SUMMARY
Lung cancer is the leading cause of cancer mortality in industrialized countries. Despite of extensive researches to optimize the available treatments, the overall survival at 5 years is around 16%. Consequently, research aimed to identify new therapies for this disease is very active. The identification of the role of epidermal growth factor receptor (EGFR) in the processes leading to cell proliferation in lung cancer, for example, has allowed the development of EGFR inhibitors currently approved for clinical use (gefitinib and erlotinib).
Chemokines are small basic cytokines with chemotactic activity. In addition to controlling the recruitment of leukocytes in inflammatory processes, new roles have been attributed more recently to chemokines in other biological processes including, for example, angiogenesis and hematopoiesis. Chemokines have also been implicated in the pathogenesis of different diseases, such as bronchial asthma, multiple sclerosis, post-viral myocarditis, atherosclerosis.
In the recent years the potential role of chemokines in the cancer progression has been the subject of much research. Most tumors produce at least some chemokines and express chemokine receptors on
the cell surface. Since chemokines are able to attract inflammatory leukocytes, the initial interpretation of this observation was that the role of these molecules was to stimulate the immune response. On the other hand, it should be noted that tumors are considered as 'Darwinian' systems in which the selection pressure favours features such as loss of contact inhibition, independence from growth factors produced by the host, the ability to induce angiogenesis. It 's so hard to imagine that the tumor cells retain their ability to produce chemokines if the role of these proteins was only to induce the host immune response. Based on these considerations it has been postulated that chemokines play a role in favoring the development of cancer. And it is possible to imagine that they can act either directly, through interaction with their receptors through an autocrine mechanism, and indirectly through the recruitment of leukocytes, which could provide growth factors and angiogenesis.
The potential role of interleukin-8 (IL-8) in the development and progression of lung cancer has been extensively investigated. Clinical studies have shown a positive correlation between the amount of mRNA for IL-8 and the microvessel count in samples of lung cancer, and a negative correlation between these markers and survival, suggesting
that indeed IL-8 promotes tumor growth, at least in part through the stimulation of angiogenesis. More recent studies have also confirmed the presence of a correlation between the expression of IL-8 and prognosis in lung cancer. A direct role of IL-8 in the proliferation of cancer cells was also observed. Luppi et al. have shown that IL-8 acts as a growth factor on two cell lines of lung cancer, A549 and NCI-H292. The authors have shown that IL-8 acts through the transactivation of EGFR, a mechanism by which a soluble ligand, that is IL-8, through binding to its receptor on the cell surface, activates a metalloprotease which, after cutting a membrane-bound EGF, make it available to interact with EGFR and cause the subsequent release of intracellular signals downstream.
Monocyte chemotactic protein-1 (MCP-1) was initially identified for its chemotactic activity towards monocytes. MCP-1 is expressed by many tumors, and it is interesting to mention that one of its discoverers had originally named it “tumor derived chemotactic factor”. Considerations similar to those expressed about the chemokines can apply to this protein. Ueno et al. have divided a group of 135 patients with breast cancer into two groups based on the level of expression of MCP-1 in the surgical specimen, showing that patients with low levels
of MCP-1 (values below the median calculated in that population) had a disease-free survival significantly longer than patients with high levels of MCP-1. This observation was correlated with the number of tumor-associated macrophages. A more recent study showed that MCP-1 is an autocrine growth factor for prostate cancer cells. Our knowledge of the possible role of MCP-1 in lung cancer is very limited.
The present study has been done ex vivo on samples of lung cancer in humans. We enrolled patients related to our Cardio-Thoracic and Vascular Department with primary lung cancer, regardless of the histotype. During bronchoscopy performed for diagnostic purposes, biopsy sample of tumor tissue was taken and on this MCP-1 has been dosed with immunochemical techniques (ELISA). The amount of MCP-1 has been corrected for the total protein content measured by the method of Bradford. For patients unsuitable for surgery this was the only material available. For patients brought to surgery, the surgical specimen of the tumor has been sent to the laboratories of Pathology for histological evaluation. On this sample, the expression of MCP-1 has been measured through an immunohistochemical method, leading to a semiquantitative score, measured as +1/+2/+3. The values of MCP-1 are then compared, after dividing patients by histologic type (small cell and
non small cell) and stage of the disease, with the overall survival, to evaluate the any correlation between the presence of MCP-1 and evolution of disease.
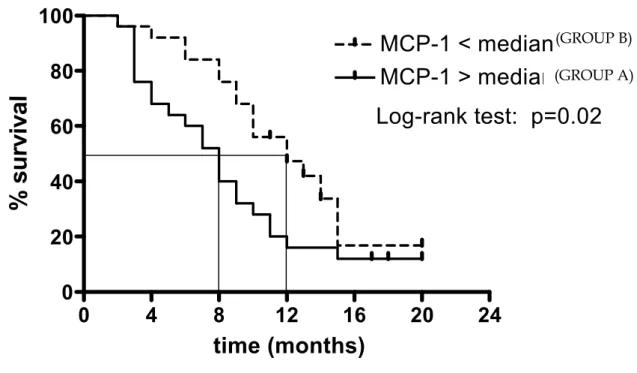
As regards the first group of patients, we enrolled 50 patients with NSLC (male/female: 44/6, mean age: 66.5 yrs) who performed bronchoscopic evaluation. Because of an inoperable staging of the disease, they were treated with chemotherapy and followed in the progression of the disease. The histologic type of lung cancer was: adenocarcinoma and squamous. The mean duration of follow-up was 9.52 months (range: 2-20), and at the end of the follow-up 11 patients were still alive.
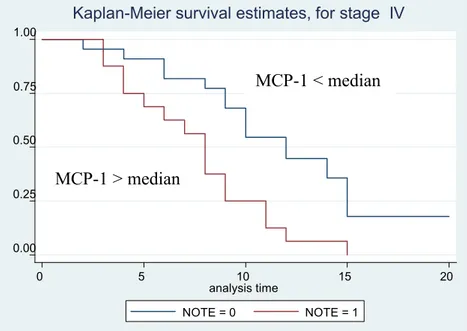
MCP-1 was detectable in all lung cancer samples taken by bronchoscopy (mean value: 33.04 pg/μg total proteins). When patients were divided according to median of MCP-1 value, patients with high 1 levels had a lower survival than patient with low levels of MCP-1 (5 vs 8 mts, respectively). The difference in the survival Kaplan-Meier curves of the two groups was statistically significant (p=00.2). When patients were divided according to the histotype and the stage, the difference in survival in relationship with MCP-1 tissue concentration was significant for the adenocarcinoma histotype and for stage IV.
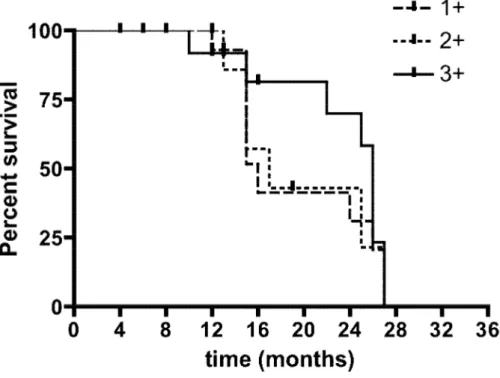
In the second group of patients with operable lung cancer, we were able to obtain the analysis of MCP-1 on a fragment of lung cancer tissue in 40 patients (male/female 37/3, mean age 72.8,). The histologic type of lung cancer was adenocarcinoma and squamous. All these patients were followed in order to evaluate for the progression of the disease. The mean follow-up time was 16.9 months (range 4-27), and at the end of the follow-up 15 of them were died. MCP-1 was measurable in all tissue samples, with the following expression: + 1, +2, +3. When we divided the patients in three categories according to the expression of MCP-1 in the lung cancer tissue, there weren’t differences in term of survival among patients with difference immunohistochemical expression of MCP-1.
