CAPITOLO 2
INTRODUZIONE
2.1 IL METABOLISMO DEL GLUCOSIOIl glucosio è un substrato usato ubiquitariamente nell’organismo umano. (1)
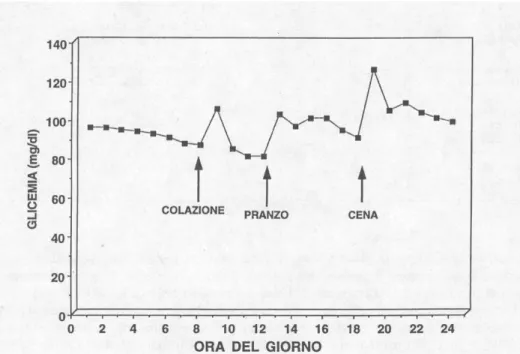
A differenza di altri substrati, la concentrazione del glucosio nella circolazione sanguigna è mantenuta entro limiti molto ristretti. (Fig.1)
Figura 1- Profilo diurno della glicemia in un gruppo di soggetti sani. Dati tratti da Reaven et al.1987
Nello stato di digiuno non prolungato, l’unica fonte di glucosio circolante è il fegato e, in minor misura, il rene (solo il 10%) (1).
Nella fase di assorbimento di un pasto contenente carboidrati, l’intestino è una seconda fonte di glucosio, che in certi intervalli di tempo può divenire quantitativamente preponderante rispetto al fegato.
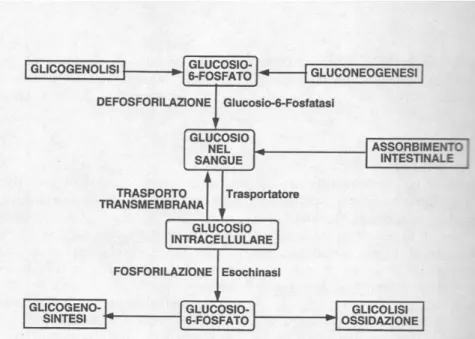
Da un punto di vista biochimico, il glucosio nel sangue è il risultato dell’equilibrio dinamico fra la reazione di defosforilazione del glucosio 6-fosfato a glucosio, negli organi produttori di glucosio, e la relazione di fosforilazione del glucosio a glucosio 6-fosfato nelle cellule utilizzatrici del glucosio (5). Alla generazione del glucosio 6-fosfato sono preposte due vie metaboliche, la glicogenolisi e la
gluconeogenesi; all’utilizzazione di glucosio 6-fosfato concorrono altre due vie metaboliche, la glicogeno-sintesi e la glicolisi. (2)
Figura 2- Reazioni e vie metaboliche cellulari responsabili dell'immissione di glucosio nel sangue e dell'utilizzazione di glucosio circolante
Il cervello e i globuli rossi sono definiti “glucosio-dipendenti” in quanto il glucosio rappresenta l’unico, o di gran lunga il principale, substrato utilizzabile. La sopravvivenza del sistema nervoso centrale e degli eritrociti, e quindi dell’intero organismo, dipende in modo critico dal glucosio. In assenza di assorbimento di carboidrati, il rifornimento di glucosio è assicurato in quantità preponderante dal fegato. Vi sono però momenti della giornata in cui l’assorbimento di glucosio eccede la necessità del cervello e dei globuli rossi e quindi tale eccedenza deve essere metabolizzata da altri organi tra cui il muscolo ed il tessuto adiposo (3).
Il livello di glucosio nel sangue è detto glicemia. A digiuno, la glicemia, varia generalmente in un intervallo piuttosto ristretto compreso tra i 70 e i 110 mg/dl mentre, a due ore dopo un pasto il valore può salire a 150 mg/dl, rimanendo comunque nella norma. Valori a digiuno, superiori ai 126 mg/dl e valori dopo un pasto superiori a 200 mg/dl potrebbero essere sintomo di diabete.
2.2 IL PANCREAS: ANATOMIA ED ISTOLOGIA
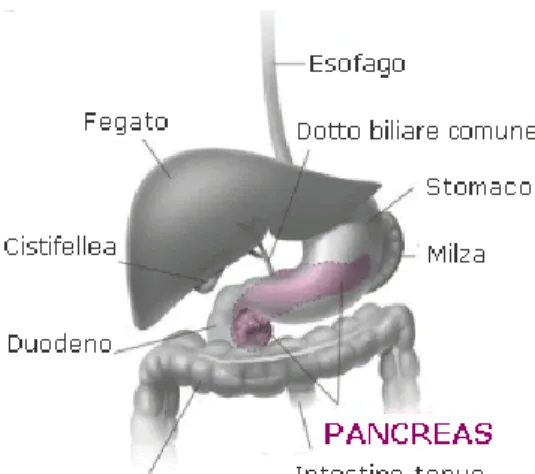
Il pancreas è una ghiandola voluminosa, lunga e piatta, situata trasversalmente nella parte superiore e posteriore della cavità addominale. Nei soggetti giovani raggiunge un peso di circa 80-100 grammi, che tende a ridursi con l'avanzare dell'età. La lunghezza complessiva si colloca intorno ai quindici centimetri.
Dal punto di vista anatomico, il pancreas viene normalmente suddiviso in tre porzioni, che prendono il nome di testa, corpo e coda del pancreas. La testa rappresenta la sua parte più grossa e spessa ed è in contatto con l'ansa duodenale. Il corpo, leggermente obliquo dal basso verso l'alto, rappresenta il segmento intermedio ed è disposto frontalmente rispetto all'aorta e alla vena cava. La coda del pancreas, infine, è in rapporto con l’ilo della milza e rappresenta il tratto assottigliato con cui termina quest'organo ghiandolare.
Figura 4 Anatomia del Pancreas
Il pancreas è dotato di una duplice funzione, endocrina da un lato ed esocrina dall'altro. La funzione esocrina consiste nella produzione di enzimi digestivi da immettere nel tubo digerente mentre, la funzione endocrina si occupa della produzione di ormoni quali l’insulina, il glucagone, la somatostatina e il polipeptide pancreatico.
