Capitolo 1
Capitolo 1
Capitolo 1
1
1.1 Introduzione
La proteina p53 (Figura 1), anche conosciuta come proteina tumorale 53, è un fattore di trascrizione che regola il ciclo cellulare e ricopre la funzione di soppressore tumorale. Fin dalla sua scoperta, 30 anni fa, è stata oggetto di intenso studio: l'elevato interesse per questa molecola è dovuto al fatto che p53 è la proteina più frequentemente alterata nei tumori umani di varia origine. Circa il 50% di tutte le neoplasie maligne umane porta mutazioni o delezioni nel gene TP53 che disabilitano la funzione di soppressore tumorale della proteina codificata. A differenza di molti geni oncosoppressori che si comportano da recessivi, TP53 si comporta come gene dominante. Infatti, mutazioni che interessano anche solo uno dei due alleli del gene (condizione di eterozigosi) possono compromettere la funzionalità della proteina. La spiegazione di tutto ciò sta nel fatto che p53 risulta idonea ad espletare la sua funzione di fattore di trascrizione solo in forma tetramerica. I tetrameri composti di alcune subunità di p53 mutanti e da altre wild type possono non essere più funzionali come fattori di trascrizione1.
Parte generale
2
Come soppressore dei tumori, p53 controlla una via di trasduzione del segnale evoluta per proteggere organismi pluricellulari dallo sviluppo del cancro che potrebbe essere promosso da sollecitazioni diverse, tra cui il danneggiamento del DNA. Normalmente, la proteina p53 nelle cellule di mammifero, in assenza di stress sia genotossici che non, è presente a bassi livelli ed ha un’emivita molto breve (6-20 minuti). In seguito a stress, si ha accumulo marcato e attivazione della proteina p533. Lo stress genotossico che danneggia il DNA induce arresto del ciclo cellulare, l'attivazione di meccanismi di riparazione, e in caso di danni irreparabili, induzione di apoptosi. La decisione da parte delle cellule di riparare le lesioni del DNA e continuare attraverso il ciclo cellulare oppure indurre l’apoptosi è rilevante per l'incidenza di mutagenesi e, successivamente, carcinogenesi. Infatti l’incompleta riparazione del danno al DNA prima di riprodursi o della mitosi può far si che insorgano mutazioni genetiche ereditabili. Tuttavia, la natura della risposta di segnalazione cellulare che determina la sopravvivenza o la morte cellulare è lungi dall'essere compresa.4
1.2 p53: struttura e funzioni.
La proteina p53 umana contiene 393 aminoacidi ed è stata suddivisa strutturalmente e funzionalmente in quattro domini:
• un dominio ammino-terminale (aa 1-43), che attiva ulteriori fattori di trascrizione; • un dominio centrale o ‘core’, sequenza specifico di legame al DNA (aa 100-300); • un dominio di tetramerizzazione (aa 324-355): la p53 nativa è infatti costituita, in soluzione, da un tetrametro e soltanto in questa forma può agire come oncosoppressore5. • un dominio regolatore carbossi-terminale (aa 363-393), ricco di amminoacidi basici, che sembra regolare il nucleo di legame al DNA. (Figura 2).
3 Figura 2. Struttura della proteina p53 che mostra i diversi domini, e punti principali di mutazioni nel cancro umano. La proteina p53 è rappresentata dal sito ammino-terminale (1) al carbossi-terminale (393) identificando con colori differenti i diversi domini. Sotto viene riportato l'elenco delle proteine cellulari e virali che riescono a legarsi alle diverse regioni di p536.
Il dominio di attivazione N-terminale interagisce con i fattori di trascrizione TFIID (TBP, TAF) e TFIIH che regolano positivamente l'espressione genica. In particolare i residui amminoacidici Phe19, Leu22, e Trp23 di p53 sono risultati necessari per l’attivazione trascrizionale in vivo e sono anche coinvolti nell’interazione con i fattori TATA-associati. La proteina Murine Double Minute 2 umana, MDM2, è un’ubiquitina ligasi E3 che regola negativamente l'attività trascrizionale di p53 attraverso il legame al dominio di attivazione N-terminale, cosi come l’adenovirus E1B-55 kDa. L'importanza della regolazione negativa di p53 da parte di MDM2 è stata evidenziata dal fatto che la delezione omozigote di MDM2 nei topi, riscontrata nei casi di letalità embrionale, comporta una delezione omozigote simultanea di p53.
Il dominio centrale di p53 contiene la regione sequenza specifica di legame al DNA indicato anche come p53 C (dove C sta per “core”). Ci sono quattro regioni altamente conservate all'interno di tale nucleo centrale, dove sono state trovate la maggior parte delle mutazioni non-senso di p53 che rendono la proteina incapace di legarsi alle specifiche sequenze di riconoscimento sul DNA, impedendo la trascrizione del gene. Le sei mutazioni più frequenti associate al cancro sono raggruppate in “hot-spots” ed interessano le posizioni degli amminoacidi 175, 245, 248, 249, 273 e 282.
Parte generale
4
Il dominio C-terminale è ricco di residui basici e regola la capacità di p53 di legare specifiche sequenze di DNA al suo dominio centrale. Questo dominio può legarsi non specificamente anche alle diverse forme di DNA come le rotture o le discordanze interne. Un cambiamento strutturale a questo livello è necessario per attivare p53 e il suo legame a sequenze specifiche del DNA. E’ stato inoltre constatato che la delezione di questo dominio, la sua fosforilazione in specifici residui da parte della proteina chinasi C (PKC) o della caseina chinasi II o il legame con anticorpi PAB421 possono attivare il legame al DNA tramite il core centrale6.
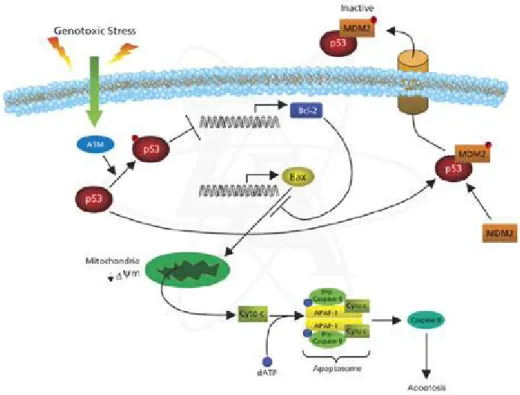
La proteina p53 è un potente fattore di trascrizione in grado di attivare più geni bersaglio, causando l'arresto del ciclo cellulare, apoptosi o senescenza (Figura 3). Il suo ruolo biologico è infatti quello di garantire l'integrità del genoma delle cellule. p53 può stimolare i processi di riparazione, meccanismi di protezione, o la cessazione della divisione cellulare e l'induzione di una morte cellulare programmata. Per raggiungere i suoi obiettivi, p53 può utilizzare un ampio spettro di attività, come la sua capacità di funzionare come fattore di trascrizione, inducendo o reprimendo geni diversi o come proteina regolatrice all'interno di numerose vie di segnalazione. L'attività di p53 è stimolata in risposta al danno al DNA. In risposta allo stress, p53 determina il destino delle cellule inducendo arresto della crescita, riparazione del DNA o l’apoptosi in caso in cui i danni al DNA siano gravi. La perdita delle funzioni di p53 e delle sue attività regolatorie è coinvolta non solo nello sviluppo di tumori, ma è anche relazionabile a malattie cardiovascolari, neurodegenerative, infettive e metaboliche. La proteina p53 ha la capacità di legare specifiche sequenze reattive di DNA e l’attivazione trascrizionale dipende dalla capacità del dominio centrale di legarsi al DNA e dalla capacità di p53 di interagire con le regioni regolatorie di certi geni. L'attivazione trascrizionale è determinata dal dominio N-terminale di p53, che contiene varie regioni che interagiscono con il macchinario di trascrizione e fattori di reclutamento che modificano la struttura della cromatina locale4.
5 Figura 3. p53 regola la risposta cellulare allo stress7.
1.3 Stabilizzazione di p53 in risposta al danno al DNA.
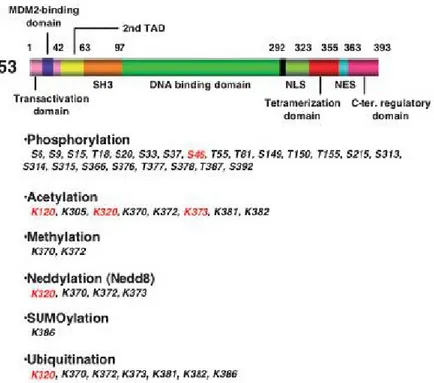
La proteina p53 è regolata principalmente attraverso modificazioni post-traduzionali, specialmente fosforilazione, e l'accumulo di p53 è il primo passo in risposta allo stress cellulare (Figura 4). Il dominio N-terminale viene in genere fosforilato, mentre il C-terminale contiene residui che vengono fosforilati, acetilati, metilati e sumoilati. Modificazioni post-traduzionali sul dominio N-terminale sono importanti per la stabilizzazione di p53, mentre quelli sul C-terminale inibiscono la capacità di regolare il legame sequenza-specifico con il DNA, lo stato di oligomerizzazione, il processo di importazione/esportazione nucleare, e l'ubiquitinazione4.
Parte generale
6 Figura 4. Domini funzionali e modifica dei residui in p53. TAD indica dominio di transattivazione. NLS e NES significano rispettivamente segnali di localizzazione e di esportazione nucleare. Modifiche correlate all'apoptosi sono evidenziate in rosso4.