These results suggest that MCP-1 might contribute to the poor prognosis of the lung cancer, particularly in patients observed in the inoperable stage of the disease. The difference between the results we obtained in the two groups of patients (50 vs 40 patients) may be due to the different method of measurement of MCP-1 in the inoperable and operable patients including the different methods of expression of the results, the different stages of the disease, and the different mortality observed during the follow-up in the two groups of patients.
Further analyses will be necessary in order to better define whether MCP-1 is significantly involved in NSCLC behavior. MCP-1 could be a novel target in cancer treatment once its role in immune regulation and angiogenesis is better understood.
INTRODUCTION
EPIDEMIOLOGY OF LUNG CANCER
In industrialized countries, lung cancer is the most common form of cancer in males and it has become the second in cancer incidence in women (1). It represents approximately 25-30% of all cancers, with incidence rates of 60-76 per 100,000 inhabitants (94 per 100,000 in Italy, approximately 40,000 new cases per year), with an overall 5-year survival of 13% which remained almost unchanged over the past 20 years (2, 3). The incidence ratio between men and women has decreased in recent years from an initial 7:1 to a current 2.5:1 in the U.S. (5:2 in Europe), also as a result of the spread of smoking among women (2). The international statistical analysis by age group, referring to both sexes, shows a higher incidence in the age range between 35 and 75 years (3), with a peak between the 5th and the 6th decade of life, and over a third of new cases involves patients aged over 70 years (2). Regarding the increase of overall mortality for all cancers in industrialized countries, we can also observe a greater increase in lung cancer mortality (29% of all cancer deaths in men and 8% in women),
reaching, in Italy, the first cause of death in man and the fourth cause of death in women, in whom, however, it has recorded in recent years a dramatic increase in incidence.
ETIOLOGY AND RISK FACTORS
Because of the increased incidence and mortality of this cancer, it is useful to describe the possible etiological factors in order to prevent it. We distinguish endogenous and exogenous factors (3).
The exogenous factors are well known. The most important is the cigarette smoke: in the world, statistics show that lung cancer is caused by smoking in 84% of cases reported in males and in 46% of women (1, 4) . The risk of death is associated with duration of smoking and the number of cigarettes smoked each day. It is estimated that in Italy, smoke cigarette is the cause of 85% of deaths from lung cancer (5, 6). Cigarette smoke contains many chemicals products with mutagenic properties, or benzopyrene, benzoantracene, fluoranthene, nitrosamines, or chromium, nickel and arsenic, polonium-210 and toxic oxygen radicals. Exposure to cigarette smoke is also able to affect the balance between the different enzyme activities of their lung tissue, leading to
activating enzymes, i.e. aryl hydrocarbon hydroxylase, inhibiting and inactivating enzymes such as glutathione S- transfrerasi (7). The DNA presence in cells isolated from the smokers’ lung of carcinogens irreversibly linked to the bases, called "abducted" confirms that these phenomena induced by tobacco smoke are involved in lung carcinogenesis (8). The amount of “abducteds” correlates with the number of cigarettes smoked per day, but not with the amount of cigarettes smoked during lifetime. In addition, “abducteds” are found in individuals who stopped smoking less than six months, and are not found in non smokers, or in people who had stopped smoking for more time (9). Regarding the risk of developing lung cancer with age starting in smoking, the highest risk is reached by starting to smoke before age 15 (1). Finally, considering the effects of passive smoking, it is estimated that 25% of non-smokers it can be the determining factor for the onset of disease (10).
It is important to underline the risk to which non smokers are exposed because of inhalation of passive smoke in a family.
There are, however, other exogenous factors which have been given a etiopathogenic role in lung cancer. Among them, we must remember environmental factors, work-related factors and dietary
factors. As for work-related factors, best known risks are those derived from exposure to ionizing radiation, asbestos, to complex mixtures of polycyclic aromatic hydrocarbons, radon, the nitrogen mustards, the clorometiletere bisclorometiletere, cadmium, chromium compounds and some nickel and some products of fossil fuels (11). An increased risk of lung cancer was found in miners and workers of quarries (12), in subjects exposed for many years to inhalation of exhaust gases of internal combustion engines (13, 14) and especially in workers exposed to inhalation of silica (12). In any case, the risk is higher for those who, in addition to the above risk factors, are smokers (7). With regard to dietary factors, some studies have shown a possible association between the risk of developing lung cancer and poor dietary intake of vitamin A, B-carotene (7, 15).
Despite the many attempt to reduce the burden of these risk factors, the results obtained in terms of reduction in the incidence of lung cancer are disappointing.
More emphasis has been done recently to the endogenous factors, in order to better explain the development of the disease and to have new targets for intervention.
Among the endogenous factors, epidemiologic studies show an association between family history and an increased risk of lung cancer, providing the first evidence of host susceptibility. Lung-cancer susceptibility and risk are also increased in inherited cancer syndromes caused by rare germ-line mutations in p53, retinoblastoma, and other genes, as well as a germ-line mutation in the epidermal growth factor receptor (EGFR) gene. More recently, three large genomewide association studies identified an association between single-nucleotide polymorphism (SNP) variation at 15q24–15q25.1 and susceptibility to lung cancer. The region of the SNP variation was recently linked to lung carcinogenesis and includes two genes encoding subunits of the nicotinic acetylcholine receptor alpha, which is regulated by nicotine exposure.
Lung-cancer susceptibility and risk also increase with reduced DNA repair capacity (particularly when accompanied by exposure to tobacco smoke) that results, for example, from germ-line alterations in nucleotide excision repair genes such as ERCC1. Increased expression of DNA synthesis and repair genes, including RRM1 (the regulatory subunit of ribonucleotide reductase) and ERCC1, in non small cell lung
cancer correlates with a better prognosis overall but no benefit from platinum-based chemotherapy.
Changes in certain genes (e.g., proinflammatory interleukin-8 [IL8] and some DNA-repair genes) occur in non malignant lung tissue of smokers and patients with lung cancer, a finding consistent with diffuse tissue injury. These changes probably precede epithelial clonal evolution, an important element of the molecular origins of lung and other cancers. Patches of clonally related cells, or clonal patches containing 40,000 to 360,000 cells, have been mapped in the lung. The size and number of subclones in a clonal patch may contribute to the cancer risk. Early events in the development of non small cell lung cancer include loss of heterozygosity at chromosomal region 3p21.3 (site of RASSF1A, a member of the Ras association domain family, and FUS1), 3p14.2 (FHIT, a fragile histidine triad gene), 9p21 (p16), and 17p13 (p53). All these genes are tumor-suppressor genes. Loss of heterozygosity patterns in squamous-cell carcinoma and adenocarcinoma differ (e.g., chromosome 3p deletions are much more extensive in squamous cell carcinoma). Mutations in the EGFR kinase domain occur early in the development of adenocarcinoma that is generally unrelated to smoking, and KRAS mutations occur early in the development of smoking-related
adenocarcinoma (1). Clonal patches with methylation of promoter regions of genes (epigenetic changes), p53 mutation, EGFR mutation,
c-Myc amplification, loss of heterozygosity, and microsatellite instability can occur in normal tissue surrounding non–small-cell lung tumors (1, 5, , 8, 9, 11, 13) and may be associated with a greater risk of recurrence and second primary tumors. These findings suggest that in the future molecular analyses of surgical margins may help identify patients most likely to benefit from adjuvant therapy.