Gli ormoni prodotti dal pancreas endocrino regolano molteplici aspetti della nutrizione cellulare: dalla velocità di assorbimento degli alimenti, ai depositi intracellulari, al metabolismo dei nutrienti. L’alterazione del pancreas endocrino, o l’alterata risposta degli ormoni pancreatici da parte dei tessuti bersaglio, provoca rilevanti disordini nell’omeostasi dei nutrienti che includono le importanti sindromi cliniche raggruppate sotto il nome di diabete mellito.
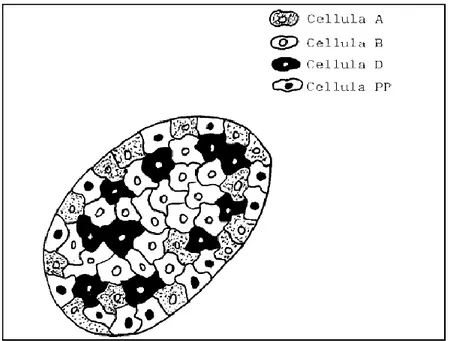
Il pancreas endocrino è costituito da circa 1 milione di microscopiche ghiandole endocrine che prendono il nome di isole di Langerhans le quali occupano circa l’1-1,5% della massa complessiva del pancreas.
Questa struttura multicellulare costituisce l’unità endocrina del pancreas responsabile della regolazione del glucosio nel sangue.
Nelle isole di Langerhans sono stati individuati quattro tipi cellulari: Cellule α che secernono il glucagone
Cellule β che secernono l’insulina
Cellule δ che secernono la somatostatina
Cellule F che producono il polipeptide pancreatico (PP)
L’architetture delle cellule è stata individuata inizialmente nei roditori. In questa specie animale, essa è caratterizzata da una zona centrale costituita dalle cellule β circondate da cellule non- β. Questa distribuzione suggereisce che vi sono sicuramente delle interazioni cellulari (anche in termini di attività elettrica) tra le cellule β e le cellule adiacenti le quali invece non sono funzionalmente accoppiate e lavorano come unità indipendente. (4)
La disposizione delle cellule pancreatiche nell’uomo è leggeremente differente rispetto ai roditori: il numero di cellule δ e PP è simile in entrambe le specie mentre nell’uomo le cellule β sono meno abbondanti. Le cellule α, raggiungono il 33-46% della popolazione rispetto ai roditori, ipotizzando che la secrezione del glucagone giochi un ruolo maggiore nelle cellule dell’uomo. (5) Le isole pancreatiche sono riccamente vascolarizzate e questo è importante al fine di garantire una rapida rilevazione dei livelli di glucosio nel plasma da parte delle cellule pancretaiche e un’adeguata risposta secretoria.
E’ stato inoltre ipotizzato che la direzione del flusso all’interno dell’isola di Langerhans sia un fattore importante nel veicolare l’insulina secreta nella regione centrale dell’isola verso la zona periferica, dove essa modula ed inibisce il rilascio di glucagone dalle cellule α. (6)
Figura 5 Struttura delle isole di Langerhans
2.3 GLI ORMONI DEL PANCREAS ENDOGENO
La porzione endocrina del pancreas, costituita dalle isole del Langehrans, produce due ormoni importantissimi: l’insulina che viene prodotta dalle cellule β che rappresentano, quantitativamente, circa 3/4 delle isole del Langehrans e il glucagone che viene prodotto dalle cellule α. A questi ormoni pancreatici se ne associa un terzo, chiamato somatostatina prodotta dalle cellule δ situate alla periferia dell’isola, responsabile, dell’inibizione del rilascio di insulina e glucagone.
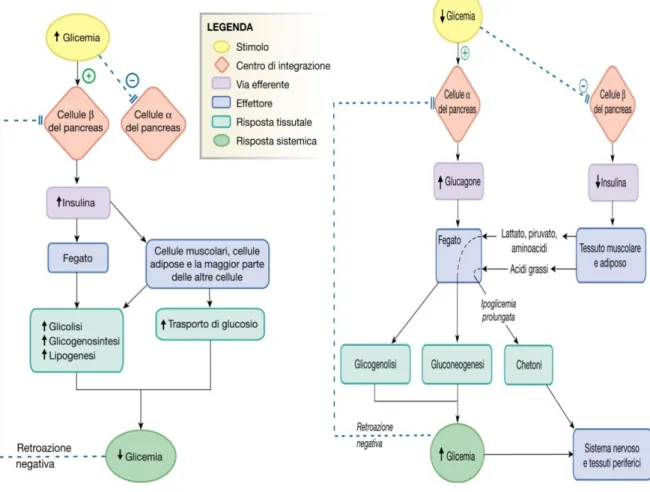
Gli ormoni insulina e glucagone hanno un’importanza notevole nel controllo della concentrazione di glucosio nel sangue, indispensabile per mantenere in funzione diversi organi. Esso si basa sulla coordinazione nella secrezione di glucagone e insulina i quali rispondono in maniera opposta ai cambiamenti della concentrazione sanguigna di glucosio: condizioni ipoglicemiche stimolano la secrezione di glucagone e condizioni iperglicemiche stimolano la secrezione di insulina (7) [Nadal et al. 1999, Quesada et al. 2006].
L’insulina e il glucagone hanno effetti opposti non solo sulla glicemia ma anche sul metabolismo dei nutrienti. L’insulina agisce prevalentemente sul muscoli, fegato e tessuti adiposi con effetti anabolici, inducendo l’up-take di glucosio, inibendo la produzione epatica e promuovendo l’accumulo sottoforma di grasso o glicogeno. Al contrario, il glucagone induce effetti catabolici principalmente attivando la glicogenolisi e la gluconeogenesi nel fegato che induce il rilascio di glucosio epatico nel flusso sanguigno.
Attualmente, il diabete è associato ad una irregolarità nei livelli normali di insulina e glucagone. Un livello eccessivo di glucagone nel plasma rispetto ai livelli dell’insulina può causare una produzione epatica di glucosio superiore al normale che potrebbe risultare critica nel mantenimento dell’iperglicemia nei soggetti diabetici. (8)
2.4 L’INSULINA: BIOSINTESI E BIOCHIMICA
L’insulina fu isolata la prima volta dal pancreas da Banting e Best nel 1922.