Il gene MDM2 è un bersaglio trascrizionale di p53, e una volta sintetizzata, la proteina MDM2 può legarsi a p53 nel dominio N-terminale, portando alla sua rapida degradazione attraverso il meccanismo ubiquitina-proteosoma. In risposta a un danno al DNA, la chinasi ATM fosforila rapidamente p53 a livello della Ser15. La chinasi serina/treonina Chk2 agisce a valle di ATM fosforilando p53 a livello della Ser20. Questi siti di fosforilazione del dominio N-terminale di p53 sono vicini alla regione di legame alla proteina MDM2 e riescono in tal modo a bloccare l'interazione tra p53/MDM2, portando alla stabilizzazione di p53, che sfugge dalla degradazione del proteosoma. Recenti scoperte suggeriscono che la fosforilazione costitutiva di p53 da parte di PKC nel dominio C-terminale contribuisce alla sua degradazione attraverso la via ubiquitina-proteosoma4.
7
1.4 Pathway della p53.
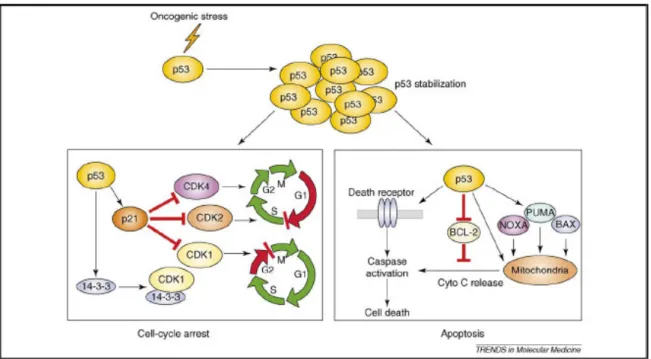
Come sopra detto, p53 è un fattore di trascrizione che controlla la risposta cellulare allo stress genotossico, oncogenico e ipotossico attraverso l'induzione dell’arresto del ciclo cellulare e l'apoptosi (Figura 5). Questa proteina agisce in diversi modi:
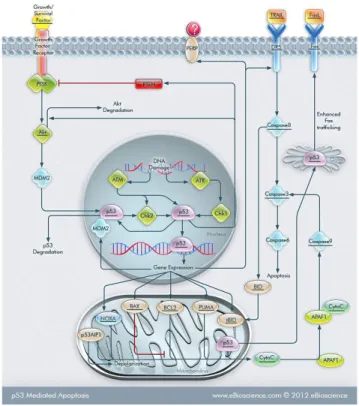
• inducendo l'arresto del ciclo cellulare attraverso la stimolazione della trascrizione del gene che codifica per p21 e la proteina 14-3-3σ;
• inducendo apoptosi attraverso due differenti vie: la via estrinseca e la via intrinseca (attraverso l'attivazione della trascrizione di geni bersaglio, come ad esempio PUMA e NOXA, o interagendo con proteine anti-apoptotiche, come Bcl-2 e Bcl-X, o inducendo l'attivazione di caspasi).
Figura 5. Il soppressore del tumore p53 è una potente molecola con attività antiproliferativa e pro-apoptotica8.
Il circuito p21.
L’arresto del ciclo cellulare in risposta all'attivazione di p53 è mediato da due proteine principali: p21, che arresta le cellule principalmente nella fase G1 del ciclo cellulare, e 14-3-3σ, che arresta le cellule nella transizione G2/M. La proteina p21, target trascrizionale diretto di p53, è un potente inibitore delle chinasi ciclina-dipendenti (CDK), che blocca efficacemente la progressione del ciclo cellulare in fase G1.
Parte generale
8
La proteina p21 è regolata in diversi punti, dalla sua trascrizione alla degradazione, e questi eventi di regolazione caratterizzano l’arresto del ciclo cellulare p53-dipendente9. La proteina p53 è uno dei molti regolatori trascrizionali ben documentati del locus p21. La regolazione positiva e negativa di questi altri fattori detta la misura in cui p53 transattiva p21. Ad esempio, MIZ1 (MYC interacting zinc-finger protein-1) e SP1 (specificity protein 1) regolano p21 in modo sia p53-dipendente che p53-indipendente. Recentemente è stato dimostrato che ZAC1 (zinc-finger protein), che regola l'apoptosi e l’arresto del ciclo cellulare, transattiva sinergicamente p21 attraverso un’interazione diretta con SP1 e un’interazione funzionale con p5310. Al contrario, l’induzione di p21 p53-dipendente è soggetta ad una miriade di fattori antagonisti. Ad esempio, il fattore di trascrizione MYC reprime l'attivazione di p21 in risposta a molteplici forme di danno al DNA attraverso la sua interazione con MIZ1. È interessante notare che p53 attiva l'espressione di p21 sia direttamente, a livello trascrizionale, che indirettamente, regolando fattori che aumentano la stabilità del mRNA12. L’ultimo segnale integratore di eventi post-trascrizionali è il ribosoma, che sintetizza la proteina, e che a sua volta può subire una regolazione post-traduzionale. La proteina p21 è soggetta ad una serie di modificazioni post-traduzionali che alterano la sua attività, localizzazione e/o stabilità subcellulare. La proteina p21 può essere fosforilata in almeno sette residui di Ser/Thr con effetti variabili sulla sua localizzazione e stabilità. Ad esempio, AKT/PKB può fosforilare p21 su Thr145 o Ser146. Questo è un caso interessante perché la fosforilazione di Akt a livello della Thr145 aumenta la stabilità di p21 e blocca la sua associazione con PCNA (proliferating cell nuclear antigen), promuovendo l’arresto del ciclo cellulare, mentre la modifica a livello della Ser146 attiva un complesso ciclina D1-CDK4 che guida la proliferazione.13. Inoltre, la fosforilazione della Thr145 da parte dell’oncogeno PIM1 chinasi è in grado di alterare la localizzazione di p21, sequestrandola nel citoplasma dove non riesce a bloccare la progressione del ciclo cellulare. Inoltre, la stabilità di p21 può essere alterata sia positivamente che negativamente dalla fosforilazione della Ser130 da parte della p38α/JNK1 e della Ser114 da parte della GSK3β. Questi esempi rappresentano solo un piccolo sottoinsieme delle chinasi e dei siti di fosforilazione che regolano p21, ma sicuramente servono a dimostrare come un unico tipo di modificazione post-translazionale sia in grado di modulare l'attività di p21 ben dopo la transattivazione p53-dipendente. Un’ulteriore regolazione di questo circuito si ha a livello della degradazione del proteosoma. La degradazione di p21 ubiquitina-mediata avviene in diverse fasi del ciclo cellulare, ad opera di diverse ligasi E3, tra cui SCF-Skp2, APC-Cdc20 e CRL4Cdt2. E'
9
noto che p21 può essere degradata dal proteosoma indipendentemente dal suo stato di ubiquitinazione. La proteina p21 può essere stabilizzata senza modificazioni post-traduzionali attraverso la formazione di complessi con Ciclina D1, mediata da una sovraespressione di RAS, o legandosi a WISP39/Hsp90, entrambi i quali impediscono l'associazione di p21 con il proteosoma. Quest’ultimo step di regolazione può amplificare o smorzare il segnale iniziato da p53 attiva per determinare il livello e l'attività di p21 nella cellula, come pure il suo effetto finale sul ciclo cellulare9.
Proteina target 14-3-3σ.
La proteina 14-3-3σ è un target trascrizionale diretto di p53 che si lega alla fosfatasi mitotica Cdc25 sequestrandola nel citoplasma e impedendole così di attivare i complessi CDK1-ciclina B. Le proteine p21 e 14-3-3σ lavorano in modo coordinato per far rispettare l’arresto del ciclo cellulare in risposta a vari agenti dannosi per il DNA e proteggere la cellula dall’apoptosi. Relativamente poco si sa circa ulteriori co-regolatori trascrizionali di 14-3-3σ; tuttavia, è stato dimostrato che BRCA1 aumenta i livelli di mRNA di 14-3-3σ in maniera p53-dipendente. Nelle cellule embrionali di topo, BRCA1 è necessaria per la piena espressione di 14-3-3σ, e questa constatazione ha portato alla scoperta che in molte linee cellulari di cancro umano con p53 wild-type, una sovraespressione di BRCA1 causa una sovraregolazione di 14-3-3σ. La regolazione negativa della trascrizione di 14-3-3σ invece è dovuta ad una ipermetilazione del promotore del DNA e avviene in molti tessuti normali, ma è anche molto diffusa nei tumori umani, anche se i meccanismi con cui essa è stabilita rimangono in gran parte sconosciuti. Degna di nota, la difesa di 14-3-3σ da parte delle chinasi IKKα attraverso il silenziamento nei cheratinociti, tramite il legame al promotore e il blocco dell'azione della istone-metiltransferasi SUV39H1 e della DNA-metiltransferasi DNMT3A9.
La via apoptotica estrinseca.
L'induzione della via apoptotica estrinseca avviene mediante l’attivazione di recettori di morte, definiti TNF-R, mediante il legame con i loro ligandi specifici. In breve, i segnali provenienti dall'esterno della cellula, che determinano che essa dovrebbe sottoporsi ad apoptosi, sono mediati attraverso tali recettori, come ad esempio il recettore Fas. Il legame del Fas-ligando al recettore Fas (Cd95/Apo1), componente chiave della via estrinseca, si traduce in un raggruppamento di recettori che portano al reclutamento di FADD e pro-caspasi 8 al complesso.
