1
Aggregation Induced Emission of Gold(I)
complexes in water or water mixtures
Andrea Pinto,a Noora Svahn,a João Carlos Lima,b and Laura Rodrígueza,c,* a Departament de Química Inorgànica i Orgànica, Secció de Química Inorgànica,Universitat de Barcelona, Martí i Franquès 1-11, E-08028 Barcelona, Spain. E-mail: [email protected]. Tel.: +34 93 4039130
b LAQV-REQUIMTE, Dep. Química, Fac. Ciências e Tecnologia, Univ. Nova de
Lisboa, Monte de Caparica, Portugal.
c Institut de Nanociència i Nanotecnologia (IN2UB). Universitat de Barcelona, 08028
Barcelona (Spain)
Abstract
Gold(I) complexes are an increasing area of investigation due to the possibility of giving rise to supramolecular aggregates with particular and morphologies that can be modulated together with their luminescent properties. A detailed study has been carried out for gold(I) complexes that self-assemble in aqueous medium (in pure water or in mixtures of water with organic solvents in different proportions). The majority of the examples reported until now are found in mixtures of water and DMSO, acetone, DMF or acetonitrile. The addition of cations to a solution of gold(I) complexes has been observed to show a direct impact on the resulting process of aggregation.
The use of perhalogenated ligands together with isocyanide moieties should be denoted to promote the resulting self-organization. Nevertheless, other ligands like alkynyls or carbenes also promote self-assembly.
A careful analysis of the data shows that aurophilic interactions have a key role in the formation of the resulting aggregates and in the enhancement of the luminescence (aggregation induced emission, AIE).
2
Introduction
The development of fluorescent organic materials is of great interest for both fundamental research and practical applications such as in sensors, displays, photoelectronic devices, biological labelling and memories.1 Controlling or tuning the photoluminescence properties of these compounds by external stimuli is a particularly attractive way to obtain photofunctional materials. Transition-metal complexes (TMC) are great candidates to this field and increasing attention is paid in this way.2-5 In particular, organometallic complexes represent a fascinating class of TMC, since they exhibit a rich chemistry due to the formation of inter- and intramolecular metal···metal bonds (so-called metallophilic interactions). Additionally, other well-known non-covalent intermolecular interactions such as hydrogen bonding, , and van der Waals interactions may also be involved. These kind of interactions have been observed to be responsible for the formation of micro- and nano-sized supramolecular assemblies.6-11 The formation of large aggregates can induce a quenching effect of the luminescence of the complexes (called Aggregation Induced Quenching, ACQ12) or, on the contrary, an increase on their displayed luminescence (Aggregation Induced Emission, AIE13-15). Focusing on AIE, this photophysical phenomenon is shown by a group of luminogenic materials that are non-emissive when they are dissolved in good solvents but become highly luminescent when they are clustered in poor solvents or in solid state. This concept has been widely used and there are numerous examples of a variety of applications for AIE materials.16-20
Most of the AIE active molecules in literature are organic and this phenomenon has been less explored for metal complexes. Similar to organic compounds, TMC also exhibit rich optical and luminescent properties usually associated with metal centers and their stability in common organic solvents. However, differently from conventional fluorescent compounds, which are singlet-state emitters, d6, d8 and d10 complexes containing heavy-metals are triplet emitters and as such display phosphorescence. Among these metals, one of the challenging fields of development of TMC-based phosphorescent sensors is the exploration and development of gold(I) luminophores.21-23
Due to the high spin−orbit coupling constant of gold, the access to the low-lying
3 the well-known establishment of aurophillic interactions, made gold(I) complexes as a promising future in the supramolecular interactions needed for AIE.
Taking into consideration all these facts, in this review article we will report on gold(I) complexes that display AIE. Moreover, due to the evident interest on water-based applications, such as sensing of biologically relevant species, we have decided to focus our study on AIE processes involving water. Although the vast majority of aggregated structures reported in the literature are found in organic solvents, there is an increasing number of reports in pure water or water mixtures with organic solvents in the last years. Gold(I) AIE systems are herein differentiated and described based on their relative water to organic solvent contents.
Aggregation in mixtures of water and organic solvents
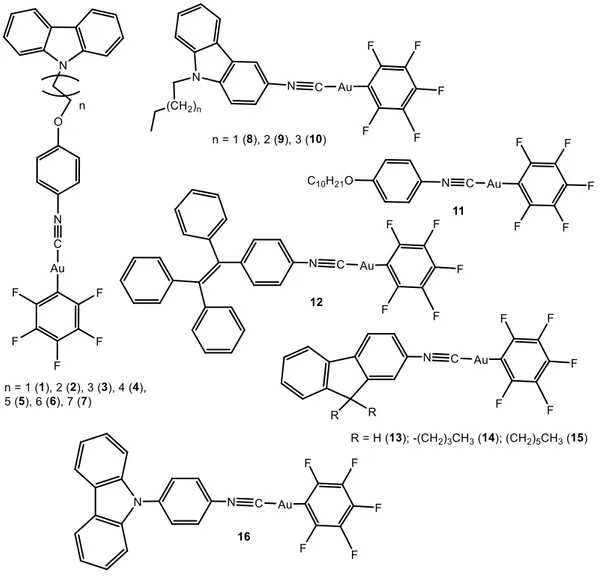
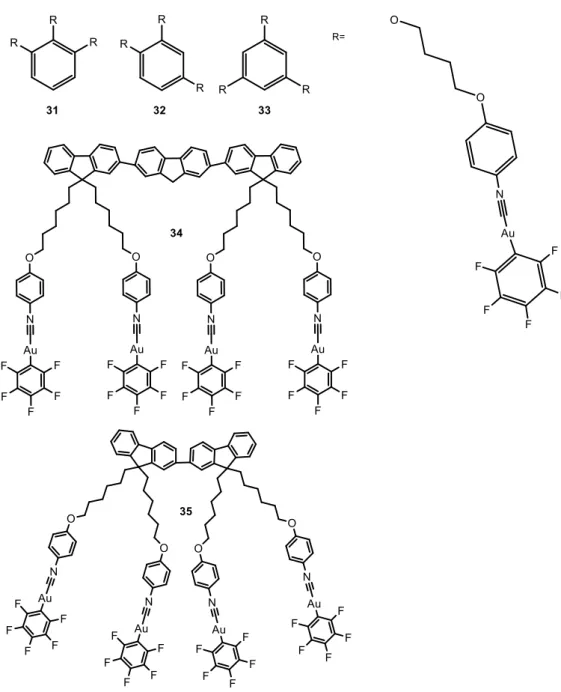
The larger number of examples found in the literature of this field are based on gold(I) perhalogenated cyanide complexes, extensively described by the group of Liu, although some examples with other ligands such as alkynyl and carbene moieties are also presented. The particular choice and synthesis of the ligands coordinated to the metal atom will determine the nuclearity of the resulting complexes. They have been divided in this work according to this, as mononuclear complexes (Figure 1), dinuclear complexes (Figure 4) and complexes with higher nuclearity (Figure 6). Reported luminescence quantum yields are collected in Table 1.
Compounds 1-1624-29 were obtained by the reaction of C
6F5Au(tht) (tht = tetrahydrothiophene) with the corresponding isocyano ligand in CH2Cl2 at room temperature under inert atmosphere in good yields.
The AIE behavior of complexes 1-10 and 13-16 was evidenced in DMF/H2O mixtures, or in THF/H2O mixtures for 12, with different water fractions (fw) and following the corresponding changes in their UV absorption spectra and in photoluminescence (PL). The absorption spectra of the compounds are red-shifted with increasing fw. Meanwhile, level-off tails could clearly be observed in the visible region as the fw values were increased, which result from the scattering effect of nanoscopic aggregates.30
4
Table. 1. Reported luminescent quantum yields for the gold(I) complexes presented in
this review.
