Structure of fully protonated proteins by
proton-detected magic-angle spinning NMR
Loren B. Andreasa, Kristaps Jaudzemsa, Jan Staneka, Daniela Lallia, Andrea Bertarelloa, Tanguy Le Marchanda, Diane Cala-De Paepea, Svetlana Kotelovicab, Inara Akopjanab, Benno Knottc, Sebastian Wegnerc, Frank Engelkec, Anne Lesagea, Lyndon Emsleya,d, Kaspars Tarsb, Torsten Herrmanna, and Guido Pintacudaa,1
aCentre de Résonance Magnétique Nucléaire à Très Hauts Champs, Institut des Sciences Analytiques (UMR 5280– CNRS, Ecole Normale Supérieure de Lyon, Université Claude Bernard Lyon 1), Université de Lyon, 69100 Villeurbanne, France;bBiomedical Research and Study Centre, LV-1067 Riga, Latvia;cBruker Biospin, 76287 Rheinstetten, Germany; anddInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
Edited by Adriaan Bax, National Institutes of Health, Bethesda, MD, and approved June 21, 2016 (received for review February 18, 2016) Protein structure determination by proton-detected magic-angle
spinning (MAS) NMR has focused on highly deuterated samples, in which only a small number of protons are introduced and observa-tion of signals from side chains is extremely limited. Here, we show in two fully protonated proteins that, at 100-kHz MAS and above, spectral resolution is high enough to detect resolved correlations from amide and side-chain protons of all residue types, and to
reliably measure a dense network of1H-1H proximities that define a
protein structure. The high data quality allowed the correct identi-fication of internuclear distance restraints encoded in 3D spectra with automated data analysis, resulting in accurate, unbiased, and fast structure determination. Additionally, we find that narrower proton resonance lines, longer coherence lifetimes, and improved magnetization transfer offset the reduced sample size at 100-kHz spinning and above. Less than 2 weeks of experiment time and a single 0.5-mg sample was sufficient for the acquisition of all data necessary for backbone and side-chain resonance assignment and unsupervised structure determination. We expect the technique to pave the way for atomic-resolution structure analysis applicable to a wide range of proteins.
NMR spectroscopy
|
magic-angle spinning|
protein structures|
proton detection|
viral nucleocapsidsD
espite tremendous progress in the analysis of biomolecular samples over the last two decades (1–7), routine application of magic-angle spinning (MAS) NMR in biology is still limited by the inherently low sensitivity. The direct detection of proton reso-nances is a straightforward way to counter this problem, but entails a trade-off with resolution due to the strong homonuclear dipolar interactions among proton nuclei. High-resolution proton-detected methods were first demonstrated with modest spinning frequencies by today’s standards (∼10 kHz) and relied on a reduction of1H-1H couplings by high levels of dilution with deuterium, typically perdeuteration, and complete (8, 9) or partial (10–12) protonation at exchangeable sites. The need for narrow proton resonances without such extreme levels of deuteration has motivated a con-tinuous technological development, resulting in a dramatic increase in the available spinning frequency (13–20).At MAS frequencies of 40–60 kHz, deuteration and 100% reprotonation at exchangeable sites, primarily amide protons, re-sult in resolved and sensitive spectra, similar in quality to the case of higher dilution levels and lower spinning frequencies (21–23). This opens the way to rapid sequential assignment of backbone resonances (24–27), as well as to the unambiguous measurement of detailed structural and dynamical parameters (28–32). A further increase in the MAS frequency to 100 kHz allows resonance as-signment (20), a structure determination of a model protein (16), and interaction studies (15) with as little as 0.5 mg of sample.
However, a high deuteration level severely limits observation of side-chain signals, which are essential for the determination of a protein structure at high resolution. The redundancy of information intrinsic to spectra of side-chain protonated proteins, which leads to
a mutually supportive network of distance restraints, is also crucial to an unbiased and robust spectral analysis by unsupervised algo-rithms (33–36).
One approach to access side-chain proton resonances is the fractional labeling of side chains with2H and1H, which has the advantage of protonation at many sites, but with a dramatic re-duction in sensitivity and potential loss of resolution due to the presence of multiple isotopomers (37–39). These problems have limited the application of these strategies for protein structure determination.
Alternatively, ILV-methyl labeling with suitable precursors has been used for structure determinations (16, 28, 40), and other tailored labeling approaches have also been proposed (41). How-ever, these methods still introduce only a limited set of side-chain protons. Furthermore, they are implemented with deuterated growth media, which can reduce or even eliminate protein ex-pression. Moreover, complete amide reprotonation in the interior of the protein can be problematic for systems that lack a refolding protocol (42), such as investigated in the present study. Extremely narrow aliphatic lines were reported for leucine residues in a stereo-array isotope-labeled (SAIL) protein at 80-kHz MAS (43). Unfortunately, the high cost of SAIL has prevented its widespread application.
All of these drawbacks are overcome if resolved side-chain proton resonances are available from a fully protonated sample. Fully protonated samples are by far the simplest to produce, and
Significance
Protein structure determination is key to the detailed description of many biological processes. The critical factor that would allow general application of magic-angle spinning (MAS) solid-state NMR to this end is improvement in sensitivity and resolution for as many nuclear spins as possible. This is achieved here with detection of resolved1H resonances in protonated proteins by
increasing MAS rates to frequencies of 100 kHz and above. For large proteins and assemblies, ultrafast spinning narrows spectral resonances better than Brownian motion on which solution NMR relies, removing a fundamental barrier to the NMR study of large systems. This is exploited here to determine the de novo struc-ture of a 28-kDa protein dimer in a 2.5-MDa viral capsid assembly.
Author contributions: L.B.A., K.J., J.S., D.L., A.L., L.E., K.T., T.H., and G.P. designed research; L.B.A., K.J., J.S., D.L., A.B., T.L.M., D.C.-D.P., S.K., I.A., K.T., T.H., and G.P. performed research; D.C.-D.P., S.K., I.A., B.K., S.W., and F.E. contributed new reagents/analytic tools; L.B.A., K.J., J.S., D.L., A.B., T.L.M., T.H., and G.P. analyzed data; and L.B.A., K.J., J.S., D.L., A.B., A.L., L.E., K.T., T.H., and G.P. wrote the paper.
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and restraints have been deposited in the Pro-tein Data Bank,www.wwpdb.org/(PDB ID codes5JXVand5JZR), and chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank,www.bmrb.wisc.edu/ (accession codes30094and30088).
1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online atwww.pnas.org/lookup/suppl/doi:10. 1073/pnas.1602248113/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1602248113 PNAS | August 16, 2016 | vol. 113 | no. 33 | 9187–9192
CHEMISTR Y BIOPHYSIC S AND COMPUTAT IONAL BIOLOGY
their effective use could be applied to a much wider array of molecules including biomolecules labeled in mammalian systems (44). However, although amide proton assignment is possible for fully protonated proteins above 40-kHz MAS (45–48), proton resonances remain significantly dipolar broadened at 40–60 kHz, limiting the applicability of this spinning regime for side-chain assignment and structure determination (41, 45, 49–51).
Here, we investigate the resolution at an increased MAS fre-quency of 100 kHz and above, and find that it enables rapid structure determination in fully protonated proteins. To our knowledge, we present the first two examples of such structure determinations, the small model protein GB1 (52) and the de novo determination of Acinetobacter phage 205 (AP205) coat protein (AP205CP), a 28-kDa dimer in a 2.5-MDa viral capsid assembly (53). In both cases, we used a single 0.5-mg sample of uniformly 13C,15N–labeled protein for backbone and side-chain resonance assignment, and for collection of a dense network of proton–proton distance restraints. Each structure was calculated
from data acquired in less than 2 weeks, and making use of unsupervised analysis algorithms as implemented in UNIO (54). Results and Discussion
NMR Structures. Fig. 1 shows structural ensembles for
micro-crystalline GB1 and sedimented micromicro-crystalline nucleocapsids of AP205CP, determined by solid-state NMR with MAS at fre-quencies of 100 kHz and above.
GB1 is a small and well-characterized globular protein of 56 residues. The bundle of NMR conformers calculated here (Fig. 1A) has a backbone heavy-atom root-mean-square deviation (rmsd) of 0.48 Å, an all-heavy-atom rmsd of 1.04 Å, and deviates from the X-ray structure (PDB ID code 2QMT) by 1.45 Å (see full statistics inTable S1).
AP205CP forms a homodimer of 2× 130 residues, which is the basic subunit of the icosahedral AP205 capsid comprised of 90 dimers (53). The bacteriophage encodes for four genes, which are packed as single-stranded RNA (ssRNA) in a 28- to 30-nm icosahedral protein capsid. Because of its size, the AP205 capsid
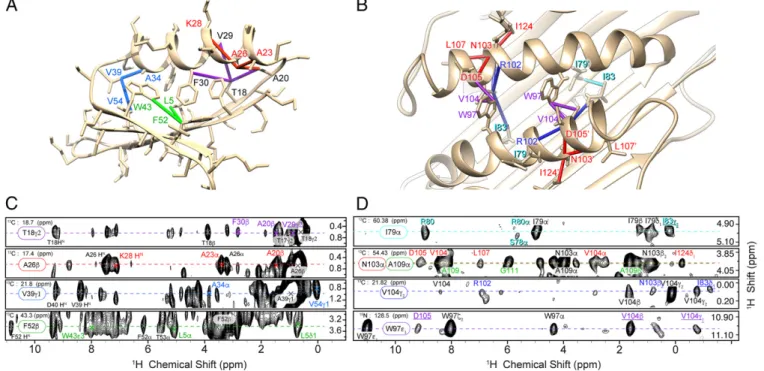
Fig. 1. MAS NMR structures of GB1 (PDB ID code 5JXV; Top) and AP205CP (PDB ID code 5JZR; Bottom). Ribbon diagram of the 10 lowest-energy conformers for GB1 (A) and dimeric AP205CP (E), with monomers colored in cyan and tan. The approximate location of 1 of the 90 dimers in the cryo-EM electron density is also illustrated (53). The long-range proton–proton contacts measured in this study are depicted as dark blue lines onto the lowest-energy conformers of the two pro-teins: contacts between amide protons (B and F), con-tacts among amide and ILV-methyl–labeled protons (C and G), and all proton–proton contacts (D and H).
