato subisce una reazione di desolforazione ad opera della 3-mercaptopiruvato solfo transferasi (MPST) per cedere piruvato e H2S.
Quando invece sono presenti ioni solfito (SO32-), il 3-mercaptopiruvato può essere convertito
tramite CAT in piruvato e tiosolfato (S2O32-) che a sua volta reagisce con il glutatione (GSH)
per produrre H2S, SO32- e glutatione ossidato (GSSG).
CAT
L-CISTEINA 3- MERCAPTOPIRUVATO
+ +
αααα
-CHETOGLUTARATO L-GLUTAMMATO
VIT B6+
MPST
SO
3CAT zinco
PIRUVATO + H
2S TIOSOLFATO + PIRUVATO
GSH
H
2S +H
2SO
3+ GSSG
Infine l’ultimo processo biosintetico è catalizzato dalla cisteina liasi che converte il solfito (SO32-) ed L-cisteina in L-cisteato e H2S.(Li et al, 2009).
cisteina liasi
L-CISTEINA + SO
3
L-CISTEATO +
L’attività di CBS e CSE è influenzata da diversi fattori sia esogeni che endogeni.
Ad esempio nel sistema nervoso centrale Ca2+e calmodulina sono in grado di regolare l’attività di CBS e, in particolare, fattori che comportano un aumento di Ca2+ intracellulare favoriscono la produzione di H2S.
Ciò può essere determinato da agenti come gli agonisti glutamatergici che attivano recettori NMDA (N-metile-D-aspartato) e AMPA (a-ammino-3-idrossi-5-metile-4-isossazolo propionato); al contrario si pensa che NO renda inattivo CBS.
Trattando il sistema cardiovascolare, la formazione di H2S mediata da CSE è migliorata da
NO-donatori in modo dipendente da cGMP; sono stati inoltre trovati inibitori di NO-sintetasi che riducono la produzione di H2S. (Zhao et al, 2003).
Il solfuro di idrogeno è un gas con una struttura molto simile a quella dell’acqua ed è, infatti, solubile in essa e nel plasma; è un acido debole e si dissocia in soluzione acquosa in idrogenione (H+) e anione idrosolfuro (HS-) e successivamente può decomporsi in H+ e ione solfuro (S2-) (H2S↔HS- + H+↔S2- + 2H+; pKa1=7.04, Ka1:1.3∗10-7 e pKa2=11.96 Ka2:1∗10-19
a 25°C) (Myers, 1986).
L’atomo di zolfo non è elettronegativo come l’ossigeno e questo fa rendere l’H2S molto meno
polare dell’acqua.
SCHEMA 1. Dissociazione del solfuro di idrogeno
E’ una molecola altamente lipofila e si diffonde liberamente attraverso le membrane cellulari. A causa di ciò, esistono forze intermolecolari relativamente deboli per l’H2S e i punti di
fusione ed ebollizione sono molto più bassi di quanto lo sono in acqua.
Le temperature di ebollizione di H2S e di H20 sono rispettivamente -60,7°C e 100,0°C.
FIGURA 1. Similitudini nella struttura molecolare tra H2O e H2S
Caliendo et al, 2010
A temperatura e pH fisiologico circa il 18,5% del solfuro totale esiste come H2S e l’81,5%
come HS-. (Dombkowski et al, 2004); la forma chimica S2-, invece, non è presente in quantità apprezzabili poiché la dissociazione HS- si verifica solo ad alti valori di pH.
Non è chiaro se è lo stesso H2S, o un derivato, che media gli effetti biologici; tuttavia è stato
riportato che l’HS- può rilassare gli anelli aortici dei ratti (Ondrias et al, 2008).
L’H2S inoltre, presenta alcuni effetti tossici negli esseri umani (a concentrazioni <100 ppm)
come irritazione agli occhi, mal di gola, vertigini, nausea, mancanza di fiato, oppressione toracica (Reiffenstein et al, 1992).
Esposizioni all’H2S a concentrazioni > 1000 ppm, può causare gravi effetti avversi,
specialmente per il sistema nervoso centrale (CNS) e depressione respiratoria che variano dalla perdita di coscienza alla morte (Kage et al, 2002).
C’è una controversia riguardo alla concentrazione di H2S nel sangue e nel plasma.
Tutti gli studi recenti hanno usato metodi indiretti e chimicamente aggressivi per misurare l’H2S e hanno rilevato concentrazioni tra 30 e 300 µM (Whitfield et al, 2008); concentrazioni
indicanti H2S nel sangue dovrebbero essere individuabili dall’odore.
Al momento, sebbene il meccanismo d’azione per questi effetti tossici non sia chiaro, si ritiene che il nuovo mediatore agisca sui mitocondri a basse concentrazioni (micromolari) mediante inibizione reversibile di citocromo c ossidasi (Reiffenstein et al, 1992) ed è difficile immaginare come i mitocondri sarebbero in grado di produrre sufficienti quantità di ATP per le necessità metaboliche a queste alte concentrazioni di H2S (Petersen, 1977).
Whitfield e colleghi (2008) hanno recentemente misurato concentrazioni di H2S nel plasma e
nel sangue umano da una varietà di mammiferi in tempo reale usando un elettrodo polarografico e hanno trovato che l’H2S è inavvertibile alla risoluzione del sensore (≈15 nM)
e quando l’H2S esogeno viene aggiunto al sangue, viene consumato rapidamente.
Rimane da accertare se l’H2S in compartimenti cellulari possa raggiungere concentrazioni
abbastanza alte da evocare reazioni vascolari in vivo.
Pertanto, mentre l’H2S può essere una molecola di segnale paracrino o autocrino
4 CATABOLISMO
L’H2S presenta un’emivita piuttosto breve, al di sotto di 30 minuti, pertanto viene
rapidamente metabolizzato tramite diverse vie responsabili (Li et al, 2009).
Un primo importante processo di degradazione, che sembra essere il principale, consiste nell’ossidazione in tiosolfato nei mitocondri forse mediante una via non enzimatica collegata ad un trasporto di elettroni.
A sua volta il tiosolfato, grazie all’enzima rodanese e all’aiuto di cianuro convertito in tiocianato, viene biotrasformato in solfito; quest’ultimo poi viene ossidato ad opera di solfito ossidasi (SO) a solfato (Hildebrandt e Grieshaber, 2008).
Il solfato comunque non può essere definito un marcatore affidabile per stimare la quantità di H2S prodotta; ioni solfato infatti possono derivare anche dall’ossidazione diretta di cisteina
diossigenasi oppure dall’ossidazione di solfiti prodotti da altre fonti (Li et al, 2009).
mitocondri:
ossidazione
2HS
-+ 2O
2S
2O
32-+ H
2O
rodanese
S
2O
32-+ CN
-SCN
-+ SO
32- solfito ossidasiSO
3SO
42-Un’altra via catabolica risiede nel citosol delle cellule dove una minor parte dell’H2S viene
metilato da tiolo S-metiltransferasi (TSMT) a metantiolo e poi a dimetilsolfuro (Furne et al, 2001).
citosol:
TMST TMST
H
2S CH
3-SH CH
3-S-CH
3Infine un altro processo catabolico avviene nel sangue e comporta la reazione tra H2S e
metaemoglobina per formare solfoemoglobina (Volkel et al, 2000).
sangue:
H
2S + metaemoglobina solfoemoglobina
Sono state trovate altre vie di ossidazione di H2S a solfito nei neutrofili attivati (Mitsuhashi et
al, 2005).
Va inoltre ricordato che questo mediatore endogeno è una specie riducente e pertanto può essere consumata facilmente da una serie di agenti ossidanti circolanti (Whiteman et al, 2005; Chang et al, 2005).
5 MECCANISMI DELLE AZIONI BIOLOGICHE DELL’H
2S
Studi recenti riportano due meccanismi fondamentali che spiegano l’attività biologica dell’H2S:
uno di questi riguarda l’interazione del mediatore con i sistemi redox. L’H2S
infatti reagisce con almeno quattro diverse specie reattive: anione superossido radicale (Mitsuhashi et al, 2005), perossido di idrogeno (Geng et al, 2004), perossinitrito e ipoclorito (Whiteman et al, 2005). Sono tutti composti molto aggressivi che l’H2S è in grado di bloccare esplicando così un’azione protettiva
nei confronti di lipidi e proteine (Whiteman et al, 2005).