Methylated genes in premalignant squamous cell lung lesions (e.g., metaplasia and dysplasia) are p16 and FHIT (frequently and very early) and O-6-methylguanine-DNA methyltransferase (MGMT), death-associated protein kinase (DAPK), and RASSF1A (less frequently or rarely and in advanced precancers) (16). The early methylation of p16 in the development of squamous cell lung cancer (e.g., in normal lung tissue in approximately 50% of smokers)(17) exemplifies differences with that in the development of adenocarcinoma, in which p16 methylation occurs very rarely and only late in precursors (e.g., high-grade atypical adenomatous hyperplasia). Methylation markers in sputum are associated with the risk of lung cancer (e.g., methylated p16) (9, 17) and the recurrence of lung cancer (methylated ASC-TMS1,39 also
called PYCARD). Recent data show that promoter methylation of various genes, including p16 in stage I non–small-cell lung cancer, is associated with recurrence after resection (11, 12). Agents that reverse epigenetic changes have shown promise in a mouse model of lung carcinogenesis and are being tested in humans with lung cancer (11-13).
Bronchoalveolar stem cells, which may be precursors of lung adenocarcinoma, were identified recently in studies in mice (12). The KRAS, Pten, phosphoinositide 3-kinase (PI3K), and cyclin dependent kinase pathways have been implicated in the proliferation of these stem cells. The potential role of bronchoalveolar stem cells and other tumorigenic stem-cell populations in the development and prognosis of human lung cancer and its resistance to drugs is an important area of future investigation (13-24).
Role of EGFR in the susceptibility to lung cancer
EGFR regulates important tumorigenic processes, including proliferation, apoptosis, angiogenesis, and invasion, and, along with its ligands, is frequently overexpressed in the development and progression of non–small-cell lung cancer (1, 2, 5). Clinical trials of the EGFR tyrosine kinase inhibitor erlotinib for second-line or third line
treatment of such tumors and of the monoclonal antibody against EGFR, cetuximab (combined with chemotherapy), for treatment of previously untreated, advanced disease (22-24) validated EGFR as a molecular target for therapy. EGFR mutations that were discovered during clinical trials led to extensive studies of the roles of these mutations and EGFR amplification in the pathogenesis of the disease and its prognosis and sensitivity to treatment.
Several groups of investigators independently identified somatic mutations in the kinase domain of EGFR in lung adenocarcinoma in approximately 10% of specimens from patients in the United States and in 30 to 50% of specimens from patients in Asia (25). The mutations occur with increased frequency in women and non smokers and are tightly associated with sensitivity to the EGFR tyrosine kinase inhibitors gefitinib and erlotinib and so appear to explain most of the dramatic responses to these agents.
More than 80% of these mutations in lung cancer involve in-frame deletions within exon 19 or the L858R mutant within exon 21. EGFR mutations are associated with an improved prognosis in non–small-cell lung cancer, even when treated with cytotoxic chemotherapy. EGFR amplification is detected in dysplasia (especially of a high grade), which
is associated with lung-cancer risk when detected in the sputum of smokers, and is associated with a poor prognosis but also with sensitivity to EGFR inhibitors (25). The epidemiologic links highlight three factors — whether the patient is a nonsmoker, Asian, and female — that are associated independently and collectively with an improved response to EGFR tyrosine kinase inhibitors. However, erlotinib appears to prolong survival in virtually all subgroups of patients with non– small-cell lung cancer (24, 25). There are major differences in clinical, pathological, sex-related, and molecular factors between smokers and lifelong non smokers in whom lung cancer develops. The vast majority of patients who have an initial response to erlotinib and gefitinib eventually have a relapse (25, 26, 27). Recent studies have identified
EGFR T790M mutations (in exon 20) in tumors before drug treatment (25, 27, 28) and in tumors of patients who had a relapse after therapy with standard reversible EGFR tyrosine kinase inhibitors. The binding kinetics of the mutant EGFR appear to be altered by the T790M mutation. Irreversible EGFR inhibitors suppress T790M-mutant tumor cells in vitro and are promising treatments for T790M-mutant tumors. Amplification of the met proto-oncogene (MET), another major mechanism of acquired resistance to EGFR tyrosine kinase inhibitors,
marks a poor prognosis. Other proposed resistance mechanisms include activation of other receptor tyrosine kinases, such as insulin-like growth factor 1 receptor, which can bypass EGFR to activate critical downstream signaling pathways, KRAS mutations, and the epithelial-to-mesenchymal transition (27, 29).
The epithelial-to-mesenchymal transition is a program of cell development involving loss of cell adhesion, repressed E-cadherin expression, and increased cell mobility. Preclinical and clinical data suggest that EGFR mutations are early events in the development of non–small-cell lung cancer (29). EGFR mutations, including those involving exons 18, 19, and 20 and L858R, can transform fibroblasts and lung epithelial cells. Furthermore, in transgenic mice with lung-specific expression of exon 19 deletion or the L858R mutation, atypical adenomatous hyperplasia, which is considered to be a precursor lesion of peripheral adenocarcinoma, was followed by lesions resembling bronchioalveolar carcinoma at 5 to 6 weeks of age and invasive adenocarcinomas at 8 to 10 weeks (29). Deinduction of mutant EGFR expression led to regression of tumors, suggesting the need for persistent mutant EGFR activity for continued tumor survival. Lung tumors also developed in transgenic mice with lung-specific expression
of EGFR variant III mutation (in-frame deletion of exons 2–7 from the extracellular domain). Mutations of the region that encodes the tyrosine kinase domain of EGFR have been detected in specimens of atypical adenomatous hyperplasia from Asian patients with no history of smoking. These mutations also occur in normal epithelium within and adjacent to tumors with EGFR tyrosine kinase mutations (a localized-field effect, possibly reflecting stem-cell expansion) and before EGFR amplification, a change associated with tumor progression and metastasis.
HER2 mutations and amplification have been identified in patients with lung adenocarcinoma. The frequency of such mutations is less than 5%, and the frequency of such amplification is 5 to 10%. HER2 kinase domain mutations (in-frame insertions in exon 20) and EGFR kinase domain mutations have similar associations with female sex, nonsmoking status, and Asian background in patients with adenocarcinoma (30, 31). HER2 amplification is associated with sensitivity to inhibitors of the EGFR tyrosine kinase; HER2 mutations are associated with resistance to such inhibitors but also with sensitivity to HER2-targeted therapy. HER3 kinase domain mutations have not been detected in patients with non–small-cell lung cancer (30). Mutations in
the HER4 kinase domain were found in 2 to 3% of Asian patients with this disease, with a possible association with male sex and smoking.
