PhD Course
Translational Medicine
Hématologie, Oncogenèse et Biothérapies
GALECTIN-3 AND ALDOSTERONE PROFIBROTIC PATHWAYS IN THE
FAILING HEART
Author
Giuseppe Vergaro, MD
Tutors
Prof. Michele Emdin, MD, PhD
Prof. Claudio Passino, MD
Prof. Alain Cohen Solal, MD, PhD
Supervisor
Prof. Fabio A. Recchia, MD, PhD
Academic Year
2016/2017
A
BSTRACTBackground
Natriuretic peptides represent, nowadays, the most widely used biomarkers for heart failure (HF) management and risk stratification, though they do not reflect directly a specific pathway of cardiac damage. Differently, galectin-3 (Gal-3), a soluble β-galactoside binding lectin which holds relevant prognostic value as well, is biologically linked to the process of inflammation and fibrosis and may thus be a marker of a peculiar mechanism of cardiac damage and remodelling, and a potential therapeutic target.
In past years, the renin-angiotensin-aldosterone system (RAAS) has been largely demonstrated to exert profibrotic, prohypertrophic and proinflammatory effects in the pathophysiology of HF, and the elevation of circulating levels of its effectors (in particular plasma renin activity and aldosterone) is associated with a worse outcome in HF patients, despite optimal pharmacological and non-pharmacological therapy. Both Gal-3 and RAAS effectors may thus be considered as culprit biomarkers. Further, there is initial experimental evidence that Gal-3 and RAAS may interplay in the development of cardiovascular damage in hypertensive models. However, such interaction has not been investigated in the setting of left ventricular (LV) dysfunction and HF, nor there is clinical demonstration that Gal-3 is associated with cardiac fibrosis or with the evolution of cardiac remodeling.
With the present translational project, we aimed therefore to test the following hypotheses: 1. Gal-3 participates in the mechanisms of aldosterone mediated cardiac fibrosis and tissue remodeling in a murine model of LV dysfunction;
2. Gal-3 level is associated with the development of LV remodeling and fibrosis and can predict the effects of neurohormonal antagonism in patients with chronic HF due to non-ischemic dilated cardiomyopathy (NIDCM).
Materials and Methods
Animal study. Adult male mice with cardiac-specific hyperaldosteronism (AS) underwent isoproterenol subcutaneous injections (200 mg/kg x 2/day over two consecutive days), to be then randomized to receive placebo, a Gal-3 inhibitor (modified citrus pectin, MCP), an aldosterone antagonist (potassium canrenoate), or MCP+canrenoate for 14 days. The effects of different pharmacological approaches were assessed by serial echocardiographies, analysis of gene expression and protein content of key determinants of inflammation and fibrosis, and by histology.
Clinical study. We prospectively enrolled 150 consecutive patients referred for HF management, with a diagnosis of NIDCM. Patients with malignancies, acute or chronic inflammatory diseases, hematological and autoimmune disorders and severe chronic kidney disease (estimated glomerular filtration rate <30 ml/Kg/min) were excluded. All patients underwent a comprehensive clinical evaluation, a biohumoral characterization, including the assay of Gal-3, 2-D echocardiography, and a contrast-enhanced cardiac magnetic resonance (CMR) for the assessment of LV volumes and fibrosis by the late gadolinium enhancement (LGE) technique. A smaller cohort of 70 patients also received soluble suppression of tumorigenicity protein 2 (sST2) assay at baseline as well as a follow-up CMR after 24 months for the evaluation of LV reverse remodeling, defined as a >10 percentage units increase in LV ejection fraction or a >10% decrease in LV end-diastolic volume indexed.
Results
Isoproterenol induced a rapid and persistent decrease in LV systolic function which was markedly improved by treatment with either MCP or canrenoate. MCP and canrenoate also reduced cardiac hypertrophy and fibrosis and the expression of genes involved in fibrogenesis (Coll-1 and Coll-3) and macrophage infiltration (CD-68 and MCP-1). Further, after treatment with isoproterenol, Gal-3 gene expression and protein levels were decreased by both canrenoate and MCP. The combined use of antagonists of Gal-3 and aldosterone resulted in enhanced effects on cardiac hypertrophy, inflammation, and fibrosis, when compared to MCP or canrenoate alone.
In our cohort of NIDCM patients, median Gal-3 value was 14.4 ng/mL (IQR 11.7–19.0 ng/mL); LGE was detected in 106 (71%). Patients with LGE had higher Gal-3 than those without (15.4, 11.8–21.0 vs 13.1, 11.7–16.4 ng/mL, p=0.006). Among univariate predictors of LGE presence (Gal-3, male sex, disease duration, arterial hypertension, left and right ventricular ejection fraction, left ventricular stroke volume), Gal-3 maintained its predictive value at multivariate analysis, together with sex, hypertension, disease duration and right ventricular ejection fraction. At follow-up CMR, 35 patients (50%) showed reverse remodeling. Gal-3, but not sST2 resulted as an independent predictor of LV reverse remodeling at multivariate analysis.
Conclusion
Data from animal experiments suggest that Gal-3 participates in mechanisms of aldosterone-mediated myocardial damage in murine model of HF. Likewise, in the clinical setting of NIDCM, Gal-3 seems to be associated with LV fibrosis and to the evolution of cardiac remodeling, possibly identifying the subset of patients with a more pronounced response to pharmacological neurohormonal antagonism.
On a whole, our data may support the use of Gal-3 as a biomarker for the management of HF and as a potential therapeutical target.
R
ÉSUMÉIntroduction
Les peptides natriurétiques représentent aujourd'hui les biomarqueurs les plus utilisés pour la gestion de l'insuffisance cardiaque (IC) et la stratification du risque, bien qu'ils ne reflètent pas directement une voie spécifique de lésion cardiaque. De manière différente, la galectine-3 (Gal-3), une lectine soluble de liaison au ß-galactoside, qui possède également une valeur pronostique pertinente, est biologiquement liée au processus d'inflammation et de fibrose. Elle peut ainsi être considérée comme un marqueur de mécanisme particulier de lésions cardiaques et remodelage et également une cible thérapeutique potentielle.
Au cours des dernières années, le système rénine-angiotensine-aldostérone (SRAA) a été largement démontré comme exerçant des effets profibrotiques, prohypertrophiques et pro-inflammatoires dans la pathophysiologie de l’IC. L'élévation des taux circulants de ses effecteurs (en particulier l'activité rénine plasmatique et l'aldostérone) est associé à un mauvais pronostique chez les patients atteints d’IC, malgré une thérapie pharmacologique et non pharmacologique optimale. Les effecteurs de Gal-3 et de SRAA peuvent donc être considérés comme des culprit biomarkers. En outre, il existe des preuves expérimentales initiales que Gal-3 et SRAA peuvent interagir dans le développement des dommages cardiovasculaires dans des modèles d’hypertension. Cependant, une telle interaction n'a pas été étudiée dans un contexte de dysfonction ventriculair gauche (VG) et d’ IC, ni la démonstration clinique que Gal-3 est associée à la fibrose cardiaque ou à l'évolution du remodelage cardiaque.
Avec le présent projet translationnelle, nous avons donc cherché à tester les hypothèses suivantes:
1. La Gal-3 participe aux mécanismes de la fibrose cardiaque et du remodelage tissulaire médiés par l'aldostérone dans un modèle murin de dysfonction du VG;
2. Le niveau de Gal-3 est associé au développement du remodelage du VG et de la fibrose et peut prédire les effets des l'antagonistes neurohormonaux chez les patients atteints d'IC chronique due à une cardiomyopathie dilatée non ischémique (CMDNI).
Méthode
Études animales. Des souris mâles adultes atteintes d'hyperaldostéronisme cardiaque spécifique ont été soumises à des injections sous-cutanées d'isoprotérénol (200 mg/kg x 2/jour pendant deux jours
consécutifs), puis randomisées pour recevoir un placebo, un inhibiteur de Gal-3 (modified citrus pectin, MCP), un antagoniste de récepteurs aux minéralocortïcoides (canrenoate de potassium) ou l’association de deux (MCP + canrenoate) pendant 14 jours. Les effets des différentes approches pharmacologiques ont été évalués par échocardiographie en série, l’analyse de l'expression génique et protéique des principaux déterminants de l'inflammation et de par la mesure de la fibrose cardiaque en histologie.
Études cliniques. Nous avons recruté de façon prospective 150 patients consécutifs référés pour le traitement de l’IC, avec un diagnostic de CMDNI. Les patients présentant des tumeurs malignes, des maladies inflammatoires aiguës ou chroniques, des troubles hématologiques et auto-immuns et une maladie rénale chronique sévère (taux de filtration glomérulaire estimé <30 ml/Kg/min) ont été exclus. Tous les patients ont bénéficié d'une évaluation clinique complète, d'une caractérisation biohumorale, incluant le dosage de Gal-3, l'échocardiographie bidimensionnelle, et une imagerie par résonance magnétique cardiaque (IRM) contrastée pour l'évaluation des volumes et de la fibrose par le late gadolinium enhancement (LGE). Une plus petite cohorte de 70 patients a également reçu un dosage de soluble suppression of tumorigenicity protein 2 (sST2) à l’inclusion, et une IRM de suivi à 24 mois pour l'évaluation du remodelage inverse du VG, définie comme une augmentation de >10% de la fraction d’ejection du VG ou une diminution de 10% du volume diastolique du VG indexé.