Fig. 2. High-resolution1H-detected spectra recorded on fully protonated microcrystalline GB1 and AP205CP. (A)15N-1H CP-HSQC of GB1 at 60 kHz (black), and at 111.111-kHz MAS (red). (B) Hα-Cα (Bottom) and methyl regions (Top) of a13C-1H CP-HSQC of GB1 at 111.111-kHz MAS. (C)15N-1H CP-HSQC of AP205CP acquired on a fully protonated sample at 100-kHz MAS (red), and on a perdeuterated sample, exchanged in 100% H2O, at 60-kHz MAS (black). The exchange-protected residues are labeled in the spectrum and colored on the NMR structure in the Inset. (D) Hα-Cα (Bottom) and methyl regions (Top) of a13C-1H CP-HSQC of AP205CP at 100-kHz MAS.
is inaccessible to solution NMR. Its crystals can be obtained easily in many different conditions, but despite our best efforts they do not diffract beyond 15-Å resolution, and similarly the cryo-EM structure (53) is resolved to only about 20 Å. However, sedimented microcrystals of AP205 nucleocapsids provide resolved MAS NMR spectra (27). The bundle of NMR conformers of the AP205CP dimer calculated here (Fig. 1E) has a backbone heavy-atom rmsd of 1.23 Å and an all-heavy-atom rmsd of 1.84 Å over structured regions (Table S1). The structure reveals a chain-intertwined dimer, typical for all ssRNA phages, with an extendedβ-sheet formed from both monomers and a pair of long helices engaged in a multitude of intermolecular contacts. The NMR structure is in agreement with the X-ray structure of an assembly-deficient AP205CP mutant dimer (PDB ID code 5FS4) to a backbone heavy-atom rmsd of 2.35 Å.
Each of the two structures is held together by a dense network of long-range (between residues i and j,ji − jj > 4)1H-1H contacts, also shown in Fig. 1, which encode distances up to about 5.5 Å. This amounts to 236 and 410 meaningful long-range contacts for GB1 and AP205CP, respectively, including 104 intermolecular contacts that define the dimer interface of AP205CP (Table S1). For these two structures, the total information content is described by∼13.7 and 5.3 meaningful, nonredundant distance restraints per residue, approaching the criteria established for solution NMR determina-tions at high resolution (55). For AP205CP, the restraints per residue increase to 6.2 by considering only the assigned residues, and to 7.7 by considering only residues in structured regions.
Previous structural approaches in1H-detected MAS NMR relied on extensive deuteration, which resulted in the observation of contacts only among amide protons and ILV-methyl sites (16, 23, 28, 31, 56).
However, amide–amide contacts (Fig. 1 B and F), or even contacts between ILV-methyl and amide protons (Fig. 1 C and G), represent a small number of the total, the large majority of which involve side-chain protons (Fig. 1 D and H). Although in principle a small set of long-range restraints could define a high-quality structure, a large set indicates a high-quality determination in the presence of limited pre-cision and assignment ambiguity, which are typical of many systems of interest. As shown in Fig. 1, a large set of proton–proton contacts can be effectively acquired and automatically assigned in fully protonated proteins. This set includes side-chain protons, which are essential to constrain secondary structure elements (helix–sheet contacts in GB1, and helix–sheet and helix–helix contacts in AP205CP). The key for obtaining these sets of distance constraints lies in the possibility of directly recording and assigning resolved resonances for proton sites throughout the backbone and side chains of the two proteins.
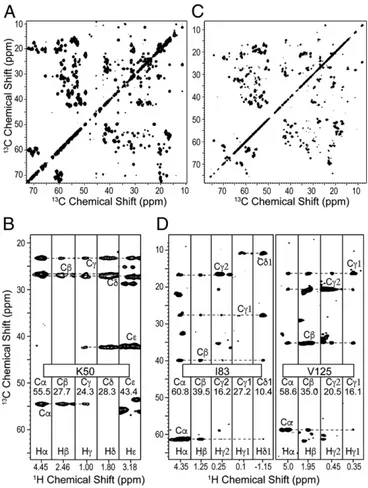
Resolution of1H-Detected Spectra.GB1 is a 6.2-kDa model
micro-crystalline system known for its high spectral quality in solid-state NMR (52). Fig. 2 A and B shows its amide and aliphatic 2D cor-relation spectra [cross-polarization heteronuclear single-quantum correlation (CP-HSQC)] at 60 and 111 kHz, which at the faster spinning condition demonstrate amide proton linewidths averaging about 100 Hz (0.1 ppm at 1 GHz) and aliphatic and aromatic proton linewidths of 100–200 Hz (0.1–0.2 ppm).
The 28-kDa AP205CP dimer represents a challenging case of biological interest. The sample exhibits increased heterogeneity, manifested in linewidths of about 150–200 Hz (0.15–0.20 ppm) for amide, Hα, and methyl protons, at 100-kHz MAS (Fig. 2 C and D). A successful structure calculation in the presence of this inhomogeneity suggests that the method will be applicable to a range of biological targets of similar spectral quality, such as membrane-embedded proteins, fibrils, and macromolecular as-semblies (27, 57, 58).
Fig. 2C also compares the amide spectrum of AP205CP to that of a perdeuterated sample purified in H2O. Although most amide protons exchange during purification, as indicated by the obser-vation of a cross-peak in both spectra, several weak or missing signals for the perdeuterated sample indicate incomplete exchange, a phenomenon observed for a variety of proteins (24, 59). For AP205CP, amide groups engaged in hydrogen bonds in the middle of the twoα-helices, in the innermost β-strands, or at the dimer interface, are inaccessible to proton exchange. In general, nonexchangeable protons lie in the most structured regions of a protein, where the density of potential internuclear contacts is higher. The possibility of studying samples directly expressed in 100% H2O is an advancement that extends1H-detected struc-tural determinations to key exchange-protected regions.
Backbone and Side-Chain Resonance Assignment.1H,13C, and15N
backbone and side-chain resonances were assigned using a suite of proton detected 3D spectra correlating backbone and side-chain chemical shifts.
For GB1,1HN,13Cα,13C′, and15N backbone resonances were first assigned using both13C-based interresidue matching (27), amide15N matching (48), and automated analysis using the UNIO-MATCH algorithm (60, 61). These assignments were then extended by mea-surement of the Hα shifts using a (H)NCAHA spectrum (46). For AP205CP, previous backbone 1HN, 13Cα,13Cβ, 13C′, and 15N as-signments (27) were manually extended to 78% of the sequence and to the assignment of Hα resonances with a set of experiments linking Cα/Hα pairs in sequential residues through15N or13C′ resonances. Next, Hα and Cα chemical shifts were used as anchors to propagate resonance assignment from backbone to side chains with a (H)CCH spectrum (62). This was implemented here with a CP-based sequence and WALTZ-16 mixing (63), applied at a nutation frequency of one-quarter of the rotor frequency (25 or 27.8 kHz), to induce isotropic mixing of13C magnetization. This spectrum correlates multiple 13C resonances of an aliphatic side chain to each of its side-chain 1H resonances, resulting in the robust identification of the1H and13C shifts. This is in contrast with ILV-labeled samples, in which it is difficult to unambiguously
Fig. 3. (A and C)13C-13C projection and (B and D) selected strips of the (H)CCH spectrum of GB1 (A and B) and AP205CP (C and D). GB1 and AP205CP spectra were run at a MAS frequency of 111 or 100 kHz, respectively, with WALTZ-16 mixing applied for 15 ms at 27.8 kHz or 14.4 ms at 25 kHz of radiofrequency field.
Andreas et al. PNAS | August 16, 2016 | vol. 113 | no. 33 | 9189
CHEMISTR Y BIOPHYSIC S AND COMPUTAT IONAL BIOLOGY
connect a single methyl proton at the end of a side chain to an amide1H resonance (in a1H-detected approach) or to a backbone 13C resonance (in a13C-detected approach). Fig. 3 shows the 13C-13C planes from the 3D (H)CCH spectrum for the two pro-teins, and strips corresponding to the13C-1H resonances of selected residues.
Overall, for GB1, the completeness of assignment of proton resonances was 94.4%, and the all-atom completeness was 85.9%, with the missing resonances being primarily aromatics. For AP205CP, the completeness of assignment of proton resonances was 64.9%, and the all-atom completeness was 62.6%.
1H-1H Contacts and Structure Calculation. For protein fold
de-termination, 3D radiofrequency-driven recoupling (RFDR) spectra (64) were recorded that directly probe 1H-1H proxim-ities. These spectra contain two 1H dimensions and therefore benefit doubly from the narrow backbone and side-chain proton resonances. In this specific implementation, 1H-1H contacts were resolved using the shift of15N, aliphatic13C, or aromatic13C, and acquired in 72 h for GB1 and 103 h for AP205CP.