Parte generale
10
La caspasi 8 si auto attiva, e su di essa si attiva a sua volta la pro-caspasi 3, l’effettore caspasi principale di apoptosi. La proteina p53 può promuovere l'apoptosi attraverso l'attivazione trascrizionale di geni che codificano per i recettori di morte localizzati a livello della membrana plasmatica, tra cui Fas, DR4, DR5 e PERP14. Inoltre, per migliorare la trascrizione Fas, la sovraespressione di p53 può aumentare i livelli di recettore sulla superficie cellulare, promuovendo il traffico del recettore Fas dal Golgi. Due altri membri della famiglia dei recettori di morte che sono trascrizionalmente indotti da p53 sono DR4 e DR5/KILLER, che sono recettori contenenti un dominio di legame per il ligando TNF-correlato induttore di apoptosi (TRAIL). Sia DR4 che DR5 vengono indotti dal danno del DNA e possono innescare o aumentare l'apoptosi indotta da TRAIL e dagli agenti chemioterapici15. Un altro gene pro-apoptotico, PERP, si attiva in risposta al danno al DNA nelle cellule trasdotte sia con E2F1 o con la proteina adenovirale E1A. In topi privati di tale gene è stato dimostrato che la sua carenza porta ad una compromessa risposta apoptotica p53-dipendente in timociti e neuroni. Anche se l'espressione PERP rimane significativamente elevata in tutti i tipi cellulari testati, il requisito funzionale per PERP nell’induzione di apotosi risulta diverso in ogni tipo di cellula. La cinetica di induzione PERP causata dal danno al DNA e la presenza di p53 come elemento reattivo supportano l'idea che tale gene possa essere un bersaglio trascrizionale diretto di p53, ma l'esatto meccanismo attraverso il quale PERP media l'apoptosi rimane ancora una domanda senza risposta. Un altro modo con cui p53 inibisce i segnali pro-sopravvivenza nella cellula è tramite l'up-regolazione dell’oncosoppressore PTEN. L'attivazione di recettori di superficie attraverso il legame con i loro ligandi (mitogeni, citochine) o l'attivazione di oncogeni come Ras, attivano la via di segnalazione Akt/PKB che promuove la crescita cellulare e la resistenza all'apoptosi.I segnali vengono trasdotti dalla fosfatidilinositolo 3-chinasi (PI3K) che fosforila il fosfatidil-inositolo 2-fosfato (PIP2) formando PIP3. PIP3 è necessario per alcune chinasi, come PDK1, che fosforilano e attivano Akt/PKB.
Tuttavia, al momento dell'attivazione della fosfatasi lipidica PTEN, che converte PIP3 a PIP2, PDK1 non è più attiva, e in tal modo viene compromessa la via di segnale Akt/PKB; quindi l'attivazione di PTEN arresta la divisione cellulare e porta all'induzione di apoptosi. È interessante notare che l'attivazione della trascrizione del gene PTEN da parte di p53 è fortemente stimolato dalla fosforilazione di Ser4615.
11
La via apoptotica intrinseca.
La via apoptotica intrinseca è prevalentemente controllata dalle proteine della famiglia Bcl2, che incidono direttamente sulla permeabilizzazione della membrana esterna mitocondriale (MOMP, mitochondrial outer membrane permeabilization).
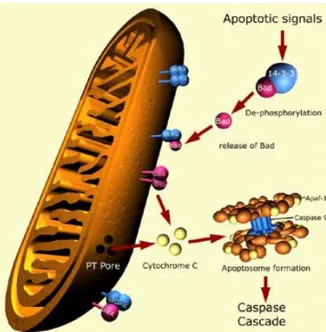
Sebbene non sia chiaro l’esatto meccanismo della MOMP, sembra che le proteine pro-apoptotiche della famiglia Bcl-2 (come Bax e Bak) abbiano un ruolo cruciale come molecole effettrici formatrici di pori. Le proteine anti-apoptotiche della famiglia Bcl-2 (come Bcl-2 e Bcl-XL) probabilmente si oppongono al fenomeno della MOMP mediante un processo di eterodimerizzazione con le proteine pro-apoptotiche “Bax-like”. Le proteine pro-apoptotiche formatrici di pori, inducono inizialmente soltanto la permeabilizzazione della membrana mitocondriale esterna lasciando intatta l’ultrastruttura della membrana interna. Quindi l’attivazione di Bax e Bak e la risultante MOMP non comporta direttamente la perdita irreversibile della funzionalità mitocondriale e la morte cellulare, ma porta all’attivazione di varie vie di morte programmata, tra cui l’apoptosi classica. Dopo tale processo, il citocromo c viene rilasciato dallo spazio mitocondriale intermembrana e si lega alla proteina citosolica Apaf-1 formando una grande struttura multi-proteica nota come apoptosoma. Una volta formato, l’apoptosoma recluta l'iniziatore inattivo caspasi-9 e lo attiva rimuovendo il pro-dominio. Una volta attiva, la caspasi-9 innesca una cascata di attivazione delle caspasi esecutive quali la pro-caspasi-3 e -7, che possono attaccare diversi bersagli cellulari e alla fine portano alla morte cellulare, con i caratteristici tratti distintivi di apoptosi, come la condensazione della cromatina e il blebbing cellulare16.
Parte generale
12 Figura 6. Via di apoptosi mitocondriale17.
La famiglia Bcl2 è costituita da classi di proteine ad attività anti-apoptotica (prosurvival) e attività pro-apoptotica. La classificazione è basata su somiglianze strutturali dei quattro domini omologhi caratteristici di Bcl2 (domini BH, da Bcl2 Homology domain), chiamati BH1, BH2, BH3 e BH4. I domini BH sono fondamentali per il corretto funzionamento della proteina; infatti, la delezione di questi domini attraverso tecniche di biologia molecolare, influenza il tasso di sopravvivenza/apoptosi cellulare. Le proteine Bcl2 anti-apoptotiche, come Bcl2 e Bcl-xL, presentano tutti e quattro i domini BH, mentre le proteine pro-apoptotiche possono a loro volta essere suddivise in base al numero di domini BH che possiedono; ad esempio, proteine con più domini BH, come Bax (Bcl2-associated X protein) o Bak (Bcl2 homologous antagonist killer), oppure proteine che possiedono solo il dominio BH3, come Bid, Bim and Bad (chiamate anche BH3 only). Il dominio BH3 è il requisito strutturale minimo per la funzione pro-apoptotica ed è risultato essenziale per l’eterodimerizzazione tra i membri della famiglia Bcl2. Molte delle proteine della famiglia Bcl2 hanno inoltre anche domini transmembrana. Curiosamente, alcuni membri della famiglia Bcl2 sono stati associati all’apoptosi p53-dipendente (Figura 7).
Il primo membro ad essere identificato come un bersaglio p53-inducibile è stata la proteina pro-apoptotica Bax, ma gli elementi specifici che all'interno del gene Bax sono sensibili alla p53 sono stati identificati più tardi. In un modello animale in cui l’apoptosi p53-dipendente attenua la crescita del tumore del cervello, è stato osservato che l'assenza di Bax aumenta la crescita tumorale, mentre i livelli di apoptosi risultano diminuiti.
13
Successivamente è stato dimostrato che Bax interagisce con un'altra proteina p53-indotta: ASC (apoptosis-associated speckle-like protein), che contribuisce al passaggio di Bax ai mitocondri15.
Un gene bersaglio molto importante regolato da p53 è quello che codifica la "BH3 only" proteina PUMA (p53 upregulated modulator of apoptosis), che regola l'apoptosi a monte di Bak e Bax. Negli esseri umani il gene PUMA codifica per due isoforme PUMA-α e PUMA-β contenenti il dominio BH3, che sono entrambe sovra regolate in risposta all'attivazione di p53. PUMA è un potente attivatore della cascata apoptotica in quanto modula l'attività di Bax facilitando il rilascio del citocromo c dai mitocondri18. La rilevanza fisiologica del ruolo di PUMA nell’apoptosi p53-mediata è stata dimostrata in topi privati del gene stesso. I topi in assenza PUMA presentano diverse carenze in punti chiave del processo apoptotico che sono le stesse osservate nei topi privati del gene di p53.
Sono stati osservati anche diversi effetti indipendenti da p53: infatti recentemente si è dimostrato che la proteina PUMA può essere indotta anche dalla proteina p73, e potrebbe quindi avere un ruolo nell’apoptosi indotta da p73, indicando che PUMA potrebbe intervenire in vie apoptotiche indipendenti da p53. Più recentemente è stato dimostrato che in cellule ematopoietiche l'espressione PUMA è inibita da Slug, un altro gene p53 indotto in seguito a γ-irradiazione. Qui, gli autori hanno osservato che l'espressione di Slug impedisce ai progenitori emopoietici di subire danni al DNA che inducono apoptosi, e hanno identificato PUMA come obiettivo chiave15.
Un nuovo membro della famiglia di proteine "BH3 only" ad azione pro-apoptotica regolata da p53 è Noxa. I topi privati del gene Noxa hanno mostrato notevole resistenza alla proteina adenovirale E1A e ai danni al DNA che inducono apoptosi. Allo stesso modo per il meccanismo con cui PUMA attiva il rilascio del citocromo c dai mitocondri, si crede che Noxa agisca come sensibilizzante interagendo e interferendo con i membri pro-apoptotici della famiglia Bcl2.