Oltre a ciò l’H2S svolge la sua attività mediante l’attivazione di canali al potassio
ATP sensibili, (KATP); diversi effetti di H2S sono riprodotti da altre sostanze che
stimolano l’apertura di suddetti canali, come il pinacidil o diazossido, altre sostanze invece come glibenclamide inibiscono gli effetti del mediatore bloccando i canali al potassio.
I canali KATP sono un fattore fondamentale che regola numerose funzioni biologiche come
l’attività cardiaca, il tono della muscolatura liscia, la secrezione di insulina, il rilascio di neurotrasmettitori (Nichols, 2006); ciò perchè sono ubiquitari e si trovano, quindi, nelle cellule pancreatiche beta, nei neuroni e nelle cellule della muscolatura liscia scheletrica e miocardica.
Sono dei complessi di proteine transmembrana in grado di collegare lo stato metabolico delle cellule con la loro eccitabilità (Nichols, 2006).
Nello specifico, in condizioni di ridotto metabolismo dinamico, collegato ad un aumento di ADP e una riduzione del rapporto ATP/ADP, il canale viene attivato; questo comporta l’uscita di ioni potassio dalla cellula e conseguente iperpolarizzazione della membrana.
Il canale KATP ha una struttura piuttosto complessa; si parla di un’architettura etero-ottamerica
formata dalla combinazione di due tipi di sotto unità transmembrana:
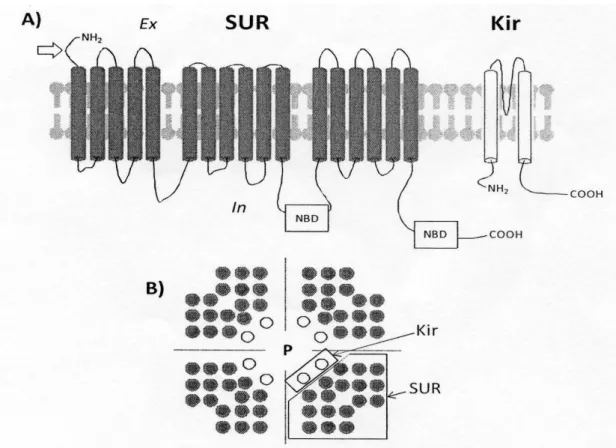
a) proteine che formano pori dette KIR, costituite da due segmenti transmembrana ed appartenenti alla famiglia dei canali al potassio inward rectifying;
b) proteine regolatrici conosciute come sotto unità SUR (recettore sulfonilurea) costituite da 17 segmenti transmembrana appartenenti alle “ATP-binding cassette proteins” che possono essere considerate come sensori del rapporto ATP/ADP.
FIGURA 6. Architettura generale dei canali KATP.
A. La struttura delle sotto unità SUR è composta da 17 domini trans membrana (in grigio) e mostra due domini nucleotidici intracellulari (NBD); le frecce indicano il sito extracellulare per H2S. La sotto unità Kir è composta da due domini trans membrana (in bianco). In: lato intracellulare; Ex: lato extracellulare
B. 4 sotto unità Kir circondano la regione del poro (P), formando il canale selettivo per il potassio (P); ciascuna di loro è associata ad una sotto unità SUR.
Martelli et al, 2010
I canali KATP sono formati da 4 proteine KIR appartenenti alla sottofamiglia 6 (KIR6) e
ciascuna di loro è associata ad una sotto unità SUR (Miki e Seino, 2005).
Due sottotipi di proteine KIR6 (KIR6.1 e KIR 6.2) e tre sottotipi di SUR (SUR1, SUR 2A e SUR 2B) sono coinvolte nella struttura dei canali KATP (Matsuo et al, 2005).
Il canale KATP ha una struttura estremamente complessa formata da 96 domini transmembrana
e 12 siti nucleotidici leganti e per questo sia composti endogeni che diverse classi di farmaci agiscono come agonisti o antagonisti di suddetti canali esercitando le loro azioni su differenti zone.
Scoperte piuttosto recenti hanno dimostrato che attraverso l’espressione eterologa di sotto unità KIR 6.1 e SUR 1 nella linea cellulare HEK-293 il canale di attivazione KATP indotto da
H2S richiede obbligatoriamente la co-espressione di entrambe le sotto unità mentre l’H2S è
inefficace quando viene espressa soltanto la sotto unità KIR 6.1.
Il sito di interazione tra H2S e il canale KATP viene localizzato nella sotto unità SUR e in
particolare nel suo sito N-terminale extracellulare (Jiang et al, 2009).
Ci sono diverse condizioni patologiche e fisiologiche in cui l’H2S è coinvolto; i vari articoli
pubblicati a riguardo hanno affrontato questo argomento di ricerca usando una fonte esogena di H2S o inibitori della sua sintesi agendo su CBS o CSE.
Gli inibitori farmacologici della biosintesi di H2S includono DL-propargilglicina (PAG) e
beta-cianoalanina (BCA).
PAG BCA
FIGURA 2. Struttura di PAG (DL-propargilglicina) e di BCA (β-cianoalanina)
Sebbene caratterizzati da bassa potenza, scarsa selettività e limitata permeabilità della membrana cellulare, questi composti sono stati usati in diversi studi allo scopo di verificare gli effetti di inibizione della produzione di H2S. Essi sono non specifici ed è probabile che
interagiscano con altri enzimi piridossal-fosfato-dipendenti.
6 PRINCIPALI EFFETTI E RUOLI REGOLATORI DELL’H
2S
6.1 SISTEMA NERVOSO CENTRALE
Uno dei tanti distretti in cui l’H2S esercita importanti funzioni è il sistema nervoso centrale e
proprio a questo livello è presente a livelli relativamente alti.
Innanzitutto grazie alla sua azione antiossidante e all’attivazione di canali KATP (Kimura et al,
2004) questo importante mediatore è responsabile, nell’uomo, della protezione dei neuroni; ecco quindi che viene visto un possibile ruolo dell’H2S nella protezione da malattie
neurodegenerative (Whiteman et al, 2005).
L’H2S aumenta la concentrazione di Ca2+ nelle cellule gliali e promuove onde citosoliche di
Ca2+ in astrociti mediando così la trasduzione del segnale gliale (Lee et al, 2006) e dato che la CBS è un enzima calcio-calmodulina dipendente, la biosintesi del mediatore dovrebbe essere accuratamente controllata dalla concentrazione intracellulare di calcio.
In aggiunta, è regolata anche da S-adenosilmetionina che agisce come attivatore allosterico di CBS.
Abe e Kimura nel 1996 osservarono un’alta espressione di CBS nell’ippocampo e nel cervelletto dei ratti; in conseguenza di ciò fu scoperto che gli inibitori CBS riducevano la concentrazione di H2S nel cervello e allo stesso modo i livelli di H2S nel cervello erano ridotti
anche in topi con deficit di CBS (Eto e Kimura, 2002).
Anche nei pazienti affetti da Alzheimer è stata individuata una diminuzione dell’attività di CBS e una drastica riduzione del livelli di H2S (Eto et al, 2002).
L’H2S è coinvolto anche in patologie come il colpo apoplettico (Qu et al, 2006) e l’Alzheimer
(Beyer et al, 2004).
Nel colpo apoplettico, esso pare agire come mediatore di lesioni ischemiche e pertanto si è suggerito che l’inibizione della sua produzione sia un potenziale approccio di cura nella terapia del colpo apoplettico (ictus).
Le potenziali azioni fisiologiche di H2S nel cervello includono omeostasi del calcio, long-term
potentiation (LTP) a livello dell’ippocampo, soppressione di stress ossidativo e modulazione di neurotrasmissione (Qu et al, 2008).