Ras–Raf–Mek
The Ras–Raf–Mek pathway is involved in signalling downstream from EGFR and in other pathways leading to the growth of cancer cells and tumor progression. Activating KRAS mutations are limited to non– small-cell lung cancer (predominantly adenocarcinomas), virtually mutually exclusive of mutations in the EGFR and HER2 kinase domains, and associated with resistance to EGFR inhibitors (tyrosine kinase inhibitors and cetuximab) and chemotherapy. Most KRAS mutations in lung adenocarcinoma are smoking-related G→T transversions (substitutions of a purine for a pyrimidine) and affect exon 12 (in 90% of patients) or exon 13 (1, 2, 30, 31). A distinct KRAS mutational profile consisting of G→A transition mutations was recently detected in patients with adenocarcinoma who had never smoked; its functional significance is unclear (31). Transversions (smokers) and transitions (nonsmokers) also have been reported for p53 mutations in lung adenocarcinoma (1). KRAS mutations appear to be an early event (e.g., detectable in the preinvasive lesions of atypical adenomatous
hyperplasia and bronchoalveolar carcinoma (3, 8) that precedes smoking-related lung adenocarcinoma. They generally mark a poor prognosis. Further evidence supporting this gene’s role in the pathogenesis of lung cancer comes from transgenic mice bearing a mutated KRAS and in which multifocal atypical adenomatous hyperplasia and adenocarcinoma develop (31). MET activation occurs early in KRAS-induced carcinogenesis in this model. BRAF mutations have also been detected in non–small-cell lung cancer (6, 9) and may be an early event in lung tumorigenesis (31).
PI3K–Akt–mTOR
The pathway consisting of PI3K, Akt, and mammalian target of rapamycin (mTOR), which is downstream of EGFR, is activated early in lung carcinogenesis (31). Akt is also over expressed in bronchial dysplasia. Inhibition of Akt can induce apoptosis of human premalignant and malignant lung cells and prevent lung carcinogenesis in an animal model. An mTOR inhibitor can block malignant progression of atypical adenomatous hyperplasia lesions in the KRas mouse model (31). Since mTOR drives tumorigenesis in part through macrophages, a prominent component of the tumor microenvironment,
the antitumor effect of mTOR inhibition requires the tumor microenvironment.
There is mutation or amplification of PIK3CA, which encodes the PI3K catalytic subunit, in a subgroup of non–small-cell lung tumors, especially squamous-cell carcinoma, in association with increased PI3K activity and Akt expression.
LKB1
LKB1 (also called STK11) is frequently mutated in non–small-cell lung tumors and is thought to act as a tumor-suppressor gene through interactions with p53 and CDC42, modulating the activity of AMPK (a multifunctional protein kinase) and other possible mechanisms that are just beginning to be studied (31, 32). LKB1 is thought to function in early tumorigenesis, subsequent differentiation, and the development of metastases (32).
Results in transgenic KRas-mutant mice in which LKB1 was inactivated suggest that the gene plays a role in the differentiation and invasive behaviour of such tumors (31). The presence of LKB1 mutations alone (i.e., without KRas mutations) was not associated with the development of lung cancer in mice. Low levels of LKB1 protein were
associated with high grades of dysplasia in atypical adenomatous hyperplasia lesions, suggesting that LKB1 has an early role in the development of premalignant lesions in the lung (32). LKB1 mutations (including point mutations and deletions) were found in 34% of adenocarcinomas and 19% of squamouscell carcinomas from 144 human specimens of non–small-cell lung cancer (31, 32). LKB1 mutations are associated with smoking and with KRAS mutations and are virtually exclusive of EGFR mutations (32).
TITF1
Amplification of thyroid transcription factor 1 (TITF1, also called
NKX2-1) in the 14q13.3 region was the most common focal event in a high-resolution analysis of gene copy numbers in human lung adenocarcinoma (33). This study used an array with the capacity to genotype many SNPs. As a result, the investigators also identified amplification in regions containing KRAS, Myc, vascular endothelial growth factor (VEGF), and several cell cycle genes in the tumor specimens. TITF1 encodes a lineage-specific transcription factor that is essential for the formation of cells lining lung alveoli (type II pneumocytes). In vitro, transfection of immortalized normal human
lung epithelial cells with at least two of the three genes TITF1, NKX2-8, and PAX-9 in the 14q13.3 region caused increased growth of the cells (33) suggesting that these three genes may work cooperatively in the pathogenesis of lung cancer in which there is amplification at 14q13.3 (detected by high-resolution comparative genomic hybridization array). Recent data indicate that squamous-cell carcinoma also exhibits TITF1 amplification, as detected on fluorescence in situ hybridization, but not TITF1 protein, in contrast to adenocarcinoma (32, 33).
Among other endogenous factors such as sex (persists more frequently in males), age (higher incidence between 35 and 65) and precancerous lesions (epithelial metaplasia and dysplasia), to define the role of the immune system remains controversial and not clearly (1).
Angiogenesis as a mechanism of lung cancer progression
VEGF levels in bronchial epithelial cells of smokers increase in association with the progression of bronchial dysplasia from low grade to high grade (34, 35). Bronchial hyperplasia, metaplasia, and carcinoma in situ are associated with increased microvessel density, and a distinctive pattern known as angiogenic squamous dysplasia can occur
(36). Factors associated with increased tumor angiogenesis correlate with the development and prognosis of lung cancer (34, 35, 36). Circulating VEGF levels may predict the clinical benefit of VEGF inhibitors in patients with this disease. Many angiogenic factors are regulated at least in part through the hypoxia-regulated pathways, such as hypoxia-induced factor (HIF) 1α and 2α (37, 38). In addition to hypoxia, VEGF and other angiogenic factors are also regulated by EGFR through HIF dependent and independent mechanisms (39) and by oncogenes such as KRAS and p53. VEGF has recently been validated as a therapeutic target on the basis of the results of a phase 3 trial, which led the Food and Drug Administration (FDA) to approve the VEGF monoclonal antibody bevacizumab in combination with standard chemotherapy for previously untreated, advanced non–small cell lung cancer (41). Interactions between the VEGF and EGFR pathways and an association between acquired resistance to EGFR blockade and increased VEGF expression in preclinical models led to the hypothesis that dual blockade of VEGF and EGFR might be more effective than either approach alone.
HISTOLOGY OF LUNG CANCER
The current histologic classification of lung cancer is based on the idea of providing for the various types of lung cancer criteria identification based on microscopic characters demonstrated in areas of better differentiated tumor. The various histological types are as follows, followed by their impact:
A. Squamous cell carcinoma (epidermoid carcinoma) (30-50%) which include also spindle cell carcinoma
B. Adenocarcinoma (25-30%) which includes acinar adenocarcinoma, Papillary adenocarcinoma, Bronchiole-alveolar carcinoma, Solid carcinoma with mucus production,
C. Adenosquamous carcinoma (1-4%),
D. Large cell undifferentiated carcinoma (10-20%), which includes Giant cell carcinoma, Clear cell carcinoma,
E. Small cell undifferentiated carcinoma (SCCL), (20-25%) which includes Oat small cell lung cancer, Intermediate cell, Combined with other small cell histologies,
F. Carcinoma of the bronchial glands which includes Adenoid cystic carcinoma and Mucoepidermoid carcinoma.
Briefly, squamous cell carcinoma is recognized for the histological features of intercellular bridges, the formation of horny pearls and keratinization of individual cells. In 70%of cases it has a central origin, at the bifurcation of segmental bronchi or sub-segmental, often undergoes necrosis with large cavities, and it often causes atelectasis of lung parenchyma with areas of atypical pneumonia or bronchopneumonic outbreaks, which tend to abscess. In 30% of cases, however, it has peripheral origin and spread over areas of sclerosis, scarring of the parenchyma, tending to invade the controlateral lung, diaphragm, pleura or pericardium.
Adenocarcinoma is presented as a mucoid mass which may extend to an entire lobe; those forms that originate from areas juxta-hilar infiltrate the bronchi and bronchial wall and can cause blockage. The pleura that surrounds the peripheral mass is thickened and corrugated. These tumors, however, can also be small, but cause a marked response dermoplasty, they may resemble small gashes with brown center, small digital processes to infiltrate the parenchyma near the periphery (29).