Compound Quantum Yield Sample state Reference
36 0.05 Dichloromethane, RT 39 37 0.06 Dichloromethane, RT 39 39 0.05 Dichloromethane, RT 39 46 0.04 Dichloromethane, RT 45 47 0.04 Dichloromethane, RT 45 48 0.04 Dichloromethane, RT 45 49 0.03 Dichloromethane, RT 45 50 0.05 Dichloromethane, RT 45 51 0.04 Dichloromethane, RT 45 52 0.044 Benzene, RT 48 53 0.05 Benzene, RT 48 54 0.05 Benzene, RT 48 55 0.06 Benzene, RT 48 61 0.10/0.15 Water, RT/Solid 56 62 0.01/0.02 Water, RT/Solid 57 63 0.05 Water, RT 61 64 0.03 Water, RT 62 65 0.04 Water, RT 62 66 0.01 Water, RT 62 67 0.26 Water, RT 62 75 0.01 Water, phosphate buffer, NaCl 67 20 0.50 Thin Film 34 21 0.65 Thin film 34,37 22 0.05 Thin Film 34 31 0.15 Thin film 38 32 0.08 Thin film 38 33 0.08 Thin film 38 34 0.11 Thin Film 34 35 0.06 Thin Film 34 12 0.21 Crystals 27 30 0.41 Crystals 36 16 0.22 Solid 29 26 0.68 Grounded Solid 1 27 0.45 Grounded Solid 1 28 0.31 Grounded Solid 1 58 0.52 PAA matrix 52
5
Figure 1. Chemical structures of mononuclear perhalogenated complexes that aggregate
in water mixture of solvents.
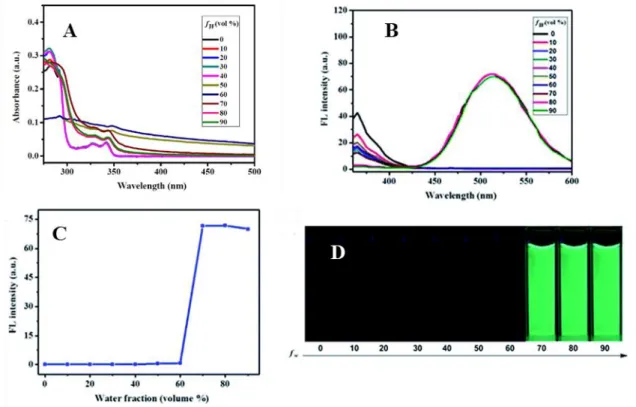
Weak blue emission bands are observed in pure DMF. However, when the water contents in the DMF solution was increased between 40-70%, a new green emission band was observed with max at 515 nm, accompanying the aggregation, which is observed in absorption spectroscopy. When the fw value in the DMF solution reached 90%, the emission intensity of the luminogens at 510-550 nm was clearly higher than that in pure DMF, and the resulting strong green fluorescence was observed under UV light upon excitation at 365 nm (Figure 2). This was because water is a non-solvent for these complexes and induces aggregates’ formation, with 40-50 nm size or even smaller (between 3-10 nm size) for 13-16 according to DLS measurements. There seems to be
6 no correlation between the length of the long alkyl chain in complexes 1-7 and the resulting aggregates’ size.
The formation of the nano-aggregates could result in a variation in the intermolecular gold···gold interactions, which are expected to be responsible for the resulting green fluorescence. 11,31
A particular case is compound 12, which displays a clear increase on the emission intensity with emission maximum at 492 nm (blue-shifted with respect to the others) and gives rise to the formation of very large structures between 100 and 457 nm (depending on water composition) when aggregated.
Figure 2. (A) UV spectra of complex 1 (1.0 × 10-5 M) in DMF-water mixtures with various water contents (0-90%); (B) PL spectra of the dilute solutions of 1 (1.0 × 10-5 M) in DMF–H2O mixtures with different water fractions (fw). Excitation wavelength: 330 nm. (C) Changes in the emission intensity of 1 at 515 nm in DMF–H2O mixtures with various volume fractions of water (0–90%). (D) The fluorescence images of 1 (concentration: 1.0 × 10-5 M) in diverse DMF–H
2O mixtures with various fw values (90%) under 365 nm UV irradiation. Adapted from ref. 25 with permission from The
Royal Society of Chemistry.
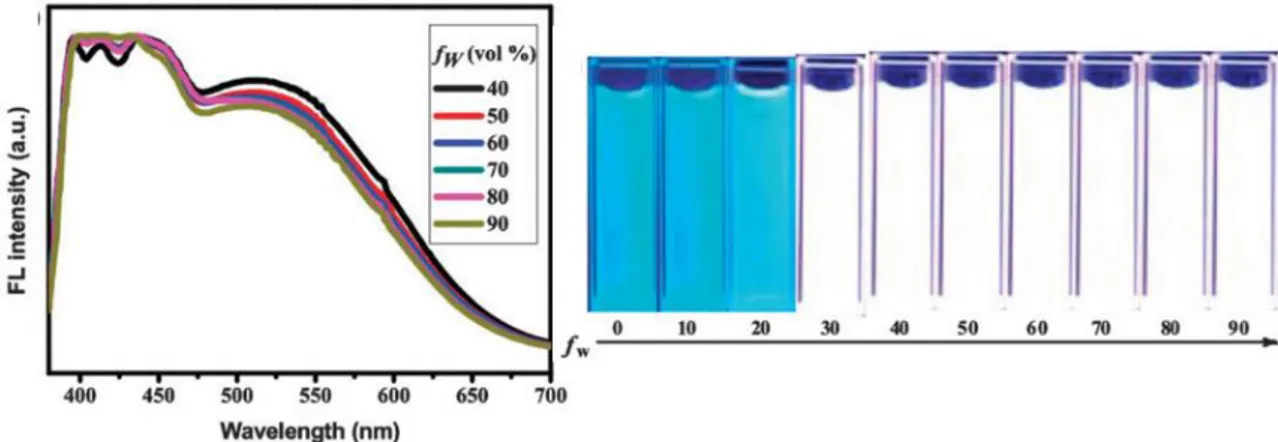
Distinctly, compound 11 exhibits AIE characteristics and additional thermochromic behavior, where its fluorescent properties show reversible changes from blue to
yellow-7 green in response to temperature stimuli.24 The thermochromic behavior is an indication of polymorphism and the change in luminescence was attributed to changes on the intermolecular packing with temperature. In this case, the AIE behavior was examined in EtOH–H2O mixtures with various water compositions. Interestingly, the PL intensity was not only significantly enhanced with the addition of water, but the emission color also changed from blue (402 nm and 425 nm) to yellow-green (559 nm), as shown in Figure 3.
Figure 3. (a) Luminescence spectra of the dilute solutions of 11 (2.0·10-5 M) in EtOH– H2O mixtures with different volume fractions of water (excitation wavelength = 310 nm). The inset shows the emission images of 11 (2.0·10-5 M) in pure EtOH as well as 40%, and 60% water fraction. (b) Luminescence spectra of compound 11 (2.0·10-5 M)
in water–EtOH mixtures with different volume fractions of water (40%–50%). Excitation wavelength = 310 nm (right). Reproduced from ref. 24 with permission from
The Royal Society of Chemistry.
A vibronical resolution band was observed centered at ca. 425 nm, assigned to the intra-ligand localized -* excited state.32 It is expected that two kinds of nano-aggregates might be formed, displaying blue and yellow emission caused by the difference in molecular packing and the change in the intermolecular gold···gold interactions.
The decrease in fluorescence intensity observed in Figure 3 upon aggregation, according to the authors, results from the fact that only the molecules on the surface of the nanoparticles emit light and contribute to the fluorescence intensity, even if the intermolecular gold···gold interactions enhance light emission. The net outcome of
8 these antagonistic processes depends on which process plays the dominant role in affecting the fluorescent behavior of the aggregated molecules.