Alte concentrazioni di H2S (50-160µM) inducono un long-term potentiation (LTP)
nell’ippocampo migliorando così il meccanismo dell’apprendimento e della memoria (Kimura et al, 2005). Questo effetto è svolto grazie ad una maggiore sensibilizzazione dei recettori NMDA nei confronti del glutammato (Kimura, 2000) tramite un’aumentata produzione di AMPc ad opera di H2S.
species and tissues effect in vitro or in vivo and effect of inhibitor
rat, brain NaHS down-regulated the expression of c-fos and increased the expression of
γ-aminobutyric acid Breceptor subunits 1 and 2 (GABA(B)R2) in recurrent febrile
seizures. Hydroxylamine (an inhibitor of CBS) up-regualtes c-fos and down-regulates GABA(B)R2.
rat, primary hepatocyte H2S protects neurons from oxidative stress rat primary hepatocytes.
human, cultured human SH-SY5Y cells
H2S inhibits HOCl-mediated inactivation of α(1)-antiproteinase and protein
oxidation, HOCl-induced cytotoxicity, intracellular protein oxidation, and lipid peroxidation.
mouse, hippocampal cell line H2S protects from oxidative glutamate toxicity through ATP-dependent K+ and
Cl- channels and increases glutathione levels. mouse, brain endothelial cells
(bEnd3)
H2S protects from oxidative stress on brain endothelial cells.
glia cells H2S has a protective effect on oxidative stress on brain endothelial cells.
rat, primary cultures of astrocytes and hippocampal slices
H2S increases the influx of Ca2+ and causes the release of stored intracellular Ca2+ ,
La3+, and Gd3+. Inhibitors of Ca2+ channels block Ca2+ waves induced by neuronal excitation or from exogenously applied H2S.
rat, primary cultured microglial cells Hydrogen sulfide regulates calcium homeostasis in microglial cells. PAG and BCA significantly decrease [Ca2+]i.
mouse, primary cultured microglia and immortalized murine BV-2 microglial cells
Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated kinase (MAPK) in microglia.
TABELLA 3. Effetti di H2S nel Sistema Nervoso Centrale in modelli di animali affetti da malattia
Caliendo et al, 2010
6.2 SISTEMA GASTROINTESTINALE
Nell’intestino dei ratti sono state trovate concentrazioni di CSE e CBS; da qui è stato visto l’H2S come un possibile mediatore di motilità gastrointestinale (Li et al, 2009).
A questo livello l’H2S mostra un duplice effetto, sia antinocicettivo sia rilassante.
E′ stato dimostrato che il mediatore ha un effetto antinocicettivo sulla percezione del dolore viscerale grazie ad una distensione colorettale sia nei ratti sani che in ratti con colite e inoltre tramite l’attivazione dei canali KATP induce un rilassamento dose-dipendente dell’ileo e della
muscolatura liscia del colon (Distrutti et al, 2006); si è anche dimostrato che esso attiva neuroni sensoriali primari.
Riguardo agli effetti sulla muscolatura liscia intestinale, è probabile che l’H2S endogeno sia
fisiologicamente coinvolto nel controllo della motilità intestinale e nella funzione secretiva dato che la somministrazione del mediatore esogeno stimola la secrezione di cloruro nel colon sia delle cavie sia di quello umano (Schicho et al, 2006).
Inibisce inoltre le risposte contrattili spontanee o indotte per reazione dell’ileo e del colon di differenti specie di mammiferi (Distrutti et al, 2006).
Infine sono stati trovati CBS e CSE anche nella mucosa gastrica, dove si pensa che l’H2S
svolga un’importante azione protettiva contro lesioni delle mucose.
Sempre a questo livello è stato dimostrato che la somministrazione di idrosolfuro di sodio (NaHS), donatore di H2S, può ridurre lo sviluppo di lesioni nella parete gastrica indotte dai
farmaci anti-infiammatori non steroidei.
Studi hanno dimostrato che l’H2S induce anche un forte aumento del flusso ematico nella
mucosa gastrica e la vasodilatazione indotta è contrastata dalla glibenclamide; da ciò si capisce ancor più il coinvolgimento dei canali KATP (Fiorucci et al, 2006).
In aggiunta, l’H2S può avere un ruolo come mediatore pro-infiammatorio in sepsi addominale,
endotossiemia e pancreatite in contrasto con i suoi effetti antinfiammatori in modelli animali di gastrite e colite.
species and tissues effect in vitro or in vivo and effect of inhibitor mouse, human, rat; Jejunum,
colon, muscular strips
H2S inhibits in vitro intestinal and colonic motor patterns. This effect is dependent
on SK channels and glybenclamide-sensitive K(ATP)channels.
mouse, small intestine H2S prevents in ischemia-reperfusion, leukocyte rolling, and leukocyte adhesion
through an eNOS- and p38 MAPK-dependent mechanism.
mouse intestine Intraperitoneal injections of lysine acetyl salicylate increased concentration of
endogenous hydrogen sulfide.
rat, liver In normal hyperhomocysteinemic and cirrhotic rat livers, endothelial dysfunction
caused by homocysteine was reversed by perfusion of the livers with sodium sulfide.
mouse, liver H2S protects the murine liver against ischemia-reperfusion injury through an
up-regulation of intracellular antioxidant and antiapoptotic signaling pathways. mouse, cecal ligation and
puncture (CLP) induced sepsis
Inhibition of H2S formation by PAG significantly reduced the phosphorylation of
ERK1/2 in lung and liver 4 h after CLP, coupled with decreased degradation of IκBα
and activation of NF-κB.
rat, hepatic arteria CSE-derived H2S contributes to hepatic arterial buffer response and partly mediates
vasorelaxation of the hepatic artery via activation of K(ATP) channels. PAG
significantly reduced the buffer capacity. human, erythrocytes and
colonic mucosa
No evidence of defective enzymic detoxication of sulfide by Rhodonese or thiol methyltransferase was found in patients with ulcerative colitis or Crohn’s disease.
mouse, colon H2S-releasing mesalamine derivate, ATB-429, reduces colitis-associated leukocyte
infiltration and expression of several proinflammatory cytokines.
mouse, pancreatic acini The prinflammatory effect of H2S may be mediated by substance P neurokinin-l
receptor (SP-NK-lR) related pathway in mouse pancreatic acinar cells. mouse, cecal ligation and
puncture induced sepsis
H2S acts as an important endogenous regulator of leukocyte activation and trafficking
during an inflammatory response (in cecal ligation and puncture-induced sepsis) rat, gastric mucosal epithelial cells NaHS/H2S protects gastric mucosal epithelial cells against oxidative stress through
stimulation of MAP kinase pathways.
rat, colorectal distension (CRD) ATB-429, a novel H2S-releasing derivative of mesalamine inhibits hypersensitivity
induced by CRD in both healthy and postcolitic allodynic rats by a K(ATP)
channel-mediated mechanism.
rat and mouse, pancreas NaHS/H2S targets T-type Ca
2+
channels, leading to nociception. Endogenous H2S
produced by CSE and possibly T-type Ca2+ channels are involved in pancreatitis-related pain.
rat and mouse, gastrointestinal tract
The capacity for H2S synthesis varies throughout the rodent gastrointestinal tract, as
does the distribution and contribution of the two key enzymes (CSB and CSE). guinea pig, gastric
antrum muscle strips
H2S has multiple actions during the regulation of gastric motility in the guinea pig.
An excitatory effect is mediated via inhibition of the voltage-gated K+ channel.
rat H2S synthesis is markedly up-regulated during experimental colitis. Inhibition of H2S
synthesis interfered with resolution/healing of the colitis, while administration of H2S donors accelerated resolution/healing.
TABELLA 5. Effetti di H2S nel Sistema gastrointestinale in modelli animali affetti da malattia
6.3 SISTEMA ENDOCRINO
Anche nel sistema endocrino l’H2S svolge il ruolo di un fondamentale mediatore tant’è che
studi recenti suggeriscono che un suo deficit può essere un rilevante fattore patogenetico nello sviluppo di malattie cardiovascolari associate al diabete.