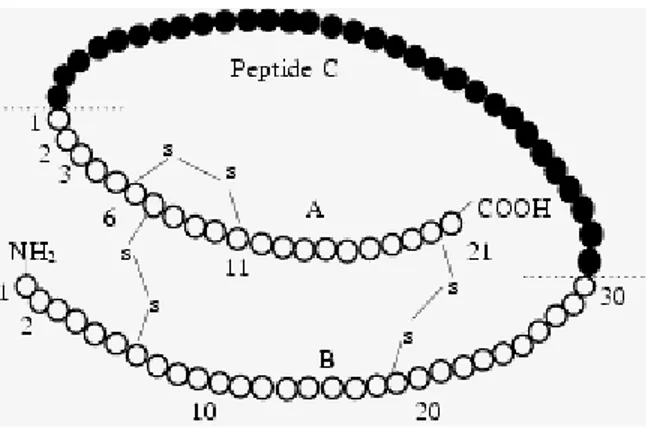
Essa è una proteina di piccole dimensioni con un peso molecolare di circa 5808 Da nell’uomo, ed è formata da due catene di 51 aminoacidi (una catena A costituita da 21 aminoacidi e un catena B costituita da 30 amino acidi) connesse tra loro da ponti disolfurici, la cui separazione porta alla perdita dell’attività funzionale della molecola ormonale.
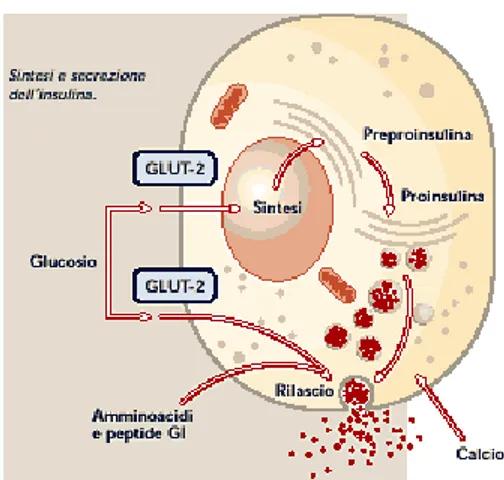
La sintesi proteica dell’insulina inizia con la traduzione dell’RNA insulinico e con la formazione, da parte dei ribosomi e del reticolo endoplasmatico, del pre-proormone insulina. Questo ha un peso molecolare di circa 11.500 Da e viene scisso nel reticolo endoplasmatico formando proinsulina, del peso molecolare di circa 9000 Da. La proinsulina viene trasportata nell’apparato del Golgi fino a formare insulina, impacchettata in granuli secretori rivestiti di clatrina. La maturazione dei granuli secretori è caratterizzata dalla perdita del rivestimento di clatrina e dal clivaggio (separazione) della proinsulina ad insulina e ad un piccolo peptide di connessione detto Peptide C ad opera di proteasi in quantità equimolare con l’insulina. Quando viene riversata nel sangue, l’insulina, circola quasi interamente non legata a molecole di trasporto e ha un emi-vita di soli 6 min, per cui la maggior parte viene rimossa dalla circolazione cardiaca entro 10-15 minuti. Tranne la quota che si combina con le cellule bersaglio, il resto dell’insulina viene degradato da enzimi in prevalenza nel fegato e in misura minore nei reni. Per iniziare ad agire sulle cellule bersaglio, l’insulina deve dapprima combinarsi con una proteina recettrice della membrana e attivarla. Il recettore per l'insulina è una glicoproteina transmembrana costituita da 4 catene: 2α esterne alla cellula e 2β interne alla cellula, unite fra loro da ponti di solfuro.
Figura 6 Struttura della pro-insulina umana
La principale funzione dell’insulina è quella di promuovere il deposito dei nutrienti assunti con l’alimentazione. Sebbene l’insulina, direttamente o indirettamente, influenzi la funzione di quasi tutti i tessuti, possiamo dire che essa esercita la sua azione in modo preponderante sul fegato, muscolo e tessuto adiposo.
Il fegato è il primo organo raggiunto dall’insulina attraverso il ciclo ematico e qui essa effettua la sua azione attraverso due vie:
- Anabolica: stimola la sintesi del glicogeno e al tempo stesso inibisce la degradazione del glicogeno;
- Catabolica: inibisce la glicogenolisi e la gluconeogenesi.
2.5 MECCANISMO DI SECREZIONE DELL’INSULINA
Le cellule β sono dotate di un grosso numero di trasportatori di glucosio (GLUT-2) i quali fanno si che l’entità dell’ingresso di zucchero nella cellula sia proporzionale al valore della glicemia, sempre che questa resti nei limiti fisiologici. Una volta all’interno della cellula, il glucosio viene fosforilato a glucosio 6-fosfato dalla glicochinasi. Il glucosio 6-fosfato viene poi ossidato con formazione di adenosina trifosfato (ATP) che inibisce i canali di potassio sensibili all’ATP. La chiusura di questi canali depolarizza la membrana cellulare, provocando l’apertura dei canali del calcio voltaggio-dipendenti. Il calcio entra cosi nella cellula e, stimolando la fusione della membrana cellulare con le vescicole contenenti insulina, permettendo all’ormone di essere secreto per esocitosi nel liquido extracellulare. Alcuni ormoni, come il glucagone e il peptide inibitorio gastrico, nonché l’acetilcolina, possono far aumentare il calcio intracellulare mediante altre vie di trasduzione del segnale e possono cosi potenziare gli effetti del glucosio. Altri ormoni ancora, tra i quali la somatostatina e la noradrenalina, inibiscono invece l’esocitosi dei granuli contenenti insulina.
Figura 7 Meccanismo di secrezione dell'insulina
2.6 CONTROLLO DELLA SECREZIONE DELL’INSULINA
La stimolazione della secrezione insulinica avviene in risposta a stimoli esogeni. In vivo ciò è costituito dalla risposta delle cellule β all’ingestione di pasti ed il glucosio rappresenta il più potente stimolatore del rilascio di insulina.