Bronchiole-alveolar carcinoma always appears as distinct clinicopathologic entity. Its apparent origin by pneumocytes II, the growth along the alveolar septa, its low tendency to develop aspects
dermoplasty or glandular, the peculiar ability to spread by Aerogen and low tendency to distant metastases are all factors that contribute to differentiate from histotype adenocarcinoma (1).
The large cell undifferentiated carcinoma often has a peripheral origin, far from the bronchi, in the form of mass with extensive necrotic and hemorrhagic foci (29). It is, however, very difficult to distinguish from undifferentiated adenocarcinoma and squamous cell carcinoma. With the use of electron microscopy and histochemical-immunity, an increasing number of cases is attributed to these histotypes, with a progressive reduction of incidence of large cell undifferentiated carcinoma. Its behavior is extremely aggressive (1).
The small cell undifferentiated carcinoma or small cell is localized more frequently in the lobar or segmental bronchi (30). Is also characterized by having a local and systemic spread rapidly (1).
DIAGNOSIS AND STAGING
The locoregional and distant extent of lung cancer should be defined as precisely as possible, because it is of fundamental importance in the therapeutic strategy. The "staging" of the disease must be
performed both at diagnosis to indicate more effective therapeutic solution and after any treatment, to evaluate the response to it or not and the presence of recidivism (restaging). Physicians have available standardized staging criteria to define the lung cancer extension. They are:
- Physical examination, - Chest X-ray (2 proiections),
- CT of the lung parenchyma and PET total body, - Bronchoscopy with brushing and bronchial biopsies,
- Blood-chemical tests and other examinations (abdominal and brain CT, MRI, skeletal scintigraphy) to highlight the presence of distant metastases.
With the help of all these methods we can classify lung carcinomas in the TNM system, which is the classification system adopted worldwide, where T indicates the size of the primary tumor, N lymph node sharing, regional and remote, M is the presence or absence of metastases in the contralateral lung or other organs. The TNM system gives the lung cancer stage. New TNM classification was published in 2009 (ninth edition).
Based on the IASLC multicenter data collected worldwide on 81495 patients diagnosed or registered with lung cancer between 1990 and 2000 (7, 44), the T, N, and M descriptors were analyzed and recommendations for changes in the seventh edition of the TNM classification were proposed on the basis of differences in survival. This new TNM classification replaces the old UICC classification and the staging system according to C. Mountain 1997. The changes principally affect the T and M classifications. For the T component, tumor size was found to have prognostic relevance and its analysis led to recommendations to subclassify T1 into T1a and T1b, and T2 tumors into T2a and T2b and to reclassify T2 tumor >7 cm into T3 tumors. In the M category, M1 was recommended to be subclassified into M1a (contralateral lung nodules and pleural dissemination) and M1b (distant metastasis). There is no change in the N category. The proposed changes for the new stage grouping are to upstage T2b N0 M0 from stage IB to stage IIA and to downstage T2a N1 M0 from stage IIB to stage IIA and T4 N0-N1 M0 from stage IIIB to stage IIIA.
TREATMENT OF LUNG CANCER
The division in NSCLC and SCLC is important also from a therapeutic point of view. While in the case of non-small cell cancer the best treatment is surgery, in the case of small cell cancer the standard of care is chemotherapy (47).
For small cell lung cancer, especially if the patient is in good physical condition and in the case of disease confined to the chest, the chemotherapy associated with radiotherapy directed to the chest is the main choice; instead, if the disease has spread (if the patient is good physical condition) the choice is high-dose chemotherapy and palliative radiotherapy directed to the metastasys point. The most successful schemes are those containing cisplatin (37), in relation to its optimal toxicity profile and demonstrated the synergistic action with several other drugs. The two schemes are the most commonly used drugs cisplatin and vindesine (PV system) and Cisplatin and Etoposide (PE regime) in the case of three-drug regimens, always containing cisplatin, there was no superiority over a two schemes drugs. Other
chemotherapy regimens are available, among which use of Irinotecan. The chemotherapy should be administered for 2-3 cycles in the absence of severe toxicity, before assessing the response, if positive, may lead to the continuation of the therapy up to a maximum of 4-6 cycles. In case of stationarity, the decision to continue or not must be individualized. However, if there is progression or the presence of severe toxicity, chemotherapy should be discontinued (47, 48).
For NSCLC, stages I-II-IIIA are treated with surgery with 5-year survivals respectively 60-80%, 20-40%, 10-30%. Within the stage III, group IIIA is still proposed for surgery, while in IIIA N2 the combination of radiation and neoadjuvant chemotherapy followed by surgery may be recommended if there is partial response (PR) or stable disease (SD). However, for these three stages, adjuvant chemotherapy may be useful after surgery. Stages IIIB and IV are considered inoperable and the treatment is mainly chemotherapy, although the 5-year survivals are very low (respectively 5% and 1%) (47) .
Advanced non small cell lung cancer
In 1995, a landmark meta-analysis of randomized clinical trials in patients with advanced and metastatic non-small cell lung cancer (NSCLC), compared chemotherapy to supportive care. This showed that cisplatin-based chemotherapy resulted in increased patient survival and resulted in the widespread use of chemotherapy in patients with good performance status. The NSCLC Collaborative Group meta-analysis compared chemotherapy to best supportive care and used individual and updated patient data from 1190 patients and 11 trials (47). It established that cisplatin based chemotherapy prolonged patient survival with a hazard ratio of 0.73 and a P value of 0.001. This was in contrast to regimens based on alkylating agents, which had a detrimental effect on patient survival. A series of drugs so-called third generation cytotoxics, were subsequently introduced for the treatment of NSCLC. The most prominent of these were docetaxel, gemcitabine, paclitaxel and vinorelbine. All the agents except for gemcitabine were active on the cell spindle. Gemcitabine however had a different mechanism of action and functioned as an anti-metabolite which was associated with prolonged cell uptake and cell potentiation (2, 14). Its activity as a single agent was first reported in 1994 (2, 11, 41) and it was subsequently shown to be active in association with cisplatin (41). The
ECOG 1594 study was undertaken to compare different cytotoxic regimens (2, 5). The test regimens were gemcitabine, cisplatin-docetaxel and carboplatin-paclitaxel. The reference regimen in that study was cisplatin and Paclitaxel, given over 24 h. The study accrued 1167 patients. Overall, the median survival varied between 7.4 and 8.2 months, but none of the test arms resulted in increased survival rate, when compared to the reference arm. The most active of the test arms was the gemcitabine-cisplatinum combination which had a statistically significantly longer time to progression (4.5 compared to between 3.3 and 3.6 months in the reference and other regimens). The gemcitabine-cisplatin combination was also associated with a numerically higher 2-year survival rate of 15.7% compared to between 10.5% and 11.5% with the other regimens.
In conclusion, in advanced-stage (stage IIIB or IV) non–small-cell lung cancer (NSCLC), doublet combinations of platinum compounds (cisplatin or carboplatin) with gemcitabine, vinorelbine, or taxanes (paclitaxel or docetaxel) are reference regimens (2, 49). Cisplatin plus gemcitabine, in a 3-week schedule, is an effective widely used regimen for first-line treatment of NSCLC.