For each of these spectra, signals were manually identified and converted into unassigned peak lists, containing the frequency coordinates and the intensity of each signal. For GB1, the NMR structure was calculated with the UNIO software package using as inputs these unrefined peak lists, the assigned backbone and side-chain chemical shifts, and backbone dihedral angles pre-dicted from the chemical shifts using TALOS+ (65). The stan-dard unsupervised protocol of iterative cross-peak assignment, conversion into distance restraints, and structure calculation was applied as implemented in the program UNIO-CANDID (66). The chemical-shift–based assignment tolerances were set to the corresponding experimental linewidths, namely 0.15 and 0.4 ppm for proton and heavy-atom dimensions, respectively. Despite the fivefold increase in the proton tolerances compared with typi-cally used solution NMR parameters, the average initial
as-signment ambiguity per peak of ∼16 remained modest. Not
surprisingly, due to the high quality of the input data, an efficient network-anchored assignment of the contacts (66) resulted in a defined fold of the protein already in the first cycle of the
iterative protocol. This is a crucial criterion certifying the re-liability of a result in an unsupervised data analysis run (35).
AP205CP represents a particular challenge for structure de-termination due to the larger size and the dimeric protein topology. Using assignment tolerances corresponding to the experimental linewidths, the initial chemical-shift–based assignment ambiguity per peak was∼56, which corresponds to a computational ambiguity of 112, due to the need to distinguish between intrasubunit and intersubunit contacts in the dimer. To improve convergence, the UNIO-CANDID protocol described for GB1 was supplemented with four 1H-1H distance restraints and 27 hydrogen bond re-straints. Namely, two intermolecular helix–helix restraints (Val104 Hγ1-Trp97 He1 and Asp105 Hα-Trp97 He1), one intermolecular helix–strand restraint (Val104 Hγ1-Ile83 Hδ1), and one intra-molecular helix–loop restraint (Asn103 Hα-Ile124 Hδ1) were applied. These spectrally unambiguous distance restraints were manually identified based on unique chemical shifts of involved side-chain1H nuclei. Two helix–helix contacts were entered as intermolecular because they cannot be satisfied for a monomer structure, whereas the calculation converges to the correct fold when the other two manually entered restraints are defined as ambiguous, i.e., as intermolecular or intramolecular (Supporting Information). Based on the observed chemical shifts that indicatedβ-sheet sec-ondary structure, and on the observation of cross-strand Hα-Hα, HN-HN, and Hα-HNRFDR contacts, 6 intermolecular and 21 intramolecular hydrogen bonds were imposed in the calculation betweenβ-strands as detailed inFig. S1. By using these additional restraints, a defined fold for the dimer was found after the first iteration of the automated UNIO-CANDID cross-peak assign-ment ensuring a reliable final result (Supporting Information).
It should be emphasized that relative alignment of two AP205CP monomers could be determined from the NMR data without the need for mixed isotope labeling that is often applied to filter intramolecular contacts (67). This is particularly important because there is no protocol for disassembly and reassembly of the AP205CP complex that would be needed for such an approach.
Statistics of the experimental restraints and structure calcula-tion are summarized inTables S1–S3, and further details can be found inSupporting Information. Although it was not possible to
Fig. 4. (A and B) Representative restraints from the (H)CHH spectra are displayed on the lowest-energy NMR structures of GB1 (A) and AP205CP (B). The color of these restraints indicates the corresponding strip from the spectra shown in C and D. In the case of AP205CP (D), the labels of intermolecular cross-peaks are underlined. The RFDR mixing time was 0.5 ms for GB1 and 1.0 ms for AP205CP.
unambiguously identify any interdimer contacts from the present 3D RFDR spectra, the determination of the AP205CP dimer structure paves the way to a future joint analysis of the NMR data with the cryo-EM density maps (53) to assemble 90 dimers into an atomic-level description of the global capsid architecture.
Sensitivity of 1H-Detected Spectra. The high-resolution spectra
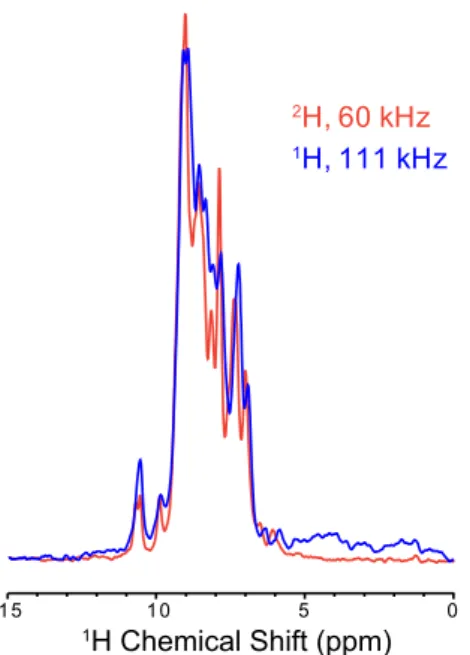
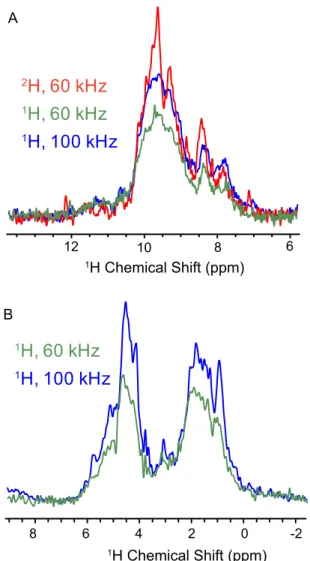
demonstrated in Figs. 2–4 were acquired with no compromise in sensitivity compared with the previous state of the art at 60-kHz MAS in 1.3-mm rotors. An increase in the spinning rate requires reduced sample dimensions. This provides increased detection sensitivity associated with improved inductive coupling of smaller coils (almost a factor of 2 with respect to 1.3-mm rotors) but invariably entails a reduction in the sample volume (here by a factor of 4–5 with respect to 1.3-mm rotors) (18, 68). Combined, we therefore expect a theoretical 2.2- to 2.7-fold loss in the sensitivity of a single-pulse spectrum. Surprisingly, we measured approximately equal sensitivity for15N-1H and13C-1H CP-HSQC spectra in a 0.7- and a 1.3-mm probe, as shown by the comparison of the total area in the 1D proton spectra ofFigs. S2andS3. This observation points to an improvement in other factors, such as radiofrequency (RF) homogeneity, matching of RF field profiles along the rotor axis, and probe electronics.
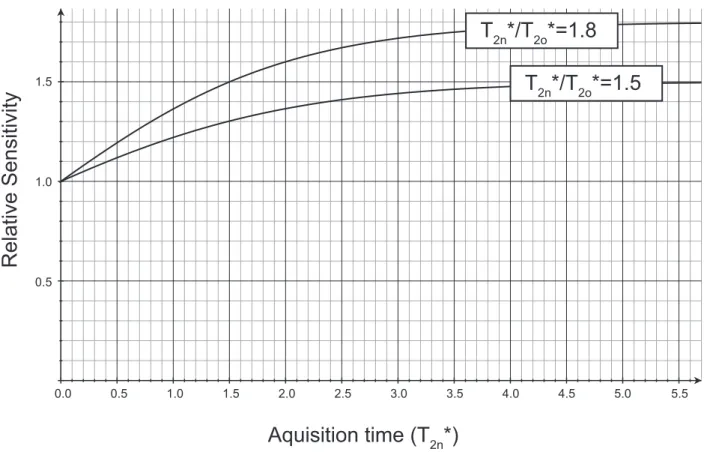
Additionally, loss in sample volume is offset by narrower proton lines, longer coherence lifetimes, and improved transfer efficiencies (69). For GB1, the amide1H refocused transverse coherence lifetime (T2′) at ∼111-kHz MAS increased to 4.6 ms compared with 1.8 ms at the previous state-of-the-art of 60-kHz MAS. This corresponds to a nonrefocusable linewidth of about 70 Hz, indicating that about one-half of the contribution to the linewidth is still homogeneous. We observed improvement in1H linewidths by about a factor of 1.5–1.8 for GB1 and dramatic improvements in T2′ of13C′,13Cα, and15N, when increasing the MAS rates from 60 to∼111 kHz (Supporting Information). This results in improved J-transfer efficiency and therefore higher sensitivity or reduced experiment time for sequences that depend on the T2′, producing for GB1 an additional sensitivity gain of about 2.1 for an out-and-back Cα → Cβ transfer, and 1.6 for a full C′ → Cα transfer (48). Finally, for the RFDR-based 3Ds that encode two proton dimensions, there is also a sensitivity benefit that depends on the indirect sampling. For example, when sampling extends to 1.5T2* at 111-kHz MAS, as in the case of GB1, this amounts to a factor of about 1.3–1.5, as detailed in
Fig. S4. Gains due to linewidth and T2′ are modest for AP205CP for backbone resonances due to significant inhomogeneity in this sample. The exact sensitivity gain depends on the details of the sample and the pulse sequence, but is more significant for ex-periments with many transfers on which the majority of the data acquisition time is spent. This bodes well for the extension to higher dimensional spectra for fully protonated samples once higher sensitivity is available from cryoprobes or higher field magnets.
Conclusions
In conclusion, to our knowledge, we have presented here the first examples of protein structure determination by MAS NMR
using fully protonated samples and1H detection. The narrowed 1H linewidth, which arises due to fast sample spinning, allows the successful application of efficient protocols for resonance as-signment and automated structure determination involving backbone and side-chain protons from all residue types. Notably, we have demonstrated that backbone and aliphatic side-chain assignment and high-resolution protein structure determination can be achieved using less than 2 weeks of instrument time, a single 0.5-mg sample, and rapid and unsupervised analysis of internuclear contacts.
The straightforward1H-based structure determination under fast sample spinning removes a fundamental barrier to the effi-cient NMR study of large systems, because the high resolution operated by MAS does not rely on stochastic Brownian motion, and therefore is independent of the size of the molecule, as demonstrated here in the de novo atomic-resolution structure of AP205CP in an intact 2.5-MDa capsid assembly. This approach overcomes the need for deuteration, and not only allows access to side-chain protons, but also circumvents potential problems of incomplete proton backexchange and difficulty of protein ex-pression in deuterated media.
