CHAPTER 2
ADENOSINE RECEPTOR
ANTAGONISTS
93
2.1 INTRODUCTION
Adenosine (40), a major constituent of nucleic acid, consists of the purine base adenosine linked to the ribose moiety and it is an endogenous nucleoside presents in all tissues in the mammalian organisms where it modulates many important physiological functions.89
N N N N NH2 O OH OH H H H H HO Adenosine 40
It is formed by degradation of adenosine monophosphate (AMP) and is an integral part of the cellular energy system deriving within the cells from the hydrolysis of AMP through the action of the enzyme ecto-5’-nucleotidase. In particular, intracellular adenosine concentrations are low under normoxic condition, when the prime source is probably S-adenosylhomocysteine (SAH), by the action of SAH-hydrolase. During hypoxia, adenosine concentration may be bootsed up to 50-fold, due to the degradation by cytosolic 5’-nucleotidases of excessive amounts of AMP, which is generated from the metabolic degradation of Adenosine-5'-triphosphate (ATP) via Adenosine diphosphate (ADP). The adenosine that is formed is released from the cell by a concentration-dependent, bi-directional nucleoside transporter, and may then exert its extracellular effects. Adenosine produced from extracellular AMP by ecto-5’-nucleotidases may also contribute to extracellular adenosine levels.89 Extracellular adenosine is inactivated by rapid reuptake into the cell, through the action of specialized transporter proteins. At intracellular level, adenosine may then be degraded by adenosine deaminase (ADA) to inosine, which is consequently degradeted in several steps to uric acid. Figure 38
94
Figure 38. Metabolic pathways of adenosine.89
Adenosine posses several properties in common with the classic neurotransmitters: i) it exerts its actions via receptors; ii) its actions can be blocked by specific antagonists; iii) adenosine-producing enzymes are present in synapses; iv) its actions are terminated by an efficient reuptake system and a metabolizing system. Adenosine is usually designated as a neuromodulator because there is no evidence that it is stored in or released by specific purinergic nerves. The most impressive changes of adenosine metabolism occur under conditions leading to states of hypoxia or hypoglycemia revealing that adenosine levels rise rapidly after interruption of blood flow in a variety of animal species. Elevated levels of adenosine influence biochemical processes, e.g. excitatory amino release or Ca2+ influx, resulting in neuroprotective actions. High metabolic rates and insufficient oxygen supply determine local adenosine formation via an increase in ATP breakdown. The effect counteracts the situation that has generated suggesting that adenosine has an important function in the adjustment of energy or oxygen supply to local demand. Adenosine’s role appears to be a homeostatic regulation under normal physiological condition and a protective role in emergency situations. In the central nervous system, that is under a permanent inhibitory purinergic tone, adenosine has a function in synaptic homeostasis controlling the level of synaptic excitability. Analogously, it has been proposed that adenosine plays a homeostatic role in the regulation of artery
95 coronary, cerebral and skeletal muscle blood flow, and in renal, gastric and immune system function. During emergency situation as cardiac ischemia or stroke, elevated adenosine levels exert a protective effect, reducing neuronal overexcitability, increasing local blood supply and decreasing Ca2+ influx into the cell and finally preventing cell death. The emerging picture of the adenosine’s role involves the interaction with different adenosine receptors (ARs) residing in proximity with either presynaptic calcium and/or potassium channels and communicating with them by some membrane-associated mediators. ARs activation indirectly influences the action of neurotransmitters and other neuromodulators, behaving as a modulator of modulators. In addition, adenosine could be considered also as a fine-turner and in this way contributes to a very sophisticated interplay between its own receptors and the receptors for other neurotransmitters or neuromodulators.89
2.1.1 Classification of Adenosine Receptors
Adenosine mediates many of its physiological effects via cell surface receptor. The receptors for adenosine and adenine nucleotides were first proposed to be classified as the P1 and P2 purinergic receptor.90
This classification was based on:
(i) the relative potencies of adenosine and adenine nucleotides; (ii) the sensitivity to antagonism by methylxanthines;
(iii) modulation of activity of adenylate cyclase
In addition, the further P2 receptor classification was characterized by: (i) the rank order of potency: adenine nucleotides > adenosine (ii) insensitivity to antagonism by methylxanthine
(iii) induction of prostaglandin synthesis
The existences of two subtypes of P1 receptors was independently proposed.91, 92 Evidences for one proposal was based on the observation of either increased stimulation or increased inhibition of cAMP formation by adenosine analogues compared to adenosine.91 The receptor subtypes which mediated inhibition of formation of cAMP was termed the A1 subtypes and the receptor subtypes which mediated stimulation of formation of cAMP was termed the A2
96
subtype. Evidence for the other proposal was based on the observation of two profiles for the relative effects of adenosine, N6-(phenyl isopropyl)adenosine and 5’-N-ethylcarboxamidoadenosine on adenylate cyclase activity.92
These receptor subtypes were termed Ra (activation of adenylate cyclase activity) and Ri (inhibition of adenylate cyclase activity), respectively.