Grosse quantità di CSE e CBS sono state trovate nel pancreas e ciò ha fatto pensare che l’H2S
sia coinvolto nel controllo della secrezione di insulina ed inoltre è stato dimostrato che un valore alto di glucosio, stimolo di per sè pro-apoptotico per le cellule beta pancreatiche, aumenta l’espressione di CBS e la formazione di H2S in cellule beta di animali con glicemia
normale.
L’H2S esogeno inibisce l’apoptosi delle cellule beta provocata da un elevato valore di
glucosio e per di più l’aumentata produzione di H2S endogeno indotta da alto glucosio
protegge le cellule beta da glucotossicità attenuando anche l’apoptosi di queste ( Kaneko et al, 2009).
E’ noto il meccanismo d’azione dell’H2S di attivare i canali KATP; un inibitore di CSE, la
propargil-glicina (PAG), aumenta il rilascio di insulina dalle cellule di insulinoma dei ratti (Yang et al, 2005); di contro l’H2S esogeno riduce il rilascio di insulina indotto da glucosio in
queste cellule (Yang et al, 2005) e in cellule pancreatiche isolate di topi.
Infine un altro studio (Brancaleone et al, 2008) ha riportato che in un modello di diabete di tipo 1 nei topi è compromessa la biosintesi enzimatica di H2S e ciò è correlato ad
species and tissues effect in vitro or in vivo and effect of inhibitor H2S Role on Insulin Metabolism
Zucker diabetic fatty (ZDF) rats, pancreatica β-cells
H2S significantly increases KATPchannel activity rat pancreatic β-cells. Insulin release
is impaired in ZDF by a high pancreatic production of H2S. PAG increased serum
insulin level, lowered hyperglycemia, and reduced hemoglobin A1c level.
mouse, mouse pancreas High glucose increased CSE expression in β-cells. L-Cys or NaHS suppressed islet cell apoptosis with high glucose and increased glutathione content in MIN6 β-cells.
The CSE inhibitor PAG antagonized L-cysteine effects. rat, INSI-E cells derived from
a rat insulinoma
H2S at physiologically relevant concentration induced apoptosis of insulin-secreting
β cells by enhancing ER stress via p38 MAPK activation. H2S Role System
rainbow trout; chromaffin cells (posterior cardinal vein and anterior kidney)
H2S elicits catecholamine secretion via membrane depolarization followed by
Ca2+-mediated exocytosis. H2S-induced catecholamine secretion is unaltered
by the nicotinic receptor blocker hexamethonium H2S Role on Hypothalamo-Pituitary
rat, hippocampus NaHS caused a concentration-dependent decrease in KCl stimulated
corticotropin-releasing hormone release. S-Adenosylmethionine mimics the effect of NaHS but did not affect hypothalamo-pituitary-adrenal function under resting conditions while inhibiting stress-related glucocorticoid increase.
TABELLA 4. Effetti di H2S nel Sistema Endocrino in modelli animali affetti da malattia
Caliendo et al, 2010
6.4 H
2S E INFIAMMAZIONE
L’H2S è coinvolto in diverse situazioni infiammatorie; diverse evidenze sperimentali
species tissues/cells effect in vitro effect in vivo effect of inhibitor/mediator involvement Proinflammatory Effects for H2S
mouse lung liver proiflammatory in septic shock
cecal ligation and puncture (CLP)induced sepsis
PAG reduced lung and liver mycloperoxidase activity.
PAG reduced leukocyte rolling and adherence
significantly in mesenteric venules coupled with decreased mRNA and protein levels of adhesion molecules in lung and liver. guinea pig airways tackykinin-mediated
neurogenic
inflammatory responses in guinea pig airways
intratracheal instillation of NaHS increased the total lung resistance and airway plasma protein extravasation
Capsazepine and SR140333 or SH48968 inhibit NaHS effect.
mouse lung H2S involvement in the generation
of substance P (SP) via NK-l receptor PAG pretreatment or post-treatment significantly decreased the production of SP in lung.
mouse pancreas H2S mediates pancreatitis induced
by caerulein hyperstimulation
PAG treatment reduced pancreatic and lung edema, diminished acinar-cell injury/necrosis.
rat, mouse H2S proinflammatory: paw
edema, hemorrhagic shock
Pretreatment with PAG significantly reduces proinflammatory effects. human IFNγ-primed human U937 H2S in proinflammatory proinflammatory cytokines, Involvement ERK-NF- κB pathway
rat nociceptive in formalin assay PAG attenuated both
nociceptive flinching and hind paw edema. Anti-Inflammatory Effects of H2S
rat mesenteric venules leukocyte adherence in
mesenteric venules, leucocyte infiltration in the air pouch carrageenan-induced hindpaw edema activation of ATP-sensitive K+(KATP) channels mouse caerulein-trated acinar cells
H2S inhibits the production
of proinflammatory cytokines by activation of the (PI3K)/AKT pathway PI3K inhibitor in LY294002 abolished the H2S-mediated
activation of AKT and increases TNF- α and IL- lβ levels
mouse lung H2S exerts protective effects
in acute lung injury
activation of
anti-inflammatory and antioxidant pathways
rat stomach H2S promotes GI healing
in the rat. mouse LPS-stimulated microglia
and astrocytes
H2S produced an
antiinflammatory effect.
inhibition of inducibile nitric oxide synthase and p38 MAPK signaling pathways
rat lung H2S attenuates the development
of pulmonary hypertension.
elevating IκBα
expression, down-regulating NF-κB p65 expression
rat myocardium cardioproctetive effect of NaHS antiapoptonic and anti-inflammatory effects macrophage RAW2647 LPS-stimulated anti-inflammatory H2S can inhibit NO
production and NF-κB activation. BCA enhanced NO production.
Yorkshire swine
myocardium protection in response to
ischemia-reperfusion injury
Therapeutic sulfide reduces tissue levels of IL-6, IL-8, TNF-α, and MPO activity.
rat colon Inhibition of H2S synthesis in rats
resulted in inflammation and mucosal injury.
Intracolonic
administration of H2S
donors reduced the severity of colitis and reduced colonic expression of mRNA for TNF-α
TABELLA 2. Attività pro ed anti-infiammatoria di H2S
Caliendo et al, 2010
7 SISTEMA CARDIOVASCOLARE
7.1 EFFETTI VASCOLARI DELL’H
2S NELLA CIRCOLAZIONE
SISTEMICA
Una conoscenza sempre più accurata sul ruolo biologico dell’H2S nel cuore e nei vasi
sanguigni sta facendo emergere l’importanza che questo mediatore ha nel controllo dell’omeostasi cardiovascolare.
In animali integri, le iniezioni endovenose di donatori H2S, come NaHS, suscitano una
riduzione dose-dipendente di breve durata nella pressione arteriosa media che evidenzia un effetto vasodilatatore dell’H2S (Li et al, 2008).
Studi recenti hanno infatti dimostrato che un deficit di H2S endogeno contribuisce alla
patogenesi dell’ipertensione; in effetti l’attività e l’espressione di CSE sono più basse in ratti spontaneamente ipertesi (SHR).
Inoltre la somministrazione cronica di NaHS abbassa la pressione sanguigna nei SHR ma non nei ratti normotesi (Yan et al, 2004) mentre la somministrazione di inibitori di CSE riduce la concentrazione di H2S nel plasma ed eleva la pressione del sangue in ratti normotesi ma non
in SHR indicando che l’H2S è coinvolto nella regolazione del tono vascolare in condizioni
normali e che la produzione del mediatore si riduce nell’ipertensione.
L’H2S può causare vasodilatazione tramite un effetto centrale (Dawe et al, 2008; Ufnal et al,
2008) ma ha effetti dilatatori diretti nei vasi mesenterici perfusi (Cheng et al, 2004) ed epatici dei ratti (Siebert et al, 2008).
Rilassa anche il corpo cavernoso isolato di coniglio e uomo e può svolgere un ruolo importante insieme all’NO nel controllo della funzione erettile (Villa Bianca et al, 2009).