The emission spectra of crystals of 11 show bands at 407 and 428 nm, and intriguingly, after gentle heating (> 55 ºC) of the crystals in a slender piece of glass, a broad emission band, with a maximum wavelength of 530 nm, was observed. Its luminescence intensity and maximum wavelength was reversibly changed during consecutive heating and cooling cycles over the temperature range 25 ºC–59 ºC, from yellow-green (ca. 530 nm band, at high temperature) to blue (ca. 425 nm band, at lower temperatures).
According to the authors, aurophilic interactions are responsible for the spectral changes, with the contribution of new ligand-to-metal–metal charge transfer (LMMCT) excited state at higher temperatures [C6F5-Au···Au], together with weak and other intermolecular interactions (such as the C–H···F and C···F) found in the corresponding X-ray crystal structure.
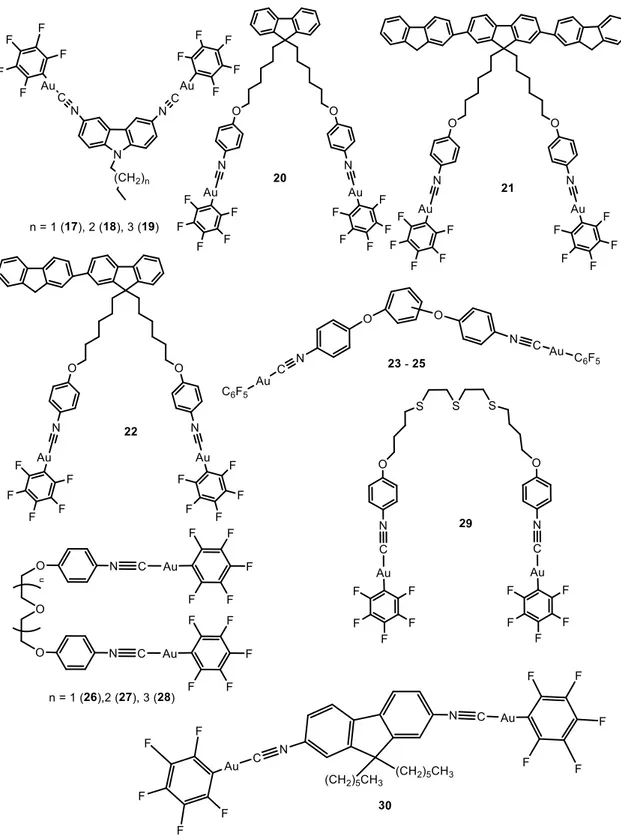
Dinuclear complexes 17-30 presenting the same Au-C6F5 group were synthesized in a similar way after the previous synthesis of the corresponding isocyanide organic ligand, in some cases, following different steps.1,26,33-36 AIE of 17-22 and 30 were analyzed in DMF:H2O; in the case of 23-25, in DMSO:H2O mixtures; 26-28 in acetone: H2O and
29, in acetonitrile: H2O. In some cases, the compounds present a weak blue emission at
ca. 400 nm when they are perfectly dissolved but a clear change on the emissive
properties occurs in mixtures containing ca. 30-40% fw where a new and very intense emission peaks around 500-570 nm. Absorption spectra also display a red-shifted with increasing fw.
X-ray crystal structure of 19 indicates the presence of strong intermolecular aurophilic interactions, with shortest Au···Au distances of 3.040 Å. These interactions hinder the rotation of carbazole units, indicating a possible involvement of charge transfer from metal-metal to ligand (MMLCT). It was not observed any specific trend or effect of the alkyl length chain of these complexes on the resulting AIE properties. On the contrary, X-ray crystal structure of 30 does not show particularly short Au···Au distances but close C–H···F and C–H··· contacts responsible for the organization of the compound in the crystal. Nevertheless, additional aurophilic interactions are expected to be favoured in the presence of water, due to the formation of nanoaggregates or a metastable state phase.36
9
Figure 4. Chemical structures of dinuclear perhalogenated complexes that aggregate in
10 The particular AIE properties of 21 should be highlighted.37 This complex exhibits blue-green fluorescence in pure DMF solution under 365 nm UV light due to the presence of three fluorene units. However, when the water fraction in the DMF solution was increased to 30%, two fluorescence bands were observed with max at around 408 nm and 510 nm, which combine in a notable white emission, (Figure 5). The same white emissive properties were reported both in thin films and in solid state for the same compound, being a new class of white light-emitting material with a good deal of interest.
Figure 5. Emission spectra of the dilute solutions of 21 (2.0·10-5 M) in DMF-H2O mixtures with different volume fractions of water (40–90%). exc = 365 nm (left); The fluorescence images of 21 (concentration: 2.0·10-5 M) in different DMF-H
2O mixtures with different water fractions under 365 nm UV irradiation (right). Adapted from ref. 37
with permission from The Royal Society of Chemistry.
In the case of 26-28, all the emission peaks of the mixtures are red-shifted at first, then blue-shifted as the fw values increase. This was attributed to the effect of the solvent polarity on the ICT transition, as usually observed in donor-acceptor (D-A) molecules, decreasing the emission efficiency and red-shifting the emission.1 At higher fw values aggregation comes into play, the intermolecular Au···Au interactions become significant and the local environment becomes less polar, causing an increase in the emission efficiency and a blue shift in the emission. These studies were performed with other water/co-solvent mixtures with THF, acetonitrile, DMF, DMSO and acetone. It was observed that the polarity and viscosity of the solvent have a clear impact in the
11 luminescent properties, both in the observed shift upon and in the resulting luminescent lifetimes.
Tri- and tetra-nuclear AuC6F5 (Figure 6) were also synthesized by the same researchers and their AIE behaviour analysed in DMF:H2O mixtures.34,38
Figure 6. Chemical structures of tri- and tetranuclear perhalogenated complexes that
aggregate in water mixture of solvents.
Similar green AIE bands were observed in all cases when increasing fw in the mixtures with DMF. The emission of complexes 31-33 in thin films formed from solutions of the complexes in dichloro- or trichloromethane were also analyzed. Interestingly, although
12 the recorded emission band was observed in the same wavelength as in DMF:H2O (ca. 500 nm), the corresponding quantum yields were affected by the position of the gold(I) groups in the benzene unit, (see Table 1). That is, the higher value of luminescence quantum yield was recorded for 31, where the three gold(I) atoms are in close vicinity, thus favoring aurophilic and other weak interactions between the chains.
The higher nuclearities and extended fluorene moieties in 34 and 35 give them interesting AIE profiles. In pure DMF 34 displays similar emission to the dinuclear complex 22, exhibiting two emission bands with max at 353 nm and 441 nm, responsible for a blue emission under a 365 nm UV light. When fw reaches 20%, a new emission band peaks at 516 nm with the correspondingly change on the emission colour from blue to green, in agreement with the formation of aggregates. On the other hand, the emission of 35 exhibits a blue-green fluorescence under 365 nm UV illumination even in pure DMF, due to the presence of three fluorene units. However, when the water fraction in the DMF solution is increased to 20%, two fluorescence bands are observed: a higher energy band, with vibronic resolution centred at 418 nm and a broad band at 538 nm. Correspondingly, 35 displayed white light emission when the water fraction exceeded 20%, and the very broad emission bands cover the whole visible range. In this case, the white emissive behaviour starts at lower fw with respect to the previous dinuclear complex reported by the same authors (20% instead of 30% previously observed in complex 21). Naturally, the higher number of gold(I) units favour the intra- and intermolecular interactions and the formation of emissive aggregates.