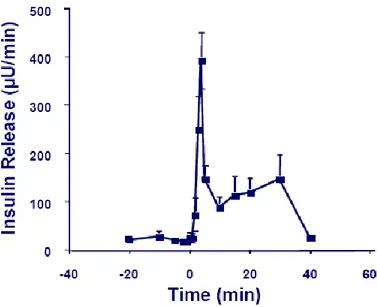
Quando la glicemia a digiuno è ai suoi valori normali di 80-110 mg/mL, la secrezione dell’insulina è minima, dell’ordine di 25 ng/min pro Kg. Se però la glicemia basale sale bruscamente ad un livello di 2-3 volte superiore a quello normale e si mantiene su questi valori viene inviato un segnale al pancreas che aumenta nettamente la secrezione di insulina, passando per due fasi distinte:
1. Un primo aumento della secrezione ormonale, fino a 10 volte, si manifesta entro 3-5 min dalla brusca impennata della glicemia ed è dovuto all’insulina già formata che viene immediatamente immessa in circolo dalle cellule β. Questo forte aumento iniziale non è durevole, anzi, si riduce a circa la metà nei successivi 5-10 min (fase precoce).
2. Se la concentrazione di glucosio si mantiene elevata, la secrezione si innalza una seconda volta, raggiungendo, in 2-3 ore un nuovo tetto, ad un livello di solito anche più alto della fase iniziale. Questo aumento è dovuto sia alla liberazione di altra insulina preformata sia all’attivazione del sistema enzimatico che sintetizza l’insulina e ne promuove la liberazione dalle cellule pancreatiche (fase tardiva).
Figura 8 Rilascio di insulina in seguito ad un aumento di glucosio
2.7 IL GLUCAGONE: BIOSINTESI E BIOCHIMICA
Il glucagone, scoperto nel 1960 è considerato l’ormone antagonista dell’insulina in relazione alla glicemia.
Ad oggi, le conoscenze sul glucagone sono ancora poche paragonate a quelle sull’insulina e ciò è attribuibile a diversi fattori tra cui l’esiguo numero di cellule α nei modelli animali presi in considerazione.
Il glucagone pancreatico, il cui gene nell’uomo è localizzato nel cromosoma 2, è un polipeptide a singola catena costituita da 29 amino acidi con un peso molecolare di 3485 Da. E’ sintetizzato nelle cellule pancreatiche α delle isole di Langerhans e deriva dal pro-glucagone, un precursore del glucagone composto da 160 aminoacidi. All’interno della molecola del pro-glucagone vi sono altri peptidi connessi a coppie tra cui: il glucagone, il GLP-1 (glucagon-like peptide 1), il GLP-2 (glucagon-like peptide 2).
L’emivita del glucagone è di circa 3-6 min e viene rimosso principalmente dal fagato e dal rene (9). Il ruolo principale della secrezione del glucagone, allo stato basale, è quello di mantenere un rate di produzione epatica sufficiente a rimpiazzare l’utilizzazione del glucosio (10)
Al contrario dell’insulina infatti, che promuove il deposito di substrati energetici in diversi distretti, il glucagone rende i substrati energetici disponibili per i tessuti nelle ore lontane dai pasti (in cui non si ha assorbimento diretto di glucosio). (10)
Scissione del glicogeno epatico (glicogenolisi). Il glucagone, contrariamente all’insulina stimola la sintesi epatica di glucosio in modo da aumentare la concentrazione di glucosio nel sangue quando richiesto. (11)
Tutto ciò avviene attraverso questa serie di eventi in cascata:
1. Il glucagone attiva l’adenil ciclasi nella membrana dell’epatocita; 2. L’adenin ciclasi provoca la formazione di AMP ciclico;
3. Questo attiva la proteina regolatrice della protein-cinasi; 4. Questa proteina a sua volta attiva la proteinchinasi; 5. La proteinchinasi attiva la chinasi della fosforilasi b; 6. Questa converte la fosforilasi b in fosforilasi a;
7. La fosforilasi a promuove la degradazione del glicogeno in glucosio 1-fosfato; 8. Questo viene infine defosforilato e il glucosio liberato dall’epatocita.
Aumento della gluconeogenesi a livello epatico
Promozione del rilascio, da parte del fegato, di chetoni a partire da acidi grassi (chetogenesi). In condizioni di digiuno prolungato il glucagone aumenta la capacità, da parte del fegato, di produrre chetoni in modo da garantire il normale apporto di nutrienti al cervello con un costo minimo per l’organismo stesso. Il fegato, a causa della sua contiguità con il pancreas, rappresenta il principale organo bersaglio per l’azione del glucagone.
Il glucagone svolge un importantissimo ruolo nell’indurre una costante disponibilità di glucosio per il cervello e per gli altri organi stimolando la produzione di glucosio epatico ad una velocità che impedisce sempre la comparsa di una ipoglicemia
Sono i recettori del glucagone, situati sulle cellule epatiche, ad inviare al fegato il segnale di secrezione epatica. Si tratta di recettori proteici di 62 kDa di massa attivati direttamente dall’ormone appartenenti alla famiglia dei recettori accoppiati alle proteine G.
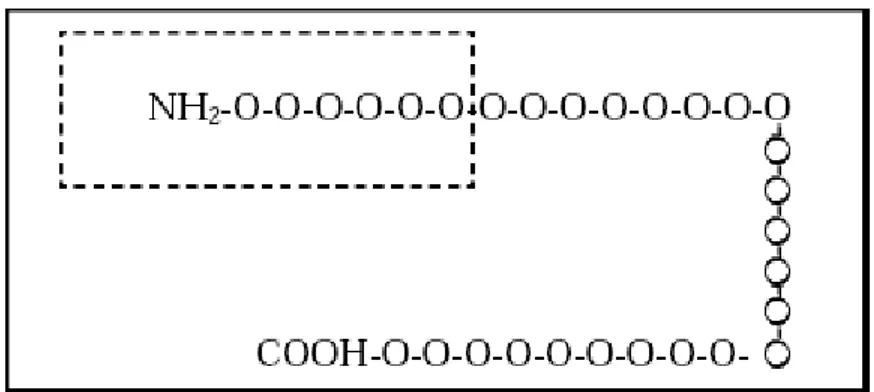
La parte della molecola di glucagone che sembra esserecoinvolta nel legame con il recettore comprende i primi sei aminoacidi nella parte N-terminale dell’ormone.(figura 6)
Quando il glucagone si lega al recettore, si ha una variazione conformazionale del recettore stesso che viene così attivato. La sua attivazione scatena la serie di eventi in cascata che si concludono con la secrezione di glucosio dal fegato e la sua immissione nella circolazione sanguigna.