Parte generale
14 Figura 7. Apoptosi mediata da p5320.
1.5 Processi di feedback positivi e negativi di p53.
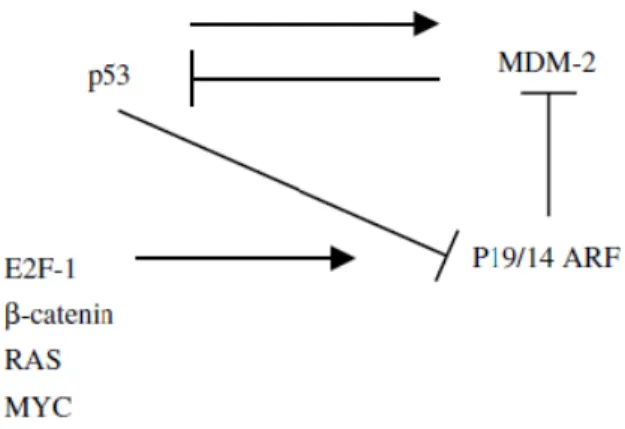
Numerosi studi in letteratura hanno individuato nel pathway di p53 dieci cicli di feedback positivi o negativi. Ciascuno di questi cicli crea un circuito composto di proteine la cui attività o velocità di sintesi sono influenzate dall'attivazione di p53, e questi a loro volta provocano un'alterazione dell’attività di p53 stessa nella cellula. Tra questi, sette sono processi di feedback negativo (MDM2, COP1, Pirh2, p73 delta N, ciclina G, Wip-1 e Siah-1) e tre sono i processi di feedback positivo (PTEN-AKT, p14 / 19 ARF e Rb) che modulano l'attività di p53. Tutte queste reti o circuiti sono autoregolati, in quanto sono indotti dall’attività di p53 a livello trascrizionale, dalla repressione trascrizionale di p53 (p14/19 ARF) o regolati da proteine p53-indotte. Sei di questi circuiti di feedback agiscono attraverso MDM2 (MDM2, ciclina G, Siah-1, p14/19 ARF, AKT e Rb) per modulare l’attività di p5321. Un dato interessante è che la via di p53 è intimamente legata ad altre vie di trasduzione del segnale che giocano un ruolo importante nelle origini del cancro. Uno dei primi collegamenti studiati coinvolge P14/P19 ARF e MDM2. La proteina p14/19 ARF si lega alla proteina MDM2 e modula la sua attività di ubiquitina ligasi, aumentando i livelli della proteina p53.
15
La trascrizione del gene P14/19 ARF è positivamente regolato da E2F1 e dalla beta-catenina mentre è negativamente regolata da p53 stessa. Inoltre, i livelli di proteina p14/19 ARF sono aumentati dall’attività di Ras e Myc in una cellula (Figura 8)22.
Figura 8. Circuito p53/MDM2/p14/19 ARF21.
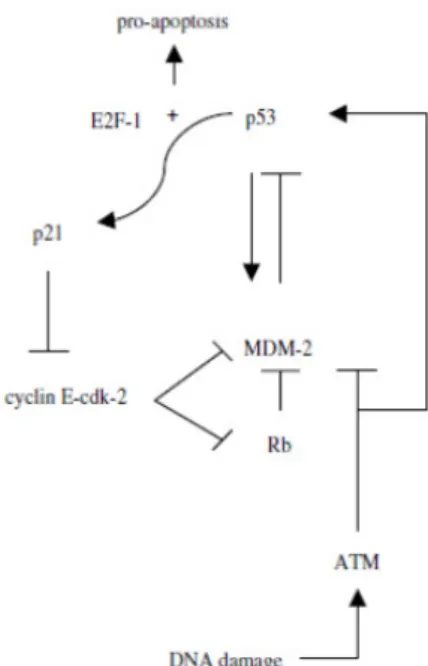
La proteina Rb, proteina del retinoblastoma, può essere trovata nelle cellule in un complesso con MDM2 e p53, con conseguente aumento dell’attività di p53 e una maggiore attività apoptotica, in quanto il legame della proteina con MDM2 ne inibisce l’attività. Rb lega e inibisce fattori di trascrizione appartenenti alla famiglia E2F che giocano un ruolo centrale nella transizione dalla fase G1 alla S nel ciclo cellulare.La normale funzione di Rb è quindi quella di bloccare la cellula in uno stadio del ciclo cellulare prevenendone errate o dannose divisioni. Alti livelli di E2F-1 attivo, non legato a Rb, traslano la risposta di p53 dall’arresto in G1 all’apoptosi. Sia Rb che MDM2 sono fosforilate e inibite dalla ciclina E-cdk2. Quando p53 è attivata, stimola la sintesi della proteina p21, che inibisce l'attività della ciclina E-Cdk2, e questo, a sua volta agisce sul complesso Rb-MDM2 che promuove l'attività di p53 e l’apoptosi. Dopo il danno al DNA, sia la proteina MDM2 che la proteina p53 vengono modificate dalla proteina chinasi ATM. Questo aumenta l'attività di p53 nello stesso modo in cui il complesso p53/MDM2-Rb aumenta la funzione di p5323 (Figura 9).
Parte generale
16 Figura 9. Circuito ciclina/Cdk/Rb/MDM-221.
Parte dell’attivazione della proteina p53 comporta la sua fosforilazione a livello dei residui di serina situati in 33 e 46 da parte della p38 MAP chinasi (Figura 10). La p38 MAP chinasi è essa stessa attivata tramite fosforilazione (regolata dalla via Ras-Raf-Mek-Erk) e può essere invertita o inattivata ad opera della Wip-1 fosfatasi. Il gene Wip-1 è un gene p53-reattivo e p53-regolato cosi che forma un circuito di feedback negativo collegando i percorsi di Ras e p5324.
17
Una proteina attivata positivamente da p53 regola la trascrizione della ubiquitina ligasi Siah-1, che a sua volta agisce degradando la proteina catenina. Dai livelli di beta-catenina dipende l’attività del gene p14/19 ARF, che a sua volta regola negativamente MDM-2 e si traduce in elevati livelli di p5325 (Figura 11).
Figura 11. Circuito Siah-1/beta-catenina/p14/p19 ARF21.
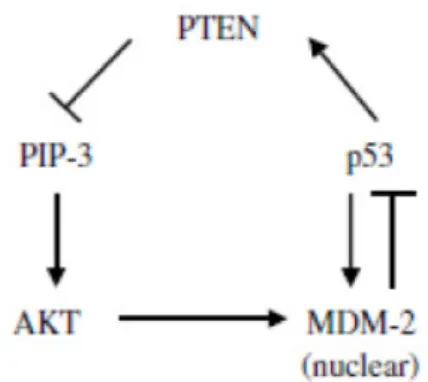
In alcuni tipi di cellule, la proteina p53 induce la trascrizione del gene PTEN. La proteina PTEN è un PIP-3 fosfatasi. PIP-3 attiva la chinasi AKT, che ha diversi substrati proteici anti-apoptotici compresa la proteina MDM-2. La fosforilazione comporta la traslocazione di MDM-2 nel nucleo dove inattiva p5321 (Figura 12).
Figura 12. Circuito PTEN/AKT/MDM-221.
Questi processi di feedback positivi e negativi comportano due eventi: modulano l'attività di p53 nella cellula e coordinano la sua attività con altre vie di trasduzione del segnale che regolano l'ingresso della cellula nel ciclo cellulare21.
Ci sono due ulteriori circuiti di autoregolazione negativa di p53. Uno dei geni p53-reattivi più attivi è il gene della ciclina G. Esso viene rapidamente trascritto ad alti livelli dopo l'attivazione di p53 in un'ampia varietà di tipi cellulari.
Parte generale
18
La proteina ciclina G forma un complesso con la fosfatasi PP2A, che rimuove un residuo fosfato da MDM-2 il quale viene aggiunto alla proteina MDM-2 da una chinasi CDK. La fosforilazione di MDM-2 da parte della ciclina A/cdk2 inibisce la sua attività, quindi la ciclina G-PP2A fosfatasi aumenta l’attività di MDM-2 e inibisce p5326 (Figura 13).
Figura 13. Circuito Ciclina G/MDM-221.
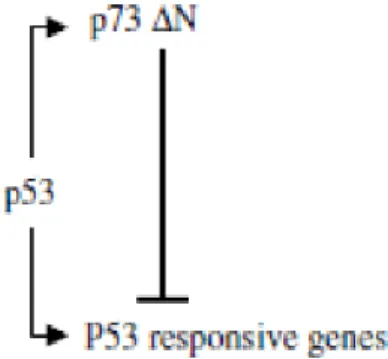
Il secondo processo di feedback negativo coinvolge un membro della famiglia dei fattori di trascrizione di p53, che includono p53, p63 e p73, i quali sono legati strutturalmente e funzionalmente e si sono evoluti da un precursore comune.
In seguito a stress il gene p53 viene attivato e a sua volta stimola la trascrizione dal gene 73 di un particolare m-RNA sottoposto a splicing, chiamato p73 delta N. Questo si traduce in una proteina p73 senza il suo dominio ammino-terminale21 (Figura 14).
Figura 14. Circuito p73 delta N21.
Le tre proteine della famiglia di p53 hanno struttura simile composta da un dominio di attivazione trascrizionale N-terminale, collegato a un dominio centrale che si lega ad una specifica sequenza di DNA.
19
Tutti e tre i fattori di trascrizione della famiglia di p53 riconoscono la stessa sequenza di DNA, anche se p53, p63 e p73 sono in grado di avviare processi trascrizionali distinti. Così, quando p53 attiva la trascrizione di p73 delta N, questa può legarsi a molti dei geni regolati da p53, ma l'assenza di un dominio di transattivazione comporta che essa agisca come un repressore o concorrente dell’attivazione trascrizionale di p5321.