Other drugs are pemetrexed which is a potent inhibitor of thymidylate synthase and other folate-dependent enzymes, including dihydrofolate reductase and glycinamide ribonucleotide formyl transferase. Pemetrexed is currently approved in combination with cisplatin for first-line treatment of malignant pleural mesothelioma, for the first line treatment of istotype adenocarcinoma and as a single agent for second-line treatment of advanced NSCLC (48, 49). For instance, cisplatin– gemcitabine is the standard regimen in Europe and other parts of the world, but carboplatin–paclitaxel is preferred in the United States. The inclusion of third-generation chemotherapy agents such as paclitaxel, docetaxel, gemcitabine, vinorelbine, irinotecan, and pemetrexed in platinum-based doublets is more effective in terms of response rates and survival, and also improves tolerability compared with cisplatin alone or older platinum-based combinations (47, 49). The overall benefit obtained by modifying chemotherapy regimens has been small and has yielded no tangible improvement in overall survival (OS). Maximum median OS reached with chemotherapy plateaus at 8–10 months, even with third-generation chemotherapy agents such as pemetrexed. In a large phase III study (48) comparing platinum doublets, first-line cisplatin–pemetrexed provided efficacy similar to
that for cisplatin–gemcitabine, with a median os of 10.3 months for each treatment arm. In a pre-specified analysis, the median OS was significantly longer for cisplatin–pemetrexed than for cisplatin– gemcitabine in patients with histology [n = 847; 12.6 months vs. 10.9 months; hazard ratio (hr): 0.84; p = 0.03] and large-cell carcinoma histology (n = 153; 10.4 months vs. 6.7 months; hr: 0.67; p = 0.03). However, the median survival of patients with squamous histology assigned to cisplatin–pemetrexed (n = 244) was 9.4 months; it was 10.8 months for patients assigned to cisplatin– gemcitabine (n = 229; hr: 1.23;
p = 0.05) (49). For patients having NSCLC without further subtype classification (n = 252), no significant difference was observed between the two arms. These outcomes support the use of cisplatin–pemetrexed in non-squamous tumours only (48). Carboplatin–pemetrexed demonstrated efficacy similar to that of carboplatin–gemcitabine in the first-line treatment of metastatic NSCLC. Further trials are required to fully elucidate the role of platinum doublets in NSCLC based on histology.
In the recent years, two new concepts have been introduced to the field of NSCLC (2): maintenance therapy and targeted biologic agents. Maintenance therapy, with either a chemotherapeutic or biologic agent,
is given to patients after first-line therapy. Choice of therapy may include drugs included in the induction regimen or different agents (early second-line treatment) with the aim of preventing progression and prolonging progression-free survival (PFS). The optimal treatment duration for NSCLC patients remains a matter of discussion. A number of studies have evaluated regimens using either sequential or maintenance chemotherapy as post-first-line treatment for NSCLC patients who have not experienced disease progression. A review of those studies suggests that the optimal timing and duration of maintenance therapy (or immediate compared with delayed second-line therapy) remain unclear (2, 30).
Targeted agents modulate events in the cancer cells and provide several advantages over chemotherapeutics, including fewer toxicities and the possibility of a longer duration of therapy (15, 24). Two main groups of targeted agents for NSCLC are the inhibitors of epidermal growth factor receptor (EGFR) and vascular endothelial growth factor (VEGFR) (46). Erlotinib and bevacizumab (41) are the respective representatives of these groups, and both are approved for use in the United States, Canada, and Europe based on their safety and efficacy profiles. A wealth of available clinical data supports the use of these
agents in the treatment of metastatic NSCLC (22-30). Other EGFR inhibitors include cetuximab, which is not currently approved in the United States, Canada, or Europe for the treatment of NSCLC, and gefitinib, which was recently granted marketing authorization by the European Medicines Agency (EMEA) the United States, and Canada for the treatment of EGFR mutation–positive NSCLC.
Erlotinib is an EGFR tki (tirosin-kinasi inhibitor) that suppresses intracellular signalling pathways, which normally promote cell growth and proliferation (2, 24, 26, 28, 30).
The anti-VEGF monoclonal antibody bevacizumab (41, 46) was the first targeted agent to increase efficacy in first-line NSCLC when added to a platinum doublet. The key angiogenic factor VEGF plays multiple roles in tumour angiogenesis and has become a target for anticancer drug development. Specifically, VEGF has been shown to promote survival and to increase permeability of existing tumour vasculature while stimulating the growth of new tumour vessels. In addition to its effects on tumour vasculature, VEGFf is known to have a direct effect on tumour cells, including survival, migration, and invasion (30, 48). Anti-VEGF therapy such as bevacizumab has been proposed to exert a “dynamic” anti-angiogenic effect on tumour vasculature throughout the
course of its use, with important effects observed early and continued later in treatment (24, 40). Two early effects of antiVEGF therapy include regression of existing tumour microvasculature and normalization of remaining tumour vasculature. A third effect is the continued inhibition of new tumour vasculature that may contribute to additional benefits observed over longer periods of time.
Based on results from the E4599 trial, bevacizumab plus carboplatin–paclitaxel became the Eastern Cooperative Oncology Group (ECOG) reference standard and received approval from the U.S. Food and Drug Administration for first-line treatment of non-squamous advanced NSCLC (41). Similarly, based on STUDY AVAIL and E4599, bevacizumab in combination with platinum-doublet chemotherapy received EMEA approval. In April 2009, bevacizumab in combination with carboplatin–paclitaxel was approved in Canada. A meta-analysis of more than 13,000 bevacizumab-treated patients provided reassurance that the risk of CNS bleeding in patients with brain metastases is not unduly increased with the use of bevacizumab, which led to an update of the EMEA label to allow patients with untreated CNS metastases to receive the drug. The United States has never had any label restriction on bevacizumab for patients with cns metastases; however, new data
regarding the safety of bevacizumab in patients with CNS metastasis is included in the “warnings and precautions” section of that country’s monograph (41, 47).
Second Line Chemotherapy
It can be used in those patients with advanced NSCLC eventually relapse or become refractory to first-line treatment (47). Acceptable toxicity and improved quality of life are especially important for those patients (although efficacy remains the main goal of therapy). Several chemotherapy agents, including docetaxel and pemetrexed, have demonstrated efficacy and have been approved by the U.S. Food and Drug Administration for second-line treatment of patients with locally advanced or metastatic NSCLC. In Canada, approved second-line chemotherapy agents are intravenous docetaxel and intravenous pemetrexed, the latter for non-squamous histology only (41, 48, 49, 50). Docetaxel has been reported to achieve response rates of 15%– 20% (7, 44), an os of 8.3 months, and 1-year survival rates of up to 37% . However, docetaxel is associated with serious toxicities. Pemetrexed offers a similar median os of 7.9 months, but with a milder toxicity profile than is seen with docetaxel. Erlotinib is the only approved targeted biologic agent for NSLCL in the second-line setting (26, 47).
Unlike chemotherapy, erlotinib has no cumulative hematologic toxicities, allowing for a longer treatment duration. In contrast, the toxicities associated with chemotherapy allow for only a limited number of cycles (approximately 4 cycles median). A retrospective practice review found that second-line erlotinib treatment is efficacious and well-tolerated, and does not diminish the benefit of third-line chemotherapy (40). Furthermore, erlotinib and docetaxel can be given for third-line treatment of nsclc. Pemetrexed is approved for use in second line in the United States, Canada, Europe and it remains a strong candidate for patients who experience disease progression after bevacizumab and erlotinib in previous lines of therapy (40, 41).