We expect the approach to enable structure determination for a wide range of molecules such as membrane proteins and macromolecular complexes.
Methods
Sample Preparation. Uniformly 13C,15N-labeled GB1 and AP205CP samples were expressed in Escherichia coli, purified, and precipitated as described previously (26, 52). A full description is provided inSupporting Information.
Structure Calculation. For both proteins, the standard UNIO protocol was used, which consists of seven cycles of cross-peak assignment, conversion into a meaningful, nonredundant set of distance restraints, and structure calcu-lation by simulated annealing. In the second and subsequent cycles, the in-termediate protein structures were used to guide the process of cross-peak assignment. Further details are discussed inSupporting Information, and calculation statistics are reported inTables S2andS3.
NMR Spectroscopy. All spectra were recorded atω0H/2π = 1 GHz and a MAS rate of 100 kHz (AP205CP) or 111.111 kHz (GB1) using a Bruker 0.7-mm HCN probe. Spectrometer settings, as well as acquisition and processing param-eters specific for each 2D and 3D spectrum, are discussed inSupporting In-formationand summarized inTables S4andS5. Simulations of the isotropic 13C-13C mixing were performed using the software package SIMPSON (70) and are reported inFig. S5.
ACKNOWLEDGMENTS. We thank Lénaïc Leroux for technical assistance with the NMR spectrometers. We acknowledge financial support from CNRS (IR-RMN FR3050 and Fondation pour la Chimie des Substances Naturelles), from the People Programme of the European Union’s Seventh Framework Pro-gramme (FP7) (FP7-PEOPLE-2012-ITN 317127“pNMR”), and from the Euro-pean Research Council under the EuroEuro-pean Union’s Horizon 2020 Research and Innovation Programme (Grant 648974“P-MEM-NMR”). L.B.A., K.J., and J.S. are supported by three Marie Curie incoming fellowships (Research Ex-ecutive Agency Grant Agreements 624918“MEM-MAS,” 661175 “virus-DNP-NMR,” and 661799 “COMPLEX-FAST-MAS”). J.S. received support from a European Molecular Biology Organization fellowship (ALTF 1506-2014, LTFCOFUND2013, and GA-2013-609409).
1. Castellani F, et al. (2002) Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 420(6911):98–102.
2. Lange A, et al. (2006) Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature 440(7086):959–962.
3. Wasmer C, et al. (2008) Amyloid fibrils of the HET-s(218-289) prion form a beta so-lenoid with a triangular hydrophobic core. Science 319(5869):1523–1526. 4. Jehle S, et al. (2010) Solid-state NMR and SAXS studies provide a structural
basis for the activation of alphaB-crystallin oligomers. Nat Struct Mol Biol 17(9): 1037–1042.
5. Loquet A, et al. (2012) Atomic model of the type III secretion system needle. Nature 486(7402):276–279.
6. Wang S, et al. (2013) Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat Methods 10(10): 1007–1012.
7. Xiao Y, et al. (2015) Aβ(1-42) fibril structure illuminates self-recognition and replica-tion of amyloid in Alzheimer’s disease. Nat Struct Mol Biol 22(6):499–505.
8. Chevelkov V, et al. (2003)1H detection in MAS solid-state NMR spectroscopy of
bio-macromolecules employing pulsed field gradients for residual solvent suppression. J Am Chem Soc 125(26):7788–7789.
9. Paulson EK, et al. (2003) Sensitive high resolution inverse detection NMR spectroscopy of proteins in the solid state. J Am Chem Soc 125(51):15831–15836.
10. Zheng L, Fishbein KW, Griffin RG, Herzfeld J (1993) 2-Dimensional solid-state1H-NMR
and proton-exchange. J Am Chem Soc 115(14):6254–6261.
11. Chevelkov V, Rehbein K, Diehl A, Reif B (2006) Ultrahigh resolution in proton solid-state NMR spectroscopy at high levels of deuteration. Angew Chem Int Ed Engl 45(23):3878–3881.
12. Akbey U, et al. (2010) Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. J Biomol NMR 46(1):67–73.
13. Kobayashi T, et al. (2013) Study of intermolecular interactions in the corrole matrix by solid-state NMR under 100 kHz MAS and theoretical calculations. Angew Chem Int Ed Engl 52(52):14108–14111.
Andreas et al. PNAS | August 16, 2016 | vol. 113 | no. 33 | 9191
CHEMISTR Y BIOPHYSIC S AND COMPUTAT IONAL BIOLOGY
14. Nishiyama Y, Malon M, Ishii Y, Ramamoorthy A (2014) 3D15N/15N/¹H chemical shift
correlation experiment utilizing an RFDR-based ¹H/¹H mixing period at 100 kHz MAS. J Magn Reson 244:1–5.
15. Lamley JM, et al. (2014) Solid-state NMR of a protein in a precipitated complex with a full-length antibody. J Am Chem Soc 136(48):16800–16806.
16. Agarwal V, et al. (2014) De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem Int Ed Engl 53(45):12253–12256.
17. Zhang R, Pandey MK, Nishiyama Y, Ramamoorthy A (2015) A novel high-resolution and sensitivity-enhanced three-dimensional solid-state NMR experiment under ultrafast magic angle spinning conditions. Sci Rep 5:11810.
18. Andreas LB, Le Marchand T, Jaudzems K, Pintacuda G (2015) High-resolution proton-detected NMR of proteins at very fast MAS. J Magn Reson 253:36–49.
19. Böckmann A, Ernst M, Meier BH (2015) Spinning proteins, the faster, the better? J Magn Reson 253:71–79.
20. Penzel S, et al. (2015) Protein resonance assignment at MAS frequencies approaching 100 kHz: A quantitative comparison of J-coupling and dipolar-coupling-based transfer methods. J Biomol NMR 63(2):165–186.
21. Zhou DH, et al. (2007) Solid-state protein-structure determination with proton-detected triple-resonance 3D magic-angle-spinning NMR spectroscopy. Angew Chem Int Ed Engl 46(44):8380–8383.
22. Lewandowski JR, et al. (2011) Enhanced resolution and coherence lifetimes in the solid-state NMR spectroscopy of perdeuterated proteins under ultrafast magic-angle spinning. J Chem Phys Lett. 2(17):2205–2211.
23. Knight MJ, et al. (2011) Fast resonance assignment and fold determination of human superoxide dismutase by high-resolution proton-detected solid-state MAS NMR spectroscopy. Angew Chem Int Ed Engl 50(49):11697–11701.
24. Ward ME, et al. (2011) Proton-detected solid-state NMR reveals intramembrane polar networks in a seven-helical transmembrane protein proteorhodopsin. J Am Chem Soc 133(43):17434–17443.
25. Zhou DH, et al. (2012) Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. J Biomol NMR 54(3):291–305. 26. Barbet-Massin E, et al. (2013) Out-and-back13C-13C scalar transfers in protein
reso-nance assignment by proton-detected solid-state NMR under ultra-fast MAS. J Biomol NMR 56(4):379–386.
27. Barbet-Massin E, et al. (2014) Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc 136(35):12489–12497.
28. Huber M, et al. (2011) A proton-detected 4D solid-state NMR experiment for protein structure determination. ChemPhysChem 12(5):915–918.
29. Knight MJ, et al. (2012) Structure and backbone dynamics of a microcrystalline metalloprotein by solid-state NMR. Proc Natl Acad Sci USA 109(28):11095–11100. 30. Ma P, et al. (2014) Probing transient conformational states of proteins by solid-state R1ρ
relaxation-dispersion NMR spectroscopy. Angew Chem Int Ed Engl 53(17):4312–4317. 31. Linser R, et al. (2014) Solid-state NMR structure determination from diagonal-compensated,
sparsely nonuniform-sampled 4D proton-proton restraints. J Am Chem Soc 136(31): 11002–11010.
32. Good DB, et al. (2014) Conformational dynamics of a seven transmembrane helical protein Anabaena Sensory Rhodopsin probed by solid-state NMR. J Am Chem Soc 136(7):2833–2842.
33. Guerry P, Herrmann T (2011) Advances in automated NMR protein structure de-termination. Q Rev Biophys 44(3):257–309.
34. Rosato A, et al. (2012) Blind testing of routine, fully automated determination of protein structures from NMR data. Structure 20(2):227–236.
35. Guerry P, Duong VD, Herrmann T (2015) CASD-NMR 2: Robust and accurate un-supervised analysis of raw NOESY spectra and protein structure determination with UNIO. J Biomol NMR 62(4):473–480.
36. Buchner L, Güntert P (2015) Systematic evaluation of combined automated NOE as-signment and structure calculation with CYANA. J Biomol NMR 62(1):81–95. 37. Asami S, Schmieder P, Reif B (2010) High resolution1H-detected solid-state NMR
spectroscopy of protein aliphatic resonances: Access to tertiary structure information. J Am Chem Soc 132(43):15133–15135.
38. Asami S, Szekely K, Schanda P, Meier BH, Reif B (2012) Optimal degree of protonation for ¹H detection of aliphatic sites in randomly deuterated proteins as a function of the MAS frequency. J Biomol NMR 54(2):155–168.
39. Mance D, et al. (2015) An efficient labelling approach to harness backbone and side-chain protons in1H-detected solid-state NMR spectroscopy. Angew Chem Int Ed Engl
54(52):15799–15803.
40. Andreas LB, et al. (2015) Structure and mechanism of the influenza A M218-60 dimer of dimers. J Am Chem Soc 137(47):14877–14886.