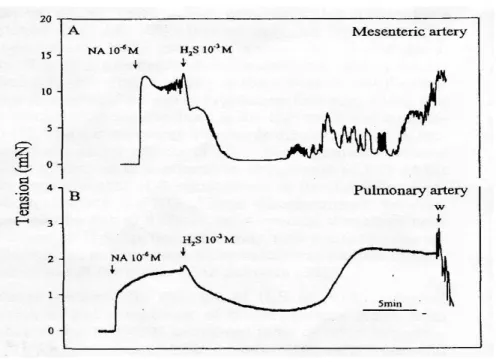
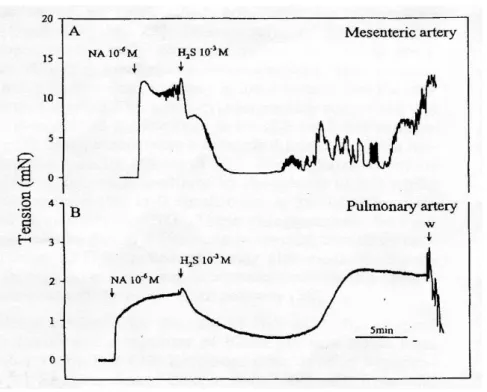
FIGURA 1. Registrazioni originali di tensione che mostrano gli di H2S (10-3M) in arterie di resistenza polmonari e mesenteriche di ratti precontratte con noradrenalina (NA, 10-6M)
L’H2S rilassa l’aorta pre-contratta, le arterie coronarie e la vena porta dei ratti come anche le
arterie mesenteriche dei topi (Cheang et al, 2010).
Provoca inoltre reazioni rilassanti nella muscolatura liscia vascolare e questa azione è stata osservata sia nei grandi vasi come, ad esempio, l’aorta toracica e la vena porta dei ratti sia nei vasi di resistenza periferica che svolgono un ruolo più significativo delle grandi arterie nella regolazione di resistenza vascolare e pressione sanguigna (Cheng et al, 2004).
Si deduce nuovamente da ciò che l’H2S provoca diminuzioni dose-dipendenti della pressione
arteriosa in modelli sperimentali di ipertensione (Li et al, 2009).
Studi più recenti mostrano che gli effetti vascolari di H2S dipendono dalla sua concentrazione
in quanto il mediatore contrae le aorte di ratti e topi e le arterie mammarie a basse concentrazioni (10-100 µM), ma rilassa gli stessi vasi sanguigni a più alte concentrazioni (100-1000µM) (Webb et al, 2008).
In vertebrati non mammiferi, gli effetti vascolari dell’H2S possono essere complessi in quanto
esso può causare dilatazione, costrizione o reazioni multifasiche in vasi sanguigni isolati (Dombkowski et al, 2005).
Tuttavia, in uno studio in vivo su tartarughe, le iniezioni di NaHS hanno condotto ad un restringimento dose-dipendente della circolazione sistemica (Stecyk et al, 2010).
Non sono chiari i meccanismi che sono alla base del rilassamento indotto da H2S, ma
contrariamente al rilassamento indotto da NO e CO, sembra che non coinvolga un aumento sGC-dipendente nella concentrazione di GMPc (Li et al, 2009).
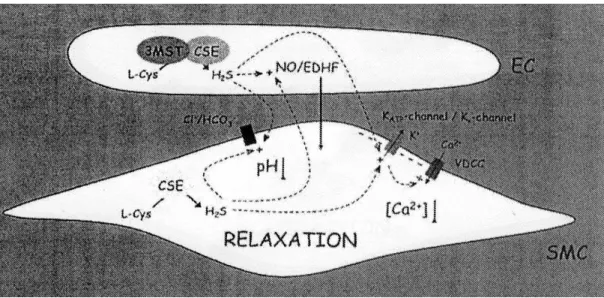
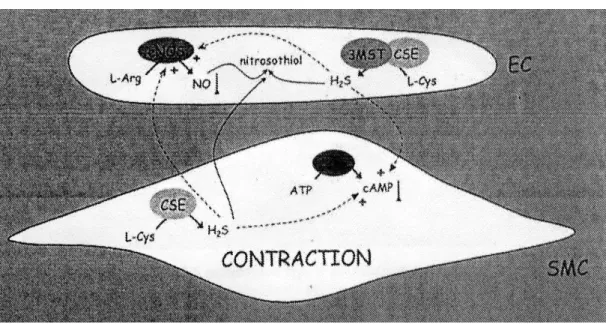
FIGURA 2. Una sintesi dei potenziali meccanismi mediati da H2S nel rilassamento vascolare. L’H2S viene sintetizzato a partire da cisteina (L-Cys) mediante la cistationina-gamma-liasi (CSE) nelle cellule della muscolatura liscia (SMC) e mediante 3-mercaptopiruvato solfo esterasi (3MST) e CSE nella cellula endoteliale (EC). L’H2S rilassa SMC attraverso un effetto diretto sui canali KATP o i canali Kν. Questo iperpolarizza SMC e chiude i canali Ca2+ voltaggio-dipendenti (VDCC), diminuendo così la concentrazione intracellulare di calcio [Ca2+]. L’H2S rilassa SMC anche attraverso il rilascio di un fattore iperpolarizzante endotelio-derivato (EDHF) ed ossido nitrico (NO) dall’endotelio e mediante un possibile aumento nell’attività dello scambiatore Cl-/HCO3-, che fa diminuire il pH intracellulare.
Skovgaard et al, 2011
E’ correntemente riconosciuto che l’effetto primario dell’H2S è sui canali del potassio ATP
sensibili della muscolatura liscia ed esso rilassa proprio i vasi sanguigni aprendo questi canali. Questa conclusione si è basata su esperimenti in vivo e in vitro i quali mostrano che il pinacidil, un agonista dei canali KATP, riproduce gli effetti dell’H2S e che la glibenclamide,
un antagonista dei canali KATP, blocca il rilassamento riducendo l’attività ipotensiva del mediatore in vivo e i suoi effetti vasodilatatori in vitro (Li et al, 2009); l’effetto dell’H2S
inoltre è fortemente ridotto negli anelli aortici esposti ad alto K+ extracellulare (Cheng et al, 2004).
I canali KATP svolgono un ruolo importante nel rilassamento mediato da H2S ma è chiaro che
il vaso-rilassamento è più complesso e che possono contribuirvi anche altri meccanismi. Studi con la tecnica di patch-clamp su cellule della muscolatura liscia (SMC) mesenterica e dell’aorta di ratto hanno dimostrato un effetto diretto dell’H2S sui canali KATP che si
manifesta con un aumento della corrente al K+ e iperpolarizzazione delle SMC (Tang et al, 2005). L’iperpolarizzazione delle SMC è abilitata ad inibire i canali Ca2+- voltaggio- dipendenti e ciò è dimostrato dal ridotto rilassamento all’H2S negli anelli di aorta dopo
l’applicazione di nifedipina, un inibitore dei canali Ca2+, o la rimozione di Ca2+ extracellulare (Zhao e Wang, 2002).
Molti studi sul ruolo dei canali KATP nel rilassamento da H2S sono stati condotti su tessuti di
ratti.
Uno studio sulle arterie mammarie umane ha mostrato che l’H2S rilassa parzialmente i vasi
attraverso un’apertura dei canali KATP, al contrario, in uno studio sull’aorta dei topi, i canali KATP non erano coinvolti nel rilassamento (Webb et al, 2008).
Inoltre, nelle arterie coronarie dei ratti l’effetto rilassante dell’H2S appare essere mediato
attraverso canali K+ (Kνcanali) voltaggio-sensibili (Cheang et al, 2010).
In tessuti non vascolari, inoltre, altri canali come canali K+ attivati da Ca2+ a larga conduttanza (BKCa), canali del calcio del tipo T e tipo L e canali al cloro intracellulari sono
coinvolti nella reazione all’H2S (Telezhkin et al, 2009; Malekova et al, 2009).