Résultats
Concernant la partie expérimentale, l'isoprotérénol a induit une diminution rapide et persistante de la fonction systolique du VG chez la souris, qui a été nettement améliorée par le traitement au MCP ou canrenoate. Le MCP et le canrenoate ont également prévenue l'hypertrophie et la fibrose cardiaques ainsi que l’augmentation de l'expression de gènes impliqués dans la fibrogénèse (Coll-1 et Coll-3) et l'infiltration de macrophages (CD-68 et MCP-1). En outre, après traitement à l'isoprotérénol, l'expression génique et protéique de Gal-3 ont été diminués par le canrenoate et le MCP. L'utilisation combinée d’un d'un inhibiteur de Gal-3 et d'aldostérone a entraîné des effets additifs sur l'hypertrophie cardiaque, l'inflammation et la fibrose, comparativement au MCP ou au canrenoate seul.
Dans notre cohorte de patients avec CMDNI, la valeur médiane de Gal-3 était de 14,4 ng/mL (IQR 11,7-19,0 ng/mL); le LGE a été observée chez 106 (71%) patients. Les patients ayant un LGE positif avaient un taux de Gal-3 plus élevée que ceux qui n'en avaient pas (15,4; 11,8-21,0 vs 13,1; 11,7-16,4 ng/mL, p=0,006). Après analyse univariée, la Gal-3, le sexe masculin, l’ancienneté de la cardiopathie, l’hypertension artérielle, la fraction d'éjection ventriculaire gauche et droite et le volume ventriculaire gauche, sont des paramètres significativement associès à la présence de LGE. En revanche après analyse multivariée suel la Gal-3, le sexe, l’ancienneté del la cardiopathie et la fraction d’éjection du
ventricule droit sont des facteurs prédicteurs indépendants de la présence de LGE. L’IRM de suivi a montrée que 35 patients (50%) présentaient un remodelage inverse. L’analyse multivariée met en evidence Gal-3 comme un prédicteur indépendant du remodelage inverse contrairement au sST2.
Conclusion
Les données expérimentales faites sur l’animal suggèrent que Gal-3 participe à des mécanismes de lésions myocardiques médiées par l'aldostérone dans un modèle murin de IC. De même, dans un cadre clinique de CMDNI, Gal-3 semble être associée à la fibrose du VG et à l'évolution du remodelage cardiaque, en identifiant éventuellement le sous-ensemble de patients avec une réponse plus prononcée à l'antagonisme neuro-hormonal pharmacologique. Dans l'ensemble, nos données peuvent soutenir l'utilisation de Gal-3 comme un biomarqueur pour la gestion de l’IC et également l’envisager comme une cible thérapeutique potentielle.
D
ECLARATION OFA
UTHENTICITYThe present thesis is the result of my own work; third-party intellectual property has been properly disclosed and referenced.
My contribution to each of the Chapters already object of (or submitted for) publication is as follows: - Chapters 2,3 and 4: review of the literature and manuscript writing;
- Chapter 7: patients’ recruitment, data analysis and interpretation;
- Chapter 8: patients’ recruitment, data collection, analysis and interpretation, manuscript writing;
- Chapter 9: development of the experimental model, animal experiments, molecular biology, data collection, analysis and interpretation, manuscript writing;
- Chapter 10 and 11: patients’ recruitment, data collection, analysis and interpretation, manuscript writing;
- Chapter 12: data analysis and interpretation, manuscript writing.
T
ABLEOFC
ONTENTSPart 1 Biomarkers in heart failure: from pathophysiology to clinical management
Chapter 1 An introduction to chronic heart failure 17
1.1 Epidemiology and classification 17
1.2 Pathophysiology 18
1.3 References 19
Chapter 2 Biomarkers of heart failure with preserved and reduced ejection fraction 23
2.1 Introduction 23
2.2 Biomarkers of heart failure with reduced ejection fraction 24 2.2.1 B-type natriuretic peptides in heart failure with reduced ejection fraction (HFrEF):
standing on the shoulder of giants
24
2.2.2 B-type natriuretic peptide-guided therapy 26
2.2.3 Back to the De Bold’s “atrial natriuretic factor”: MR-proANP 27
2.2.4 Cardiac troponins 28
2.2.5 Candidate biomarkers of HFrEF 29
2.3 Biomarkers of heart failure with mid-range and preserved ejection fraction 36
2.3.1 Natriuretic peptides 37
2.3.2 Other biomarkers in HFpEF 38
2.3.3 Directions 39
2.4 Conclusions 39
2.5 References 40
Chapter 3 Markers of fibrosis, inflammation, and remodeling pathways in heart failure 59
3.1 Abstract 59
3.2 Introduction 59
3.3 Cardiac remodeling 62
3.4 The pathophysiological role of cytokines in myocardial fibrosis 63
3.5 Biomarkers of ECM 68
3.6 Renin-angiotensin-aldosterone system, transforming growth factor beta and cardiac
fibrosis 70
3.7 Markers of ongoing myocardial damage: troponins 72
3.8 “Novel” biomarkers of myocardial fibrosis and remodeling 73
3.8.1 ST2 73
3.8.2 Galectin-3 74
3.8.3 Biglycan and decorin 75
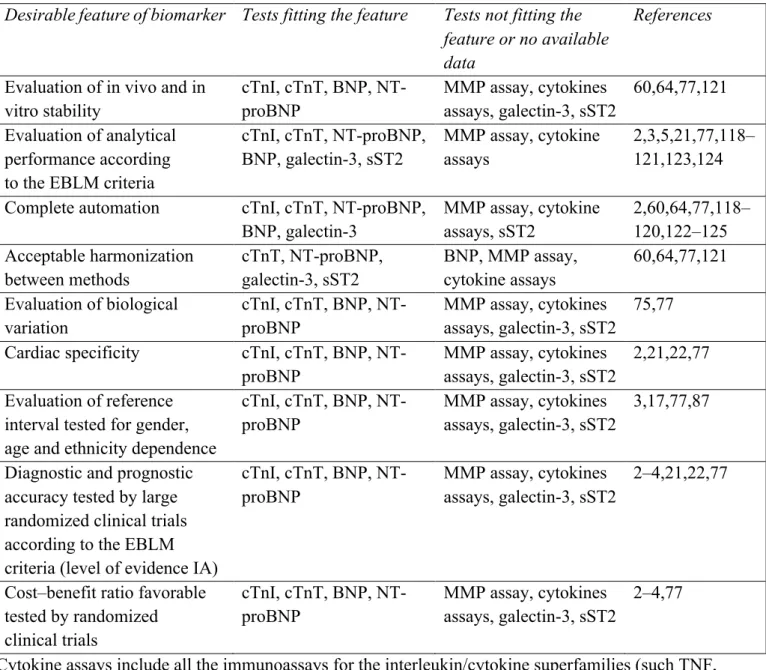
3.9 Analytical performance of biomarkers for heart failure: some general considerations 75
3.10 Perspectives 76
3.11 References 77
Chapter 4 Biomarkers of activation of renin-angiotensin-aldosterone system in heart failure: how useful, how feasible?