Figura 9 Parte della molecola di glucagone interessata nel legame con il recettore di membrana
2.8 MECCANISMO DI SECREZIONE DEL GLUCAGONE
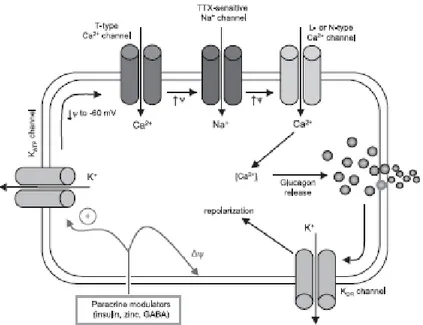
Le cellule pancreatiche α sono equipaggiate di specifici set di canali per il Na+ e il Ca2+ che generano potenziali d’azione per basse o nulle concentrazioni di glucosio (12) Questa attività elettrica modula i segnali del Ca2+ e la secrezioe del glucagone.
Nei topi è stato osservato che la secrezione di glucagone richiede l’avvio di una completa depolirizazione a cascata della membrana cellulare che inizia quando i canali T del Ca2+ sono attivati. Tali canali consentono una depolarizzazione della membrana attravero l’ingresso degli ioni Ca nella cellula che portano il potenziale ad un valore intermedio. In conseguenza di ciò i canali del Na+ sensibili alla tetrodoxina (TTR) si aprono e consentono un afflusso di ioni Na che depolarizzano ulteriorente la membrana cellulara delle cellule α portando all’attivazione dei canali L del Ca2+ .Tali canali consentono un sostanzioso ingresso di Ca2+ nella membrana cellulare che innesca l’esocitosi dei granuli contenenti il glucagone nello spazio extra-cellulare.
La membrana cellulare è ripolarizzata per mezzo dell’apertura dei canali KDR e il processo può quindi ricominciare. (13)
L’azione combinata dei canali KATP e del glucosio influenza la riuscita di questo meccanismo. Ad una bassa concentrazione di glucosio infatti un certo numero di canali KATP presenti sulla cellula α si aprono e portano il potenziale di membrana a -60 mV causando l’attivazione dei canali T del Ca2+ e quindi l’inizio della fase di depolarizzazione a cascata. Al contrario, un aumento della concentrazione di glucosio ed del rapporto intracelulare ATP/ADP porta ad una chiusura dei canali KATP provocando l’inattivazione dei canali ionici T impedendo l’esocitosi e quindi il rilascio di glucagone. (14)
Figura 10 Meccanismo di secrezione del glucagone Un modello simile è stato proposto anche per le cellule α dell’uomo (15)
Tale schematizzazione del meccanismo di rilascio del glucagone da parte delle cellule α è sostenuto in diversi reports in cui viene indicato il ruolo iperpolarizzante del glucosio. (16)
Non vi sono dati conclusivi se l’effetto del glucosio sia diretto o mediato dal rilascio di insulina o di somatostatina, ormoni di cui è stato dimostarato un effetto inibitorio diretto sulle cellule α. (17)
2.9 REGOLAZIONE DELLA SECREZIONE DEL GLUCAGONE
La concentrazione del glucosio nel sangue è di gran lunga il fattore più potente nel controllo della secrezione del glucagone.
La prova che il glucosio sia effettivamente un soppressore della secrezione del glucagone è stata condotta per la prima volta in studi sul cane e sperimentato successivamente nell’uomo. Attraverso infusioni o ingestioni di glucosio si è dimostrando che un incremento nella concentrazione plasmatica di glucagone è direttamente correlato con l’entità della diminuzione della concentrazione plasmatica di glucosio.
Bisogna tener presente che l’effetto della glicemia sulla secrezione del glucagone è esattamente l’opposto di quello esercitato dal glucosio sulla secrezione dell’insulina. Infatti, una diminuzione della glicemia dal suo normale valore a digiuno, circa 90 mg/100 ml, verso livelli più bassi fa aumentare la concentrazione plasmatica di glucagone di diverse volte. Invece, l’aumento della glicemia al di sopra dei livelli normali riduce il glucagone plasmatico ma favorisce il rilascio di insulina da parte delle cellule β.(figura 11)
Diversi studi di dose-risposta hanno dimostrato che le cellule α sono molto più sensibili al glucosio delle cellule β per cui la soglia di soppressione del glucagone da parte del glucosio è molto più bassa rispetto alla soglia di stimolazione dell’insulina (2-3mMdi glucosio per il glucagone contro i 4-5 mM per l’insulina).Concentrazioni di glucosio intorno ai 6-10 mM provocano l’inibizione massima della secrezione da parte delle cellule α, mentre concentrazioni superiori ai 20 mM causano la stimolazione massima della secrezione di insulina.
L’effetto predominante del glucagone si ha, in ogni caso, nella fase di ipoglicemia in cui esso viene secreto in elevate quantità, provocando cosi un forte aumento della liberazione di glucosio da parte del fegato in modo da riprostinare la normale condizione fisiologica (normoglicemia).