La rimozione dello strato di cellule endoteliali o l’inibizione della NO sintasi endoteliale (eNOS) per mezzo di L-NAME (L-nitro-arginina metil-estere) riducono l’effetto dell’H2S in
Canali K+ attivati da Ca2+ a piccola conduttanza sono coinvolti nel rilascio di un fattore iperpolarizzante endotelio-derivato (EDHF) e di NO (Dalsgaard et al, 2010), e l’inibizione di questi canali tramite una combinazione di apamina e caribdotossina riduce il rilassamento da H2S (Cheng et al, 2004).
Pertanto una piccola ma significativa parte del rilassamento è endotelio-dipendente mediata dal rilascio di NO e/o EDHF.
Come l’H2S faccia da mediatore per il rilascio di NO non è chiaro, tuttavia è stato dimostrato
che l’H2S rilascia NO da nitrosotioli (Ondrias et al, 2008); in particolare sta emergendo che
l’H2S condivide quasi tutti gli aspetti benefici di NO senza essere la fonte di metaboliti tossici
(Lefer, 2007).
Recentemente è stato suggerito che il ruolo dell’H2S nell’omeostasi cardiovascolare diventa
più importante e cruciale quando il controllo mediato da NO viene compromesso, come nel caso della disfunzione endoteliale.
Sebbene il vaso-rilassamento indotto da H2S possa essere ottenuto da effetti diretti
endotelio-dipendenti su cellule della muscolatura liscia (Li et al, 2009), gli inibitori di NO sintasi o la rimozione endoteliale possono attenuare il rilassamento indotto da H2S suggerendo che l’NO
endoteliale fornisce un parziale contributo all’effetto vaso-rilassante dell’H2S (Zhao et al,
2003).
Tale componente endotelio-dipendente che contribuisce al vaso-rilassamento indotto da H2S
non è attribuibile a canali KATP (Cheng et al, 2004), ed è stato ipotizzato il potenziale coinvolgimento di altri canali ionici, a parte quelli KATP.
Curiosamente, sembra che le arterie di resistenza siano molto più sensibili all’H2S rispetto ad
arterie più grosse (Cheng et al, 2004).
In aggiunta, la parte endotelio-dipendente del rilassamento da H2S è più pronunciata in arterie
Ulteriori possibili meccanismi di rilassamento da H2S includono una riduzione nella
produzione di ATP attraverso un’inibizione metabolica (Kiss et al, 2008) e una riduzione nel pH intracellulare dovuta ad un effetto diretto sullo scambiatore.
Cl/HCO3 (Lee et al, 2007). I meccanismi che sottintendono al restringimento mediato
dall’H2S a basse concentrazioni sono scarsamente compresi ma non coinvolgono i canali
KATP (Lim et al, 2008).
FIGURA 3. Una sintesi dei potenziali mediati da H2S nella contrazione vascolare. L’H2S contrae SMC mediante una riduzione della disponibilità di ossido nitrico (NO), in quanto reagisce con NO stesso formando un composto nitrosotiolo e attraverso l’effetto inibitorio sulla sintasi endoteliale di ossido nitrico (eNOS). Inoltre, H2S contrae SMC tramite una riduzione della concentrazione di cAMP.
L-Arg: L-arginina; ATP: adenosin trifosfato; AC: adenilato ciclasi; cAMP: adenosina monofosfato ciclica.
L’interazione tra H2S e NO non è chiara; sembra che l’NO endoteliale contribuisca agli effetti
vaso-rilassanti dell’H2S, e quest’ultimo potrebbe potenziare o attenuare l’effetto rilassante
dell’NO nell’aorta dei ratti (Geng et al, 2007).
Le interazioni tra H2S e NO potrebbero anche verificarsi ad altri livelli dato che i donatori NO
regolano la produzione di H2S in tessuti vascolari di ratti aumentando l’espressione e l’attività
di CSE (Zhao et al, 2003).
Per contro, i livelli di H2S circolanti, così come l’espressione e l’attività di CSE nel sistema
cardiovascolare, si riducono dopo un’inibizione cronica di NO-sintasi.
L’H2S potrebbe sopprimere l’effetto vasodilatatore di NO tramite un’inibizione diretta di
eNOS o ridurre la disponibilità di NO attraverso la formazione di un non identificato composto nitrosotiolo non vasoattivo (Kubo et al, 2007).
Inoltre studi recenti affermano che l’H2Spuò contrastare non solo l’eccesso di NO prodotto in
presenza di infiammazione (Whiteman et al, 2006), ma anche la quantità di NO generato in condizioni fisiologiche (Kubo et al, 2007).
E′interessante che nei ciclostomi, un gruppo di vertebrati filogeneticamente primitivi, l’aorta dorsale, dove l’H2S è un vasocostrittore monofasico, sia carente di NO vascolare; è quindi
improbabile in questi vasi che la contrazione sia mediata da un’interazione H2S-NO e ciò
suggerisce che altri meccanismi sono coinvolti nella contrazione mediata da H2S (Olson et al,
2008).
Perciò il ruolo di NO nel mediare gli effetti vascolari dell’H2S è controverso.
Alcuni studi concludono che il rilassamento da H2S è mediato dal rilascio di NO, mentre altri
concludono che la costrizione di H2S viene mediata da una ridotta disponibilità di NO.
Tutte queste divergenze tra i vari studi possono essere dovute a differenze nelle specie, nei vasi o nelle concentrazioni di H2S applicato.
L’H2S potrebbe anche mediare la contrazione attraverso un meccanismo
endotelio-indipendente che comporta l’inibizione di AMP ciclico (Lim et al, 2008).
Prove dirette del ruolo dell’H2S come vaso-rilassante fisiologico e come regolatore della
pressione sanguigna nasce dall’osservazione che topi in cui viene inibito CSE sviluppano ipertensione (Yang et al, 2008).
La presenza di CSE in cellule endoteliali di topo è stata confermata nello stesso studio in cui è stato anche dimostrato che la metacolina, un agonista del recettore muscarinico, suscita un rilascio di H2S da cellule endoteliali associato alla concentrazione (Yang et al, 2008).
Questo effetto dipende dall’influsso di calcio e da una regolazione calcio-calmodulina di CSE. Inoltre, in arterie mesenteriche di topi con deficienza di CSE, il rilassamento indotto da metacolina è fortemente compromesso e ciò suggerisce che l’H2S è un fattore rilassante
endotelio-derivato (EDRF)(Yang et al, 2008).
Il ruolo che l’H2S svolge nel controllo vascolare in stati fisiologici o di malattia è ancora poco
chiaro e attende di essere ulteriormente studiato; inoltre le conoscenze attuali riguardo le reciproche influenze tra NO e H2S non sono ancora ben chiare e sono necessari ulteriori sforzi
nelle ricerche per comprendere l’esatta dinamica di tali interazioni in modelli sperimentali cardiovascolari sia fisiologici che patologici.
Insieme all’attività vasorilassante, l’H2S (come l’NO) ha un’ampia gamma di ruoli biologici;
ad esempio, inibisce l’aggregazione piastrinica indotta da ADP, collagene, adrenalina, acido arachidonico, trombossano, trombina.
Attenua inoltre il rimodellamento vascolare in modelli sperimentali di ipertensione (Yan et al, 2004), inibisce le proteine chinasi attivate da mitogene e sopprime la proliferazione di cellule della muscolatura liscia aortica riducendo efficacemente il progredire di lesioni arteriosclerotiche (Du et al, 2004).
L’H2S può anche avere effetti sulla reazione infiammatoria vascolare dove svolge un ruolo
importante nella destabilizzazione e rottura della placca arteriosclerotica; tuttavia, riguardo a questo punto ci sono delle controversie: l’H2S sembra esercitare effetti antiinfiammatori sui
macrofagi (Oh et al, 2006), ma effetti pro-infiammatori sulle cellule della muscolatura liscia vascolare (Jeong et al, 2006).
Prove recenti, inoltre, hanno dimostrato una diretta interazione tra H2S e prostaglandine
vasoattive nella regolazione del tono vascolare (Koenitzer et al, 2007); sono tuttavia necessari ulteriori studi per chiarire questo punto.