There is an obvious need for consistency in nomenclature and classification of receptors in order to minimize confusion in the literature. This need was recognised by the International Union of Pharmacology (IUPHAR) Committee on Receptor Nomenclature and Drug Classification, which has suggested a set of rules for nomenclature of receptors and receptor subtypes.93 They state that a “completely” defined receptor would include both molecular and pharmacological information about that receptor. This definition includes a known endogenous ligand, a unique pharmacological profile (based on agonist and antagonist data), a distinct amino acid sequence, structural type (e.g., G protein-coupled, gated ion channel, etc.) and effector system.94 Based on these nomenclature and classification rules P1 receptors are also named “adenosine receptors” (reffering to the endogenous ligand), while the subtypes are named the A1, A2A, A2B and A3 subtypes.95,96 Each of the subtypes has been characterised by molecular cloning, agonist activity profile, antagonist activity profile, G protein-coupling and effector systems.94 Figure 39
97 The four AR subtypes have now been characterized from pharmacological, structural and functional point of views. The A1, A2A and A2B subtypes were initially discovered and classified in the classical manner (i.e., by a study of agonist pharmacology). Evidence from recent cloning, sequencing and expression of each of these subtypes has provided structural and functional confirmation of their original classification as distinct adenosine receptor subtypes. In contrast, the A3 AR subtype was discovered by molecular biology studies, then followed by classical pharmacological studies. All four AR subtypes have recently been cloned from a variety of mammals, including humans.94 Analysis of the amino acid sequences of the cloned receptors demonstrates that they all fit the seven transmembrane spanning domain structural motif, which is the model for all G protein-coupled receptors.97,98,99 This structural motif encompasses seven domains, each ~22-26 hydrophobic amino acids, which traverse the cell membrane. These domains possess alpha helical secondary structure. The extracellular domains of the receptor protein comprise the N-terminus, often containing one or more glycosylation sites, and three extracellular loops connecting the transmembrane domains (EI-III), while the cytoplasmic or intracellular domains of the receptor protein comprise the C-terminus and similarly three intracellular loops (CI-III). The C-terminus contains phosphorylation and palmitoylation sites, which are involved in regulation of receptor desensitization and internalization. All ARs, with the exception of the A2A, contain a palmitoylation site near the C-terminus. The A2A AR is the only subtype with an extraordinary long C-terminus, 122 amino acids versus 36 amino acids in e.g. the A1 AR. Figure 40
98
Figure 40
Heterotrimeric G proteins are guanine-nucleotide regulatory protein complexes composed of α and a dimer βγ subunits. They are responsible for transmitting signals from GPCRs to effectors, e.g. adenylyl cyclase (AC). Until now, 16 α, 5 β and 14 γ isoform have been reported.100
G proteins are divided into several subclasses with a specific activity profile: Gs proteins stimulate adenylyl cyclase, Gi proteins inhibit adenylyl cyclase and stimulate G protein-coupled inwardly-rectifying potassium channels (GIRK), G0 proteins stimulate K+ ion channels, Gq/11 proteins activate phospholipase C (PLC), G12 proteins activate Rho guanine-nucleotide exchange factors (GEFs) and the olfactory G protein, Golf, stimulates adenylyl cyclase. When a ligand activates the G protein-coupled receptor, it induces a conformational change in the receptor that allows the receptor to function as a guanine nucleotide exchange factor (GEF) that exchanges GDP for GTP on the Gα subunit. In the traditional view of heterotrimeric protein activation, this exchange triggers the dissociation of the Gα subunit, bound to GTP, from the Gβγ dimer and the receptor. However, models that suggest molecular rearrangement, reorganization, and pre-complexing of effector molecules are beginning to be accepted. Both Gα-GTP and Gβγ can then activate different signaling cascades (or second messenger pathways) and effector proteins, while the receptor is able to activate the next G protein. The Gα subunit will eventually hydrolyze the attached GTP to GDP by its inherent enzymatic activity, allowing it to re-associate with Gβγ and starting a new cycle. A group of proteins called Regulator of G protein signaling (RGSs), act as GTPase-activating
99 proteins (GAPs), specific for Gα subunits. These proteins act to accelerate hydrolysis of GTP to GDP and terminate the transduced signal. Figure 41
Figure 41. Activation cycle of G-protein by G-protein coupled receptors.