41. Sinnige T, Daniëls M, Baldus M, Weingarth M (2014) Proton clouds to measure long-range contacts between nonexchangeable side chain protons in solid-state NMR. J Am Chem Soc 136(12):4452–4455.
42. Lian LY, Middleton DA (2001) Labelling approaches for protein structural studies by solution-state and solid-state NMR. Prog Nucl Magn Reson Spectrosc 39:171–190. 43. Wang S, et al. (2015) Nano-mole scale side-chain signal assignment by1H-detected
protein solid-state NMR by ultra-fast magic-angle spinning and stereo-array isotope labeling. PLoS One 10(4):e0122714.
44. Chow WY, et al. (2014) NMR spectroscopy of native and in vitro tissues implicates polyADP ribose in biomineralization. Science 344(6185):742–746.
45. Marchetti A, et al. (2012) Backbone assignment of fully protonated solid proteins by
1H detection and ultrafast magic-angle-spinning NMR spectroscopy. Angew Chem Int
Ed Engl 51(43):10756–10759.
46. Zhou DH, et al. (2007) Proton-detected solid-state NMR spectroscopy of fully pro-tonated proteins at 40 kHz magic-angle spinning. J Am Chem Soc 129(38): 11791–11801.
47. Wang S, et al. (2015) Nano-mole scale sequential signal assignment by1H-detected
protein solid-state NMR. Chem Commun (Camb) 51(81):15055–15058.
48. Andreas LB, et al. (2015) Protein residue linking in a single spectrum for magic-angle spinning NMR assignment. J Biomol NMR 62(3):253–261.
49. Barbet-Massin E, et al. (2014) Insights into the structure and dynamics of measles virus nucleocapsids by1H-detected solid-state NMR. Biophys J 107(4):941–946.
50. Vasa SK, Rovo P, Giller K, Becker S, Linser R (2016) Access to aliphatic protons as reporters in non-deuterated proteins by solid-state NMR. Phys Chem Chem Phys 18:8359–8363. 51. Xiang S, Biernat J, Mandelkow E, Becker S, Linser R (2016) Backbone assignment for
minimal protein amounts of low structural homogeneity in the absence of deutera-tion. Chem Commun (Camb) 52(21):4002–4005.
52. Franks WT, et al. (2005) Magic-angle spinning solid-state NMR spectroscopy of theβ1 immunoglobulin binding domain of protein G (GB1):15N and13C chemical shift
as-signments and conformational analysis. J Am Chem Soc 127(35):12291–12305. 53. van den Worm SH, Koning RI, Warmenhoven HJ, Koerten HK, van Duin J (2006) Cryo
electron microscopy reconstructions of the Leviviridae unveil the densest icosahedral RNA packing possible. J Mol Biol 363(4):858–865.
54. Guerry P, Herrmann T (2012) Comprehensive automation for NMR structure de-termination of proteins. Methods Mol Biol 831:429–451.
55. Montelione GT, et al. (2013) Recommendations of the wwPDB NMR Validation Task Force. Structure 21(9):1563–1570.
56. Linser R, Bardiaux B, Higman V, Fink U, Reif B (2011) Structure calculation from un-ambiguous long-range amide and methyl1H-1H distance restraints for a microcrystalline
protein with MAS solid-state NMR spectroscopy. J Am Chem Soc 133(15):5905–5912. 57. Linser R, et al. (2011) Proton-detected solid-state NMR spectroscopy of fibrillar and
membrane proteins. Angew Chem Int Ed Engl 50(19):4508–4512.
58. Eddy MT, et al. (2015) Lipid bilayer-bound conformation of an integral membrane beta barrel protein by multidimensional MAS NMR. J Biomol NMR 61(3-4):299–310. 59. Chill JH, Louis JM, Miller C, Bax A (2006) NMR study of the tetrameric KcsA potassium
channel in detergent micelles. Protein Sci 15:684–698.
60. Volk J, Herrmann T, Wüthrich K (2008) Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J Biomol NMR 41(3):127–138. 61. Dutta SK, et al. (2015) APSY-NMR for protein backbone assignment in high-throughput
structural biology. J Biomol NMR 61(1):47–53.
62. Agarwal V, Reif B (2008) Residual methyl protonation in perdeuterated proteins for multi-dimensional correlation experiments in MAS solid-state NMR spectroscopy. J Magn Reson 194(1):16–24.
63. Shaka AJ, Frenkiel T, Freeman R (1983) NMR broadband decoupling with low radio-frequency power. J Magn Reson 52:159–163.
64. Paulson EK, et al. (2003) High-sensitivity observation of dipolar exchange and NOEs between exchangeable protons in proteins by 3D solid-state NMR spectroscopy. J Am Chem Soc 125(47):14222–14223.
65. Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: A hybrid method for pre-dicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44(4):213–223.
66. Herrmann T, Güntert P, Wüthrich K (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319(1):209–227.
67. Loquet A, Habenstein B, Lange A (2013) Structural investigations of molecular ma-chines by solid-state NMR. Acc Chem Res 46(9):2070–2079.
68. Peck TL, Magin RL, Lauterbur PC (1995) Design and analysis of microcoils for NMR microscopy. J Magn Reson B 108(2):114–124.
69. Nieuwkoop AJ, et al. (2015) Sensitivity and resolution of proton detected spectra of a deuterated protein at 40 and 60 kHz magic-angle-spinning. J Biomol NMR 61(2):161–171. 70. Bak M, Rasmussen JT, Nielsen NC (2000) SIMPSON: A general simulation program for
solid-state NMR spectroscopy. J Magn Reson 147(2):296–330.
71. Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273(1):283–298. 72. O’Donoghue SI, King GF, Nilges M (1996) Calculation of symmetric multimer
struc-tures from NMR data using a priori knowledge of the monomer structure, co-monomer restraints, and interface mapping: The case of leucine zippers. J Biomol NMR 8(2):193–206.
73. Klovins J, Overbeek GP, van den Worm SHE, Ackermann HW, van Duin J (2002) Nu-cleotide sequence of a ssRNA phage from Acinetobacter: Kinship to coliphages. J Gen Virol 83:1523–1533.
74. Bertini I, et al. (2011) Solid-state NMR of proteins sedimented by ultracentrifugation. Proc Natl Acad Sci USA 108(26):10396–10399.
75. Böckmann A, et al. (2009) Characterization of different water pools in solid-state NMR protein samples. J Biomol NMR 45(3):319–327.
76. Zhou DH, Rienstra CM (2008) High-performance solvent suppression for proton de-tected solid-state NMR. J Magn Reson 192(1):167–172.
77. Bax A, Clore GM, Gronenborn AM (1990)1H-1H correlation via isotropic mixing of13C
magnetization, a new 3-dimensional approach for assigning1H and13C spectra of13
C-enriched proteins. J Magn Reson 88:425–431.
78. Baldus M, Meier BH (1996) Total correlation spectroscopy in the solid state. The use of scalar couplings to determine the through-bond connectivity. J Magn Reson A 121(1):65–69. 79. Hardy EH, Verel R, Meier BH (2001) Fast MAS total through-bond correlation
spec-troscopy. J Magn Reson 148(2):459–464.
80. Bennett AE, Ok JH, Griffin RG, Vega S (1992) Chemical-shift correlation spectroscopy in rotating solids: Radio frequency-driven dipolar recoupling and longitudinal exchange. J Chem Phys 96(11):8624–8627.
Supporting Information
Andreas et al. 10.1073/pnas.1602248113
Structure Calculation DetailsThe NMR structure of GB1 was calculated with the UNIO software package (version 2.6.0), by taking as inputs the backbone chemical shifts (determined using UNIO–MATCH), the ali-phatic side-chain chemical shifts [manually assigned from the (H)CCH total correlation spectroscopy (TOCSY) spectrum], the assignment of the characteristic aromatic1H and13C spins (for Trp-43), TALOS+ predictions (65) of 108 backbone dihedral angles based on13CO,13Cα,13Cβ,1HN, and1Hα chemical shifts, and three unrefined 3D peak lists that probe proton–proton con-tacts. The three peak lists were obtained by manual signal identi-fication in the corresponding 3D NMR spectra. In total, 703, 1,335, and 59 peaks were identified in the (H)NHH, aliphatic (H)CHH, and aromatic H(H)CH spectra, respectively. Note that only peaks giving rise to contacts to assigned resonances of Trp-43 were selected in the latter spectrum. Peak amplitudes, not vol-umes, were used to prevent integration errors due to signal overlap. Table S1 summarizes the identified cross-peaks and conformational restraints used in the structure calculation, and the structure quality in terms of root-mean-square deviation (rmsd) within a bundle of 20 conformers used to represent the results.
The standard unsupervised UNIO protocol was used, which consists of seven cycles of cross-peak assignment, conversion into meaningful distance restraints, and structure calculation using simulated annealing. The chemical-shift–based assignment tol-erances were set to the corresponding experimental linewidths, namely 0.15 and 0.4 ppm for proton and heavy-atom dimensions, respectively. At the outset of the spectral analysis, UNIO– CANDID used highly permissive criteria to assign a compre-hensive set of cross-peaks. Only the knowledge of the covalent polypeptide structure and the chemical shifts were initially ex-ploited to guide NOE cross-peak identification and NOE as-signment. In the second and subsequent UNIO–CANDID cycles, the intermediate protein 3D structures were used as an additional guide for the interpretation of the unrefined input peak lists. In each cycle, 80 initial random structures were gen-erated in UNIO, and the 20 conformers with the lowest CYANA (71) target function values were selected as an additional filter for the subsequent cycle. Because the precision of the calculated protein structures normally improves with each subsequent cycle, the criteria for NOE assignments were successively tightened during the iterations. In each UNIO–CANDID cycle, the output consisted of an updated list of assigned NOE cross-peaks for each input peak list and a final set of meaningful upper-limit distance restraints, which constituted the input for the torsion angle dynamics algorithm of CYANA (71) for 3D structure calculation. Default calibration of peak intensities, assuming a median of deduced distances of 4 Å, was used. r−6 scaling of peak intensities was assumed. In addition to the distance re-straints, torsion angle restraints for the backbone dihedral angles ϕ and ψ were automatically generated by UNIO from all back-bone chemical shifts and added to the input for each cycle of structure calculation. Also, tighter torsion angle restraints from TALOS+ (65) were used during simulated annealing. During the first six UNIO–CANDID cycles, ambiguous distance restraints (72) were used. For the final structure calculation in cycle 7, only those distance restraints were retained by UNIO that are unambiguously valid based on the protein 3D structure from cycle 6.