7.2 EFFETTI VASCOLARI DELL’H
2S NELLA CIRCOLAZIONE
POLMONARE
Riguardo agli effetti dell’H2S nella circolazione polmonare, soltanto pochi studi hanno
affrontato questo argomento: un unico studio ha infatti trattato il meccanismo insito negli effetti vascolari dell’H2S nella circolazione polmonare mostrando un effetto sinergico tra H2S
e NO.
Nelle arterie polmonari di ratti il rilassamento indotto da H2S viene attenuato dall’inibizione
della produzione di NO tramite L-NAME mentre il rilassamento indotto da NO è attenuato dall’inibizione di H2S grazie al blocco di CSE (Wang et al, 2008); tuttavia non è noto se ci sia
un effetto combinatorio dei due inibitori sugli enzimi.
Altri studi hanno mostrato una varietà di risposte all’H2S nei vasi polmonari di differenti
Nelle arterie polmonari di anatre e bovini, l’H2S induce rilassamento a basse concentrazioni
(10-8-10-5M), risposte multifasiche a concentrazioni intermedie (10-4-10-3M) e contrazioni a concentrazioni più alte (10-3-10-2M) (Olson et al, 2006).
In arterie polmonari precontratte di ratti, una dose singola di H2S ha indotto una risposta
multifasica contrazione-rilassamento-contrazione (Olson et al, 2006).
FIGURA 1. Registrazioni originali di tensione che mostrano gli di H2S (10-3M) in arterie di
resistenza polmonari e mesenteriche di ratti precontratte con noradrenalina (NA, 10-6M)
Al contrario, un diverso studio sulle arterie polmonari di ratti ha riportato un rilassamento monofasico concentrazione dipendente mediato da H2S (25,50,100µM) (Wang et al, 2008).
Arterie polmonari di alligatori americani hanno evidenziato una contrazione dose-dipendente (Dombkowski et al, 2005).
Per contro, in uno studio in vivo su tartarughe, l’NaHS, donatore di H2S, ha aumentato la
conduttanza della circolazione polmonare (Gpul), il che suggerirebbe che l’H2S sia un
vasodilatatore (Stecyk et al, 2010).
Tuttavia è molto più probabile che l’aumentato Gpul rifletta una distensione passiva dei vasi dovuta ad un aumentato flusso sanguigno polmonare causato da un aumento della gittata cardiaca e da una forte vasocostrizione sistemica che smista il sangue verso la circolazione polmonare.
7.3 H
2S COME SENSORE VASCOLARE DI OSSIGENO
L’ossigeno è essenziale per la vita e l’abilità di rilevare ossigeno è necessaria per rispondere a cambiamenti nella disponibilità di ossigeno o ad alterate esigenze metaboliche.
Quando i livelli di ossigeno sono bassi, cioè in condizioni ipossiche, i vasi sanguigni sistemici si dilatano per aumentare il flusso sanguigno e la distribuzione di ossigeno ai tessuti ipossici. Per contro, i vasi polmonari si restringono per ridurre la perfusione di zone ipossiche scarsamente ventilate; ciò migliora lo scambio di gas e preserva la PO2 arteriosa (Brimioulle
Un meccanismo che coinvolge il metabolismo di H2S può spiegare sia la vasocostrizione
ipossica sia la vasodilatazione ipossica (Wang et al, 2008).
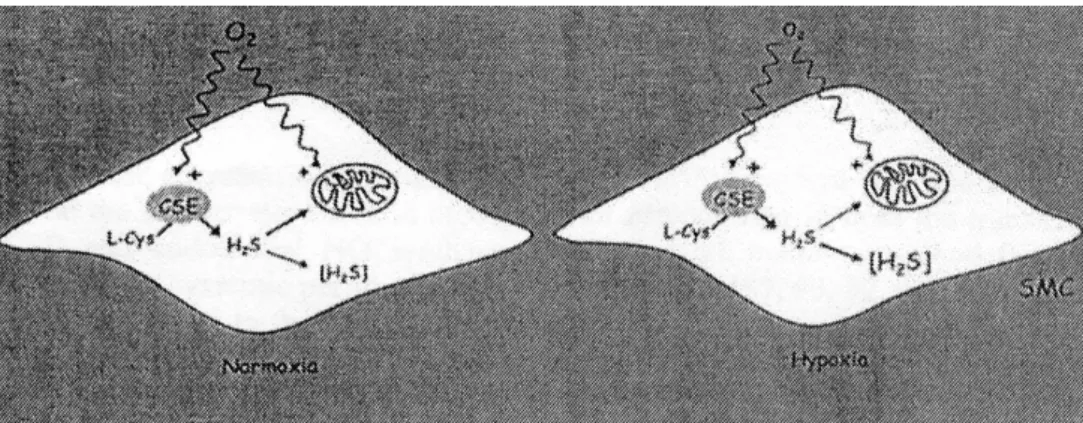
FIGURA 4. Meccanismi proposti riguardanti la rilevazione di O2 attraverso il metabolismo di H2S. L’H2S è prodotto dal metabolismo di cisteina (L-Cys) nelle cellule muscolari lisce (SMC). Durante le condizioni di normossia, H2S è continuamente ossidato nei mitocondri, mantenendo, di conseguenza, una bassa concentrazione di H2S. Una caduta nella disponibilità di ossigeno durante l’ipossia fa diminuire l’ossidazione mitocondriale di H2S portando così ad un incremento di [H2S] biologicamente attivo e ad un inizio di reazioni ipossiche. L’enzima che produce H2S dalla cisteina, cistationina-gamma-liasi (CSE), è sensibile all’O2 e ciò permette di regolare la velocità del metabolismo di H2S e quindi la sua concentrazione.
Skovgaard et al, 2011
Il modello si basa sul rapporto inverso esistente tra [O2], di per sè non vasoattivo, e [H2S], che
lo è.
L’H2S è essenzialmente sintetizzato nel citoplasma cellulare e ossidato nel mitocondrio e
equilibrio tra produzione di H2S e quantità di O2 disponibile per la sua ossidazione, ad
esempio PO2 dei tessuti.
Inoltre, la CSE è sensibile alla PO2, allo stato di ossidoriduzione intracellulare e a NO, il che
potrebbe fornire un meccanismo di modulazione di produzione di H2S (Banerjee e Zou,
2005).
Le risposte vascolari all’ipossia sono identiche a quella dell’H2S in vasi sanguigni di tutti i
vertebrati studiati fino ad ora, e l’inibizione della sintesi di H2S attenua le risposte vascolari
ipossiche (Stecyk et al, 2010).
Gli effetti vascolari di H2S sono inoltre ossigeno-dipendenti e l’H2S e l’ipossia costituiscono
stimoli competitivi: ciò suggerisce che H2S e ipossia condividono un comune effettore a valle
nella via segnalatrice mediando il tono vasomotore (Stecyk et al, 2010).
Recentemente è stato proposto che la produzione aumentata o diminuita di una specie reattiva dell’ossigeno (ROS) dalla respirazione mitocondriale svolge un ruolo importante nella vasocostrizione polmonare ipossica dei mammiferi (Waypa e Schumacker, 2005); è stato dimostrato che l’H2S cattura ROS (Geng et al, 2004) ma, talvolta, ne induce anche la
produzione (Eghbal et al, 2004), ed è quindi facile speculare che ROS potrebbe essere un collegamento comune tra vasocostrizione ipossica e vasocostrizione mediata da H2S.