The A1 AR mediates a broad range of signaling responses, which may be caused by its coupling to different G proteins.89 Figure 42
Figure 42. Intracellular pathways coupled to the adenosine A1 receptor.89
The most widely recognized signaling pathway of A1 ARs is the inhibition of adenylate cyclase causing a decrease in the second messenger cAMP.89 This in turn modulates the activity of cAMP dependent protein kinase, which phosphorylates different protein targets. Another signaling mechanism of A1 ARs
100
is activation of phospholipase C (PLC) leading to membrane phosphoinositide metabolism and increased production of inositol 1,4,5-triphosphate (IP3), diacylglicerol (DAG) and Ca2+ mobilization. Activation of phospholipase D (PLD) via A1 ARs has been described although, as in the majority of cell systems, this may be downstream of phosphoinositide hydrolysis and may require the intermediate activation of protein kinase C (PKC) or Ca2+. Stimulation of A1 ARs can activate several types of K+ channels, described principally in cardiac muscle and neurons.
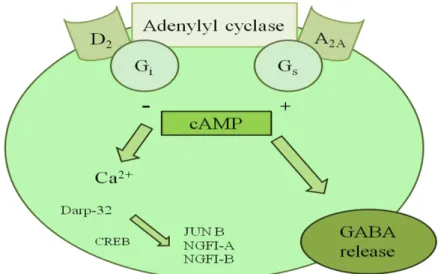
The A2A ARs transduce their signal by activation of intracellular heterotrimeric G-protein and activate adenyl cyclase leading to accumulation of cyclic AMP and induction of protein kinase A (PKA)-dependent phosphorylation of P-type Ca2+ channels. Figure 43 A2A ARs are known to activate Gs, but receptors in striatum may interact predominantly with Golf (first identified in the olfactory epithelium) because Golf is much more highly expressed in the striatum than Gs. Activation of A2A ARs has also been shown to induce a cAMP dependent phosphorylation of the dopamine- and cAMP-regulated protein kinase (DARP-32), which upon phosphorylation is converted into a potent protein phosphatase inhibitor. It has been suggested that adenosine A2A AR modulation of neurotransmitter release in striatal cholinergic and hippocampal glutamatergic nerve terminals may involve either the activation of PKA and P-type Ca2+ channels or the activation of cholera toxin-insensitive G-protein, PKC and N-type Ca2+ channels. The modulating activity of A2A ARs on neurotransmitter release has often been used to explain the neuroprotective properties associated with A2A AR blockade in different models of neurodegeneration.
101
Figure 43. Adenosine-dopamine interactions in the central nervous system.89
A2B AR stimulates adenylyl cyclase by directly coupling to Gs intracellular proteins. However, additional intracellular signaling pathways have been found to be functionally coupled to A2B ARs.89 The stimulation of A2B AR results in accumulation of intracellular Ca2+ a process that is thought to be mediated via different mechanisms as potentiation of P-type channels in pyramidal neurones of guinea pig hippocampus via coupling with Gs protein that is blocked by cAMP dependent protein kinases. Another mechanism involved could be independent of cAMP requiring Gs protein coupling or through PLC activation.89 Figure 44
102
Consequently, it has been proposed that A2B ARs are also coupled to phosphatidylinositol-specific PLC via G protein of the Gq family and the activation of this pathway results in an increase of diacylglycerol which activates PKC and IP3.89 Moreover, intracellular signaling of A2B ARs can be modulated by interaction with other receptor systems. For example, agents that increase intracellular calcium or activate protein kinase C significantly potentiate A2B -mediated cAMP production in various cells. On the other hand, bradykinin-stimulated calcium entry caused inhibition of A2B AR-stimulated adenylyl cyclase in astrocytoma cells, but direct stimulation of protein kinase C enhanced the A2B response. It is of interest that, as far as intracellular pathways are concerned A2B receptors have as much in common with A1 or A3 ARs (activation of phospholipase C), as with A2A ARs (activation of adenylyl cyclase). Unfortunately, the lack of highly selective agonists and antagonists of the A2B receptors has precluded the pharmacological characterization of this receptor system and remains to be elucidated whether all the mechanisms described herein pertain to every cell type expressing A2B ARs.89
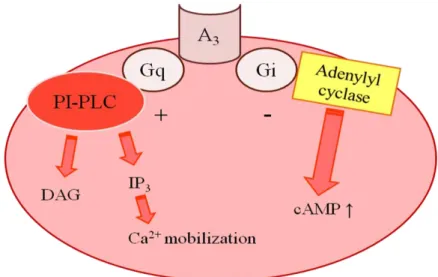
The A3 AR is coupled to inhibitory G-protein and the N-terminus of this subtype has one (rat and sheep) or two (human) potential N-linked glycosylation sites. It has been observed that none of the other adenosine receptor subtypes have glycosylation sites in the N-terminal region. Consistent with all adenosine receptors, the first extracellular loop of all the A3 ARs also contains a potential N-linked glycosylation site at amino acid 42 of the consensus sequence.89 In addition, in the C-terminal region, the Cys302 of the consensus sequence is a potential palmitoylation site that may be necessary for the formation of a fourth intracellular loop and for interaction of receptor with G proteins.89 The A3 receptors are able to cause inhibition of forskolin stimulated cAMP accumulation, to increase phosphatidylinositol specific phospholipase C and D activity and to elevate IP3 levels and intracellular Ca2+ pools. Figure 45