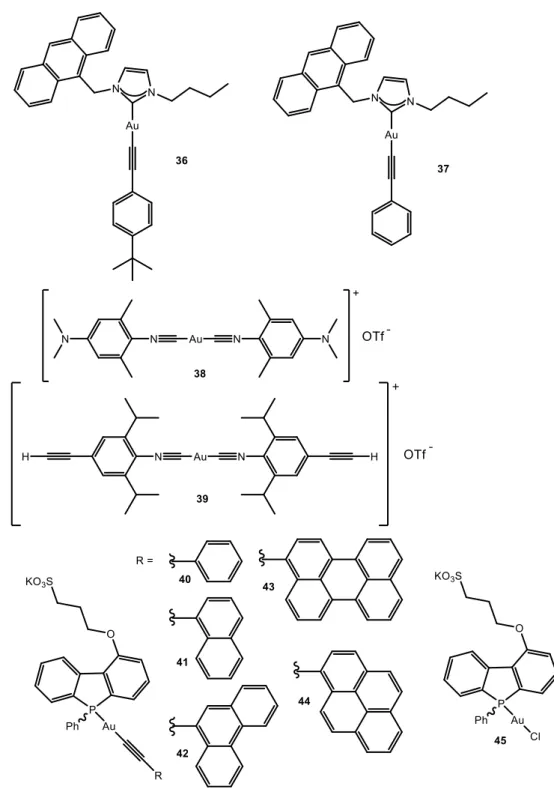
A different family of gold(I) complexes which are observed to aggregate in water/organic solvent mixtures explore the use of polyaromatic rings (non-perfluorinated) linked to gold(I) either through alkynyl or isocyanide. There are examples found in the literature of both mononuclear and polynuclear complexes, such as in the previous family. Starting from mononuclear complexes (Figure 7) notable examples were obtained in C.M. Che group39 where the correct choice of ligands gives origin to the formation of supramolecular nanowires/nanobelts in the cases of complexes 36-39 (Figures 7-8). The compounds were synthesized from the corresponding gold(I) carbene chloride in the presence of the alkynyl group previously deprotonated in the presence of NaOH. The corresponding absorption spectra of 36 and
37 display the typical pattern of the * transitions of the anthracene ligand centered
13 attributed to the * absorption band of the alkynyl moiety. In the case of 38 and 39 the corresponding absorption spectra show an intense band at 305 nm assigned to a * transition of 4-cyano-phenylacetylide.
Figure 7. Chemical structures of mononuclear alkynyl complexes (and precursor) that
14 Compounds 36, 37 and 39 are phosphorescent (in agreement with recorded lifetimes in the order of microseconds) at room temperature and 38 phosphoresces only at 77 K in dichloromethane and in solid state. The emission maxima peaks at ca. 420 nm for 36 and 37, assigned anthracene moieties vibronically resolved and at ca. 460 nm for 39 assigned to arylacetylide (broad band). In the solid state the emission bands are red-shifted due to intermolecular Au···Au and interactions. Interestingly, when 36-39 were dissolved in THF/H2O mixtures (1/9) and sonicated it was found to form nanowires of 5.6 (36), 2.8 (37) and 0.8 (38, 39) m length in the freshly prepared dispersions as evidenced by TEM images (Figure 8). Other mixture of solvents were not successful to achieve the formation of nanowires.
Figure 8. TEM images of complexes 36-39 freshly dispersed in THF/H2O (1:9, v/v) with a concentration of 40 mm. Adapted from ref. 39. Copyright 2010 John Wiley and
Sons.
It was suggested that the interactions between aromatic rings, C-H··· and cationic gold(I)··· interactions play a direct role to direct the anisotropic growth of the nanostructures and govern the self-aggregation process.
The group of V.W.W. Yam has a large expertise on gold(I) alkynyl complexes and some examples have been reported also regarding aggregation induced emission in aqueous media. Compounds 40-44 present a phosphane group as a second coordination position of the gold(I) atom.40 Because of the distinct pyramidal geometry of the phosphorus center, phospholes behave very differently from other planar heterocycles, and this may also give rise to irreplaceable self-assembly architecture. These complexes and the gold(I) chloride phosphole precursor (45) exhibited moderate luminescence in
15 degassed methanol solutions at 298 K. Dual emission was recorded for 40-42 and 45
with bands at 410–430 nm and ca. 470–625 nm. The former were attributed to the
spin-allowed metal perturbed * ligand-centered excited states of the phosphole moieties. In contrast, the long-lived lower-energy emissions (with larger Stokes’ shift) can be quenched upon exposure to oxygen, being indicative of their triple state origin. Authors assign the phosphorescence bands to metal-perturbed * ligand-centered excited states of the phosphole ligands for 40 and 45 and to the triplet metal-perturbed * ligand-centered excited states of the alkynyl units in the case of 41 and 42. The insensitivity to oxygen of the emission band at 440-550 nm of 343 and 44 let the authors assign the origin of this band to the singlet excited state of the pyrenyl and perylenyl moieties
In this work, the self-aggregation process was studied in 1·10-4 M aqueous solutions containing 25% DMSO for complexes 40-44. The formation of sheet-like structures
with a length and width of ca. 1–20 mm and 0.2–2 mm respectively was observed in all
cases, as evidenced by TEM images. The height of the nanosheets was determined to be
ca. 20–25 nm by tapping mode atomic force microscopy (TM-AFM). A molecular
dimension of ca. 1.84 nm has been estimated from the molecular structure of 43, giving rise to a bilayer thickness of 3.68 nm and thus, the authors suggested that the nanosheets
are probably comprised of multi-bilayer aggregates of approximately 5–7 repeating
units. The amphiphilic nature of the complexes drives self-organization, with hydrophilic sulfonate head groups pointing towards water and hydrophobic phosphole and alkynyl moieties packing closely to each other through hydrophobic interactions in order to minimize the contact with water.
An important point is that complex 45 does not aggregate. That means that the alkynyl ligand is essential for the aggregation as it provides an additional hydrophobic moiety and rigidity for the bilayer core.
NMR and UV-visible absorption spectroscopies provide additional evidence for the aggregation. 1H-NMR spectra recorded at different D2O/DMSO-d6 compositions show that the aromatic protons become broader and were silent with increasing D2O contents while they are clearly observed in 100% DMSO-d6. On the other hand, upon increasing
16 the water content in the DMSO solution, the absorption bands at ca. 320 and 335 nm decrease gradually in intensity and become less defined. The appearance of a new absorption tailing at ca. 360 nm and beyond was attributed to the formation of aggregates where the aromatic chromophores are in close proximity.41 Plotting the normalized degree of aggregation against the volume fraction of DMSO resulted in a clear sigmoidal curve, indicative of an isodesmic growth mechanism for the aggregates,
i.e. a non-nucleated pathway with gradual assembly and disassembly profiles.42,43 Based on this model, the corresponding thermodynamic parameters responsible for aggregation can be calculated and the calculated values of G0 around -35 kJ/mol for 41 and 42 and -41 and -47 kJ/mol for 43 and 44, are in the order of expectation for the establishment of aurophilic interactions in the multi-bilayer assembly.44 It was also shown that the complexes with a larger extent of the -conjugated surface at the alkynyl moiety have a higher propensity towards aggregation due to an increase in the size of the hydrophobic portion.
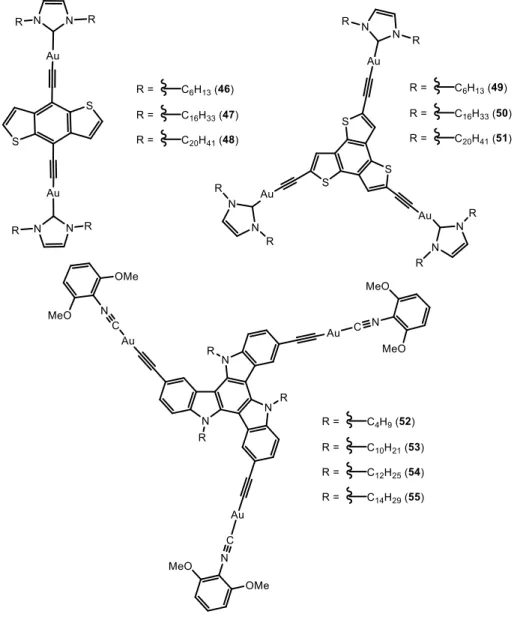
Aggregation of polynuclear alkynyl gold(I) complexes (Figure 9) was very recently reported by the same group.45 In this case, THF-water mixtures were used as co-solvents.