For the future, the main goal is to provide the best possible treatment in terms of both efficacy and safety in each line of therapy. The striking improvements in outcomes demonstrated in both first- and second-line settings with targeted therapies provide a rationale for their use. The targeting of multiple pathways using a wide range of new drugs and combinations of agents is currently under investigation. The mode of action of bevacizumab suggests that its continued use may prevent recurrence of tumour angiogenesis. The advantage of continuing bevacizumab therapy beyond progression has been
demonstrated in an observational study (BRITE) in patients with colorectal cancer (39). Compared with alternative or no post-progression treatment, bevacizumab given to patients post progression resulted in superior improvement in os. However, these data have yet to be confirmed in a prospective phase III trial in patients with NSCLC. As compared with chemotherapeutic agents, targeted agents may offer reduced toxicity, especially with prolonged use. Combinations of targeted agents may also have potential as novel treatment paradigms, perhaps even representing an alternative to chemotherapy. Bevacizumab and erlotinib combination therapy has shown promise in phase I/II 58 and phase II trials in patients with recurrent NSCLC, with reduced toxicity as compared with chemotherapy (47).
Tailoring Therapy
Predictors of response may help to guide individual treatment decisions; however, for most drugs, clinically validated markers have yet to be identified (47). Until reliable biomarkers for response and resistance (old or newly developed) are identified, differences in toxicity between chemotherapeutic and targeted agents may provide the best guide for individual treatment decisions, in view of similar efficacy.
Gefitinib recently received EMEA (39) and Canadian approval for treatment of patients with EGFR mutation– positive disease. EGFR mutations can be viewed as predictive markers of high clinical benefit with EGFR tki therapy, especially for first-line treatment in eligible patients. Histology appears to be an important consideration for optimal outcomes with both pemetrexed and bevacizumab. A trial (BRIDGE) in patients with predominantly squamous-cell histology (who are currently excluded from bevacizumab clinical trials) is ongoing to determine ways in which to safely integrate bevacizumab into the treatment of these patients (39, 40). A personalized targeted approach is the future of treatment in all lines, but tumour re-biopsy will be required for analysis of biomarkers, including not only newly developed markers of resistance to EGFR tki, but also sensitivity to agents such as BIBW 2992 (T790M mutation and c-Met amplification). Analysis of circulating tumour cells and blood biomarkers to define predictors of tumour response and treatment benefit is a great need for the future (47, 74).
Chemokines are a family of chemoattractant cytokines, which can be induced by cytokines, growth factors and pathogenic stimuli. They are small proteins (8-10 KDa) that are divided in four groups, C, CC, CXC and CX3X, according to the number and the spacing of the first two cysteine residues in the amino-terminal part of the protein. The CXC family can be further subdivided in two categories depending on the presence or absence of an “ERL motif” (glutamic acid-leucine-arginine) preceding the first cysteine residue in the protein (51).
Chemokines orchestrate cell movement during homeostatic trafficking of hematopoietic stem cells, lynphocytes and dendritic cell as well as during inflammatory responses. They develop their effects by binding to seven transmembrane domain G protein-couples receptors. Chemokines bind to the extracellular N-terminus of the chemokines receptor, this leads to phosphorilation of serine/threonine residues on the cytoplasmic C-terminus, signalling and then receptor desensitization (56, 57). Binding of a chemokine to its receptor stimulates transcription of genes involved in invasion, motility, extracellular matrix interaction and cell survival (58).
The first described action of chemokines was their ability to selectively regulate the recruitment and trafficking of leucocyte subsets
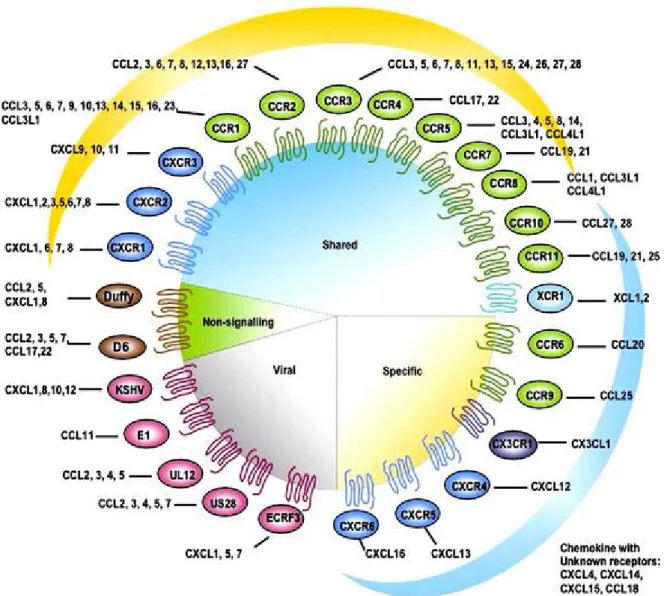
through chemoattraction. Additional roles for chemokines are now recognized including regulation of hematopoietic precursors, angiogenesis and extracellular matrix regulation. Each receptor can respond to more than one chemokines; many chemokines can use more than one receptor, and cells may express several chemokines and chemokine receptors. The profile of chemokine receptor expression of an individual cell is determined by cell lineage and stage of differentiation, expression can also be modulated by microenvironmental factors such as growth factors, hormones, other inflammatory cytokines and hypoxia (50, 52-56) (Figure 1).
Fig. 1 Chemokines and their receptor (51).
In normal physiology, chemokines have pleiotrophic effects: therefore, in cancer chemokines will also have a wide-ranging functions. Growth promotion, angiogenesis, manipulation of the local immune environment, immune invasion, cancer cell invasion and metastatis are all roles which have been attributed to chemokines in cancer biology. But it has been demonstrated that chemokines can be used to correct immune dysfunction in cancer, stimulate local immune responses or
inhibit growth of tumor cells (51, 57). Therefore, the actions and activity of chemokines in cancer biology depends on the setting in which they are expressed. However, there is no evidence that any chemokines can directly initiate a normal cell to become malignant, but there is increasing evidence that chemokines play a role in the promotion and biology of cancer (56-58).
Because of their ubiquitous expression and our detailed understanding of chemokine function in other settings, it has been tempting to infer that chemokines play an important role in tumor biology. However, this assumption should be viewed with a healthy skepticism (57, 58). For example, one could argue from first principles that chemokines are rather unlikely to be centrally important in cancer pathophysiology. This argument would point out that the single most important insight into cancer pathogenesis to emerge from the past 25 years of research is that cancer is fundamentally a genetic disease. Wholesale gene loss, inactivation through interstitial deletion or mutation, overexpression through amplification or translocation, or viral mimicry of these processes underlie cancer pathogenesis (56, 58, 67).
Furthermore, our understanding of basic processes that control cell proliferation has grown concomitantly so that we can see how most of
these abnormalities produce neoplasia by their effects on growth factor receptor signal transduction, cell cycle checkpoint controls, or DNA synthesis (64-68). A fundamental validation of these insights is the fact that reverse genetics identifies these same genes as cancer susceptibility loci. However, to date, no susceptibility gene in any cancer has been mapped to a chemokine or chemokine receptor locus. The only example that even comes close is the increased incidence of cervical carcinoma in women with WHIM syndrome. Although this disease is associated with CXCR4 mutations, its accompanying cervical cancer susceptibility reflects an immune deficiency that is permissive for high risk human papillomavirus infection rather than an inherent abnormality in cervical epithelial cell biology (55, 56, 67, 68).
While this may be a valid argument against the idea that chemokines contribute to cancer pathogenesis, it does not rule out the possibility that they play an important role in cancer pathobiology. Research in cancer biology has demonstrated that, in addition to cell autonomous abnormalities in neoplasm, various host factors can have a profound effect on cancer behavior (57). These include host inflammatory states and immune deficiencies that reduce a postulated inherent resistance to some cancers. In fact, WHIM syndrome falls into
this category. Because chemokines are involved in such a broad array of normal host activities that impact cancer, it is likely that they will have important effects on cancer pathobiology that may be amenable to therapeutic manipulation.