103
Figure 45. Intracellular pathways coupled to the adenosine A3 receptor.89
2.1.2 Physiological significance of endogenous adenosine
The initial realization of the physiological significance of adenosine was in 1929.101 Pronounced physiological effects were observed on both cardiovascular and renal function following administration of adenosine to mammals. The clinical evaluation of adenosine in man proved disappointing due to the short half-life of adenosine, and interest waned in the potential therapeutic applications.94
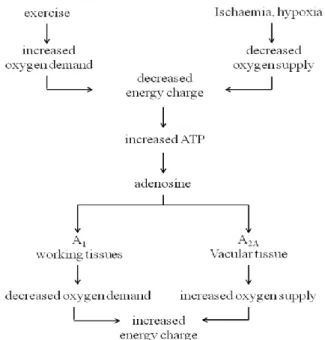
The ability to further study the physiological role of endogenous adenosine in a variety of mammalian tissues followed the discovery that the xanthines, caffeine and theophylline, were potent non selective adenosine antagonists. A cascade of publications on adenosine and adenosine receptors soon followed. Adenosine is now known to regulate a diverse range of physiological functions, to the extent that almost all mammalian organ systems are affected by adenosine. Adenosine is proposed to function as a paracrine homeostatic modulator with a global rather than specific role. The physiological responses to adenosine are complex and depend on the receptor subtype activated, the mammalian species, and the type and metabolic state of the tissue.94 The A1 and A2A AR subtypes are proposed to play complementary roles in adenosine's regulation of the energy supply demand balance of cells. A schematic representation of this homeostasis is presented in Figure 46. An increase in the oxygen demand of tissues (exercise) or a decreased oxygen supply (ischemia, hypoxia) results in an imbalance of the energy supply demand of tissue. An acute increase in the adenosine levels,
104
follows due to the metabolism of ATP. Adenosine diffuses from the cell where it acts locally to activate adenosine receptors to decrease the oxygen demand (A1) or increase the oxygen supply (A2A) and so reinstate the balance of energy supply demand within the tissue.94 The actions of both subtypes is to increase the amount of available oxygen to tissues and so protect cells against damage caused by a short term imbalance of oxygen.
Figure 46. Role of adenosine, adenosine A1 and A2A ARs in energy supply demand balance.94
The mechanism for tissue specific expression of the A1 and A2A subtypes is not known, however the receptors are present in nearly all mammalian tissue types and may be coexpressed in the same cell type. A consequence of the global homeostatic function of endogenous adenosine is the multitude of physiological responses mediated by adenosine, presented in Table 10. These responses generally lead to a decrease in the oxygen demand and/or increase in the oxygen supply, reinstating the energy supply demand balance within the tissue. One of the most important functions of endogenous adenosine is its cytoprotective effect, preventing tissue damage during traumas such as hypoxia, ischemia, hypotension and seizure activity.94 The A2B AR subtype is a low affinity receptor, adenosine exhibiting activity at this subtype at concentrations greater than 10 MM. There is little information on the physiological significance of the A2B subtype, a
105 consequence of the lack of suitably potent and selective ligands for detailed study. This deficiency is also reflected by the means in which A2B AR clones are pharmacologically identified: not by specific binding of particular ligands, as for the other receptor subtypes, but rather by the lack of specific binding of ligands. However, the expression of A2B subtype clones to give pure populations of this receptor may facilitate future pharmacological characterization. However, speculative role for the A2B AR has been proposed. It was suggested that the A2B AR functions during life threatening systems failure, reactivating the heart and brain, in order to resuscitate. This action would override the protective functions afforded by the A1 and A2A ARs. More recent studies have demonstrated that activation of A2B ARs leads to increases in intracellular calcium concentrations in cultured cells and chloride ion secretion via cAMP in intestinal epithelial cells. The latter has implications for the treatment of secretory diarrhea associated with inflammation.94
Table 10. The Physiological effects mediated by endogenous adenosine in various
mammalian tissues, attributed to activation of A1 and A2A receptor subtypes.