1.6 Interazione p53/MDM2.
Il gene MDM2 è stato originariamente identificato su cromosomi double-minute di fibroblasti 3T3 nel topo, mentre solo più tardi è stata scoperta un’associazione della proteina MDM2 con p53, chiarendo l’importante ruolo fisiologico che essa svolge nel suo controllo. Gli embrioni di topo privati del gene MDM2 muoiono presto dopo l'impianto, ma riescono a sopravvivere se vengono privati anche del gene TP53. Questo fornisce la prova genetica convincente che il ruolo più importante di MDM2 è la regolazione fisiologica della funzione di p53, almeno nel primo sviluppo.
È importante sottolineare inoltre che la proteina MDM2 stessa è il prodotto di un gene p53-inducibile, per cui le due molecole sono collegate tra loro attraverso un ciclo di feedback negativo volto a mantenere bassi i livelli cellulari di p53 in assenza di stress. Principalmente, MDM2 è una ligasi E3 e promuove la degradazione di p53 attraverso un percorso ubiquitina-dipendente in proteosomi nucleari e citoplasmatici 26S. La modificazione delle proteine tramite la coniugazione con l’ubiquitina è un meccanismo generale di targeting intracellulare e avviene mediante l’attacco di catene di poliubiquitina in modo covalente su residui di lisina delle proteine bersaglio, che poi vengono degradate all’interno dei proteosomi (Figura 15). Recenti studi, tramite l’utilizzo di MDM2 e p53 mutanti, hanno chiarito i requisiti strutturali per la distruzione MDM2-mediata di p53. Inoltre questi risultati hanno rivelato l'importanza delle diverse regioni su entrambe le proteine e dimostrato che, per l'accumulo di p53 in risposta allo stress, il meccanismo di regolazione deve essere interrotto in modo che p53 possa sfuggire agli effetti di degradazione promossi da MDM2.
MDM2 presenta un’attività E3 ubiquitina ligasi specifica per se stessa e per la p53 nel suo dominio evolutivamente conservato C-terminale RING FINGER (zinc-binding); la porzione RING FINGER è fondamentale per l'attività ligasi E3.
Parte generale
20
MDM2 trasferisce la monoubiquitina su residui di lisina contenuti principalmente nel dominio carbossi-terminale di p53. L’ubiquitinazione di p53 mediata da MDM2 avviene nel nucleo in un complesso con le p300/CBP (CREB-binding protein), proteine coattivatrici trascrizionali, che fungono da impalcatura. La maggior parte di MDM2 endogena è destinata a complessi p300/CBP nel nucleo. Tuttavia, considerando che MDM2 catalizza la monoubiquitazione di p53, che di per sé non è un substrato per la degradazione proteosomica, i complessi p300/MDM2 mediano la poliubiquitazione finale di p5310.
Figura 15. Regolazione da parte di MDM2 di p53. MDM2 e p53 formano un circuito di feedback negativo8.
1.7 Requisiti strutturali per l'interazione p53/MDM2.
La MDM2 umana è una fosfoproteina di 491 amminoacidi che interagisce attraverso il suo dominio ammino-terminale con un α-elica presente nel dominio ammino-terminale di transattivazione di p53. Ciò comporta numerosi effetti negativi su p53 stessa: infatti, MDM2 blocca direttamente la sua attività trascrizionale e, ancora più importante, data la sua funzione di ligasi E3, induce l’ubiquitinazione di p53 per la degradazione proteosomica. Nella Figura 16 si riporta la struttura cristallina del complesso p53/MDM227.
21 Figura 16. Struttura cristallina dell’interazione p53/MDM227.
La base biochimica di inibizione della funzione di p53 mediata da MDM2 è stata chiarita da dati cristallografici che hanno dimostrato che il dominio ammino-terminale di MDM2 presenta una tasca idrofobica profonda in cui si lega il dominio di transattivazione di p53, sfuggendo in tal modo all’interazione con la macchina trascrizionale. Questo fatto è stato confermato da analisi biochimiche. L'interazione diretta tra le due proteine avviene tra una tasca idrofobica relativamente piccola (aa 25-109) nel dominio ammino-terminale di MDM2 e un peptide anfipatico di 15 amminoacidi nel dominio ammino-terminale di p53. Il sito di legame minimo tra MDM2 e la proteina p53 è stato successivamente mappato sui residui 18-26. La mutagenesi sito-diretta ha mostrato l'importanza dei residui di p53 Phe19, Trp23, Leu26, Leu 14 e Leu 22, tra i quali i primi tre risultano essere i più importanti. Il legame per l'interazione p53/MDM2 consiste di soli tre legami a idrogeno e il più profondo è quello che coinvolge il residuo Trp23 di p5328. Di conseguenza, se i siti di legame di p53 per MDM2 risultano mutati questo conferisce alla proteina resistenza alla degradazione da parte di MDM2. Allo stesso modo, un’altra causa di mancato legame con la p53 può essere una mutazione su MDM2 dei residui Gly58, Glu68, Val75, o Cys77. I domini che interagiscono mostrano una configurazione a serratura stretta all’interfaccia p53/MDM2. La parte idrofoba dell’α-elica anfipatica di p53, che è formata dagli amminoacidi 19-26, si inserisce profondamente nella tasca idrofobica di MDM2. Thr18 è molto importante per la stabilità dell’α-elica di p53. La tasca di MDM2 è formata dagli amminoacidi 26-108 e si compone di due porzioni a struttura simile che si ripiegano in un solco profondo fiancheggiato da 14 residui idrofobici ed aromatici.
Parte generale
22
Misure sperimentali hanno dimostrato che la forza del legame p53/MDM2 varia da una Kd di 60 a 700 nM, a seconda della lunghezza del peptide p53. La fosforilazione di Ser15 e Ser20 non influisce sul legame mentre la fosforilazione di Thr18 lo indebolisce di 10 volte, indicando che la sola fosforilazione di Thr18 può essere responsabile dell’annullamento del legame p53/MDM2. Tuttavia, la stabilizzazione di p53, dovuta a radiazioni ionizzanti, risulta da un’inibizione del legame con MDM2 attraverso una cascata di fosforilazione che prima richiede la fosforilazione di Ser15 di p53, necessaria per la successiva fosforilazione di Thr1829. In conclusione, conformazione e idrofobicità sembrano essere due requisiti critici per l'interazione tra MDM2 e p53. Un requisito fondamentale, ma non sufficiente per la degradazione di p53 è l'interazione diretta tra p53 e MDM2 attraverso i loro domini ammino-terminali. Un’ulteriore requisito di p53 è infatti la sua regione di omo-tetramerizzazione, che migliora la degradabilità attraverso un migliore legame con MDM2, perché una p53 mutante monomerica perde sensibilità alla degradazione da parte di MDM2. Come detto sopra i residui di lisina, soprattutto nel dominio carbossi-terminale di p53, sono tasselli obbligatori per la monoubiquitazione, quindi tale dominio (aa 363-393) risulta essere fondamentale28.
1.8 p53 come bersaglio terapeutico.
La proteina p53 è un fattore di trascrizione che si attiva in risposta allo stress cellulare. A seconda della gravità della minaccia per l'integrità del genoma, p53 può indurre l’arresto del ciclo cellulare oppure l’apoptosi. Dato che p53 ha un ruolo cruciale nei processi di crescita e soppressione cellulare, deve essere strettamente regolata per consentire alle cellule normali di funzionare. Questo risultato è ottenuto in larga misura grazie all’azione della proteina MDM2. In tutti i tumori le funzioni di p53 vengono annullate come mezzo per eludere l'apoptosi e questo può avvenire sia direttamente, disabilitando l’azione di p53 attraverso la mutazione o la delezione del gene che la codifica, o indirettamente, tramite alterazione dei vari componenti che ne regolano l’attività. Circa la metà dei tumori mantiene una p53 wild-type e in questi la normale regolazione di p53 è talvolta interrotta attraverso la sovraespressione diretta di MDM2 (in circa il 7% dei tumori). Tale fenomeno di amplificazione genica è particolarmente frequente (circa 30%) in sarcomi osteogenici umani e sarcomi dei tessuti molli30.
Dato il ruolo centrale di p53 nella soppressione tumorale, le strategie terapeutiche non genotossiche che riescono ad attivare le sue funzioni sono risultate molto interessanti.
23
Molti potenziali bersagli terapeutici all'interno del pathway di p53, a valle della risposta allo stress, offrono la possibilità di attivare la sua funzione in modo non genotossico, bypassando eventuali limitazioni che potrebbero rendere un target a monte del pathway inefficace. Tra questi è stato riconosciuto il sistema regolatorio p53/MDM2. La modulazione di questo sistema tramite l’azione di piccole molecole rappresenta un campo molto attivo della ricerca30.
La domanda chiave per qualsiasi strategia terapeutica che mira ad attivare la risposta p53 è se questo si tradurrà in un effetto selettivo sulle cellule tumorali o comporterà un’azione anche sulle cellule dei tessuti sani. Tale specificità di p53 di uccidere le cellule tumorali, ma non le cellule normali, appare alla base della sicurezza di tale terapia genica, che ha ottenuto l'approvazione in Cina ed è ora in fase di sviluppo in altri paesi.