La causa più comune di ipoglicemia moderata è la mancata assunzione di nutrienti per un prolungato periodo di tempo (16-24 ore), tempo occorrente per esaurire le riserve di glicogeno epatico. Una causa di ipoglicemia iatrogena frequente nei diabetici è l'accidentale (o intenzionale) sovradosaggio di farmaci antidiabetici o di insulina, oppure il non mangiare quando necessario dopo aver preso questi medicinali. Un'altra grave causa di ipoglicemia è l'insulinoma, un tumore delle cellule beta delle isole di Langerhans del pancreas: questo tumore causa una superproduzione di insulina che si riversa nel sangue in dosi massicce. Ci sono ovviamente altre cause, sia nei diabetici che nelle persone non diabetiche. In genere, nei diabetici l'ipoglicemia è rara e non molto grave, si risolve facilmente con un moderato apporto di carboidrati, tuttavia se non si interviene può portare alla perdita di coscienza. Nei non diabetici si differenzia l'ipoglicemia in due casi: reattiva (postprandiale o comunque dopo i pasti) e digiunale (postassorbitiva). L'ipoglicemia reattiva si ha circa 4 ore dopo aver mangiato; non tutte le cause sono ben comprese, e ci sono ancora pareri discordanti su quali siano le maggiori. L'ipoglicemia digiunale invece si diagnostica con un esame del sangue che mostri una glicemia sotto i 54 mg/dl dopo una notte a digiuno, fra i pasti o dopo esercizio fisico. L'ipoglicemia reattiva può essere, se particolarmente severa, segno di prediabete in persone predisposte e con parenti malati. L'ipoglicemia digiunale invece può essere sintomo di iperproduzione insulinica o di ridotta disponibilità delle riserve di glicogeno e alcune tra le cause possono essere l'assunzione di alcuni farmaci o di tossine, gravi malattie, alcuni tipi di tumori, carenze ormonali, alcolismo o alcune condizioni ricorrenti durante l'infanzia. Si parla inoltre di ipoglicemia funzionale quando la causa è un problema del metabolismo, e di ipoglicemia idiopatica quando invece non è possibile individuare una causa fisiologica definita per il basso valore di glicemia.
Uno dei più importanti meccanismi paracrini responsabili dell’ inibizione del rilascio di glucagone è invece condotto dall’insulina.
Quando le cellule β sono stimolate secernono insulina; le prime cellule bersaglio raggiunte dall’insulina nelle isole di Langherans sono proprio le cellule α le quali, fornite di appropiati recettori rilevano la presenza di tale ormone e riducono la loro produzione di glucagone.
Tuttavia, l’insulina sembra avere un ruolo determinante anche nell’aumentare l’attività dei canali KATP presenti sulle cellule α isolate nei topi inducendo un effetto inibitorio sul rilascio del glucagone attraverso l’ iperpolarizzazione della membrana cellulare (Franklin et al., 2005).
Un eccessivo rilascio di insulina nell’isola provoca però alterazioni delle cellule α che può portare ad una diminuzione della loro capacità di sopperire all’ipoglicemia, come avviene in caso di diabete di tipo II. Questa assunzione fa chiaramente capire che la condizione necessaria affinchè le cellule α possano intervenire durante l’ipoglicemia è costituita da una rapida diminuzione dell’insulina intraisola.(Banarer et al.,2002)
Oltre al glucosio e all’insulina, che indubbiamente giocano il maggior ruolo nella secrezione di glucagone, vi sono anche altre sostanze che posso modulare la funzione delle cellule α:
Somatostatina : La somatostaina agisce localmente all’interno delle stesse isole di Langerhans deprimendo la secrezione sia dell’insulina che del glucagone (18)
Studi immonologici hanno dimostrato che fra i 5 recettori identificati della somatostatina l’SSTR2 è espresso dalle cellule α dei topi. Questi recettori sono accoppiati alle proteine G e producono diversi effetti intra-cellulari tra cui l’attivazione dei canali K+ nelle cellule α che iperpolarizzano la membrana cellulare e impediscono il rilascio di glucagone (19)
Un altro effeto negativo della somatostaina è evidente nell’interazione con l’adenilato ciclasi e il cAMP (20) mentre ha un effetto positivo agendo come messaggero extra-cellulare e attivando un meccanismo autocrino che stimola la secrezione delle cellule α dei topi attraverso un aumento dell’esocitosi associata ad un aumento nei livelli di cAMP (21) Altri messaggeri extra-cellulari: il GABA è un altro modulatore delle cellule pancreatiche α rilasciato dalle vescicole contenenti insulina.Tale messaggero influenza la corrente di polarizzazione della membrana cellulare delle cellule α per mezzo dei recettori iperpolarizzandola e facendo decrescere l’azione di rilascio del glucagone nelle cellule dei topi (Rorsman et al.1989). Insieme al GABA vi sono anche altri neurotrasmettitori come L-glutammato e l’amilina che stimolano opportuni recettori al rilascio o all’inibizione del glucagone. (11)
Attività fisica: Nell’attività fisica molto intensa il tasso ematico del glucagone spesso aumenta fino a 4-5 volte, ma non è chiaro perché ciò avvenga, dato che in questa situazione non si osserva necessariamente un abbassamento della glicemia.Perartro, questo aumento del glucagone ha quanto meno il vantaggio di prevenire una diminuzione del glucosio ematico.Uno dei fattori che potrebbero essere responsabili di questo aumento del glucagone nell’attività fisica è la maggiore concentrazione di aminoacidi in circolo anche se potrebbero essere coinvolti anche altri fattori, come una stimolazione nervosa β-adrenergica delle isole di Langerhans. (22) (vedi figura 8)
Figura 12 Effetto di un esercizio sui livelli di concentrazione di glucosio,insulina e glucagone
La secrezione del glucagone è influenzata anche dai neurotrasmettitori (acetilcolina e noradrenalina) rilasciati dai terminali nervosi presenti sulle isole di Langerhans in grado di assicurare una rapida risposta in caso di ipoglicemia e quindi proteggere il cervello da possibili danni.
2.10 EFFETTO DEL GLUCAGONE NELLA FASE POST-PRANDIALE
Durante l’assunzione di un pasto misto, ricco di carboidrati, l’azione delle cellule α è inibita a favore del rilascio di insulina da parte delle cellule β che promuovono l’accumulo di glicogeno nel fegato e nei muscoli.
Al contrario, durante lo stato di fame o di limitazione di carboidrati, l’individuo sano mostra un aumento della concentrazione di glucagone a digiuno e una riduzione nella concentrazione di insulina riducendo il rapporto molare insulina-glucagone a valori inferiori a 0.4. Questa duplice azione ormonale serve a massimizzare la produzione epatica di glucosio, inizialmente, attraverso un aumento nella glucogenolisi e successivamente, quando le scorte di glucosio finiscono, per mezzo della gluconeogenesi. Se il digiuno viene prolungato il fegato dà inizio alla produzione di chetoni mettendo a disposizione cosi un nutrimento alternativo per il sistema nervoso centrale. Quando la chetonemia diventa sufficiente a garantire il giusto apporto di nutrimenti, la gluconeogenesi si arresta risparmiando l’utilizzo di azoto e quindi garantendo la sopravvivenza dell’individuo al prolungato digiuno.