Table S2 below summarizes the convergence of the GB1 structure calculation. In particular, we report the average
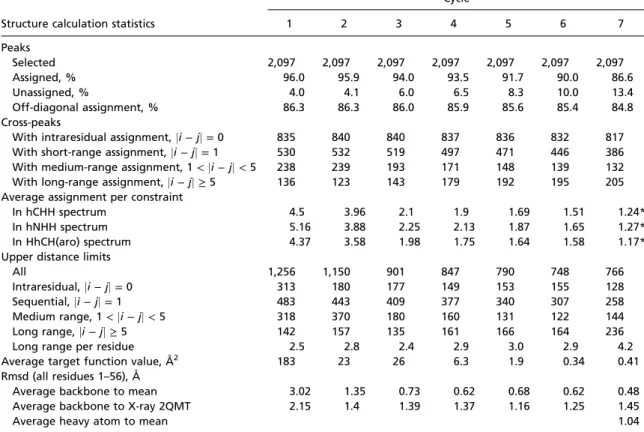
back-bone rmsd to the mean structure of the bundle, the rmsd to the X-ray reference structure (PDB ID code 2QMT), and the number of long-range restraints per residue. The reliability of the calculation is confirmed by the fact that less than 15% of cross-peaks remained unassigned in cycle 7, and backbone rmsd to mean structure in the first cycle is about 3 Å. UNIO structure calculations that meet these criteria are empirically considered as reliable and to yield the correct protein fold (35).
The NMR structure of AP205CP was calculated with the identical UNIO protocol as used for GB1. In total, 723 and 1,363 peaks were manually selected in the (H)NHH and aliphatic (H)CHH spectra, respectively. The UNIO software package (version 2.6.0) was modified to handle structural analysis of symmetric dimers, a feature that was previously not available. The evolution of UNIO cross-peak assignment and structure calculation is summarized in Table S3, again confirming the reliability of the resulting NMR structure as stated above. To improve conver-gence, the UNIO–CANDID protocol was supplemented with four1H-1H distance restraints and 27 hydrogen bonds restraints. The four distance restraints were manually identified based on unique chemical shifts of involved side-chain1H nuclei. In detail, two spectrally unambiguous intermolecular helix–helix restraints (Val104 Hγ1–Trp97 He1 and Asp105 Hα–Trp97 He1) were identified as intermolecular, because intramolecular restraints from one end of the helix to the other would distort the helix. In addition, we entered one intermolecular helix–strand restraint (Val104 Hγ1–Ile83 Hδ1), and one intramolecular helix–loop restraint (Asn103 Hα–Ile124 Hδ1) in the calculation. Based on the observed chemical shifts that indicated beta sheet secondary structure, and on the observation of cross-strand Hα-Hα, HN-HN, and Hα-HN RFDR contacts, 6 intermolecular and 21 intra-molecular hydrogen bonds were imposed in the calculation be-tween β-strands as detailed in Fig. S1. Because the β-strand arrangement could not be spectroscopically determined, test calculations were performed where intermolecular hydrogen bonds were entered as intramolecular. These calculations failed to converge and resulted in clearly distorted secondary struc-tures, such as shown in Fig. S1B.
In the future, to assemble a model of the complete capsid by NMR, interdimer contacts would be required. However, interdimer contacts are particularly challenging to identify in AP205CP, due to a low number of expected contacts for the assigned regions of the protein, compounded by signal de-generacy. Interdimer contacts could not be definitively identified from the 3D RFDR spectra, but might be resolved using more complex sample preparation, such as amino acid-type specific labeling, or with acquisition of 4D spectra. Determination of the number of dimers and their global symmetrical arrangement would be extremely challenging, or possibly impossible by NMR. Alternatively, the full capsid structure could be modeled using both the NMR data and a low- or moderate-resolution cryo-EM map.
Shimming and Magnet Stability
We were able to shim to about 20–40 Hz in1H using a sample of silicone grease or adamantane. Because the contribution of field inhomogeneity adds geometrically to the total, the imperfect shimming therefore contributes less than 13 Hz to the in-homogeneous linewidth in GB1, and less than 10% of the line overall. Data were acquired without the use of a lock. Drift in the magnetic field was minimal, generally about 10–20 1H Hz per day, and because it was fairly linear it could be largely corrected
in processing. Acquisition times were kept to a minimum to avoid problems with drift, and split into blocks when longer ac-quisitions were necessary.
Detection Sensitivity
The high-resolution spectra demonstrated in Figs. 2–4 were ac-quired with no compromise in sensitivity compared with the previous state of the art at 60-kHz MAS in 1.3-mm rotors. An increase in the spinning rate requires reduced sample dimen-sions. This provides increased detection sensitivity associated with improved inductive coupling of smaller coils (almost a factor of 2 with respect to 1.3-mm rotors) but invariably entails a reduction in the sample volume (here by a factor of 4–5 with respect to 1.3-mm rotors) (18). Combined, we therefore expect a theoretical 2.2- to 2.7-fold loss in the sensitivity of a single-pulse spectrum.
We therefore compared the sensitivity of the 0.7-mm probe with our 1.3-mm probe at a field of 1 GHz. To remove contri-butions from solvent in the detected signal, we compared15N-1H CP-HSQC or13C-1H CP-HSQC spectra. Multiple factors affect this measurement, including (i) how well the sample is packed, (ii) the fact that we use rubber spacers to seal the 1.3-mm rotor (reducing the sample volume to about 2μL), and (iii) the effi-ciency of the RF circuit. Each rotor was filled, and each CP condition was optimized to compare the best achievable signal in each case. Surprisingly, we measured approximately equal sen-sitivity for15N-1H and13C-1H CP-HSQC spectra in a 0.7- and a 1.3-mm probe, as shown by the comparison of the total area in the 1D proton spectra of Figs. S2 and S3. (This also indicates that, despite the expectation of a∼2.7 reduction in sensitivity, our 0.7-mm probe is approximately as sensitive as our 1.3-mm probe for highly deuterated proteins or disordered samples, where resolution is dominated by inhomogeneous effects, and is not significantly improved at higher spinning rates.) This ob-servation points to an improvement in other factors, such as RF homogeneity, matching of RF field profiles along the rotor axis, and probe electronics.
Coherence Lifetimes and Linewidths
For15N and1H, T2′ was measured using a 15N-1H CP-HSQC pulse sequence with a variable-time echo period on the re-spective channel. For 13C, we used a carbon-detected CP se-quence with a selective13C refocusing pulse.
We observed improvements in T2′ for 13C′, 13Cα, and 15N, when increasing the MAS rates from the previous state of the art at 60 to ∼111 kHz. Specifically for GB1, T2′ for 13C′ in-creased from 14 to 46 ms, for13Cα from 42 to 77 ms, and for15N from 60 to 120 ms. This results in improved J-transfer efficiency and therefore higher sensitivity or reduced experiment time for sequences whose efficiency depends heavily on the T2′. For ex-ample, backbone assignment spectra can involve one or more scalar-based carbon–carbon transfers in which C′ or Cα decay according to T2′. The efficiency of a scalar-based Cα → Cβ half-transfer (in-phase to antiphase) is expected to improve from 57% to 83%, and similarly, a C′ → Cα half-transfer from 81% to 89% (48). Thus, for GB1, there is a sensitivity gain of about 2.1 for an out-and-back Cα → Cβ transfer, and 1.6 for a full (re-focused, in-phase to in-phase) C′ → Cα transfer. For AP205CP at 100 kHz, we observed a proton T2′ of about 3.0, 2.2, and 2.0 for amide protons, α protons, and all side-chain protons, re-spectively. Compared with measurements at 60-kHz MAS, T2′ for13Cα increased from 8.6 to 22 ms, whereas for15N and13C′ the T2′ was relatively unchanged, changing from about 21 to 23 ms, and 25 to 27 ms, respectively.
For both GB1 and AP205CP, the homogeneous component contributes about equally to the line compared with inhomogeneous contributions (70 Hz of a 100-Hz line for GB1, and 100 Hz of a 150-Hz line for AP205CP).
Considering that the1H homogeneous linewidth is expected to decrease at higher magnetic fields (38, 45, 46), and the in-homogeneous component will increase linearly, we expect that the linewidth in hertz remains approximately constant or in-creases less than linearly. We thus expect further resolution improvement with increasing B0 field. Because the sensitivity also improves (theoretically as B03/2), there is a strong motivation for the use of high field spectrometers for proton-detected studies of fully protonated proteins.
For spectra with indirect proton acquisition, such as the RFDR spectra, there is also an improvement in sensitivity that depends on the indirect sampling. This improvement stems from the larger integral of the signal envelope at faster spinning, due an improved total linewidth (longer apparent coherence time, T2*). By simply integrating the signal for two different values of T2*, we find that the sensitivity improvement factor [Si(t)] is as follows:
SiðtÞ = Tp 2n Tp 2o 0 @1 − e− t Tp 2n 1− e− t Tp 2o 1 A,
where T2np is the new T2*, and T2op is the old T2*. As can be seen from the equation, the improvement in sensitivity reaches the ratio of T2* values in the limit of long sampling periods. For GB1, the improvement in T2* was about a factor of 1.5–1.8 when comparing 60 and 111 kHz. In Fig. S2, Si(t) is plotted for these two cases, and the x axis is shown in units of the new T2* value. At 1.5T2n*, the sensitivity improves by a factor of 1.3–1.5. Note that this analysis applies to the acquired signal, and a further difference may be observed depending on the apodization func-tion used.