E' stato dimostrato che i topi con un allele isomorfico di MDM2 producono solo il 30% del livello normale di MDM2 e presentano di conseguenza un aumento trascrizionale e funzionale di p53. Gli effetti di p53 in queste circostanze non sono letali come ci si potrebbe aspettare, sebbene gli animali siano piccoli e mostrino apoptosi p53-dipendente delle cellule linfoidi. Al contrario, essi non invecchiano prematuramente e sono resistenti alla formazione del tumore31. Allo stesso modo, la soppressione in vivo di MDM2 usando oligonucleotidi antisenso, risulta provocare effetti terapeutici antitumorali senza tossicità manifesta. Da questi ed altri risultati è evidente che il percorso p53 differisce significativamente nelle cellule normali e nelle cellule tumorali wild-type p53 e che quest’ultime sono selettivamente sensibili agli aumenti di funzioni effettrici di p53. Sono stati sfruttati diversi approcci per ridurre il controllo di p53 da parte di MDM2 compresa l'inibizione della sua espressione con conseguente annullamento dell’attività ubiquitina ligasi di MDM2 e inibizione del legame p53/MDM230.
Blocco dell’espressione di MDM-2.
Diversi studi che utilizzano oligonucleotidi antisenso per inibire l'espressione di MDM2, hanno convalidato l’utilizzo di questo approccio sia su linee cellulari che in modelli murini di cancro umano. MDM2 subisce una downregulation a causa di tali oligonucleotidi antisenso e questo comporta una stabilizzazione di p53 e un’attivazione del suo pathway nelle cellule tumorali che crescono in colture di tessuti in vitro oppure come xenotrapianti tumorali in topi vivi.
È interessante notare che, non solo le cellule p53 wild-type, ma anche le cellule che esprimono una p53 mutante hanno risposto ugualmente bene all’inibizione di MDM2.
Parte generale
24
Questa osservazione inaspettata è stata difficile da spiegare con l'inibizione selettiva dell’espressione MDM2. Uno studio recente ha suggerito che la stabilizzazione, indipendente da p53, della chinasi ciclina-dipendente inibitrice p21 a seguito della downregulation di MDM2, potrebbe contribuire all'attività antitumorale degli oligonucleotidi antisenso di MDM2. Tuttavia, questo meccanismo non è stato confermato8. Inibizione dell'attività ubiquitina ligasi di MDM2.
Poiché l’ubiquitinazione e la degradazione di p53 rappresentano il principale controllo negativo da parte di MDM2, l’attività di ubiquitina ligasi di MDM2 è stata identificata come possibile strategia di attivazione di p5332. Recentemente, sono state identificate piccole molecole inibitrici che colpiscono specificamente l'attività ligasi E3 di MDM2. Diversi composti di questa classe, denominata HL198C (Figura 18), inibiscono in vitro l’ubiquitinazione di p53 con un IC50 nel range da 20-50 mM. Nelle cellule tumorali tali
molecole attivano l'apoptosi indotta in maniera p53-dipendente, ma hanno una bassa potenza e selettività. E’ necessaria quindi un’ulteriore ottimizzazione di questi inibitori per aumentarne potenza, specificità verso la ligasi E3 e per eliminare le attività su bersagli di azione p53-indipendenti, in modo da poter valutare questa strategia8.
Inibizione del legame MDM2-p53.
L'inibizione del legame p53/MDM2 è una strategia auspicabile per la stabilizzazione e l’attivazione di p53. Tuttavia, utilizzare come target di piccole molecole un’interazione proteina-proteina è impegnativo in quanto, di solito, questo tipo di interazioni coinvolgono grandi superfici piane che sono difficili da occupare tramite l’utilizzo di composti a basso peso molecolare.
Nel caso dell'interazione p53/MDM2, tuttavia, è stato dimostrato che per il legame tra le due proteine solo un numero limitato di residui amminoacidici risultano cruciali. Infatti, solo tre amminoacidi di p53 (Phe19, Trp23 e Leu26) sono ritenuti essenziali per il legame tra le due proteine. Tali amminoacidi sono inseriti in una tasca idrofoba profonda sulla superficie della molecola MDM2. In questo modo, è stato possibile progettare piccole molecole in grado di mimare con successo questa interazione8.
Negli ultimi anni sono state scoperte e pubblicate un numero sempre crescente di piccole molecole inibitrici del legame p53/MDM2. Tuttavia, solo i composti più recenti tra quelli esaminati hanno mostrato una potenza cellulare e una selettività per il loro bersaglio molecolare apprezzabili.
25
Le prime piccole molecole antagoniste selettive e potenti di MDM2, le Nutline (Figura
18), sono state identificate da una classe di imidazoline33. Questi inibitori, in vitro, possono
spiazzare p53 da MDM2 con un valore di IC50 di 90 nM per Nutlina-3a, l'enantiomero
attivo della Nutlina-3. Studi cristallografici e strutturali hanno dimostrato che la Nutlina si lega alla tasca di MDM2 per la p53, in modo da imitare le interazioni molecolari dei residui amminoacidici cruciali di p53 (Figura 17). Le Nutline, testate su diversi tipi di colture cellulari, sono risultate in grado di inibire l'interazione p53/MDM2 in ogni contesto cellulare con un alto grado di specificità, con conseguente stabilizzazione e attivazione del pathway di p53. Le cellule tumorali proliferanti sono state effettivamente bloccate in fase G1 e G2, e condotte ad apoptosi, quando esposte a basse concentrazioni (micromolari) di Nutline33. Ulteriori studi hanno confermato che le Nutline sono altamente selettive per il loro bersaglio molecolare nella cellula. La somministrazione di Nutlina-3a per 21 giorni in topi vivi che portano xenotrapianti tumorali umani è in grado di inibire efficacemente la crescita tumorale e una riduzione del tumore a dosi non tossiche. Questi studi hanno fornito la prima dimostrazione che l'attivazione di p53 wild-type tramite inibitori farmacologici dell'interazione p53/MDM2 è fattibile e potrebbe essere sfruttata per la terapia del cancro8.
Figura 17. Aspetti strutturali dell'interazione p53/MDM2 e del legame della Nutlina. (a) MDM2 e p53 interagiscono tra loro con i loro domini N-terminali attraverso una tasca di legame p53 ben definita. La struttura cristallina del legame ha rivelato che tre residui di aminoacidi del peptide p53 (verde) sono essenziali per il legame con MDM2, e sono inseriti in una cavità abbastanza profonda sulla superficie MDM2 (giallo). La figura mostra i più importanti domini funzionali delle proteine p53 e MDM2. (b) Nutlin-2 (rosso) si lega alla tasca p53 di MDM2 simulando l'interazione dei tre residui amminoacidici cruciali del peptide p53 (verde)8.
Parte generale
26
Oltre alle Nutline, altre classi di attivatori di p53 sono riportate in letteratura, ma la loro attività non è stata ancora confermata. Una classe di antagonisti MDM2 portanti un nucleo benzodiazepinico (Figura 18) sono risultate in grado di distruggere l’interazione p53/MDM2, in vitro, con IC50 nel range del 0.5-2 mM. I primi composti pubblicati
presentavano bassa permeabilità cellulare. Tuttavia in seguito a modifiche strutturali questi composti potrebbero sopprimere la crescita di cellule p53 wild-type con IC50 nel range
10-30 mM e con una selettività 3-9 volte maggiore per cellule con p53 funzionale. Tuttavia la bassa potenza cellulare e la scarsa selettività di questi composti rende difficile valutare il loro potenziale antitumorale8.
Un’ulteriore piccola molecola, denominata RITA (riattivazione di p53 e induzione di apoptosi delle cellule tumorali) (Figura 18), è stata identificata recentemente da uno screening cellulare. RITA può attivare il patwhay di p53 attraverso un meccanismo non genotossico e indurre l’arresto p53-dipendente della crescita cellulare e l’apoptosi. E' stato presupposto che RITA attivi la p53 impedendo la sua interazione con MDM2 ma a differenza di altri inibitori, RITA sembra agire legandosi a p53 e non a MDM2. Tuttavia, altri studi con RITA non hanno confermato la capacità del composto di bloccare l'interazione p53/MDM2 in vitro, pertanto il meccanismo molecolare di attivazione di p53 tramite questa molecola attende ulteriori chiarimenti.
Basandosi sulla struttura di composti noti, Ding et al. hanno identificato composti a struttura spiro-ossoindolica (Figura 18) in grado di inibire in vitro il legame p53/MDM2 con IC50 nel range 20-2000 nM. E’ stata dimostrata inoltre una buona correlazione tra
questa attività in vitro e l’attività cellulare. I composti più potenti di questa classe hanno mostrato attività anti-proliferativa con selettività quasi 30 volte maggiore per cellule con p53 wild type rispetto a cellule p53 mutate, ma la loro attività biologica non è stata ben caratterizzata. Questi autori hanno recentemente riportato un'altra classe di antagonisti di MDM2 chinolinici (Figura 18), con una buona attività cellulare e selettività per p53 wild-type. Tuttavia, in vitro non è ancora stata segnalata nessuna attività8.