90
4.1 Abstract 90
4.2 Do we need a renin-angiotensin-aldosterone system? The biological and physiological
background 90
4.4 The genetics of the system: lesson from polymorphisms 94 4.5 When the RAAS is bad for you: the rationale for clinical use of RAAS biomarkers 96
4.5.1 Hypertension 96
4.5.2 Kidney disease 96
4.5.3 Ischemic heart disease 97
4.5.4 Heart failure 97
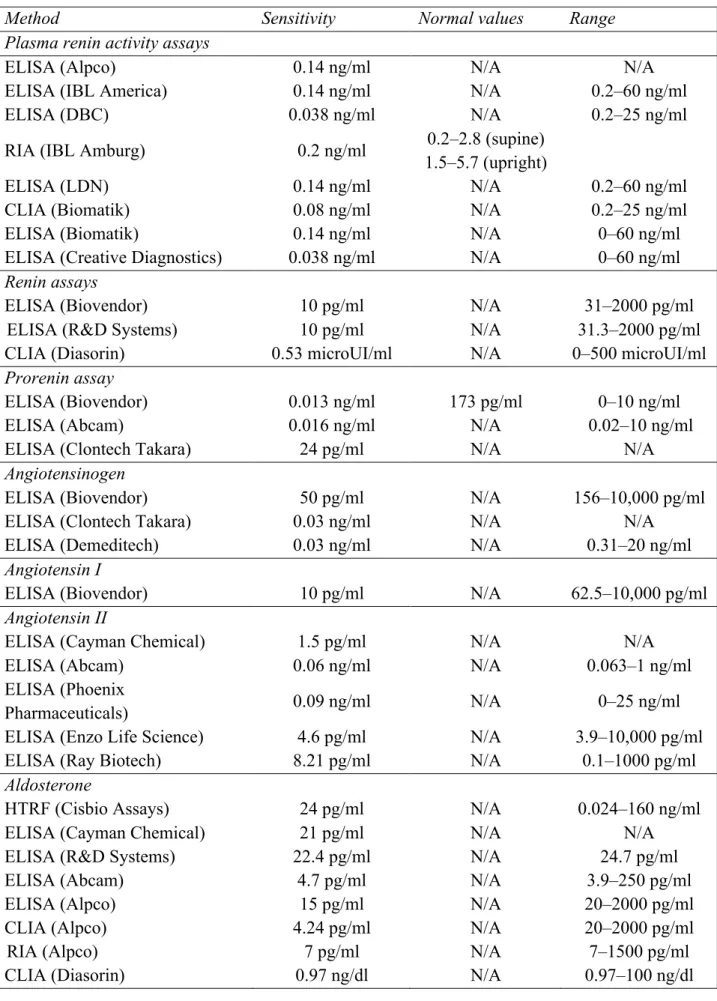
4.6 Biomarkers of RAAS: methodological aspects, and clinical implications 98
4.6.1 Plasma renin activity 98
4.6.2 Angiotensin II assays 101
4.6.3 Aldosterone assays 102
4.7 Future perspectives: could biomarkers of RAAS activation be useful for tailoring
treatment in heart failure patients? 104
4.8 References 107
Chapter 5 Galectin-3 and aldosterone interaction 116
5.1 Introduction 116
5.2 Biology of galectin-3 116
5.3 Galectin-3 in cardiac remodelling and heart failure 117
5.4 Potential role of galectin-3 in aldosterone mediated cardiac remodelling 118
5.5 Galectin-3 as a prognostic marker in heart failure 121
5.6 Pharmacological strategies of galectin-3 modulation 121
5.7 References 122
Part 2 Galectin-3 and aldosterone profibrotic pathways in the failing heart: a translational approach
Chapter 6 Project overview 127
6.1 Introduction 127
6.1.1 Original hypothesis 127
6.2.2 Project aims 127
6.2 Facilities 128
6.2.1 External collaborations 128
6.3 Expected vale of results 129
6.4 Outline of the project presentation 129
6.5 References 130
Chapter 7 Prognostic significance of myocardial extracellular volume fraction in non ischaemic dilated cardiomyopathy
131
7.1 Abstract 131
7.2 Introduction 131
7.3 Materials and methods 132
7.3.1 Study population 132
7.3.2 Cardiovascular magnetic resonance 133
7.3.3 Image analysis 133 7.3.4 Transthoracic echocardiography 134 7.3.5 Clinical follow-up 135 7.3.6 Statistical analysis 135 7.4 Results 136 7.4.1 Patients characteristics 136 7.4.2 Interstitial remodelling 137
7.4.3 Survival analysis 139 7.4.4 Reproducibility 139 7.5 Discussion 139 7.5.1 Study limitations 142 7.6 Conclusion 142 7.7 References 142
Chapter 8 Refractory hyperaldosteronism in heart failure is associated with plasma renin
activity and angiotensinogen polymorphism 148
8.1 Abstract 148 8.2 Introduction 148 8.3 Methods 149 8.3.1 Ethics statement 149 8.3.2 Patients 150 8.3.3 Biomarkers 150 8.3.4 Genotyping 150 8.3.5 Follow-up 151 8.3.6 Statistics 151 8.4 Results 152 8.5 Discussion 156 8.5.1 Study limitations 158
8.6 Conclusion and perspectives 158
8.7 References 158
Chapter 9 Inhibition of galectin-3 pathway prevents isoproterenol-induced left ventricular dysfunction and fibrosis in mice
163
9.1 Abstract 163
9.2 Introduction 163
9.3 Methods 165
9.3.1 Animals 165
9.3.2 Induction of left ventricular systolic dysfunction and treatment 165
9.3.3 Echocardiography 165
9.3.4 Organ weight and tissue analysis 166
9.3.5 Gene expression analysis 166
9.3.6 Statistics 166
9.4 Results 166
9.4.1 Galectin-3 and aldosterone inhibition reverses left ventricular remodelling and systolic dysfunction
166 9.4.2 Galectin-3 and aldosterone inhibition prevents myocardial hypertrophy and
interstitial fibrosis
167 9.4.3 Effects of aldosterone and galectin-3 inhibition on myocardial galectin-3
expression and protein content 169
9.4.4 Galectin-3 and aldosterone inhibition decreases isoproterenol-induced inflammatory and pro-fibrotic response
170
9.5 Discussion and perspectives 172
9.6 References 175
Chapter 10 Galectin-3 and myocardial fibrosis in non-ischemic dilated cardiomyopathy 178
10.1 Abstract 178
10.2 Introduction 178
10.3.1 Study population 179
10.3.2 CMR protocol 180
10.3.3 Statistical analysis 180
10.4 Results 181
10.4.1 Study population 181
10.4.2 Predictors of late gadolinium enhancement 184
10.5 Discussion 185
10.5.1 Correlates of myocardial fibrosis at cardiac magnetic resonance imaging 185
10.5.2 Galectin-3 in myocardial remodelling 186
10.5.3 Study limitation and conclusions 186
10.6 References 187
Chapter 11 Galectin-3 predicts reverse remodeling assessed by cardiac magnetic resonance in non-ischemic dilated cardiomyopathy
190
11.1 Abstract 190
11.2 Introduction 190
11.3 Methods 191
11.3.1 Study population 191
11.3.2 Cardiac magnetic resonance protocol 192
11.3.3 Statistical analysis 193
11.4 Results 193
11.4.1 Characteristics of the study population 193
11.4.2 Predictors of reverse remodelling 195
11.5 Discussion 196
11.6 References 199
Chapter 12 Clinical benefits of natriuretic peptides and galectin-3 are maintained in old dyspnoeic patients 203 12.1 Abstract 203 12.2 Introduction 203 12.3 Methods 204 12.3.1 Study population 204 12.3.2 Study design 204 12.3.3 Biomarkers quantification 205 12.3.4 Statistical analysis 205 12.4 Results 205
12.4.1 Baseline characteristics, diagnosis and outcome 205
12.4.2 Natriuretic peptide levels according to age group and origin of dyspnoea 207 12.4.3 Diagnostic performance of the 4 natriuretic peptides according to age group 208 12.4.4 Prognostic properties of NPs and novel cardiovascular biomarkers according to
age 208
12.5 Discussion 209
12.6 References 211
Results outline and interpretation 215
Clinical value of interstitial remodelling and fibrosis 215
Old (?) and new biomarkers of fibrosis in heart failure 215
Galectin-3 and myocardial remodeling in non-ischemic dilated cardiomyopathy 216
Publication list 218
L
IST OFA
BBREVIATIONSACE: angiotensin converting enzyme Ac-SDKP: N-acetyl-seryl-aspartyl-lysyl-proline
ADM: adrenomedullin AHF: acute heart failure ANP: atrial natriuretic peptide ARB: angiotensin receptor blockers AUC: area under the curve
AVP: arginine vasopressin BNP: brain natriuretic peptide CMR: cardiac magnetic resonance CRP: C-reactive protein
NIDCM: non-ischemic dilated cardiomyopathy
ECM: extracellular matrix ECV: extracellular volume ED: emergency department
EDTA: ethylenediaminetetra-acetic acid EF: ejection fraction
eGFR: estimated glomerular filtration rate ET: endothelin
Gal-3: galectin-3
GDF-15: growth differentiation factor-15 GFR: glomerular filtration rate
HCM: hypertrophic cardiomyopathy HF: heart failure
HFmrEF: heart failure with mid-range ejection fraction
HFpEF: heart failure with preserved ejection fraction
HFrEF: heart failure with reduced ejection fraction
hsTn: high sensitivity troponin
ICAM: intercellular adhesion molecules ICD: implanted cardioverter defibrillator ICTP: collagen I carboxy-terminal telopeptide IL: interleukin