Sample Preparation
Uniformly 13C,15N-labeled GB1 with the T2Q mutation was purchased from Giotto Biotech. The sample was dialyzed ex-tensively against phosphate buffer (52). The concentration was increased to about 25 mg/mL using a 3-kDa Amicon concen-trator (EMD Millipore) and microcrystallized by serial addition (an equal volume to the protein solution was added three times) of a mixture of methyl-2-4-pentane-diol and isopropanol ac-cording to a previously described protocol (52).
Uniformly13C,15N-labeled AP205 coat protein (73) was ex-pressed in Escherichia coli using a modified pETDuet vector (Novagen). Bacteria were grown in H2O medium enriched with13 C-glucose (2 g/L) and 15N-labeled ammonium chloride (1 g/L) until they reached OD600= 0.7. Isopropyl β-D -1-thiogalactopyr-anoside was added to 1 mM final concentration and cells were grown for 4 more hours before being centrifuged and frozen. Lysis buffer (40 mM Tris·HCl, pH 8.0, 300 mM NaCl, 1 mg/mL lysozyme, 10μg/mL DNase, 10 mM MgCl2) was added (3 mL per g of cells), and cells were further lysed by sonication. The re-sulting solution was centrifuged, and the supernatant was loaded on a Sepharose CL-4B (GE Healthcare) column. The resulting fractions containing the capsids were further purified on a Fractogel (Merck) ion-exchange column. The eluent was con-centrated to 10 mg/mL using a 10-kDa Amicon concentrator and crystallized in hanging drops by addition of an equal volume of precipitant solution [10 mM Hepes, pH 7.5, 0.1 M NaCl, and 10% PEG (wt/vol) 4000].
Microcrystals were harvested and packed by ultracentrifugation at 165,000× g for 15 h at 12 °C directly into the NMR rotor using a device provided by Giotto Biotech, similar to those described in literature (74, 75).
NMR Spectroscopy
All spectra were recorded atω0H/2π = 1 GHz and a MAS rate ωr/2π of 100 kHz (AP205CP) or 111.111 kHz (GB1) using a
Bruker 0.7-mm HCN probe. Sample temperature was maintained at about 10 °C using a Bruker cooling unit with regulated N2gas directed at the rotor. The temperature of this gas measured just before reaching the sample were 260 K (AP205CP) and 255 K (GB1).
The dipolar-based15N-1H and13C-1H CP-HSQC experiments follow, with little modifications, those introduced by Rienstra and coworkers (21, 46). For 1H-15N and 1H-13C CP, we opti-mized around nutation frequencies of 5/4 ωr, and 1/4 ωr, re-spectively for proton and15N (or13C), with a 10% linear ramp applied on the1H channel. For13C-15N CP, a 10% tangent ramp was applied on the15N frequency at 2/5ωrand the13C nutation frequency was about 3/5ωr. Low-power WALTZ-16 decoupling of 10 kHz was applied for heteronuclear decoupling. DIPSI-2 of γB1/2π = 20 kHz was used for13C decoupling during acquisition due to the presence of homonuclear13C-13C J-couplings in the uniformly labeled sample. Suppression of solvent signals (9) was applied using the MISSISSIPPI scheme (76) without the ho-mospoil gradient for 100–200 ms, and the interscan delay ranged from 0.8 to 1 s for GB1, and 1–1.4 s for AP205CP.
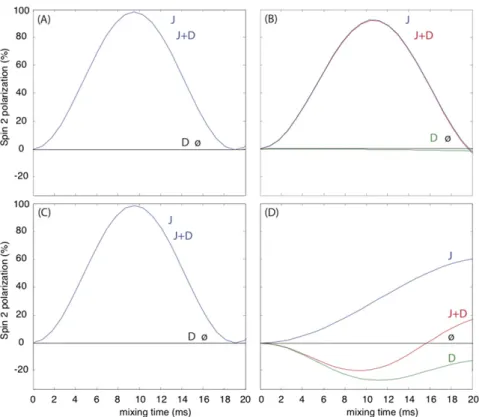
The (H)CCH pulse sequence is similar to the HCCH-TOCSY (77) as well as a related sequence (62) for MAS based on total through-bond correlation spectroscopy (78, 79). In the present implementation, composite13C pulses were applied with a low nutation frequency of one-quarter the MAS frequency, a con-dition made efficient by the fast spinning regime. Simulations using the software package SIMPSON (70) indicate that the transfer is primarily mediated by the J coupling (Fig. S5).
In 3D (H)NHH and (H)CHH spectra,1H-1H RFDR recoupling (80) was applied after the back-CP at a1H RF frequency of 100 kHz, yielding1H-1H contacts resolved using the shift of15N, aliphatic13C, or aromatic13C.
Spectra were apodized in each dimension with 50–90° shifted squared sine-bells (“qsine 3” or “qsine 2” in Bruker Topspin), and zero-filled to at least twice the number of points in the in-direct dimensions. Where linewidths are reported, no apodiza-tion was applied for the reported frequency. Acquisiapodiza-tion and processing parameters specific for each dataset are summarized in Tables S4 and S5.
Fig. S1. In A ,t h e β-strand arrangement in AP205CP. Residues identified by TALOS + as β-sheet secondary structure are labeled in red. Black arrows indicate contacts present in the (H)CHH and (H)NHH spectra. When the cross-peak could be identified from b oth directions (the majority of cases), a double-headed arrow is drawn. Dashed lines indicate the hydrogen bonds entered in the structure calculation. Intermolecular h ydrogen bonds are colored in blue. In B , the 10 lowest-energy conformers of a test calculation with an incorrect arrangement of one of the β-strands. T wo of the hydrogen bonds (residues 6– 124 and 4– 128) are entered as intramolecular instead of intermolecular. The bundle is not well defined, and the helices are severely bent, indicating disagree ment with the e xperimental data.
15 10 5 0
1H Chemical Shift (ppm)
1H, 111 kHz
2H, 60 kHz
Fig. S2. Comparison of optimized15N-1H CP-HSQC spectra acquired either in a 1.3-mm probe at 60 kHz on perdeuterated GB1 (red) or in the 0.7-mm probe at 111 kHz on protonated GB1 (blue). Both spectra were acquired on the 1-GHz spectrometer, and 100 Hz of exponential line broadening was applied.
10 12 8 6 4 6 8 2 0 -2 1H Chemical Shift (ppm) A B 1
H, 60 kHz
1H, 100 kHz
1H, 100 kHz
2H, 60 kHz
1H, 60 kHz
1H Chemical Shift (ppm)Fig. S3. Comparison of sensitivity of AP205CP in the 1.3- and 0.7-mm probes. In A, 1D15N-1H CP-HSQC spectra acquired either in a 1.3-mm probe at 60-kHz MAS with perdeuterated AP205CP (red), a 1.3-mm probe at 60 kHz with protonated AP205CP (green), or in the 0.7-mm probe at 100 kHz and protonated AP205CP (blue). Spectra were acquired on the 1-GHz spectrometer, and no line broadening was applied. In B, 1D13C-1H CP-HSQC spectra of protonated AP205CP acquired either in a 1.3-mm probe at 60-kHz MAS (green) or in a 0.7-mm probe (blue).
Aquisition time (T
2n*)
Relative Sensitivity
1.5 1.0 0.5 1.5 1.0 0.5 0.0 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5T
2n*/T
2o*=1.5
T
2n*/T
2o*=1.8
Fig. S4. Improvement in sensitivity due to improved linewidth, as a function of the acquisition time in an indirect dimension. The x axis is shown in units of the new T2* (T2n*).
Fig. S5. Simulation of the transfer efficiency of WALTZ-16 sequence at 111-kHz MAS and an external magnetic field of 1 GHz. The applied nutation frequency is one-quarter of the rotor frequency. The spin system corresponds to two13C spins. The scalar coupling constant was set to 50 Hz, and the dipolar coupling constant was chosen to correspond to a spin–spin distance of 1.5 Å. In A, the isotropic chemical shifts were 10 and −10 ppm and the CSA was neglected (aliphatic–aliphatic transfer with little offset). In B, the isotropic chemical shifts were larger, 20 and −20 ppm, and the CSA was again neglected (aliphatic– aliphatic transfer with larger offset). In C, the isotropic chemical shifts were 10 and−10 ppm, and the chemical shift anisotropy and asymmetry were set to 80 ppm and 0.9, respectively, for both spins (aromatic–aromatic transfer). Finally, in D, the isotropic chemical shifts were 80 and −10 ppm, and the chemical shift anisotropy and asymmetry were set to 80 ppm and 0.9, respectively, for the first spin only (aromatic–aliphatic transfer). In all cases, only scalar or dipolar coupling was switched on for“J” and “D” curves, respectively; both were switched on for “J + D” curves. The “ø” curves show no transfer (both J and D off). The simulations were performed by using SIMPSON software (70).