Upon excitation above 350 nm, all the complexes exhibited broad and vibronic-structured emission bands at 400-500 nm (assigned to originate from the singlet metal-perturbed * ligand-centred excited states of the alkynyl ligands) and 600-750 (for
46-48) or 500-650 (for 49-51) (attributed to the triplet ligand-centred excited states) in
dichloromethane.
Temperature dependent UV-visible spectra were recorded for 1·10-5M - 1·10-6 M solutions of the complexes containing about 60% THF in water. Upon increasing temperature, the intense and vibronically resolved absorption bands, corresponding to the ligand, decrease on intensity and while a broad absorption tailing above 400 nm increases in intensity. This was attributed to the occurrence of aggregation leading to close packing of the aromatic chromophores.46,47 As a difference with complexes 40-44, the melting curves obtained from the plots of the normalised degree of aggregation against temperature were clearly non-sigmoidal, which was indicative of a cooperative self-assembly mechanism.43 The calculated thermodynamic parameters indicate that the complexes with longer alkyl chains (48, 51 and 55) start the elongation process that
17 precedes aggregation at higher temperatures and the overall aggregation process is more exothermic. This was expected to happen since longer alkyl chains leads to a larger hydrophobic effect and therefore further favours aggregation in aqueous medium.
Figure 9. Chemical structures of polynuclear alkynyl complexes that aggregate in water
mixture of solvents.
The corresponding analysis of the resulting morphologies by TEM and SEM gave very interesting results since aggregation of the complexes display two different morphologies (Figure 10). While complexes 46, 47, 49 and 50 display spherical shape, the icosyl-containing dinuclear and trinuclear complexes 48 and 51 exhibit sheet-like nanostructures shaped like leafs or circles, in the same conditions. The morphological difference between the spherical and sheet-like aggregates is believed to result from the
18 directing effect of the lengthened hydrophobic alkyl chains. Hydrophobic–hydrophobic chain interactions are expected to undergo self-assembly in a preferred direction, driven by the long icosyl chains, forming ordered staircase-like structures in complexes 48 and
51. The shorter alkyl chains in the remaining complexes are unable to avoid that the
monomers congregate together randomly to form the spherical aggregates. This is in agreement with the same spherical morphology observed for THF:water mixtures of
52-55.48 In these cases, complexes aggregated also in a similar THF:water composition (ca. 50%) and this process was verified with changes in the UV-visible and NMR spectra, analogous to those previously reported for the previous complexes.
Figure 10. TEM images of the aggregates prepared from a) 46, b) 48, c) 49, d) 50, e) 51
and f) 52 in 50% THF–water mixture (1·10-4 M). Reproduced from ref. 45. Copyright 2015 John Wiley and Sons.
Aggregation in water
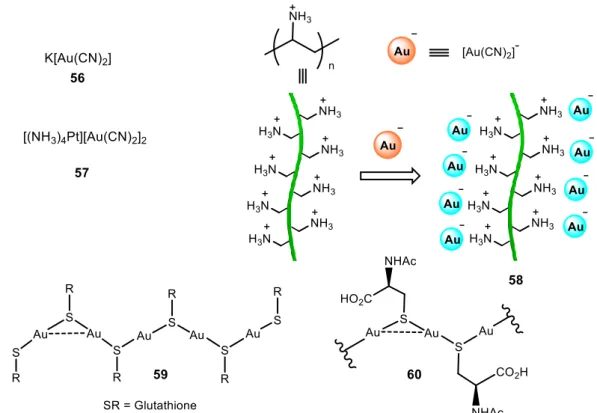
Very few reports can be found about gold supramolecular structures obtained in pure water (Figures 11 and 13). In 2000, Patternson and co-workers reported on the self-aggregation of [Au(CN)2]- (56) in water with concentration.49 This fact was recorded by means of absorption spectra with progressive red shifts following the increase in concentration up to near the saturation limits. Electronic structure calculations suggest that the observed spectral behaviour is attributed to metal···metal interactions between
19 neighboring Au(CN)2- ions. Strong photoluminescence was also recorded in aqueous solutions of K[Au(CN)2] at ambient temperature at concentrations above 10-2 M.50 The luminescence of Au(I) compounds was related for the first time to Au···Au bonded excimers and exciplexes. Two years later, Balch and co-workers reported the formation of the complex [(NH3)4Pt][Au(CN)2]2·1.5(H2O) (57) by the reaction of [(NH3)4Pt]Cl2 and K[Au(CN)2] in water.51 X-ray crystal diffraction showed the establishment of both Au···Au and Au···Pt interactions in solid state. These interactions were attributed to be the responsible for the observed emission.
Figure 11. Chemical structures of complexes containing cyanide and thioethers that
aggregate in water.
Moriuchi, Hirau and co-workers reported on the utilization of polyelectrolytes as a reliable strategy for the assembly of oppositely charged functional groups along polyelectrolytes through electrostatic interactions (58).52 In an aqueous media, emission tunability of dicyanoaurate(I) ions, [Au(CN)2]-, was attributed to the self-association through Au…Au interactions. The Au···Au intermolecular interactions are not very strong and the assembly depends critically on the concentration. At low concentrations [Au(CN)2]- exists mainly in its monomeric form. The presence of poly(allylamine hydrochloride) (PAA), which is decorated with positively charged side chains can serve
20 as a polymeric spatially aligned scaffold for the aggregation and self-association of the negatively charged [Au(CN)2]-, adding electrostatic interactions to Au···Au interactions. Lower energy absorption bands, in the region of approximately 250–320 nm, were recorded in an aqueous solution of K[Au(CN)2]with increasing amounts of the PAA, being indicative of the interaction and self-association of the gold(I) complex into the PAA matrix. The appearance of an emission band at 484 nm was observed only in the presence of PAA, as an additional proof of these interactions. This was also verified by excitation spectra that show excitation bands at ca. 290 nm.
Odriozola and co-workers found that glutathione (GSH), a naturally occurring and readily available tripeptide, was able to gelate water when mixed with Au(III) salts (59).53 GSH reacts with gold(III) chloride in water to give the corresponding GS-Au polymer (, which formed an almost transparent gel. Aurophilic interactions were used for the construction of supramolecular hydrogels for the first time, and the principle can be widely applied for the synthesis of designed hydrogels. These materials have potential biomedical applications since Au(I) thiolates have been used for more than 50 years as therapeutic agents and the variety of naturally occurring and biocompatible thiol-containing molecules is essentially infinite. SEM analysis of the lyophilized sample revealed a porous microstructure (Figure 12). The morphology of the polymeric material appeared to be in the form of platelets or flakes. Very interestingly, the GS-Au hydrogel demonstrated pH responsiveness due to the formation of neutral, anionic or cationic species at the different pH values (Figure 12 below).
A few years later, the same group reported on supramolecular systems in which the gelator species are metal-thiolates that self-assemble through metallophilic attractions, as an emerging alternative for the development of new generation drug-delivery systems.54 They presented the use of coinage metal complexes containing a thiolate ligand since many drugs contain sulphur derivatives due to the presence of cysteine residues. It was observed that the gold hydrogel (60, Figure 11) presented higher thermal stability than the silver and copper analogous, and could be heated up to the boiling point of water with no phase transition or apparent decomposition observed. A possible explanation was given by the fact that aurophilic interactions are energetically stronger compared to the argentophilic or the cuprophilic ones.55 The low pH at which these hydrogels are formed (pH < 4) limits their direct use as drug-delivery systems, but
21 still this system constitutes a novel method for easy and fast conversion of small drugs into potent hydrogelators.