KIM: kidney injury molecule LV: left ventricular
LVMI: left ventricular mass-indexed MCP: modified citrus pectin
MCP-1: monocyte chemotactic protein-1 miRNA: micro ribonucleic acid
MMP: matrix metalloproteinases
MR: mineralocorticoid receptor
MRA: mineracolorticoid receptor antagonists MR-proANP: mid-regional pro-atrial
natriuretic peptide
MR-proADM: mid-regional pro-adrenomedullin
NAG: N-acetyl β-(d)-glucosaminidase NEP: neutral endopeptidases
NGAL: neutrophil-gelatinase–associated lipocalin
NP: natriuretic peptide
NT-proBNP: amino-terminal fragment of proBNP
NYHA: New York Heart Association OMD: ongoing myocardial damage PAI-1: plasminogen activator inhibitor-1 PICP: Carboxy-terminal propeptides of collagens I
PIIICP: Carboxy-terminal propeptides of collagens III
PINP: Amino-terminal propeptides of collagens I
PIIINP: Amino-terminal propeptides of collagens III
POCT: point-of-care testing PRA: plasma renin activity proENK: proenkephalin PTX3: pentraxin-3
RAAS: renin-angiotensin-aldosterone system RIA: radioimmunologic assay
SF-1: steroidogenic transcription factor 1 SNP: single nucleotide polymorphism
sST2: soluble suppression of tumorigenicity 2 protein
sTNFR1: soluble type 1 TNF receptor sTNFR2: soluble type 2 TNF receptor TAC: transverse aortic constriction TGF β: transforming growth factor β TIMP: tissue inhibitor of matrix metalloproteinases
TNF: tumor necrosis factor URL: upper reference limit
VCAM: vascular adhesion molecules VSMC: vascular smooth muscle cell
L
IST OFF
IGURESFigure 1.1 Natural history of heart failure 19
Figure 1.2 Neurohormonal imbalance in heart failure 20
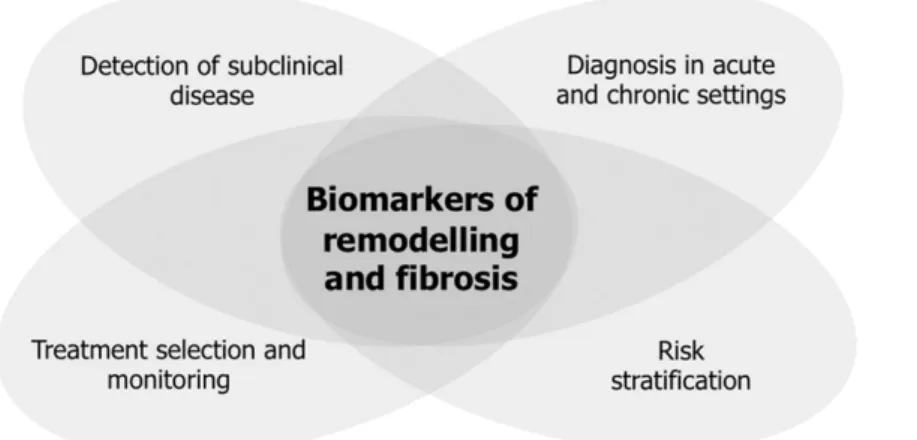
Figure 3.1 Proposed roles for biomarkers of remodeling and fibrosis in heart failure
settings 60
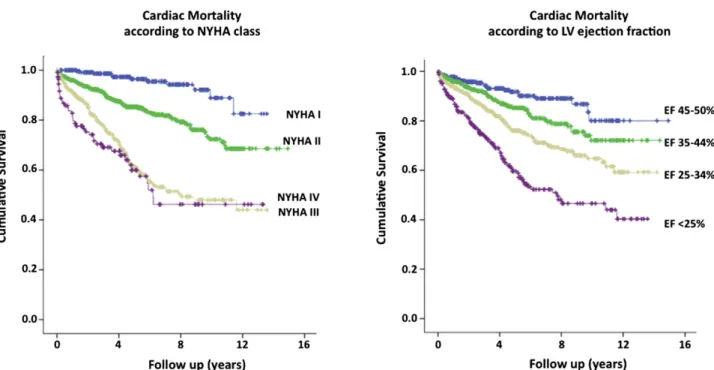
Figure 3.2 Survival trends in heart failure patients 62
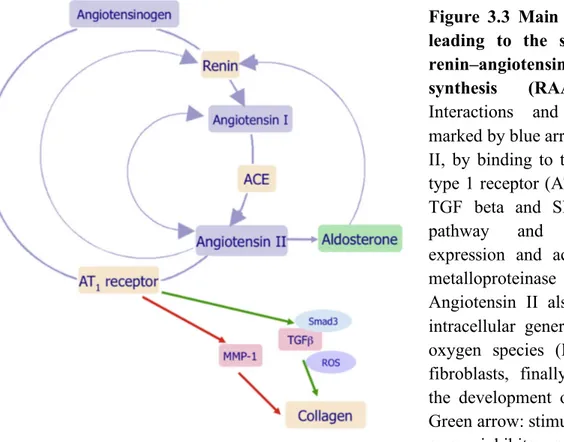
Figure 3.3 Main enzymatic steps leading to the synthesis of the renin–angiotensin–
aldosterone-synthesis (RAAS) effectors 71
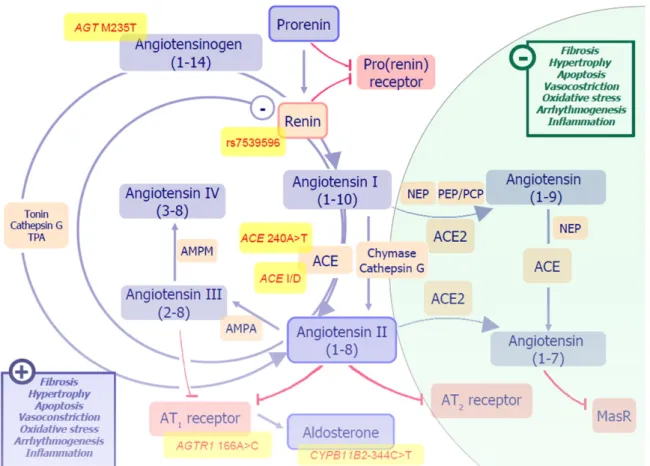
Figure 4.1 Pathways and effectors of the renin-angiotensin-aldosterone system (RAAS) 92 Figure 4.2 Increment profiles of neurohormones with regard to worsening clinical
severity in systolic heart failure 105
Figure 5.1 Immunolabeling of galectin-3 (Gal-3) and fibronectin. Quantitative RT-PCR
analysis of Gal-3 transcripts in left ventricle 120
Figure 7.1 Calculation of T1 values 134
Figure 7.2 Correlations between myocardial extracellular volume (ECV) and ventricular
remodelling 138
Figure 7.3 Correlations between myocardial extracellular volume (ECV) and arrhythmias
at 24-hour electrocardiographic monitoring 138
Figure 7.4 Kaplan-Meier survival curve according to extracellular volume (ECV) 141 Figure 8.1 Plasma renin activity (PRA) in patients with and without refractory
hyperaldosteronism 155
Figure 8.2 Genotype distribution of angiotensinogen M235T polymorphism in patients
with and without refractory hyperaldosteronism 155
Figure 9.1 Effects of aldosterone and galectin-3 antagonism on left ventricular systolic
function 168
Figure 9.2 Galectin-3 and aldosterone antagonism reduces myocardial fibrosis 169 Figure 9.3 Galectin-3 and aldosterone pharmacological inhibition reduce cardiac
galectin-3 expression and protein content 170
Figure 9.4 Effects of canrenoate and modified citrus pectin on infammatory markers 171 Figure 9.5 Effects of canrenoate and modified citrus pectin on profibrotic markers 171 Figure 9.6 Effects of canrenoate and modified citrus pectin on brain natriuretic peptide
expression 172
Figure 9.7 Putative model of aldosterone – galectin-3 interplay in the development of
myocardial fibrosis 174
Figure 10.1 Late gadolinium enhancement in patients with or without elevated plasma
galectin-3 184
Figure 11.1 Correlation between galectin-3 (Gal-3) and time course of systolic function 197 Figure 12.1 Diagnostic performance of 4 natriuretic peptides according to age groups (n =
201) 207
Figure 12.2 Prognostic performance of the NPs and 3 novel cardiovascular biomarkers
PART 1
B
IOMARKERSINH
EARTF
AILURE:F
ROMP
ATHOPHYSIOLOGY TOC
LINICALChapter 1. An introduction to chronic heart failure
1.1 Epidemiology and classification
Heart failure (HF) is a “multisystem disorder characterized by profound disturbances in circulatory physiology and a plethora of myocardial structural and functional changes that affect the systolic pumping capacity and diastolic filling characteristics of the heart” [1]. HF syndrome is a progressive disease taking place after an initial noxa, e.g. ischemia, inflammation, toxic damage, heart muscle disease, has impaired cardiac systolic and/or diastolic function. Later on, HF develops in a fashion that is largely independent from the initiating factor, through fairly standardized pathophysiological mechanisms.
During the last three decades, advances in primary and secondary prevention strategies, as well as in pharmacological and non-pharmacological therapeutic approaches have led to a dramatic decrease in deaths related to cardiovascular diseases. HF is an important exception to this trend, since HF related hospitalization have risen since 1975 and HF mortality rate still remains dramatically high, approaching 50% five years after diagnosis, worse than that of many cancers [2]. All over the world, more than 20 million people are estimated to be affected by HF, with more than 550,000 individuals newly diagnosed every year only in the United States [3].
Any structural, mechanical or electrical abnormality of the heart can cause it to fail, therefore basically each cardiovascular disease may finally end up with clinical manifestation of HF. Nonetheless, the most common cause remains coronary artery disease, with more than one-third of patients experiencing HF in the few years following myocardial infarction [4]. Although myocardial infarction is the most important individual risk factor for HF, the population attributable risk due to hypertension is probably still more important [5]. Other common causes of HF include heart muscle diseases such as idiopatic dilated cardiomyopathy, hypertrophic cardiomyopathy or infiltrative disorders, leading to restrictive cardiomyopathies (including amyloidosis), valvular or pericardial diseases, as well as toxic agents (e.g. alcohol or anthracyclines).