27 Figura 18.Piccole molecole inibitrici di MDM2: strutture chimiche8.
Parte generale 1
Capitolo 2: TSPO
Capitolo 2: TSPO
Capitolo 2: TSPO
Capitolo 2: TSPO
29
2.1 Introduzione.
La proteina traslocatrice (TSPO) è una proteina evolutivamente conservata, di 18 kDa, altamente idrofobica, costituita da 5 domini transmembranali ed espressa principalmente nella membrana esterna mitocondriale dove interagisce con StAR (steroidogenic acute regulatory protein) per trasportare il colesterolo nei mitocondri. Negli esseri umani, la proteina traslocatrice è codificata dal gene TSPO ed è stata inizialmente descritta come recettore periferico delle benzodiazepine (PBR). Solo recentemente a tale proteina è stato attribuito il nome di Proteina Traslocatrice (18 kDa), abbreviato TSPO. Le ragioni che hanno portato a questo cambiamento sono state determinate dalle nuove scoperte riguardo la localizzazione, la struttura e il ruolo fisiologico di questa proteina. E’stato osservato infatti che il TSPO non interagisce solamente con le benzodiazepine, ma con molti altri ligandi; inoltre, non è localizzato esclusivamente a livello periferico e non presenta nemmeno una struttura recettoriale nel senso tradizionale del termine. E’ stato quindi attribuito come nuovo nome TSPO in riferimento alla principale funzione svolta da questa proteina che è appunto quella del trasporto di molecole come ad esempio il colesterolo e gli intermedi per la sintesi delle porfirine34.
E’ stato dimostrato che il TSPO ha una elevata distribuzione in molti tessuti del corpo. Il cervello ed il fegato presentano livelli relativamente bassi di TSPO, mentre i tessuti e le ghiandole coinvolte nella sintesi degli steroidi ne sono particolarmente ricchi; in cuore, polmone e rene la sua presenza è invece più moderata. Il TSPO, inoltre, è espresso in concentrazioni significative anche nel sistema immunitario e nelle varie sottopopolazioni dei leucociti. Alcuni studi riportano l’espressione del TSPO sulla membrana plasmatica degli eritrociti e sulle membrane nucleari di cellule cancerose. Nei mitocondri il TSPO è un modulatore/costituente del poro di transizione di permeabilità mitocondriale
(mitochondrial permeability transition pore, MPTP), un canale localizzato sui siti di
contatto tra le membrane mitocondriali esterna ed interna, insieme ad altre proteine quali il trasportatore dei nucleotidi adenosinici (ANT, 30 kDa) ed il canale anionico voltaggio dipendente (VDAC, 32 kDa) (Figura 19)34.
Parte generale
30 Figura 19. Proteina Traslocatrice (18kDa) (TSPO)34.
Fino ad oggi sono state identificate 4 proteine citoplasmatiche capaci di legarsi al TSPO35, anche se la funzionalità di due di queste è ancora sconosciuta:
• PRAX-1 (Peripheral Benzodiazepine Receptor-associated Protein 1) è una proteina associata al TSPO, del peso di 240 kDa, costituita da 1857 amminoacidi ;
• p10, una proteina di 10 kDa che co-immunoprecipita con il TSPO;
• Steroidogenic Acute Regulatory Protein (StAR) che lega il colesterolo e ne promuove il trasferimento mitocondriale;
• PAP7, che regola anch’essa la steroidogenesi.
Per quanto riguarda la proteina PRAX-1 è stato ipotizzato un modello di interazione con il TSPO35. Dati stechiometrici evidenziano che la PRAX-1, come monomero, è in associazione con più molecole (almeno due) di TSPO (18 kDa) che si trovano nelle sue vicinanze. La porzione della proteina TSPO accessibile per il legame con la PRAX-1 è probabilmente quella C-terminale, di 14 amminoacidi, esposta verso il lato citoplasmatico della membrana esterna mitocondriale. Il ruolo della PRAX-1 è ancora sconosciuto, ma si ritiene che questa sia coinvolta nel reclutamento di siti supplementari nelle vicinanze del TSPO in maniera tale da modularne le funzioni.
Il TSPO riveste un ruolo chiave nel la funzionalità cellulare. Ad esso sono attribuite le funzioni di regolazione della proliferazione cellulare, trasporto di porfirine (protoporfirina IX, mesoporfirina IX, emina), sintesi dell’eme, immunomodulazione, regolazione della steroidogenesi e apoptosi .
31
2.2 TSPO e steroidogenesi
La biosintesi degli steroidi inizia in tutti i tessuti steroidogenici con la conversione enzimatica del precursore colesterolo in pregnenolone. Il passaggio limitante della velocità di questo processo è il trasporto del colesterolo dai depositi intracellulari, attraverso lo spazio acquoso intermembranale, verso il lato della membrana mitocondriale interna rivolta verso la matrice, a livello della quale è presente il citocromo P-450 che catalizza la reazione35 (Figura 20).
Il TSPO riveste un ruolo cruciale nel processo steroidogenico. Questo infatti, quando viene attivato da un ligando (es. PK11195), lega il colesterolo e regola il suo trasporto dalla membrana mitocondriale esterna a quella interna, aumentando la produzione di pregnenolone e la sintesi degli steroidi . Il meccanismo ipotizzato si basa su un modello sperimentale di dinamica molecolare: il TSPO accoglierebbe nelle sue 5 eliche la molecola di colesterolo formando un canale tramite il quale il colesterolo verrebbe trasportato attraverso la membrana.
Figura 20.
E’ stato inoltre osservato che i livelli sistemici di steroidi aumentano in seguito ad una lesione, ad un dolore o in caso febbre, in risposta alla stimolazione della secrezione del fattore di rilascio delle corticotropine da parte di alcune citochine. Il coinvolgimento del TSPO nella sintesi di steroidi può quindi contribuire a questo meccanismo difensivo. Inoltre la relazione presente fra i bassi livelli di steroidi e l’ansia ha suggerito l’utilizzo del TSPO come un promettente target per il trattamento dei disturbi psichiatrici che sono correlati ad una disfunzione nella sintesi di steroidi35.
Parte generale
32
Alcuni composti altamente affini al TSPO come PK11195 e la classe delle N,N-dialchil-2-fenilindol-3-il-gliossilamidi, recentemente sintetizzate nel nostro laboratorio, hanno mostrato come effetto un aumento della biosintesi del pregnenolone, e quindi dei neurosteroidi, risultando potenzialmente utili per il trattamento dell’ansia36.
2.3 TSPO e apoptosi
La cascata apoptotica che porta alla morte cellulare è un processo ben caratterizzato, e la dissipazione del potenziale transmembranale mitocondriale causata dall’apertura dell’MPTP ne rappresenta un evento critico. L’apertura di tale poro infatti, se reversibile e a basso livello di conduttanza, causa un netto influsso di ioni ed acqua dentro la matrice mitocondriale al fine di mantenere il corretto equilibrio ionico dell’organello. L’apertura prolungata, al contrario, può innescare una serie di eventi che hanno importanti conseguenze sulla fisiologia mitocondriale e sull’equilibrio bioenergetico della cellula tra cui la dissipazione del potenziale transmembrana. In seguito a questo processo viene inoltre influenzata l’omeostasi degli ioni intracellulari, e favorito il rilascio di calcio dalla matrice che determina il rigonfiamento colloido-osmotico della matrice mitocondriale e l’efflusso dal mitocondrio di proteine e molecole tra cui il citocromo c e la flavoproteina AIF (fattore d’induzione dell’apoptosi). Quest’ultima trasloca nel nucleo dove induce condensazione della cromatina e frammentazione del DNA. Nel citosol il citocromo c interagisce con Apaf-1 (fattore attivante dell’apoptosi) innescando le reazioni delle caspasi che portano all’attivazione di un complesso di enzimi fondamentale nell’indurre il riarrangiamento strutturale del nucleo, del citoscheletro e della membrana plasmatica, caratteristici dell’apoptosi35.
Il TSPO è un modulatore endogeno di questo processo, ma l’esatto meccanismo non è ancora stato definitivamente stabilito. La sua regolazione può avvenire a diversi livelli e il meccanismo proposto include la modulazione dell’MPTP o la diretta interazione con molecole pro- o anti -apoptotiche. E’ stato dimostrato che i suoi ligandi modulano l’MPTP e la risposta apoptotica. PK11195 e FGIN-1-27 sono in grado di diminuire il potenziale di membrana mitocondriale, provocando apoptosi e arresto del ciclo cellulare nella fase G1/G0 in diverse linee cellulari tumorali35.
33
2.4 TSPO e immunomodulazione.
La presenza del TSPO in un gran numero di cellule immunomodulatorie come la microglia, i monociti del sangue, i linfociti e i leucociti, ha portato a pensare ad un coinvolgimento del TSPO nella risposta immunitaria; tuttavia, il meccanismo attraverso il quale ciò avviene è ancora sconosciuto.
I macrofagi presentano molti siti di legame per il TSPO e, in particolari studi effettuati sui topi, è stato osservato che le benzodiazepine inibiscono la capacità dei macrofagi di produrre ROS e alcune citochine infiammatorie come ad esempio IL-1, TNFα e IL-6. Inoltre il TSPO è coinvolto nel metabolismo ossidativo ad opera dei fagociti, processo necessario per eliminare definitivamente antigeni esterni. La funzione immunosoppressiva di alcuni ligandi che vanno ad interagire con il TSPO ha dimostrato l’importante ruolo rivestito da questa proteina nella risposta infiammatoria.