Figure 12. SEM micrographs of a lyophilized GS-Au gel (above). pH Induced
reversibility of GS-Au hydrogels (below). Adapted from ref. 53 with permission from The Royal Society of Chemistry.
Interesting findings have been reported in our group in the last years with gold(I) alkynyl complexes containing water soluble phosphanes, mainly PTA and DAPTA (Figure 13).
In 2013, we found that the gold(I) complexes with chemical structure [Au(4-pyridylethynyl)(PR3)] (PR3 = PTA (1,3,5-triaza-7-phosphaadamantane) and DAPTA (3,7-diacetyl-1,3,7-triaza-5-fosfabiciclo[3.3.1]nonane)) give rise to the formation of luminescent hydrogelators.56,57 This behavior was quite unexpected and really exciting at the same time, since they were the first examples of gold(I) organometallic complexes, with a relatively simple chemical structure, that gave rise to the formation of very long fibers in water.
22
Figure 13. Chemical structures of complexes containing alkynyl ligands that aggregate
in water.
Until that moment, the formation of hydrogels were mainly observed in the literature with compounds that presented nucleotides, nucleosides, fatty acids, ureas, amides or amino acids among others, groups that are well known to induce gelation. In general, these ligands need to be additionally functionalized with long alkyl chains to induce noticeable gelation effects. In our work, it was observed that the establishment of aurophilic interactions (apart from other weak interactions like hydrogen bonds, and stacking)44 was relevant to form the resulting hydrogels (Figure 14).
23
Figure 14. Chemical structure of complexes 61 (up left) and 62 (up right); Image of the
hydrogelator of 61 under visible (A left) and UV (A right) light; SEM image of 62 (B); Optical microscopy image of 62 (C).
The formation of aggregated luminescent structures was evidenced by broad emission bands at ca. 500 nm due to excimeric 3IL emission (not observed in apolar solvents58) and the presence of Au···Au interactions were detected by the low absorption bands above 300 nm due to *(Au/Au)- * transitions.59 The calculation of the aggregation thermodynamic parameters were carried out from the application of Dimicoli and Hélène NMR approach of the isodesmic model.60 The results were in agreement with the presence of these Au···Au bonds.44 Changing the chromophore to a propargyloxicoumarin group (63), while maintaining the uncharged nature of the molecule, also results on the formation of very long fibers.61 The presence of a planar aromatic structure and additional atoms suitable to undergo hydrogen bonding (oxygen atoms of the coumarin) were observed to be involved in the aggregation process together with aurophilic interactions, according to X-ray crystal structure. Other changes in the molecular structure, such as the use of negatively charged phosphanes or the increase on the nuclearity of the complexes affect the resulting supramolecular structures.62 Complexes similar to 61-63 were synthesized with the triphenylphosphine-3,3’,3’’-trisulfonic acid trisodium salt (TPPTS, compounds 64-65) and a radical change on the aggregation process was observed in general. The presence of fibrillary structures was only detected for 64, the only compound containing ethynylpyridine moiety,
24 reinforcing the idea that this simple motif strongly impacts on the directionality of the resulting aggregation. In case of 65, the compound became highly soluble in water and the presence of very small aggregates could only be detected by DLS experiments.On the other hand, complexes 66 and 67 present a different pattern. PTA derivative, 66, gives rise to the formation of rod-like structures of ca. 20 m length while the higher steric hindrance of the DAPTA phosphine in 67 seem to have a direct role on the formation of smaller and spherical aggregates of ca. 100 nm (Figure 15).
Figure 15. SEM images of 64 (A), 66 (B) and 67 (C) previously dissolved in water.
Adapted from ref. 62. Copyright 2016 Elsevier B.V.
Different luminescent profiles were recorded being dependent on the nature of the chromophoric organic part of the molecules. Complex 64 displays a vibronically structured absorption band assigned to intraligand (IL) (C≡Cpy) transition. The coumarin absorption bands of 65-67 were centered at 320–350 nm and present the same profiles, although it was blue-shifted in the case of the mononuclear compound (in agreement with the effect of substituents of coumarin ligands on the resulting photophysical properties63). This fact indicated that the recorded electronic spectra correspond to * transitions of the coumarin group. Additionally, emission spectra was also able to differentiate between mononuclear and dinuclear complexes. In the case of 65, fluorescence and room temperature phosphorescence of the coumarin was observed, while only room temperature phosphorescence was displayed for 66 and 67 (stronger heavy atom effect in the dinuclear compounds). Thus, this was a nice example about how the nuclearity of the heavy atoms makes a direct influence on the intersystem crossing and induces phosphorescence of organic chromophores, being more favoured when the molecule contains two gold metal atoms.
25 Modification of compounds 64 and 65 by the introduction of a positive charge either in the phosphine or in the pyridyl moieties (68-73), through a simple methylation procedure, also impacts in the aggregated structures morphology and luminescence.10 Methylation was performed by the reaction of the complexes with CH3X (X = I-, CF3SO3-) or by the previous synthesis of the corresponding iodide or triflate 4-methylpyridylethyl ligand (68-71). The introduction of these positive charges precludes the formation of long fibrillary structures (Figure 16). When the positive charge is located at the pyridyl moiety the supramolecular assemblies behave like a lamellar byphasic system and respond to the charge at the surface, i.e., respond to the nature of the negative counterion, with a morphology transition from vesicles (in the case of water soluble iodide anion) to square-like extended structures (in the case of organic soluble triflate anion), see Figure 16. On the other hand, rod-like assemblies were detected when methylation proceeds at the phosphane with iodine counterion, revealing hexagonal packing (compound 72) while no significant aggregation could be detected with the triflate counterion (compound 73).
Figure 16. Cryogenic transmission electron microscopy image of sample of a 5·10−4 M aqueous solution of 70 (A); Scanning electron microscopy images of dried samples of
69 (B) and 72 (C). Reproduced from ref. 10 with permission from The Royal Society of
Chemistry.
Their recorded photophysical properties were also dependent on the counterion and on the phosphane. The presence of an absorption band above 300 nm was an evidence of the establishment of aurophilic interactions on the resulting aggregates. A red emission was recorded for complexes containing PTA phosphane (less soluble in water and thus, aggregated at spectroscopic concentrations, ca. 10-5 M) due to MMLCT transitions
26 while a yellow emission band was detected for DAPTA derivatives, according to 3[π– π*(alkynyl)] emission origin.
AIE in the presence of cations
There are several examples in the literature of organic molecules containing appropriate receptors that are polymerized through coordination to metal cations leading to gel formation (metalogels). Similar approach can be extended to gold(I) complexes, if suitable receptor units are added, which are able to interact with metal cations. Nevertheless the field of gold(I) supramolecular aggregates formation by the influence of the addition of external cations is underexplored. Some results are reported with gold nanoclusters64,65 but to the best of our knowledge, very few examples are reported with cation-induced aggregation of gold(I) complexes (Figure 17).
Figure 17. Chemical structures of complexes containing alkynyl ligands that aggregate
in water by the addition of cations.
Complex 29, shown before, aggregates in H2O:acetonitrile mixtures,35 however displays high selectivity and sensitivity for the presence of Hg2+ ions in this solvent mixture.
27 Experiments undertaken at 50% percentage of water, the conditions where AIE with maximum emission at 575 nm is observed, were performed in the presence of Al3+, Ca2+, Cd2+, Cu2+, Fe2+, K+, Li+, Mn2+, Na+, Ni2+, Pb2+, Zn2+, Fe3+, Ag+, and Hg2+. Although Fe3+ and Ag+ exhibited a slight quenching effect on the fluorescence of complex 29, only Hg2+ induced an effective fluorescence quenching with a concentration as low as 0.05 μmol/L. Competition experiments revealed that no obvious change of fluorescent signal could be observed in the presence of 10 equivalents of other metal ions, which suggests strong interaction of Hg2+
which confers selectivity for its detection over other metal ions. The disaggregation process of 29 in the presence of Hg2+ was verified by DLS measurements, where the 955 nm aggregates of 29 in water/acetonitrile (1:1) are strongly reduced in the presence of the cation (396 nm-size aggregates).