Table S1. Summary of identified cross-peaks and conformational restraints used in the structure calculation, and structure quality in terms of rmsd within a bundle of 20 conformers used to represent the results
GB1 AP205CP
Assigned cross-peaks NHH CHH Ar. CHH* NHH CHH
Intraresidue (i= j) 203 598 16 160 453 Sequential (ji − jj = 1) 213 170 3 202 222 Medium range (1≤ ji − jj < 5) 67 65 0 30 48 Long range (ji − jj ≥ 5) 60 123 22 42 89 Distance restraints† Total 766 1,376 Intraresidue 128 154 Sequential (ji − jj = 1) 258 562 Medium range (1≤ ji − jj < 5) 144 250 Long range (ji − jj ≥ 5) 236 410 (104‡)
No. of restraints per residue 13.7 5.3 (6.2§)
Dihedral angle restraints 108 352
Backbone rmsd 0.48 Å{ 1.23 Å#
Heavy-atom rmsd 1.04 Å{ 1.84 Å#
Backbone rmsd to PDB 2QMT 1.45 Å{ —
*Sequence-specific assignment of the aromatic side-chain resonances was made for one of six aromatic residues (Trp-43).
†Only meaningful, nonredundant distance restraints are reported, including multiple restraints generated by the UNIO protocol to resolve the multiplicity of assignment of each ambiguous peak, as described in Supporting Information.
‡Number of intermolecular restraints. §Number of restraints per assigned residue. {Calculated over the entire polypeptide chain.
#Calculated over the regions of the protein in regular secondary structure, namely, residue ranges 3–5, 7–12, 15–20, 23–38, 43–58, 74–86, 90–108, 113–115, and 125–129 of both chains in the dimer.
Table S2. Summary of the GB1 structure calculation statistics
Structure calculation statistics
Cycle 1 2 3 4 5 6 7 Peaks Selected 2,097 2,097 2,097 2,097 2,097 2,097 2,097 Assigned, % 96.0 95.9 94.0 93.5 91.7 90.0 86.6 Unassigned, % 4.0 4.1 6.0 6.5 8.3 10.0 13.4 Off-diagonal assignment, % 86.3 86.3 86.0 85.9 85.6 85.4 84.8 Cross-peaks
With intraresidual assignment,ji − jj = 0 835 840 840 837 836 832 817
With short-range assignment,ji − jj = 1 530 532 519 497 471 446 386
With medium-range assignment, 1< ji − jj < 5 238 239 193 171 148 139 132
With long-range assignment,ji − jj ≥ 5 136 123 143 179 192 195 205
Average assignment per constraint
In hCHH spectrum 4.5 3.96 2.1 1.9 1.69 1.51 1.24*
In hNHH spectrum 5.16 3.88 2.25 2.13 1.87 1.65 1.27*
In HhCH(aro) spectrum 4.37 3.58 1.98 1.75 1.64 1.58 1.17*
Upper distance limits
All 1,256 1,150 901 847 790 748 766
Intraresidual,ji − jj = 0 313 180 177 149 153 155 128
Sequential,ji − jj = 1 483 443 409 377 340 307 258
Medium range, 1< ji − jj < 5 318 370 180 160 131 122 144
Long range,ji − jj ≥ 5 142 157 135 161 166 164 236
Long range per residue 2.5 2.8 2.4 2.9 3.0 2.9 4.2
Average target function value, Å2 183 23 26 6.3 1.9 0.34 0.41
Rmsd (all residues 1–56), Å
Average backbone to mean 3.02 1.35 0.73 0.62 0.68 0.62 0.48
Average backbone to X-ray 2QMT 2.15 1.4 1.39 1.37 1.16 1.25 1.45
Average heavy atom to mean 1.04
*In previous implementations of UNIO–CANDID, only cross-peaks with highly unambiguous assignments were kept in cycle 7, and therefore the average assignment per constraint (peak) was forced to be 1 at the end of the calculation. In the current UNIO 2.6.0 version, information from unambiguous peaks is kept by splitting peak volumes after the assignment step during the conversion into distance restraints in the final cycle. This results in an average assignment per peak greater than 1. Retained peak ambiguities are ultimately converted into unambiguous distance restraints during the generation of distance restraints.
Table S3. Summary of the AP205CP structure calculation statistics
Structure calculation statistics
Cycle 1 2 3 4 5 6 7 Peaks Selected 2,086 2,086 2,086 2,086 2,086 2,086 2,086 Assigned, % 58.4 81.5 78.7 78.5 77.5 77.1 74.0 Unassigned, % 41.6 18.5 21.3 21.5 22.5 22.9 26.0 Off-diagonal assignment, % 81.7 82.5 81.9 81.9 81.6 81.5 80.8 Cross-peaks
With intraresidual assignment,ji − jj = 0 430 654 652 649 645 641 613
With short-range assignment,ji − jj = 1 343 470 463 457 447 438 424
With medium-range assignment, 1< ji − jj < 5 129 185 130 127 107 102 78
With long-range assignment,ji − jj ≥ 5 93 94 99 107 120 130 131
Average assignment per constraint
In hCHH spectrum 4.69 5.41 2.61 2.37 2.11 1.82 1.39*
In hNHH spectrum 4.24 4.40 2.27 2.29 1.97 1.7 1.36*
Upper distance limits
All 1,199 1,630 1,326 1,256 1,204 1,152 1,376 Intraresidual,ji − jj = 0 154 138 224 184 182 164 154 Sequential,ji − jj = 1 500 682 678 636 612 584 562 Medium range, 1< ji − jj < 5 329 552 250 246 208 192 250 Long range,ji − jj ≥ 5 216 258 174 190 202 212 410 Intermolecular 0 9 46 46 46 48 104
Long range per residue 0.8 0.8 0.7 0.7 0.8 0.8 1.6
Average target function value, Å2 299 38.8 32.6 13.7 8.7 10.6 8.6
Rmsd (residues 3–5, 7–9, 15–20, 23–34, 43–58, 74–86,
90–108, 113–115, 125–127), Å
Average backbone to mean 2.16 2.17 1.54 1.43 1.43 1.4 1.23
Average backbone to X-ray 5FS4 3.83 2.63 2.15 1.97 2.19 2.34 2.35
Average heavy atom to mean 1.84
*In previous implementations of UNIO–CANDID, only cross-peaks with highly unambiguous assignments were kept in cycle 7, and therefore the average assignment per constraint (peak) was forced to be 1 at the end of the calculation. In the current UNIO 2.6.0 version, information from unambiguous peaks is kept by splitting peak volumes after the assignment step during the conversion into distance restraints in the final cycle. This results in an average assignment per peak greater than 1. Retained peak ambiguities are ultimately converted into unambiguous distance restraints during the generation of distance restraints.
Table S4. Acquisition and processing parameters of used NMR spectra for GB1
Spectrum Max indirect evolution
Scans per point
Experimental time
Signal/noise
(first FID) Processing
(H)NH 50 ms (N) 4 24 min 46 60° shifted sine-bell squared
(H)CH 46 ms 8 5 h 79 60° shifted sine-bell squared
(H)NCAH 7.9 ms (CA), 19.7 ms (N) 4 25 h 3.2 90° shifted sine-bell squared
(H)CANH 7.9 ms (CA), 19.7 ms (N) 4 25 h 8.9 90° shifted sine-bell squared
(H)(CO)CA(CO)NH 3.3 ms (CA), 9.2 ms (N) 4 3.5 h 3.5 90° shifted sine-bell squared
(H)(CA)CB(CA)NH 3.3 ms (CA), 8.0 ms (N) 4 6 h 3.6 90° shifted sine-bell squared
(H)N(CA)(CO)NH 8.3 ms (CA, N) 8 7.5 h 3.3 60° shifted sine-bell squared
(H)N(CO)(CA)NH 8.3 ms (CA, N) 8 7.5 h 3.6 60° shifted sine-bell squared
(H)CCH TOCSY (15 ms) 6.8 ms 2 32.5 h 3.0 90° shifted sine-bell squared
(H)NHH (RFDR 1.8 ms) 6.9 ms (H), 13.6 ms (N) 4 15 h 14.3 60° shifted sine-bell squared
(H)CHH (RFDR 0.5 ms) 2.6 ms (H), 5.3 ms (C) 4 14.5 h 67 90° shifted sine-bell squared
H(H)CH aromatic13C filtered
(RFDR 0.5 ms)
5.7 ms (H), 10.2 ms (C) 4 38 h 4.5 90° shifted sine-bell squared
The direct proton dimension was sampled to 10 ms. Spinning rate was 111.111 kHz.
Table S5. Acquisition and processing parameters of used NMR spectra for AP205CP Spectrum Max indirect evolution Scans per point Experimental time Signal/noise
(first FID) Processing (indirect/direct)
(H)NH 15.8 ms (N) 32 1 h 17 min 83 60° shifted sine-bell squared
(H)CH 17 ms (C) 8 2 h 46 min 85 60° shifted sine-bell squared
(H)NCAH 6.36 ms (CA), 9.86 ms (N) 2 6 h 7.9 60° shifted sine-bell squared
(H)CANH 6.4 ms (CA), 9.9 ms (N) 2 6 h 5.3 60° shifted sine-bell squared
(H)(CO)CA(CO)NH 6.4 ms (CA), 9.9 ms (N) 8 25 h 6.7 60° shifted sine-bell squared
(H)(CA)CB(CA)NH — — — — —
(H)N(CA)(CO)NH — — — — —
(H)N(CO)(CA)NH — — — — —
(H)CCH TOCSY (14 ms) 7.0 ms 2 55 h 10 60/50° shifted sine-bell
H(H)NH (RFDR 1 ms) 5 ms (H), 10.3 ms (N) 4 60 h 35 72/60° shifted sine-bell squared
H(H)CH (RFDR 1 ms) 5 ms (H), 4.8 ms (C) 4 43 h 35 72/60° shifted sine-bell squared
(H)CO(N)CAH 6.4 ms (CO), 5.3 ms (CA) 24 37 h 10 90° shifted sine-bell squared
The direct proton dimension was sampled to 10 ms. Spinning rate was 100 kHz.