Historically, the classification of HF has relied on the assessment of left ventricular (LV) systolic function. HF patients having LV ejection fraction (EF) ≥50% are considered as having HF with preserved EF (HFpEF), while those with LVEF <40% as affected by HF with reduced EF (HFrEF). The latest European Society of Cardiology guidelines for the diagnosis and management of HF have recently introduced a novel clinical entity called HF with mid-range EF (HFmrEF) to classify patients in the grey area in the range 40-49% [6]. Differentiation of patients according to LV systolic function is important, given the differences in underlying etiology, pathophysiology, epidemiology, comorbid conditions and response to pharmacological therapy [7]. Specifically,
patients with HFpEF are usually older, more often women and more commonly hypertensive compared to those with HFrEF. Characteristics of HFmrEF seem to be intermediate between HFpEF and HFrEF, but further studies are still needed to better characterize this population [8].
1.2 Pathophysiology
The pathophysiological framework of HF has dramatically evolved during the last five decades. Starting from the “haemodynamic” model, focusing on the loss of contractile capacity of the failing left ventricle, other interpretative models have come in succession. One of them is the “cardio-renal model”, putting the attention on the excess in salt and water retention observed in patients affected by HF [9]. The haemodynamic and the cardio-renal models were the bases for the use of inotropes and diuretics, respectively, in the treatment of HF, but none of these strategies has ever been demonstrated to significantly modify the natural history of the disease, nor to ameliorate patients’ prognosis. It became evident, therefore, that these models could only explain some of the clinical manifestation of the HF syndrome, while they were missing some key pathophysiological element.
As stated before, HF may be viewed as a progressive disorder initiated after an index event either damages the heart muscle, with a resultant loss of functioning cardiac myocytes, or alternatively impairs the ability of the myocardium to generate force, thus preventing the heart from contracting adequately. The onset of the index event may be abrupt, as for the case myocardial infarction, gradual, as in pressure or volume overload, or it may be hereditary, as in genetic cardiomyopathies. Independently of the nature of the initial noxa, each of them finally leads to a decline in the pump function of the heart. Often, such a decline remains asymptomatic for some time (years in some cases), due to the activation of compensatory mechanisms, including the adrenergic nervous system, the renin-angiotensin-aldosterone system (RAAS), and the cytokine system. The discovery of the central role of the activation of neurohormonal systems in HF led to a novel conceptual framework, the so called “neurohormonal model,” in which HF progresses following the overexpression of biologically active molecules that are capable of exerting deleterious effects on the heart and circulation [10]. The term neurohormone reflects the original observation that many of these molecules were produced by the neuroendocrine system and acted on the cardiovascular system in an endocrine fashion. Nonetheless, some of these neurohormones have been discovered to be also synthesized directly within the myocardium, such as angiotensin II, endothelin, natriuretic peptides, and tumor necrosis factor (TNF).
These systems are initially able to restore cardiovascular function to a normal homeostatic range. Nonetheless, the chronic activation of these compensatory mechanisms finally sustains itself myocardial and target organ damage, thus further worsening cardiac function and initiating a vicious
circle that determines the onset of signs and symptoms of HF, such as reduced exercise tolerance, dyspnoea and peripheral oedema (Figure 1.1). Indeed, the RAAS, together with the activation of the adrenergic and the endothelin and vasopressin systems, promotes vasoconstriction, salt and water retention as well as myocardial necrosis, hypertrophy and fibrosis, which are only partly counterbalanced by cardiac natriuretic hormones, namely atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). Such these hormones are synthesized and released after pressure or volume overload is determined at the level of atrial or ventricular chambers (for ANP and BNP, respectively), as well as after each generic damage to the cardiovascular system (Figure 1.2).
The prominent role of neurohormonal activation in HF pathophysiology is supported by the beneficial effects of pharmacological strategies of neurohormonal antagonism. Beta-blockers, angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), mineracolorticoid receptor antagonists (MRA), all of them counteracting via direct and indirect mechanisms the adrenergic and RAAS systems, represent nowadays cornerstones of HF treatment and have significantly improved, during the last two decades, patients’ prognosis and quality of life [11]. Recently, the first-in-class angiotensin II receptor/neprilysin inhibitor LCZ696 has been introduced as a novel drug for HF. By inhibiting at the same time the RAAS and neprilysin mediated BNP degradation, LCZ696 can indeed contribute to remodulate neurohormonal activation [12].
Neurohormonal imbalance contributes to the progression of HF by promoting a complex set of changes in cardiac geometry and function, ad well as in myocardial structure, metabolism, cellular and subcellular organization which is commonly referred to as “remodelling” (Table 1.1).
Figure 1.1 Natural history of heart failure.
Starting from an index event, a progressive decline in left ventricular function is observed, which is sided be the activation of neurohomonal systems. Such systems, while being initially compensatory, finally contribute to heart failure progression and to the development of symptoms.
Table 1.1 Features of myocardial remodeling
All pharmacological and non pharmacological approaches blunting neurohormonal activation (including aerobic training and cardiac resynchronization therapy) have been also demonstrated to promote the recovery of such these functional and morphological alterations, a phenomenon termed “reverse remodeling” [11].
In the following Chapters, biomarkers of HF will be discussed, introductive to the aims and the hypotheses of the PhD project, first with a general view on “old” and “new” biomarkers of HFpEF and HFrEF, then dealing with more specific aspects of biomarker of fibrosis and of RAAS activation. Chapter 3 is part of the “Heart Failure” section of the “Handbook of Experimental Pharmacology”, planned to be edited by Springer, co-author by Dr. Vergaro. Chapters 4 and 5 are the results of an in-depth review of the literature preliminary to the conceivement of the project and published in Clinica Chimica Acta.
Figure 1.2 Neurohormonal imbalance in heart failure. According to the neurohormonal model,
activation of sympathetic and renin-angiotensin-aldosterone (RAA) system in partly conterbalanced Alterations in myocyte biology
Excitation contraction coupling
Myosin heavy chain (fetal) gene expression ß-adrenergic desensitization
Hypertrophy Myocytolysis
Cytoskeletal proteins Myocardial changes
Myocyte loss (necrosis, apoptosis) Alterations in extracellular matrix Replacement fibrosis
Alteration in left ventricular chamber geometry Dilation and increased sphericity
Wall thinning
1.3 References
1. Mann DL, Felker GM. Heart Failure – A companion to Braunwald’s Heart Disease 3rd Edition, Section 1, page 1. Elsevier, 2016.
2. Askoxylakis V, Thieke C, Pleger ST. Long-term survival of cancer patients compared to heart failure and stroke: a systematic review. BMC Cancer. 2010; 10:105.
3. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics - 2010 update: a report from the American Heart Association. Circulation. 2010; 121:e46– e215.
4. Hellermann JP, Goraya TY, Jacobsen SJ, Weston SA, Reeder GS, Gersh BJ, Redfield MM, Rodeheffer RJ, Yawn BP, Roger VL. Incidence of heart failure after myocardial infarction: is it changing over time? Am J Epidemiol. 2003; 157:1101-1107.
5. Kostis JB, Davis BR, Cutler J, Grimm RH Jr, Berge KG, Cohen JD, Lacy CR, Perry HM Jr, Blaufox MD, Wassertheil-Smoller S, Black HR, Schron E, Berkson DM, Curb JD, Smith WM, McDonald R, Applegate WB. Prevention of heart failure by antihypertensive drug treatment in older persons with isolated systolic hypertension. SHEP Cooperative Research Group. JAMA. 1997; 278: 212–216.
6. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P; Authors/Task Force Members. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016; 37:2129-2200.
7. Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JG, Cody RJ, Chioncel O, Collins SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O'Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR,
Georgiopoulou VV, Gheorghiade M. Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail. 2014; 2:97-112. 8. Lam CSP, Solomon SD. The middle child in heart failure: heart failure with mid-range
ejection fraction (40–50%). Eur J Heart Fail. 2014; 16:1049–1055.
9. Packer M. How should physicians view heart failure? The philosophical and physiological evolution of three conceptual models of the disease. Am J Cardiol. 1993; 71:3C-11C.
10. Bristow MR. The adrenergic nervous system in heart failure. N Engl J Med. 1984; 311:850– 851
11. Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005; 111:2837-2849.
12. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 371, 993-1004 (2014).
Chapter 2. Biomarkers of heart failure with preserved and reduced ejection fraction
The present Chapter is based on the following manuscript: Michele Senni, Michele Emdin, Emilia D’Elia, Giuseppe Vergaro. Biomarkers in HFpEF/HFrEF. In “Handbook of Experimental Pharmacology”. Editors: Johan Bauersachs, Javed Butler, Peter Sandner. Planned to be edited by Springer in 2017.