A clear selectivity and consequent emission enhancement was observed for 74 in the presence of Ca2+, which induces the cross-linking between Au(I)-Cys complexes and facilitates the formation of Au(I)···Au(I) contacts giving rise to the formation of strongly emissive aggregates.66 It was observed that the luminescence intensity increases continuously with higher Ca2+ contents. This behaviour was not detected for other divalent cations tested in the same conditions.
Noticeable peptidic Au(I)-metalloamphiphile complexes (75 and 76) were recently reported by Besenius’ group.67 Compound 75 was synthesized by reacting the previously prepared propargylamine-Au(I)-trisulfonated-triphenylphosphane complex [H2NCH2C≡CAu-(TPPTS)] with Fmoc–Phe–Phe–NHS under inert conditions. On the other hand, compound 76 was prepared via ligand exchange of ClAu(PTA) with Ac-Phe-Phe-HNCH2C≡CH under basic conditions, to deprotonate the alkynyl ligand. Only
75 was soluble in water at neutral conditions, due to the higher solubility of TPPTS in
this solvent, and the study was focused on water solutions of this complex. The resulting emission in conventional phosphate buffer (10 mM, pH 7.4), under physiological ionic strength and at room temperature peaks as a broad, unstructured luminescence emission band with maximum at 520 nm. This emission band was assigned to the formation of nanostructures incorporating short Au···Au distances. The large Stokes shift is indicative of phosphorescent origin of the emission suggesting a 3[(Au–P)-*(C≡C)] metal-to ligand charge-transfer or a metal-perturbed intraligand 3IL [*(C≡C)] state, where the contribution of Au(I)···Au(I) interactions is assigned
28 according to previous studies on Au(I)-alkynyl complexes.41,68 The broad emission at 520 nm disappears in CH3CN, a better solvent for the compound, that diminishes the hydrophobic contribution of weak intermolecular interactions. Instead, a new band below 400 nm appeared assigned to the Fmoc unit in the dissolved compound.
In water, the presence of cations also contribute to aggregation. The authors increased the ionic strength from 0.1 M NaCl to 1 M NaCl, and observed an increase of the emission (Figure 18). The authors assigned this observation to the fact that the addition of NaCl simultaneously screens the repulsive Coulombic interactions of the negatively charged compound and increases the hydrophobic effect originating from the apolar and aromatic moieties in the Fmoc-diphenylalanine block of amphiphile, with consequent stabilisation of the self-assembled aggregates. Nevertheless, no red-shifted emission band was detected, being indicative that the increase in ionic strength does not induce the packing by additional Au···Au interactions.
Figure 18. (A) Images of the luminescent Au(I)-metalloamphiphile 75 in 10 mM
phosphate buffer (pH 7.4) (exc = 245 nm) with 0.1 M NaCl (left) and 1.0 M NaCl (right). (B) TEM image of the Au(I)-metalloamphiphile75, deposited on carbon coated
grids from a 2 mg ml-1 solution in 10 mM Tris buffer, pH 7.5 and 1 M NaCl (negative staining was performed with 2% w/v uranyl acetate). Reproduced from ref. 67 with
permission from The Royal Society of Chemistry.
Cryo-TEM allowed the identification of 10-17 nm sized spherical objects with micelle like structures (assuming a length of 3.5 nm for the Au(I)-metalloamphiphile 75). No significant change on the morphology of the structures neither in their size was detected with increasing concentration of NaCl. Therefore, by increasing the ionic strength from 0.1 NaCl to 1 M NaCl in phosphate buffer, the formation of self-assembled micelles in
29 solution became more thermodynamically favourable, without affecting the order and size of the prepared nanostructures.
30
Conclusions
Gold(I) supramolecular aggregates are still part of an emerging area of research since the majority of the examples reported until now are quite recent. Additionally, it should be highlighted that all the results found in the literature are based on gold(I) organometallic complexes probably due to their linearity that favour the intermolecular assemblies.
It can be observed that a large investigation is made in water mixture of solvents, while the AIE behaviour in water and with the external addition of cations is increasing in the last years. In particular, AIE is widely studied for gold(I) organometallic complexes containing perhalogenated and cyanide in water mixtures. Nevertheless, it was recently observed that this process can also happen in water with relatively molecules that are simple in structure and do not contain any of the common ligands well-known to induce aggregation or elongated hydrophobic chains.
In the vast majority of the examples, the resulting self-assembly is interpreted as a process driven by the establishment of Au···Au interactions, which are directly involved in the resulting changes of the luminescent properties. In our opinion, andAu··· interactions may also be involved in these changes since the presence of water will favor hydrophobic interactions (as well as temperature will favor dispersive forces against electrostatic or H-bond type forces), and this will also contribute to a observed red shift of the emission.
Microscopic techniques are really important for the determination of the resulting morphology of the self-organized molecules. In general, it seems that for neutral complexes, molecules can grow long enough aggregates to give rise to the formation of fibrillary stages while the formation of other shapes (e.g. micelles, squares) can also be detected for cationic/anionic compounds.
Taking into consideration all these data, it has been found that gold(I) complexes are an increasing area of investigation due to the possibility of giving rise to supramolecular aggregates. Aurophilic interactions are a common motif in Au(I) supramolecular aggregation. These interactions play a key role in the resulting emission giving rise to aggregation induced emission. All in all, we can envisage that luminescent gold supramolecular structures are promising research areas in organometallic, supramolecular, and materials chemistry.
31
Acknowledgements
Authors are grateful to the Ministry of Economy, Industry and Competiveness of Spain (AEI/FEDER, UE Project CTQ2016-76120-P). This work was also supported by the Associated Laboratory for Sustainable Chemistry-Clean Processes and Technologies-LAQV, which is financed by national funds from FCT/MEC (UID/QUI/50006/2013) and co-financed by the ERDF under the PT2020 Partnership Agreement (POCI-01-0145-FEDER-007265). N.S. is also indebted to Erasmus exchange mobility Program.
32
References
1 J. Liang, Z. Chen, L. Xu, J. Wang, J. Yin, G.-A. Yu, Z.-N. Chen, S. H. Liu, J. Mater.
Chem. C, 2014, 2, 2243
2 Q. Zhao, C. Huang, F. Li, Chem. Soc. Rev., 2011, 40, 2508. 3 G.-J. Zhou, W.-Y. Wong, Chem. Soc. Rev., 2011, 40, 2541.
4 T. Torimoto, T. Kameyama, S. Kuwabata, J. Phys. Chem. Lett., 2014, 5, 336.
5 E. Aguiló, L. Soler, A. Casanovas, A.J. Moro, J.C. Lima, L. Rodríguez, J. Llorca,
Chem. Cat.Chem., 10.1002/cctc.201700518
6 A.Y.-Y. Tam, V.W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540.
7 C.A. Strassert, C.-H. Chien, M.D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz, L. de Cola, Angew. Chem. Int. Ed., 2011, 50, 946.
8 A. Kishimura, T. Yamashita, T. Aida, J. Am. Chem. Soc., 2005, 127, 179. 9 N. Lanigan, X. Wang, Chem. Commun., 2013, 49, 8133.
10 E. Aguiló, R. Gavara, C. Baucells, M. Guitart, J.C. Lima, J. Llorca, L. Rodríguez,
Dalton Trans., 2016, 45, 7328.
11 J.C. Lima, L. Rodríguez, Inorganics, 2015, 3, 1.
12 J.B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970. 13 J. Luo, Z. Xie, J.W.Y. Lam, L. Cheng, H. Chen, C. Qiu, H.S. Kwok, X. Zhan, Y. Liu, D. Zhu, B.Z. Tang, Chem. Commun., 2001, 1740.