A differenza dei carboidrati che inibiscono la produzione di glucagone, la presenza di proteine all’interno del pasto fa aumentare la concentrazione di aminoacidi nella circolazione sanguigna provocando una stimolazione delle cellule α. Lo stesso effetto stimolante avviene sulla secrezione dell’insulina anche se in quantità minori. In questo caso, perciò, la risposta del glucagone e dell’insulina non sono di segno opposto ma si raggiunge una sorta di sinergia tra i due ormoni. L’importanza della stimolazione operata dagli aminoacidi sulla secrezione del glucagone deriva dal fatto che questo ormone può così promuovere una rapida conversione degli aminoacidi in glucosio, rendendo in tal modo disponibile una quantità ancora più elevata di glucosio per i tessuti. Le modalità con cui avvengono questi meccanismi restano però ancora oscure.
Figura 13 Regolazione della secrezione di glucagone in fase post-prandiale
In diversi studi è stato osservato che i vari aminoacidi presentano effetti diversi sulla secrezione del glucagone. In particolare è stato dimostrato che l’alanina e l’arginina generano la miglior risposta alla secrezione del glucagone (10)
Figura 14 Effetto di un pasto proteico su un soggetto sano (a destra) ed Effetto dell'infusione di arginina sulle concentrazioni di insulina,glucagone e glucosio in un soggetto sano (a sinistra) [Muller et al.]
Nello studio in vitro condotto da John E.Gerich et al. è stato dimostrato anche un duplice effetto dell’arginina in preseza e assenza di glucosio. E’ stato visto che in assenza di glucosio, questo aminoacido stimola la secrezione di glucagone ma, in presenza di glucosio essa diventa un potente stimolatore del rilascio di insulina. Quindi, glucosio e arginina agiscono con meccanismi diversi sulle cellule α e β e ciò probabilmente è dovuto alla presenza di recettori separati sulle cellule α e β. (23)
2.11 DIABETE MELLITO: IPOTESI BI-ORMONALE
Il diabete mellito è una patologia che porta a turbe del metabolismo dei carboidrati, dei lipidi, delle proteine.
Esistono due tipi di diabete mellito:
1. Il diabete di tipo 1 o diabete mellito insulino-dipendente
Il diabete di tipo I è una malattia autoimmunecaratterizzata dal danneggiamento delle cellule β pancreatiche che rendono insufficiente la produzione di insulina. Si può manifestare all’improvviso, in un arco di tempo che va da qualche giorno a poche settimane, con una sequenza di eventi rappresentati da: 1) aumento della glicemia causata dalla mancanza di insulina e quindi dall’inutilizzo del glucosio da parte dei tessuti; 2) aumento dell’utilizzo dei grassi sia a fine energetici che per la sintesi del colesterolo nel fegato; 3) deplezione delle proteine corporee.
In America il diabete di tipo si manifesta di solito intorno ai 14 anni, e perciò viene spesso definito
diabete mellito giovanile.
2. Il diabete di tipo 2 o diabete mellito non insulino-dipendente
Questo tipo di diabete è di gran lunga più frequente del diabete di tipo I. Si tratta di una tipologia di malattia molto varia che si manifesta normalmente dopo la terza decade di vita, spesso tra i 50 e i 60 anni, sviluppandosi gradualmente. Ultimamente però si è assistito ad un costante aumento di casi giovanili, alcuni addirittura al di sotto dei 20 anni. Questa tendenza è da mettere in rapporto all’aumentata incidenza dell’ obesità, che costituisce, il fattore di rischio più importante per il diabete di tipo II, sia nel giovane che nell’adulto.
Contrariamente al diabete di tipo I, quello di tipo II è caratterizzato dall’aumento dei livelli plasmatici di insulina (iperinsulinemia) per compensare la diminuita sensibilità dei tessuti bersaglio all’azione metabolica dell’insulina (insulino-resistenza). Questa minore sensibilità altera l’utilizzo e l’immagazzinamento dei glicidi, facendo aumentare la glicemia e stimolando l’aumento compensatorio della secrezione insulinica. La resistenza all’insulina fa parte di una serie di disturbi a cascata conosciuti con il nome di sindrome metabolica: 1) obesità, in particolare l’accumulo di grasso a livello addominale; 2) resistenza all’insulina; 3)iperglicemia a digiuno; 4)alterazione del quadro lipidico, con aumento dei trigliceridi nel sangue e del complesso colesterolo-lipoproteine ad alta densità; 5) ipertensione. Le conseguenze più serie della sindrome metabolica colpiscono l’apparato cardiovascolare, potendo causare ateroscelrosi e lesioni in vari organi.
Nello stato di digiuno i livelli plasmatici di glucagone sia nei diabetici di tipo I che di tipo II non differiscono sostanzialmente dai valori osservati nei soggetti non diabetici. La differenza principale nella concentrazione plasmatica di glucagone di un soggetto sano ed un soggetto diabetico, sta nel suo rapporto con la concentrazione di glucosio nell’ambiente che, in assenza di terapie, è più alta nei diabetici che nei soggetti non-diabetici. Il fatto che questa differenza nella glicemia non sia direttamente associata con un livello molto basso di glucagone indica che le cellule α dei soggetti diabetici non rispondono in modo esatto all’iperglicemia.
Più di 30 anni fa,Unger e Orci proposero un’ipotesi bi-ormonale per spiegare la patofisiologia del diabete.
In accordo con questa teoria, tale malattia metabolica è il risulatato di un’insulino-deficienza o resistenza e di un eccessivo rilascio di glucagone (iperglucagonemia), che può causare un alto rate di produzione epatica di glucosio rispetto alla sua utilizzazione, favorendo l’iperglicemia. (24) Nel diabete di tipo 2, il danneggiamento nel meccanismo di rilascio di insulina e lo sviluppo
dell’insulino-resistenza è spesso accompagnato da un assoluto o relativo aumento di glucagone nello stato post-prandiale ed a digiuno (24)
In questa condizione, l’insulina non agisce come feedback negativo nei confronti della secrezione epatica di glucosio mentre il glucagone potenzia la mobilizzazione del glucosio dal fegato provocando l’iperglicemia.