2.1 Introduction
Early diagnostic and therapeutic interventions counteracting pathophysiological mechanisms of heart failure (HF) progression, such as drugs inhibiting the renin-angiotensin-aldosterone system (RAAS) and adrenergic system activation, as well as device implantation in selected patients positively influence its clinical course, impacting on the arrhythmic burden and hemodynamics. Notwithstanding, outcome is still characterized by a high morbidity and mortality. Interest is therefore growing in the optimization of HF management and in the identification and validation of novel therapeutical tools. With these premises, research has focused on the use of biomarkers not only for the purpose of screening, diagnosis and risk stratification, but also as a guide to the assessment of response to treatment in the individual patient, and finally as an entry criterion and/or a surrogate marker of efficacy in clinical trials testing novel drugs [1-3].
In 2001, a National Institute of Health working group defined a biomarker as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention”, and identified different classes of biomarkers according to their role and potential utility in disease management. In particular, biomarkers were classified as antecedent biomarkers (i.e. those identifying the risk of developing a disease), screening biomarkers (those able to screen for subclinical disease), diagnostic biomarkers (recognizing clinically overt disease), staging biomarkers (those reflecting disease severity), or prognostic biomarkers (predicting the future course of the disease and response to pharmacological and non-pharmacological approaches) [4,5] (Table 2.1).
A number of biomarkers have been proposed for HF management purposes in the last years, nonetheless only a very few of them (namely B-type natriuretic peptides) are routinely used in current clinical practice. Indeed, several characteristics are requested for a circulating biomarker to be of real clinical value, related to (pre)analytical, biological, feasibility and cost-effectiveness issues. In particular, the ideal biomarker for HF management should reflect the cardiovascular response to a specific pathogenic noxa; provide early information on cardiac involvement in preclinical stages (screening in HF stage A–B) and/or guide HF diagnosis when cardiac origin of symptoms is doubtful
Drug
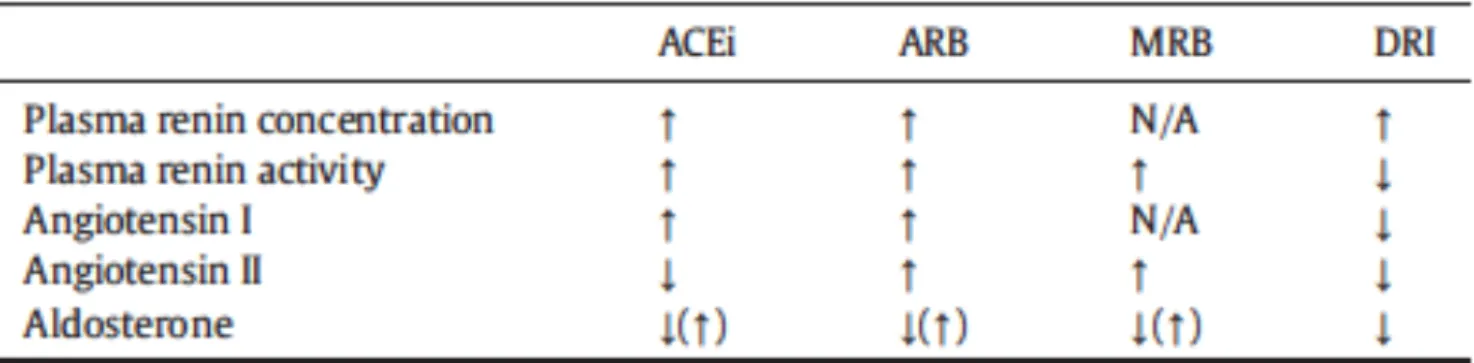
class BNP proBNP NT- PRC PRA Ang I Ang II Aldosterone Gal-3 sST2 CRP GDF-15 Beta-blockers > > = ? ACEi / = ? ARB / = / = = MRA ? ? / = ? = =? DRI / = ? ? ? ? ARNI ? = = = = ?
Table 2.1 Effects of currently recommended and previously proposed drugs for heart failure treatment on circulating levels of biomarkers. ARB: angiotensin receptor blockers; ARNI:
angiotensin receptor/neprilysin inhibitor; Ang: angiotensin; BB: beta blockers; BNP: brain natriuretic peptide; CRP: C-reactive protein; DRI: direct renin inhibitor; Gal-3: galectin-3; GDF-15: growth differentiation factor 15; MRA: mineralocorticoid receptor antagonist; NT-proBNP: amino-terminal fragment of proBNP; PRA: plasma renin activity; sST2: suppression of tumorigenicity 2.
(HF stage C); be influenced by clinical modification in the disease history; stratify patients’ risk and help in therapeutic decision-making. Still, novel biomarkers are clearly required to improve risk prediction already offered by existing models, with the mere demonstration of a statistically significant association with a pre-specified end-point not being enough [6]. Further to c-statistic and area under the curve (AUC) measure [7], providing estimate of the discriminative ability of the model (i.e. the capacity to separate subject developing/not developing the outcome), measures of calibration (indicating how close are predicted and observed risks in different groups of observations, e.g. by means of the Hosmer-Lemeshow test) and of reclassification (indicating the ability of the model to reclassify individuals into a different risk category, e.g. by calculating the net reclassification improvement, NRI) are required to adequately assess the clinical utility of novel biomarkers [8,9].
In the present Chapter, current evidences on the role of established and candidate biomarkers for HF management will be discussed, according to the main classification of HF phenotypes, based on the measurement of left ventricular ejection fraction (LVEF), including HF with reduced EF (HFrEF, LVEF <40%), HF with preserved EF (HFpEF, LVEF ≥50%) and the recently proposed category HF with mid-range EF (HFmrEF, LVEF 40-49%) [10].
2.2. Biomarkers of heart failure with reduced ejection fraction
2.2.1 B-type natriuretic peptides in heart failure with reduced ejection fraction (HFrEF): standing on the shoulders of giants
Human brain natriuretic peptide (BNP) and the amino-terminal fragment of proBNP (NT-proBNP) are produced in equimolar fashion from the cleavage of their 108-aminoacid precursor proBNP by proprotein convertases, such as corin and furin. The biologically active BNP is rapidly degraded in vivo by several peptidases, such as dipeptidyl peptidase IV and neutral endopeptidases (NEP, neprylisin) [11-13]. BNP, together with NT-proBNP, is mainly product of ventricular myocytes in response to increased myocardial wall stress due to volume or pressure overload states and plays a major role in HF pathophysiology, given its diuretic, natriuretic, vasodilator and anti-hypertrophic properties [14].
Since the beginning of the XXI century, several studies have accumulated, showing the clinical utility of B-type natriuretic peptide testing, supporting the use of a circulating biomarker to diagnose and assess severity of HF [15-17]. At present, BNP and NT-proBNP are routinely used for HF management in a large variety of clinical settings. Strong evidence exists supporting the use of natriuretic peptide testing to diagnose (and rule out) HF in patients presenting with dyspnoea. The Breathing Not Properly and the ProBNP Investigation of Dyspnea (PRIDE) trials have shown that BNP and NT-proBNP, respectively, can diagnose HF in patients admitted to the emergency department for breathlessness, with a high accuracy and a significant negative predictive value for levels of BNP <100 ng/L [16,17]. Although with lower cut-points, the diagnostic value of natriuretic peptides has been confirmed in ambulatory settings [18]. Moreover, in a cohort of around 800 patients with chronic HF, they have been shown to be associated with the presence of LV systolic dysfunction (defined as LVEF <40%) with an AUC of 0.803 (95% CI 0.757-0.849) and 0.730 (0.681-0.778) for BNP and NT-proBNP, respectively [19]. In stable patients with HFrEF, BNP circulating levels reflect disease severity, and increase with worsening symptoms (New York Heart Association, NYHA class) [20]. The assay of natriuretic peptides is currently recommended for diagnostic purposes by all major Scientific Societies, including the American College of Cardiology, the American Heart Association, the Heart Failure Society of America and the European Society of Cardiology [10,21,22], however, no single diagnostic cut-off exists and natriuretic peptide levels often fall into a “grey zone”. Further, both BNP and NT-proBNP are influenced by gender, age and comorbidities (in particular by renal function), therefore their interpretation must take into account the overall clinical assessment [23,24]. In adjunct to diagnostic properties, there is overwhelming evidence that natriuretic peptides hold prognostic value in HFrEF in both acute and chronic settings, over other clinical factors. In >19,000 patients with HFrEF form the ADHERE (Acute Decompensated Heart Failure National Registry), admission BNP levels were near-linearly associated with in-hospital mortality, independently from other major clinical and laboratory risk factors [25]. In a systematic review of studies performed in different clinical settings, Doust and colleagues have demonstrated that each
100 ng/L increase in BNP is associated with a relative 35% increase in risk [26]. NT-proBNP predicts as well both short- and long-term prognosis after hospitalization for acute HF, with values >986 ng/L predicting 1-year death [27,28]. Although most studies have investigated the prognostic role of admission natriuretic peptide levels, there is evidence that discharge BNP and NT-proBNP, and their change during hospitalization also predict outcome of patients with HFrEF. Data from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) show that the addition of discharge BNP to a clinical model significantly improved risk classification for 1-year mortality with a NRI of 5.5% [29], thus suggesting that serial natriuretic peptide testing may be useful in pre-discharge clinical decision making [30]. A post-hoc analysis of the Valsartan Heart Failure trial (Val-HeFT), enrolling patients with chronic symptomatic HF with LVEF <40% and diastolic LV diameter adjusted for body surface area ≥2.9 cm/m2, demonstrated that BNP and NT-proBNP similarly predicted all-cause mortality, while NT-proBNP outperformed BNP for the prediction of mortality and morbidity or hospitalization for HF [31]. Moreover, experience from the Val-HeFT trial indicates that changes over time in natriuretic peptides, too, hold a prognostic value in stable HF, envisaging a role for serial BNP and NT-proBNP testing in patient monitoring and in the evaluation of response to therapeutical strategies [32,33].