Nel SNC il TSPO è espresso prevalentemente nelle cellule microgliali. L’attivazione della microglia dà inizio ad una risposta infiammatoria che può aggravare la perdita neuronale in varie malattie infiammatorie neurodegenerative, come ad esempio l’Alzheimer. Poichè si è osservata una sovra espressione di TSPO nella microglia attivata, si è pensato che l’infiammazione presente a livello cerebrale durante le patologie neurodegenerative coinvolga tale recettore. Si presenta così la possibilità di utilizzare ligandi specifici per il TSPO per prevenire o limitare la neuroinfiammazione. Tuttavia il coinvolgimento della microglia attivata in varie patologie del SNC è differente a seconda del suo ruolo nella progressione e nella gravità della patologia stessa. E’ plausibile quindi ipotizzare che, attraverso l’utilizzo del TSPO come marker di tali cellule sia possibile determinare con esattezza quale ruolo la neuroinfiammazione gioca in specifiche condizioni patologiche che coinvolgono il SNC, aprendo così le porte alla cura o all’inibizione della progressione della malattia37.
2.5 Altre funzioni del TSPO.
Numerosi studi sulle funzioni del TSPO hanno dimostrato la modulazione della sua espressione in stati fisiologici e patologici. L’espressione del TSPO è sotto il controllo del sistema ormonale che colpisce l’interazione del suddetto recettore con VDAC e ANT. Anche altri fattori come le citochine (IL-1 o TNF-α), la vitamina A e le catecolamine modulano l’espressione del TSPO.
Parte generale
34
L’attività del TSPO sembra comprendere anche la modulazione della risposta cellulare nelle infezioni virali. Una delle strategie adottate dai virus per aggirare i meccanismi protettivi della cellula contro le infezioni, è quella di bloccare il processo di apoptosi. Molteplici studi hanno dimostrato che diversi virus patogeni utilizzano proprio il TSPO come target per mettere in atto il blocco dell’apoptosi. Questa scoperta offre quindi numerose prospettive per nuove strategie antivirali.
L’attività del TSPO risulta implicata anche nello stress. Infatti la sua attivazione durante l’esposizione acuta ad agenti stressogeni può essere vista come una predisposizione neuronale e metabolica per un migliore adattamento allo stress35.
Un’elevata espressione di tale recettore è stata inoltre riscontrata nelle cellule e nei tessuti neoplastici dell’ovario, nell’adenocarcinoma, nel carcinoma del fegato, del colon e nei gliomi. Questo aumento dell’espressione del TSPO è correlabile con il grado di aggressività della forma tumorale.
Il TSPO risulta sovra espresso anche nelle cellule microgliali attivate ed in generale nelle aree in cui è in corso una significativa risposta infiammatoria. Una up-regulation della proteina è osservabile anche in vari disturbi neurodegenerativi come ad esempio il morbo di Alzheimer e di Huntington, e nella sclerosi multipla. Una down-regulation di tale recettore è stata al contrario osservata in pazienti affetti da ansia, da stress post traumatico e stress cronico34,35.
Il monitoraggio dell’espressione di tale recettore, dunque, può essere una procedura rilevante in clinica per diagnosticare e/o seguire la progressione delle patologe correlate al TSPO. Per la determinazione e la caratterizzazione del TSPO si è rivelato utile l’utilizzo di probes molecolari tra cui ligandi radioattivi e ligandi fluorescenti, mediante differenti modalità e tecnologie come ad esempio la medicina nucleare (PET, CT/PET, SPECT), la risonanza magnetica nucleare o gli ultrasuoni.
La tomografia a emissione di positroni (PET), grazie all’utilizzo di radioligandi ha consentito la localizzazione e lo studio dell’espressione del TSPO nei topi, nei primati e anche nell’uomo. La PET è una delle principali tecniche di imaging nel campo della radiologia e della medicina nucleare e fornisce informazioni solo di tipo fisiologico basandosi sull’emissione di radiazioni gamma dal corpo del paziente a seguito dell’iniezione di opportuni radioligandi. Pur essendo una tecnica efficiente nella determinazione della presenza di tumori, la PET fornisce informazioni solo di tipo quantitativo.
35
Per rendere qualitativa l’indagine PET occorre una precisa mappatura dei coefficienti di assorbimento all’interno del corpo del paziente ricorrendo alla fusione dell’immagine CT (Computed Tomography) e di quella PET (CT/PET).
La tecnica PET può essere utilizzata per marcare il TSPO attraverso specifici radioligandi consentendo cosi il controllo della progressione della malattia ad esso associata e la determinazione dell’effetto prodotto dalla terapia effettuata.
Il PK11195 è stato il primo ligando radiomarcato ad essere utilizzato per il diagnostic imaging per evidenziare il TSPO in alcune malattie neurodegenerative quali il morbo di Alzheimer, di Hungtinton, il Parkinson e la sclerosi multipla.
2.6 TSPO e ligandi endogeni.
Una grande varietà di molecole endogene mostra alta affinità per il TSPO. Una delle prime molecole identificate, isolata sia a livello centrale che nei tessuti periferici (ghiandola surrenale, rene e testicoli), è un residuo di neuropeptide di 11 kDa composto da 86 amminoacidi che inibisce il legame del diazepam con il sito recettoriale delle Bz, chiamato “inibitore del legame del diazepam (DBI)”. Oltre a questa molecola e ai suoi metaboliti, sono stati isolati altri ligandi endogeni come le porfirine (protoporfirina IX, mesoporfirina IX ed emina) ed il colesterolo.
2.7 TSPO e ligandi sintetici.
Le principali classi di ligandi sintetici descritti in letteratura sono35,37 (Figura 21): • benzodiazepine (4'-clorodiazepam, Ro5-4864)
• isochinolincarbossammidi (PK11195) • imidazopiridine (Alpidem)
• derivati indolacetammidici (FGIN-1, SSR180575) • derivati fenossifenilacetammidici (DAA1097) • pirazolopirimidine (DPA714)
• N,N-dialchil-2-fenilindol-3-il-gliossilammidi.
Tutti questi ligandi, che mostrano un’alta affinità di legame per il TSPO, con valori di Ki
nell’ordine del nanomolare, sono stati utilizzati come strumenti farmacologici di ricerca per caratterizzare le proprietà e le funzioni del TSPO.
Parte generale
36 Figura 21.
I ligandi FGIN-1-27 e PK11195 hanno mostrato, in vitro, attività antitumorale, inducendo l’apoptosi e l’arresto del ciclo cellulare in linee cellulari di cancro esofageo, colon-rettale e in colture primarie di cancro. L’ipotesi che il TSPO partecipi allo sviluppo della malattia è supportata dalla sua localizzazione anche a livello nucleare e perinucleare in alcune linee cellulari cancerose: in questa sede il TSPO regolerebbe la proliferazione cellulare facilitando il trasporto del colesterolo nel nucleo. Inoltre, studi di imaging diagnostico non invasivo, mediante l’utilizzo di [3H]PK11195, hanno fornito dati di notevole interesse nella visualizzazione dei tumori mediante PET.
Ligandi come il Ro5-4864 ed il PK11195 limitano la gravità e la progressione di diversi processi infiammatori. Ciò è stato dimostrato utilizzando questi ligandi in studi sperimentali come il test della carragenina e nella terapia di patologie infiammatorie (infiammazione e iperplasia sinoviale, degenerazione del tessuto osseo e cartilagineo). SSR180575 è risultato efficace nella terapia dell’artrite reumatoide.
37
Ulteriori sperimentazioni hanno dimostrato l’efficacia di SSR180575 e PK11195 nella terapia del Lupus erythematosus (patologia autoimmune)35. Nella classe delle pirazolopirimidine, altamente affini e selettive per il TSPO, in particolare il composto DPA714 è risultato in grado di aumentare la steroidogenesi dell’ 80% rispetto ai livelli fisiologici. E’ stato osservato che i livelli sistemici di steroidi risultano significativamente aumentati a seguito di una lesione, di un dolore o in caso di febbre, a causa della stimolazione da parte di alcune citochine della secrezione del fattore di rilascio delle corticotropine. L’utilizzo di ligandi in grado di interagire con il TSPO e stimolare la sintesi di steroidi può quindi contribuire ad una risposta difensiva e antiinfiammatoria. Altri ligandi in grado di stimolare la sintesi di steroidi sono i derivati fenossifenilacetamidici37 (DAA1097).
Come descritto in precedenza, a livello cerebrale il TSPO è localizzato principalmente nelle cellule gliali. E’ stato riscontrato un marcato aumento della sua densità in caso di lesioni o situazioni patologiche a livello del SNC, in modo particolare negli stati acuti e cronici di patologie neurodegenerative. Da ciò è nata l’ipotesi che la regolazione delle funzioni del TSPO possa avere un ruolo neurotrofico e neuroprotettivo nelle lesioni neuronali. I risultati sperimentali hanno suggerito che l’espressione del TSPO poteva essere regolata da segnali provenienti da assoni in via di rigenerazione. Un ulteriore studio ha dimostrato come il ligando SSR180575 manifesta proprietà neuroprotettive in diversi modelli di patologie degenerative progressive del sistema nervoso centrale e periferico. Precisamente sia il ligando SSR180575 che il Ro5-4864 promuovono la sopravvivenza del neurone e la riparazione successiva ad assotomia. I meccanismi ipotizzati per tale azione sono: (i) regolazione del processo apoptotico in favore della sopravvivenza delle cellule gliali; (ii) produzione di mediatori come neurosteroidi, citochine o altri fattori neurotrofici che supportano la sopravvivenza del nervo.
La classe delle N, N-dialchil-2-fenilindol-3-il-gliossilammidi, rappresentata in Figura 22, è stata sviluppata e studiata nel laboratorio di ricerca presso il quale è stato svolto questo lavoro di tesi. Questi nuovi ligandi, che rappresentano una variazione della struttura base dell’alpidem, sono altamente affini e selettivi nei confronti del TSPO38.