Un’altra malfunzione riportata nei soggetti diabetici è la mancanza di soppressione del rilascio del glucagone in condizioni iperglicemiche che può contribuire all’aumento di glucosio nel sangue sia in soggetti con diabete di tipo I che in soggetti con diabete di tipo II (25). Tuttavia però questa irregolare funzionalità delle cellule α non è presente quando i livelli di insulina sono ageduati, suggerendo che l’anomalo rilascio di glucagone è un prerequisito solo di soggetti con patologia del diabete o danneggiamento dell’azione o della secrezione dell’insulina.
L’ iperglucagonemia è anche responsabile dello sviluppo di iperglicemia e diabete in pazienti con glucagonoma, una forma rara di neoplasia che interessa le cellule α del pancreas (26).
L’azione del glucagone in soggetti diabetici ha importanti conseguenze anche nella gestione dell’ipoglicemia. La risposta del glucagone a basse concentrazione di glucosio è compromessa nei diabetici di tipo I e nei pazienti con diabete di tipo II di lunga durata, aumentando il rischio di episodi di gravi ipoglicemie, specialmente in pazienti trattati con insulina (27).L’ipoglicemia iatrogena è infatti il risultato dell’azione reciproca dell’eccesso di insulina e della compromessa controregolazione glicemica che contribuisce ad aumentare il rishio di mortalità in soggetti diabetici. (27)
Infine, nei soggetti non diabetici l’ingestione di carboidraiti porta ad un significativo abbassamento dei livelli plasmatici di glucagone. Nei pazienti diabetici di tipo I e II invece, l’assunzione di carboidrati non provoca alcuna riduzione di tale ormone anzi, spesso conduce ad un paradossale aumento di glucagone e, la concomitante assunzione di insulina esogena, nei diabetici di tipo I, non è in grado di ripristinare il normale livello glicemico.
Tutti questi problemi legati alla inadeguata risposta di secrezione da parte del glucagone in soggetti diabetici , possono essere attribuiti ad una compromessa regolazione delle cellule α che include il danneggiamento del sistema di rilevazione dei livelli gi glucosio plasmatico, perdita della funzionalità delle cellule β, resistenza all’insulina o disfunzione autonomica.
Tuttavia i meccanismi che generano tale patofisiologia sono ancora largamente sconosciuti e ancora tanto c’è da scoprire per la messa a punto di una terapia veramente efficace.
2.12 OBESITA’ E DIABETE
L'Obesità è una patologia multifattoriale dovuta prevalentemente a fattori genetici, ambientali ed individuali. Si definisce obeso un individuo la cui massa di tessuto adiposo sia eccessiva, con indice di massa corporea (BMI) maggiore di 30 e una circonferenza vita maggiore di 102 cm negli uomini ed 80 cm per le donne (come dimostrato nella tabella 1 ).
Tabella 1 Classificazione dell'obesità
ll Diabete Mellito di tipo 2 (non insulino dipendente) è fortemente associato all'obesità in quanto essa è caratterizzata da insulino-resistenza.
Il rischio di sviluppare la malattia aumenta progressivamente quando aumentano l'indice di massa ed il contenuto corporeo di grassi. I soggetti con BMI> 35 Kg/m2 possono avere un rischio 40 volte più alto di quelli con un BMI< 23 Kg m2. Una disposizione centrale (al tronco) del tessuto adiposo aumenta il rischio di Diabete Mellito tipo 2.
La quantità di insulina rilasciata, nei soggetti obesi, è molto maggiore rispetto agli individui sani (iperinsulinemia compensatoria) in quanto deve compensare l’incapacità delle cellule β di produrre una risposta adeguata all’aumento di glucosio. La funzione β cellulare si trova così in una forte condizione di sovraccarico e questa situazione può provocare una diminuzione maggiore delle capacità secretorie e quindi provocare iperglicemia, ridotta tolleranza al glucosio ed infine il diabete di tipo 2.
L’obesità inoltre predispone l’individuo ad una serie di fattori di rischio cardiovascolare tra cui l’ipertensione e l’aumento del tasso di colesterolo nel sangue.
2.13 TERAPIE DEL DIABETE DI TIPO 2: Il GPL-1
Il trattamento usuale del diabete di tipo I consiste nel somministrare sufficienti dosi di insulina per riportare il più vicino possibile alla norma il metabolismo dei carboidrati, dei grassi e delle proteine.
Nei pazienti con diabete di tipo II, si raccomanda invece di seguire un alimentazione appropriata e una certa attività fisica per ridorre il peso corporeo e contrastare la resisitenza all’insulina.Se non si manifestano miglioramenti, si possono somministare farmaci che aumentano la sensibilità all’insulina o che ne stimolano la produzione epatica.In molti pazienti però, bisogna somministare direttamente insulina per poter efficacemente controllare la glicemia.
Negli ultimi anni è stato studiato con successo l’effeto prodotto dal GPL-1 (glucagon-like peptide 1). Il GPL-1 è un acido peptidico composto da 30 aminoacidi che viene rilasciato dalle cellule L dell’intestino dopo l’assunzione di un pasto.Il GPL-1 intatto, non ha effetti sugli ormoni pancreatici ma, se attiva attraverso la soppressione dei primi 6 aminoacidi della catena diventa, in relazione alla presenza di glucosio, un potente stimolatore delle cellule β e contemporaneamente un inibitore della secrezione del glucagone. E’ stato dimostrato però che il GPL-1 non blocca il rilascio di glucagone durante lo stato di ipoglicemia ma può addirittura favorirlo. (8)
Questa teoria è stata avvalorata dai diversi esami immunoreattivi che hanno mostrato la presenza di recettori del GPL-1 sulle cellule α (28)
Dato dunque, il duplice effetto del GPL-1, sono in corso molteplici studi sull’utilizzo di tale sostanza come trattamento tarapeutico in soggetti con diabete di tipo 1.