2.2.2 B-type natriuretic peptide-guided therapy
Circulating levels of B-type natriuretic peptides are influenced by most pharmacological and non-pharmacological therapeutical approaches in acute, acutely decompensated and chronic HF. Indeed, in the acute setting, treatment with furosemide and the inodilator levosimendan decreases BNP [34-36]. Moreover, a reduction >30% from baseline values following levosimendan administration has been associated with an improvement in either short- (31 days) and long-term (180 days) mortality [34]. Other therapeutic strategies acting on hemodynamics and cardiac remodeling have been demonstrated to elicit similar effects on natriuretic peptides in stable HFrEF, including angiotensin-converting enzyme inhibitors [37], angiotensin-receptor blockers [38], mineralocorticoid receptor antagonists [39], and, at a lower extent, β-blockers, whose initiation may also cause a transient increase in BNP and NT-proBNP, which subsequently decrease [37,40]. Evidence for a beneficial effect on natriuretic peptides also exists for other, non-pharmacological tools, such as aerobic training and cardiac resynchronization therapy [41,42]. More recently, the first-in-class neprilysin/angiotensin receptor inhibitor sacubitril valsartan (LCZ696), has been proved effective in reducing the risks of death and of hospitalization in patients with HFrEF compared to enalapril in the PARADIGM-HF trial [43]. Such these beneficial effects are possibly due, at least in part, to the inhibition of BNP degradation exerted by sacubitril, while its precursor and NT-proBNP are virtually resistant to
degradation [44], as also supported by the observation that after LCZ696 initiation, circulating levels of NT-proBNP decrease, while BNP slightly increases [45]. It has been therefore questioned that assessing the clinical significance of BNP in the era of LCZ696 may be challenging and that NT-proBNP may be preferred over BNP for biochemical monitoring of patients with HF, in order to assess its effectiveness on hemodynamic status [46].
As discussed before, natriuretic peptides perform well for diagnostic and prognostic purposes, are associated with disease severity and, importantly, their circulating levels change in response to evidence-based therapies for HF. Based on these premises, several studies have tested the (cost)effectiveness of strategies of treatment titration guided by natriuretic peptide circulating levels, the so called biomarker-guided therapy. In the BATTLESCARED, the first of these studies, 364 HF patients were randomly allocated to therapy guided by NT-proBNP levels (with adjustments in medications and additional follow-up visits triggered by an NT-proBNP level >150 pmol/l) or by intensive clinical management, or according to usual care [47]. NT-proBNP-guided treatment was associated with a significant reduction in primary end point of death and/or readmission with HF in younger population (age ≤75 years), who also presented more often with HFrEF (61% vs 47% in patients aged >75 years). In the same year results from the TIME-CHF trial, enrolling only patients with LVEF ≤45% were published, showing that patients with therapy titration based on NT-proBNP (target: <400 ng/L in patients aged <75 years and <800 ng/L in patients ≥75 year-old) had improved 18-month survival free of hospitalizations for HF [48]. Following these pivotal trials, other studies using either BNP or NT-proBNP have provided mixed outcomes [49]. Some systematic reviews have addressed the issue of the effects on hard end-points, such as all-cause mortality, demonstrating a benefit from natriuretic peptide-guided therapy [50], and a meta-analysis by Felker has demonstrated a 30% improvement in survival, without an increase in therapy-related adverse events [51]. As a general view, the use of natriuretic peptide to tailor HF therapy is likely to be more effective when lower target values are applied (i.e. <100 ng/L and <1,000 ng/L for BNP and NT-proBNP, respectively) [15,52,53]. Nonetheless, evidence is still scarce to support a general recommendation and some issues still need to be addressed, including the most appropriate use of biomarker with this respect (disease management vs monitoring), the choice of the end-point and the cost-effectiveness of such a strategy [54].
2.2.3 Back to the De Bold’s “atrial natriuretic factor”: MR-proANP
Although B-type natriuretic peptides represent the most widely studied cardiac hormones, both in the experimental and clinical field, atrial natriuretic peptide (ANP) was first described in early 80's, as a substance secreted from atrial granules with endocrine functions, by Adolfo De Bold [55]. ANP assay
has with time been substituted by assays of B-type natriuretic peptides both for analytical and clinical reasons [56]. More recently, the potential clinical applications of the mid-regional pro-atrial natriuretic peptide (MR-proANP), with a longer half-life than ANP, have been tested. Data from the Biomarker in the Acute Heart Failure (BACH) and the PRIDE study have demonstrated a good performance of MR-proANP for diagnostic and prognostic purposes in acute HF, even in addition to NT-proBNP [57,58]. MR-proANP also revealed a significant prognostic value in 1237 patients with chronic HF enrolled in the Gruppo Italiano per lo Studio della Sopravvivenza nell'Insufficienza Cardiaca - Heart Failure (GISSI-HF) study (proposed cut-off 278 pmol/L, AUC 0.74, 0.71–0.77), outperforming other established and candidate biomarkers (including copeptin and mid-regional pro-adrenomedullin, MR-proADM, and C-terminal proendothelin-1) and beyond NT-proBNP and a set of clinical variables (NRI=0.06) [59].
2.2.4 Cardiac troponins
Cardiac troponins are released into the circulation following the disruption of cardiomyocyte membrane due to cardiac injury, namely after cardiac necrosis, and have become the standard of care biomarker for diagnosis of myocardial infarction. Nonetheless, elevation of circulating troponins has been reported also in non-acute settings, including chronic HF, due to mechanisms still to be fully elucidated, possibly involving inflammation, neurohormonal activation, myocardial stretch, hypoxia, cytotoxicity [60]. With the improvement in troponin assays and the rise of high sensitivity troponins (hsTn), detectable circulating troponin is now observed in a large proportion of HF patients and several studies have explored its use in clinical practice as an additional biomarker for disease management. In 140 patients with acute HF, TnI was found to be increased in about 1/3 of cases and to independently predict 90-day mortality and readmission. Further, increase in TnI during hospitalization was also associated with a worse outcome [61]. In the RELAX-AHF trial, hs-TnT was elevated above the 99% upper reference limit in most of AHF patients. Baseline, peak, and peak change hs-cTnT were associated with worse outcomes, in particular with 180-day cardiovascular mortality [62]. In the ADHERE study, patient with TnI elevation had significantly higher in-hospital mortality when treated with intravenous inotropic therapy as compared with intravenous vasodilator therapy [63].
As in acute settings, circulating Tn is associated with prognosis also in chronic HF. In an analysis performed in >4,000 patients enrolled in the Val-HeFT trial, TnT was detectable in about 10% of patients using a conventional assay, while 92% showed detectable Tn when the hsTnT assay was used. In the same cohort, hsTnT was associated with the risk of death and LV remodelling, and improved prognostic discrimination when added to a baseline model including BNP [64]. The
prognostic utility of serial Tn measurement in chronic HF has been investigated in patients from both the Val-HeFT and the GISSI-HF study. In this cohort, although increase in hsTnT over 3 to 4 months was strongly associated with all-cause mortality after adjustement for clinical risk factors, baseline and NT-proBNP, it only modestly improved prognostic discrimination [65].
Previous reports show an association between the use of ACEi and betablockers with lower circulating Tn levels [65,66], thus suggesting that guideline-recommended therapy may mitigate the risk in subsets with elevated Tn. More recently, in the PARADIGM-HF trial, treatment with LCZ696 led to a significant and sustained reduction in hsTnT that was not observed in the enalapril arm [45]. Still, the issue of a Tn-guided therapy in chronic HF remains to be explored.
2.2.5 Candidate biomarkers of HFrEF
Natriuretic peptides represent nowadays a fundamental aid to the clinical management of HF, nonetheless circulating levels of either BNP or NT-proBNP increase following each generic insult to the cardiovascular system. Therefore, while they are highly sensitive in the detection of ongoing damage, they are not able to provide clinically significant information concerning the nature of the noxa. The development and progression of the HF syndrome results from a complex interplay between different pathogenic determinants sustaining the ongoing myocardial damage and (mal)adaptive mechanisms. As a consequence, biomarkers providing insights into the extent of activation of specific axes of organ damage progression may help the clinician in the process of disease (and patient) characterization and of treatment tailoring.