Just as chemokine effects in normal physiology are highly pleiotropic, their effects on cancer are likely to be multifaceted as well. Thus, chemokines might be expected to have either growth promoting or growth inhibiting influences on cancers depending on the particular setting in which they are expressed (56, 66, 67, 68). For example, because of their ability to attract and activate leukocytes, some chemokines might be expected to stimulate host anti-tumor responses. Nonetheless, some of these same chemokines are known to have angiogenic activities which could actually contribute to tumor growth and progression (67).
Pro-cancer actions of chemokines
Tumor-associated chemokines are thought to play at least five roles in the biology of primary and metastatic disease: control of the leucocyte infiltrate into the tumor, manipulation of tumor immune response, regulation of angiogenesis, action as autocrine or paracrine growth and survival factors, and direct the movement of tumor cells
themselves. The CC chemokines are major determinants of macrophage and lynphocye infiltration (51, 56, 57).
In relationship to growth and survival of tumor cells by chemokine, deregulated chemokine production by tumors may contribute directly to transformation of tumor cells by acting as growth and survival factors, in an autocrine and/or paracrine. Melanoma cells express elevated levels of the CXCR2 receptor and also constitutively produce CXCL1 and CXCL8 (IL-8) allowing autocrine stimulation by these chemokines which enhances survival, proliferation and tumor cell migration. The chemokines CXCL1 and CXCL8 also stimulate growth of pancreatic tumor cell lines. Pancreatic cancer samples also express CCR6 and proliferate in response to the CC chemokine MCP-10 (MIP-3α). In other cancers, CXCL12 can also stimulate cancer cell proliferation or survival under suboptimal conditions e.g. epithelial ovarian cancer where CXCL12 stimulation enhances tumor cell proliferation in low serum conditions.
About the role of chemokines in malignant progression, they may also regulate angiogenesis within both the primary tumor and metastatic deposits. Examples include, levels of CXCL10 in human lung cancers which were inversely related to tumor progression and elevated
levels of CXCL5 which were found in NSCLC and correlated with the vascularity of the tumors and angiogenesis. However, closer investigation revealed that these cells colonised the lungs to a similar extent as the parental cells but these micrometastases, although they survive, fail to proliferate (51, 56, 57, 65).
CXCR4 expression and action may be linked to other factors that are involved in the processes of malignancy. In breast cancer cell lines, VEGF was demonstrated to have an autocrine action and induce expression of CXCR4 expression which promoted migration/invasion towards CXCL12. VEGF and basic fibroblast growth factor also induce expression of CXCR4 on human endothelial cells which again highlights the reciprocal links between chemokines and angiogenic factors (51, 66).
Anti-cancer actions of chemokines
Examples in the literature which demonstrate that chemokines have cancer actions are limited. MCP-1 is reported to have anti-malignant properties in pancreatic cancer. Expression of CCl2 correlated positively with macrophage infiltration and reduced tumor proliferation; CCl2 has no direct effect on cancer cells but exerted its
antitumor effect through macrophages. In NSCLC patients, CCL5 expression by the cancer cells was associated with leukocyte recruitment, an active lymphocyte response and increased patient survival. Higher expression of CCL22 in lung cancer tissues was associated with increased disease-free survival time and a lower risk of recurrence after tumor resection (56, 64, 65, 68, 69).
Hence, the biological activities of chemokines in most tumors may facilitate cancer growth and spread rather than play a role in anti-tumor responses. Furthermore, some chemokines can have direct effects on tumor cells. Monocyte chemoattractant protein-1 (MCP-1, CCL2) is one of the chemokines that can have any or all of these effects on cancers (56).
Biology of MCP-1 and CCR2
MCP-1 (the systematic designation for MCP-1) is a potent chemoattractant for monocytes, memory T lymphocytes, and natural killer (NK) cells (51). Although these properties were first defined on the basis of in vitro assays using purified proteins, subsequent experiments involving injected proteins in rodents or transgenic expression in specific organs demonstrated that these properties were faithfully reproduced in vivo (55, 56, 63-66). MCP-1 is structurally and genetically
related to other chemokines (MCP-1, -2, -3, and -4 in humans, MCP-1, -2, -3, and -5 in the mouse) that share similar properties. The reason for their shared biological activities is that they all activate the same receptor, CCR2, with similar potencies. While the apparent promiscuity of CCR2 for multiple ligands was thought to imply a degree of redundancy in the MCP system, this has not turned out to be the case. Mice engineered to be deficient for MCP-1 alone have unique phenotypes, most of which match those seen in CCR2-deficient mice (51, 56). The basis for specificity in vivo appears to lie in unique patterns of expression of the various MCPs in vivo. Thus, MCP-3 (CCL7), for example, does not substitute for MCP-1 in CCL2 knockout mice because it is not expressed by the tissues that normally express MCP-1. That is, when MCP-1 is absent, the other MCPs are not upregulated to compensate for its absence (51, 57). This, of course, has therapeutic implications, namely that individual chemokines can be targeted for antagonism with some hope of success.
The unique effects of the MCP-1/CCR2 axis on mononuclear cell migration suggested that it would be an important regulator of inflammatory disease. This prediction has been validated through the
use of genetically deficient mice, antibody- or inhibitor-mediated neutralization in mice, and epidemiological studies in humans.
One of the clearest examples is cardiovascular disease. Current models of the pathogenesis of atherosclerosis suggest that endothelial cells suffer damage as a result of elevated levels of oxidized cholesterol as well as the shear stresses of hypertension or disordered blood flow (57). In response, circulating monocytes are drawn into the subendothelium where they differentiate into macrophages and attempt to repair the damage. However, ongoing hygh chronic cholesterol uptake by macrophages which become “stuck” in the arterial wall. Their chronic activation also leads to the secretion of smooth muscle cell chemoattractants and growth factors, resulting in the appearance of the mature atherosclerotic plaque. MCP-1 was thought to be an excellent candidate for the monocyte recruitment signal secreted by injured endothelial cells. This was confirmed by analysis of MCP-1- or CCR2-deficient mice. In several murine atherosclerosis models, absence of MCP-1 or CCR2 substantially reduced arterial lipid deposition (56, 64). In all cases, amelioration of disease was associated with diminished numbers of macrophages in the arterial wall consistent with a model in
which MCP-1 contributes to atherosclerosis by attracting monocytes into the subendothelium via CCR2 activation.
Again, MCP-1- or CCR2-deficient mice recruited many fewer monocytes into the site of disease (the central nervous system in this case), and the severity of disease was greatly reduced. There are clinical epidemiological data to support the notion that MCP-1 and CCR2 may play similar roles in human disease. Several studies indicate that increased plasma levels of MCP-1 following balloon angioplasty of coronary arteries predicts for early re-stenosis, which in some ways can be thought of as accelerated atherosclerosis (51, 56, 57, 68, 69). At a population level, a polymorphism in the MCP-1 promoter has been demonstrated to be associated with an increased likelihood of an individual’s having coronary artery disease (56).
Interestingly, this polymorphism has been shown to produce increased rates of MCP-1 transcription in vitro, suggesting a mechanism for this association (56, 57). On the receptor side, a CCR2 polymorphism that responds less robustly to MCP-1 in vitro compared to the more common CCR2 allele is associated with protection from coronary artery disease (57). Thus, mouse modeling and human clinical investigation support the importance of MCP-1 and CCR2 in inflammatory disease.