14 J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam, B. Z. Tang, Chem. Rev., 2015,
115, 11718.
15 J. Mei, Y. N. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang, B. Z Tang,. Adv. Mater., 2014, 26, 5429.
16 A. Qin, B.Z. Tang, Aggregation-Induced Emission: Principles, Aggregation-Induced Emission: Applications, Vol. I and II, John Wiley & Sons, 2013.
17 R. Hu, N.L.C. Leung, B.Z. Tang, Chem. Soc. Rev., 2014, 43, 4494. 18 L. Zhang, N. He, C. Lu, Anal. Chem., 2015, 87, 1351.
19 Q.-M. Fu, H. Fu, L. Hu, L. Liu, S.-Z. Liu, Z.-L. Du, W.-Y. Wong, J. Inorg.
Organomet. Polym., 2012, 22, 97.
20 M. Ganguly, J. Jana, C. Mondal, A. Pal, T. Pal, Phys.Chem.Chem.Phys., 2014, 16, 18185.
21 H. Schmidbaur, A. Schier, Chem. Soc. Rev., 2012, 41, 370. 22 X. M. He, V. W. W. Yam, Coord. Chem. Rev., 2011, 255, 2111.
33 23 S. Sculfort, P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741.
24 J. Liang, Z. Chen, J. Yin, G.-A. Yu, S. H. Liu, Chem. Commun., 2013, 49, 3567. 25 Z. Chen, L. Yang, Y. Hu, D. Wu, J. Yin, G.-A. Yu, S. H. Liu, RSC Adv., 2015, 5, 93757
26 Z. Chen, Z. Li, F. Hu, G.-A. Yu, J. Yin, S. H. Liu, Dyes and Pigm., 2016, 125, 169. 27 W.-B. Li, W.-J. Luo, K.-X. Li, W.-Z. Yuan, Y.-M. Zhang, Chin. Chem. Lett., 2017,
28, 1300.
28 Z. Chen, Y. Nie, S. H. Liu, RSC Adv., 2016, 6, 73933.
29 Z. Chen, G. Liu, S. Pu, S. H. Liu, Dyes Pigm. 2017, 143, 409.
30 C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam, B. Z. Tang, J. Am. Chem.
Soc., 2013, 135, 62.
31 J.C. Lima, L. Rodríguez, Chem. Soc. Rev., 2011, 40, 5442.
32 J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li, G. Yang, Angew. Chem., Int.
Ed., 2011, 50, 8072
33 Z. Chen, Z. Li, L. Yang, J. Liang, J. Yin, G.-A. Yu, S. H. Liu, Dyes and Pigm., 2015,
121, 170.
34 Z. Chen, X. Han, J. Zhang, D. Wu, G.-A. Yu, J. Yin, S.H. Liu, RSC Adv., 2015, 5, 15341.
35 X. Han, X. Lü, Z. Chen, G. Yu, J. Yin, S. Liu, Chin. J. Chem., 2015, 33, 1064.
36 Z. Chen, J. Zhang, M. Song, J. Yin, G.-A. Yu, S. H. Liu, Chem. Commun., 2015, 51, 326
37 Z. Chen, D. Wu, X. Han, J. Liang, J. Yin, G.-A. Yu and S. H. Liu, Chem.Commun., 2014, 50, 11033.
38 Z. Chen, P.-S. Huang, Z. Li, J. Yin, G.-A. Yu and S. H. Liu, Inorg. Chim. Acta, 2015,
432, 192.
39 A. L.-F. Chow, M.-H. So, W. Lu, N. Zhu, C.-M. Che, Chem. Asian J., 2011, 6, 544. 40 E. Y.-H. Hong, H.-L. Wong, V. W.-W. Yam, Chem. Commun., 2014, 50, 13272. 41 M. Ferrer, A. Gutiérrez, L. Rodríguez, O. Rossell, J. C. Lima, M. Font-Bardia and X. Solans, Eur. J. Inorg. Chem., 2008, 18, 2899.
42 P. A. Korevaar, C. Schaefer, T. F. A. de Greef, E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482.
43 M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning, E. W. Meijer, Chem. Eur. J., 2010, 16, 362.
34 44 R. Gavara, E. Aguiló, C. F. Guerra, L. Rodríguez and J. C. Lima, Inorg. Chem., 2015,
54, 5195.
45 E. Y.-H. Hong, H.-L. Wong, V. W.-W. Yam, Chem. Eur. J., 2015, 21, 5732.
46 L. Brunsveld, E. W. Meijer, R. B. Prince and J. S. Moore, J. Am. Chem. Soc., 2001,
123, 7978.
47 M. T. Stone, J. M. Fox, J. S. Moore, Org. Lett., 2004, 6, 3317.
48 E. Y.-H. Hong, V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 2616.
49 M. A. Rawashdeh-Omary , M. A. Omary, H. H. Patterson, J. Am. Chem. Soc., 2000,
122, 10371.
50 M. A. Rawashdeh-Omary, M. A. Omary, H. H. Patterson, J. P. Fackler, Jr., J. Am.
Chem. Soc., 2001, 123, 11237.
51 M. Stender, R. L. White-Morris, M. M. Olmstead, A. L. Balch, Inorg. Chem., 2003,
42, 4504.
52 T. Moriuchi, K. Yoshii, C. Katano, T. Hirao, Tetrahedron Lett., 2010, 51 4030. 53 I. Odriozola, I. Loinaz, J. A. Pomposo, H. J. Grande, J. Mater. Chem., 2007, 17, 4843.
54 P. Casuso, P. Carrasco, I. Loinaz, H. J. Grande, I. Odriozola, Org. Biomol. Chem., 2010, 8, 5455.
55 P. Pyykko, Angew. Chem. Int. Ed., 2004, 43, 4412.
56 R. Gavara, J. Llorca, J. C. Lima, L. Rodríguez, Chem. Comm., 2013, 49, 72.
57 E. Aguiló, R. Gavara, J. C. Lima, J. Llorca, L. Rodríguez, J. Mat. Chem. C, 2013, 1, 5538.
58 R. Gavara, J. C. Lima, L. Rodríguez, Photochem. Photobiol. Sci., 2016, 15, 635. 59 L. Rodríguez, M. Ferrer, R. Crehuet, J. Anglada and J. C. Lima, Inorg. Chem., 2012,
51, 7636.
60 J. L. Dimicoli, C. Hélène, J. Am. Chem. Soc., 1973, 95, 1036.
61 A. J. Moro, B. Rome, E. Aguiló, J. Arcau, R. Puttreddy, K Rissanen, J. C. Lima, L. Rodríguez, Org. Biomol. Chem., 2015, 13, 2026.
62 R. Gavara, E. Aguiló, J. Schur, J. Llorca, I. Ott, L. Rodríguez, Inorg. Chim. Acta, 2016, 446, 189.
63 J. S. Seixas de Melo, R. S. Becker, A. L. Maçanita, J. Phys. Chem., 1994, 98, 6054. 64 T. Shu, L. Su, J. Wang, X. Lu, F. Liang, C. Li, X. Zhang, Anal. Chem., 2016, 88, 6071.
35 65 C.-J. Liu, J. Ling, X.-Q. Zhang, J. Peng, Q.-E. Cao and Z.-T. Ding, Anal. Methods, 2013, 5, 5584.
66 Y. Guo, X. Tong, L. Ji, Z. Wang, H. Wang, J. Hu, R. Pei, Chem. Commun., 2015, 51, 596.
67 B. Kemper, Y. R. Hristova, S. Tacke, L. Stegemann, L. S. van Bezouwen, M. C. A. Stuart, J. Klingauf, C. A. Strassert, P. Besenius, Chem. Commun., 2015, 51, 5253. 68 V. W.-W. Yam, E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806.