A huge number of biomarkers have been tested or are currently under investigation for HF which may be classified according to he pathophysiological mechanism of damage they are considered to reflect. As previously suggested, biomarkers of sympathetic and RAAS activation, inflammation, fibrosis and comorbidities can be distinguished [2,3].
Sympathetic activation. Activation of the sympathetic, and inhibition of the parasympathetic systems, represent the first (mal)adaptive mechanism in disease onset and progression and the bases of the neuroendocrine model of HF, given their vasoconstrictive, profibrotic and arrhythmogenic effects [67]. Circulating norepinephrine increase with disease severity [20], and, in a seminal paper by Cohn, it was the only independent predictor of mortality in 106 patients with moderate to severe HF, although such finding was not confirmed later on in more contemporary series [68,69]. Data from the ValHeFT have shown that treatment with valsartan can blunt the increase in norepinephrine compared to placebo, while it was not affected by spironolactone [70,71].
Together with catecholamines, chromogranin A is a component of chromaffin granules in the adrenal glands and, although its biological effects on the cardiovascular system remain to be elucidated, it seems to be involved in the regulation of adrenergic system [72]. Limited evidence exists that circulating chromogranin A is increased in either acute or chronic HF and that may have some prognostic value [73,74].
Renin-angiotensin-aldosterone system activation. RAAS is a complex endocrine system participated by the kidney, liver, vascular endothelium and adrenal cortex regulating salt/water homeostasis and vasomotion. Either systemic and tissue RAAS are involved in tissue remodeling after damage, and can promote fibrosis, hypertrophy and apoptosis. All the effectors of RAAS are increased in chronic HF [20] and RAAS activation is an indirect or direct target of most effective pharmacological treatments in HF, such as beta-blockers, ACE inhibitors, angiotensin receptor blockers, direct renin inhibitors, mineralocorticoid receptor blockers and angiotensin receptor/neprilysin inhibitors. Circulating biomarkers of RAAS activation are currently available, such as plasma renin activity (PRA), renin, angiotensin II, and aldosterone, although with different feasibility and accuracy, and some of them are well recognized prognostic factors, even in patients with optimal therapy. Notably, chronic use of drugs acting on RAAS induces, per se, neurohormonal reactivation [75,76].
PRA has been demonstrated to hold an independent prognostic value in a systolic HF cohort on evidence weighted treatment, with and without significant renal comorbidity [77,78], thus potentially qualifying as a tool to identify patients with persistent RAAS activation despite adequate neurohormonal antagonism, and to select subsets at higher risk for cardiovascular events. Further, aldosterone was shown to be a predictor of all-cause mortality in ≈300 patients with HF (most of them with systolic dysfunction), independently from other clinical and biohumoral variable including NT-proBNP [79]. Specifically, the increase in circulating aldosterone following the initiation of RAAS acting therapy, a phenomenon termed “aldosterone breakthrough” may hold clinical relevance, given the growing evidence on non-mineralocorticoid receptor-mediated effects of aldosterone. Further, in a small group of patients with systolic HF, increased PRA predicted ACE-inhibitors induced natriuresis [80].
The above mentioned data support the rational for the use of biomarkers of RAAS activation as a guide for treatment monitoring and tailoring, as well as a rule-in variable for trials in HF setting, a role up-to-now discarded, as for the case of trials with the direct renin inhibitor aliskiren.
Inflammation and immunity. There is growing information on the role of inflammatory cells and pathways during acute cardiovascular injury and in the reparative process that is subsequently
activated. Elevation of inflammatory biomarkers, including C-reactive protein (CRP), members of the interleukin family (e.g. IL-1, IL-6 and IL-18) and TNF-α, is a hallmark feature of chronic ischemic and non-ischemic HF, although whether inflammation is causative to disease progression is not yet clear [81]. Moreover, viral infection is thought to participate to the development of dilated cardiomyopathy, sustaining acute and chronic inflammation [82].
The Val-HeFT study demonstrated a direct correlation between elevated CRP level and HF severity [83]; further, CRP predicts the risk of death and early readmission in acutely decompensated HF [84]. Anti-inflammatory properties have been described for statins and their effect has been tested in HF settings. Data from the CORONA study show that subjects with HF of ischemic etiology and elevated baseline hsCRP (≥2 mg/L) exhibited a greater benefit from statin therapy in terms of reduction of the primary end-point of cardiovascular death, myocardial infarction and stroke [85], while controversial results come from studies performed on patients with HF of non-ischemic origin [86,87].
Some trials have addressed TNF-α elevation in HF, which is associated with worse prognosis. Two large clinical study, the Randomized Etanercept North American Strategy to Study Antagonism of Cytokines (RENAISSANCE) and the Research into Etanercept Cytokine Antagonism in Ventricular Dysfunction (RECOVER), were stopped because of lack of clinical benefit, and patients receiving the highest dose had increased adverse cardiac outcomes [88]. Similar results were observed in the Anti-TNF-α in Congestive Heart Failure (ATTACH) trial, testing humanized neutralizing antibodies (infliximab) instead of the soluble receptor etanercept [89]. The negative results of such trials may be, at least in part, explained by the inappropriate blockade of “physiological” inflammation, that may be required for tissue reparative processes.
Fibrosis. Galectin-3. In recent years, galectin-3 (Gal-3), a soluble β-galactoside binding lectin, has been found to play an important mechanistic role in the development of cardiac fibrosis and remodeling and to identify high risk subsets in HF cohorts, thus qualifying both as a risk marker and a risk factor [90]. There is a growing amount of evidence that Gal-3 is essential for migration and phagocitic activity of macrophages. Macrophage-derived Gal-3 may then act on fibroblast proliferation and on collagen synthesis, by increasing collagen I synthesis and reversing the collagen I to collagen III ratio [91]. Together with liver and kidney fibrosis, Gal-3 has been strictly linked to the development of cardiac fibrosis, a key determinant of cardiac remodeling and HF progression, possibly by interacting with mechanisms of aldosterone mediated-damage [92]. Further, circulating levels of Gal-3 are associated with biomarkers of extracellular matrix turnover (including PIIINP,
TIMP-1 and MMP-2), after adjustment for several clinical variables in a population of 106 patients with systolic HF [93].
The experimental demonstration of a mechanistic involvement of Gal-3 in fibrotic, inflammatory and remodeling processes in heart disease led to a novel interest in the potential use of Gal-3 assayed in plasma, as a biomarker. van Kimmenade and Colleagues in 2006 first compared NT-proBNP, apelin and Gal-3 in the management of acute HF patients [94]. Out of 599 acutely dyspneic patients, 209 were later diagnosed with HF. Although Gal-3 showed a limited diagnostic accuracy in identifying acute HF, it was the strongest predictor of early events (60-day re-hospitalization for HF or all-cause mortality). Later on, Gal-3 was also demonstrated to predict long-term outcome (4 year mortality) in another cohort of acute HF patients (proposed cut-off: 14.97 ng/mL), independently of echocardiographic indices of cardiac structure and function (LV end-diastolic/systolic diameter, EF and right ventricular pressure) [94,95]. In the following years the prognostic role of Gal-3 in HF settings has been investigated in some substudies from larger clinical trials. Gal-3 levels were determined in 232 NYHA III-IV chronic HF patients enrolled in the Deventer-Alkmaar HF (DEAL-HF) study, who were then followed-up for a period of 6.5 years [96]. Baseline Gal-3 (cut-off: 17.6 ng/ml) predicted all-cause mortality after adjustment for age, gender, creatinine clearance and NT-proBNP. By dichotomizing the population according to NT-proBNP and Gal-3 levels, the Authors also demonstrated an additive prognostic power for Gal-3, since patients with elevation of both biomarkers had a 1.5- to 2-fold higher mortality rate compared to patients in other subgroups. In a larger population of 895 chronic HF patients from the HF-ACTION (Heart Failure: A Controlled Trial Investigating Outcomes of Exercise TraiNing) study with LVEF <35%, Gal-3 lost its univariate prognostic value in predicting the composite outcome of all-cause death or rehospitalisation, when corrected for peak oxygen consumption at cardiopulmonary test or NT-proBNP [97]. Despite this wide amount of data confirming its prognostic role in HF, there is still limited information on whether Gal-3 assay may help in adjusting therapeutical strategies. Current knowledge is indeed limited to a benefit from statin therapy in patients with chronic HF of ischemic cause and low Gal-3 level [98] and to the lack of power in predicting response to MRA or to cardiac resynchronization therapy [99,100].
Recently, measurement of Gal-3 has received a class IIb recommendation in acute decompensated (level of evidence A) and in chronic (level of evidence B) HF for risk stratification purposes in the 2013 ACCF/AHA Guideline for the Management of Heart Failure [21].
sST2. Suppression of tumorigenicity 2 protein (ST2) is a member of the Toll-like/interleukin-1 receptor superfamily, which is expressed, together with its ligand interleukin-33 (IL-33) following myocardial stretch and cardiovascular injury. The IL-33/ST2 interaction exerts positive effects by