Sono necessarie, tuttavia, ulteriori ricerche per stabilire l’esatta relazione tra H2S e ipossia e
species tissues effect in vitro effect in vivo effect of inhibitor rat mesenteric artery,
aorta portal vein
relaxation hypotensive KATP channel blocker
inhibits, PAG and BCA block relaxation hypertensive
rat
aorta, plasma, thoracic aorta
relaxation, regulation of excess vascular collage
reduces blood pressure
the effect was reduced by PAG
rat liver, ileum increases systolic blood
pressure (PAG)
amino oxyacetate, PAG inhibit H2S production
human internal thoracic artery
relaxation, contraction (low dose)
KATP channel blocker
inhibits mouse lacking CSE serum, heart aorta reduced H2S levels, reduced vasorelaxation
hypertension not tested
rat plasma
myocardium
reduced lipid peroxidation, increased antioxidation, inhibition oxidative stress injury
improves cardiac function
not tested
rat pulmonary artery effect on pulmonary collagen remodeling
PAG in vivo in a model of pulmonary high flow
Tabella 1. Effetti e ruoli di H2S nel sistema cardiovascolare
8 RUOLO DI H
2S NELLA PATOFISIOLOGIA VASCOLARE
8.1 H
2S E LESIONI ISCHEMICHE DEL CUORE
Si pensa che l’H2S abbia la possibilità di indurre le cellule ad avere minor bisogno di energia
e ossigeno. Questo può aiutare a prolungare il periodo di tempo in cui tessuti e cellule possono sopravvivere senza ossigeno; pertanto, l’integrazione diretta o indiretta di H2S
potrebbe ristabilire la funzione miocardica dopo una lesione ischemica. L’azione citoprotettiva del nuovo mediatore in lesioni da riperfusione ischemica miocardica è stata studiata recentemente (Elsey et al, 2010).
L’H2S non ha modificato il flusso sanguigno cardiaco nel modello sperimentale del cuore alla
Langendorff (Johansen et al, 2006) e in un modello suino di riperfusione ischemica miocardica il flusso sanguigno è rimasto inalterato anche nell’arteria discendente anteriore sinistra (Osipov et al, 2009).
Nel cuore, l’NaHS ha un effetto inotropo negativo concentrazione-dipendente (Geng et al, 2004); pertanto, fino a questo punto gli effetti cardioprotettivi devono essere considerati come un effetto diretto dell’H2S sul miocardio o un abbassamento della resistenza sistemica
vascolare e polmonare.
L’infusione di 1 µM di NaHS, ma non concentrazioni più alte (10 µM), ha limitato l’entità dell’infarto nei ratti (Johansen et al, 2006).
Anche un altro donatore di H2S, l’Na2S ha attenuato lesioni da riperfusione ischemica
miocardica nei topi, un effetto attribuito alla salvaguardia della funzione mitocondriale (Elrod et al, 2007). Anche l’S-diclofenac, che è un donatore di H2S basato sul diclofenac, medicinale
anti-infiammatorio non-steroideo, ha protetto contro lesioni da riperfusione ischemica (Rossoni et al, 2008).
Nei ratti la riduzione dell’entità dell’infarto miocardico dopo riperfusione ischemica è stata bloccata in presenza di bloccanti dei canali KATP sarcolemmali e mitocondriali, rispettivamente glibenclamide e acido 5-idrossidecanoico (5-HD). Questi bloccanti hanno anche contrastato l’effetto cardioprotettivo di NaHS in studi su lesioni da riperfusione coronarica (Zhang et al, 2007).
In studi recenti anche l’NaHS, applicata in studi di patch-clamp in cardiomiociti, sembra portare ad un’aumentata probabilità di apertura dei canali KATP (Zhang et al, 2007). Altri hanno riportato che gli effetti precondizionanti e cardioprotettivi di NaHS sono rimasti inalterati in presenza di un bloccante dei canali KATP mitocondriali ma sono stati bloccati da glibenclamide e dall’inibizione della proteina chinasi C (PKC) (Bian et al, 2006). L’attivazione dei canali KATP sembra pertanto svolgere un ruolo importante nella cardio-protezione da NaHS.
Specie radicali dell’ossigeno (ROS) si formano durante malattie cardiache ischemiche ed è stato riportato che la terapia con composti che catturano ROS contrasta lesioni cardiache (Andreadou et al, 2009). E’ stato dimostrato che l’H2S abbia effetti antiossidanti diretti
(Stasko et al, 2009), mentre altri hanno suggerito che H2S induca la sovraespressione di un
fattore di trascrizione, nfr2, con conseguente aumento di enzimi antiossidanti come eme ossigenasi e tioredossina, proteine da shock termico 90 e 70, segnali antiapoptotici (Bcl-2) e inattivazione di proapoptotici BAD (Calvert et al, 2009).
L’interazione con radicali liberi potrebbe quindi contribuire all’effetto cardioprotettivo di H2S.
IL substrato cisteina H2S infuso in ratti con lesione da riperfusione ischemica ha aumentato la
sintesi di H2S nel miocardio e ridotto l’entità dell’infarto miocardico (Elsey et al, 2010).
Questi effetti sono stati prevenuti da PAG, inibitore di CSE (Elsey et al, 2010).
Anche analoghi strutturali della cisteina, come S-allilcisteina e S-propargil-cisteina, hanno ridotto l’entità di infarti nei ratti esposti a riperfusione ischemica (Wang et al, 2009), un effetto attribuito ad una aumentata formazione di H2S e inibito da un bloccante di CSE (Wang
et al, 2009).
Altre sostanze possono direttamente influire sulla formazione di H2S nel miocardio. Pertanto,
è stato recentemente riportato che l’inibitore della fosfodiesterasi-5 (PDE 5), tadalafil, aumenta la formazione di H2S tramite un processo proteina chinasi G-dipendente e conduce
alla riduzione dell’entità dell’infarto in topi esposti a riperfusione ischemica (Salloum et al, 2009).
Attualmente è ampiamente riconosciuto che il miocardio, soggetto a episodi di ischemia sub-letale, diventa meno sensibile a successivi attacchi ischemici più severi. Questo fenomeno è conosciuto come “pre-condizionamento ischemico” (IPC), e tale effetto protettivo contro lesioni da ischemia/riperfusione miocardica è principalmente dovuto all’attivazione di canali KATP, con particolare riguardo a quelli espressi nella membrana interna mitocondriale (O’Rourke, 2000).
Prove sperimentali hanno dimostrato che il blocco della produzione di H2S endogeno tramite
PAG riduce l’effetto protettivo di IPC (Bian et al, 2006); inoltre, nel cuore perfuso isolato di ratti, la somministrazione di NaHS, al momento della riperfusione, ha ridotto l’entità dell’infarto miocardico (Ji et al, 2008), un effetto antagonizzato da bloccanti dei canali KATP, glibenclamide e 5-HD (Ji et al, 2008).
In sintesi, l’aumento della formazione di H2S nel caso di pre-condizionamento nel miocardio
riduce le lesioni da riperfusione ischemica nei roditori mentre l’effetto nel cuore dei suini è meno pronunciato (Osipov et al, 2009).
Possiamo affermare quindi che è probabile che l’H2S endogeno svolga un ruolo come
mediatore di IPC e, in accordo con questo, gli effetti protettivi di H2S esogeno, contro lesioni
da ischemia/riperfusione miocardica, sono stati evidenziati sia nel cuore perfuso di ratto sia in modelli sperimentali di infarto acuto (Zhu et al, 2007).
8.2 H
2S E IPERTENSIONE SISTEMICA
Topi privi di CSE hanno ipertensione sistemica e gli autori hanno suggerito che simili aumenti di pressione sanguigna nei topi con CSE e l’inibizione di NO sintetasi endoteliale implicano che l’H2S influenza la resistenza vascolare periferica sistemica per certi versi
paragonabile all’azione di NO (Yang et al, 2008).
Sulla base di una funzione renale normale e livelli di H2S nel cervello, gli autori suggeriscono
che l’ipertensione potrebbe essere imputata ad un’indebolita vasodilatazione endotelio-dipendente (Yang et al, 2008); tuttavia, l’ipertensione indotta dall’inibitore di NO sintetasi, L-NAME, influenza principalmente la funzione renale e ciò è dovuto ad una ridotta attività di CSE e ad un abbassamento dei livelli plasmatici di H2S (Zhong et al, 2003).
Studi recenti hanno anche rivelato che l’H2S endogeno svolge un ruolo sulla funzione renale
(Xu et al, 2009), e l’H2S è importante nella protezione contro lesioni da riperfusione