System Physiological effect of endogenous adenosine
CNS Neuromodulator mediating central inhibitory effects, inhibition of
release of neurotransmitters, neuronal firings, CNS depressant: decreases locomotor activity, anticonvulsant, sedation, hypnotic, antinociceptive, ataxic, mediates respiration
CV Vasodilatation, hypotensive, antiarrhytmic, negative chronotropic,
dromotropic and inotropic effects, inhibition of platelet aggregation, decrease of neutrophil function, stimulation of nitric oxide production by vascular endothelial cells
Kidney Biphasic modulation of rennin release, decreased glomerular filtration
rate by vasocostriction
Adipocytes Inhibition of lipolysis
Immune Immunosuppressant, inhibition lymphocyte proliferation,
antiflammatory: modulating neutrophil function
Liver Regulation of hepatic blood flow, stimulation of gluconeogenesis
Stomach Inhibition of gastric secretion
Striated/smooth muscle
106
The physiological role for the A3 AR is not adequately understood, a consequence of its relatively recent characterization and a lack of truly selective ligands for in vivo studies. Suitably selective and potent ligands (both agonists and antagonists) are being developed predominantly by radioligand binding studies, however the relationship between the radioligand binding data and selectivity in vivo is not yet established. As well, ligands which are already well characterized pharmacologically at the A1 and A2A ARs are being characterized at the A3 AR. The availability of cell lines expressing the recombinant A3 AR from different species is facilitating advancements in the study of its physiological role. So far this subtype is implicated in mediating a number of physiological responses.94 Binding of the adenosine agonist NECA to mast cells expressing the rat A3 AR resulted in increased inositol triphosphate (IP3) and intracellular Ca2+ concentrations, which potentiated antigen induced secretion of inflammatory mediators (histamine, leukotrienes, cytokines, thromboxanes and proteases). The conclusion from these studies was that the A3 AR may play a role in mediating asthmatic attacks and other allergic responses. However other adenosine receptor subtypes have now been ruled out in contributing to the observed responses. Using the A3 selective, high affinity agonist N6 -(3-iodobenzyl)-5’-N-methylcarboxamidoadenosine (3-IB-MECA), in vivo studies in mice demonstrated the A3 AR mediates a locomotor behavioural depressant effect, possibly centrally mediated. A cardio protective role for A3 ARs, activating ischemic preconditioning, has been proposed based on studies in isolated rabbit heart. N6-2-(4-aminophenyl)ethyladenosine (APNEA) produced a hypotensive response in the pithed rat, which was attributed to activation of A3 ARs. It was subsequently demonstrated that this A3 AR activation resulted in mast cell degranulation and histamine release, implicating the mast cell with a key role in A3 AR mediated hypotension in the rat.94
2.1.3 A1 Adenosine Receptor Antagonists
The A1 AR is particularly prevalent in the central nervous system (CNS), with high levels in the cerebral cortex, hippocampus, cerebellum, thalamus, brain stem and spinal cord. Numerous peripheral tissues also express the A1 AR,
107 including vas deferens, testis, white adipose tissue, stomach, spleen, pituitary, adrenal gland, heart, aorta, liver, eye and bladder. Low levels are found in the lung, kidney and small intestine.100
A number of A1 AR antagonists has been studied as diuretic agents, for the treatment of chronic lung diseases, for cardiac therapy and in dementia.102
Diuretic activity. As reported below, adenosine, acting on A1 ARs, exerts antidiuretic effects. Consequently A1 AR antagonists, blocking or reducing the adenosine action, may be effective as diuretic agents. A1 AR antagonists showed to be potassium-sparing diuretics with kidney-protecting properties, and they could be particularly helpful in fluid retention disorders, often experienced by patients suffering from congestive heart failure. In these patients A1 AR antagonists exhibit the interesting features of increased diuresis and glomerular filtration rate, whereas the diuretic furosemide increases diuresis at the expense of a decreased glomerular filtration rate. A1 AR antagonists can increase urine formation, and since they not decrease renal blood flow, are particularly useful to maintain glomerular filtration in patients having edema secondary to reduced cardiac function. Recent clinical trials confirmed their beneficial effects on renal function, even if in many studies the renal benefits disappears with higher doses of these drugs. Ongoing studies should be able to demonstrate whether A1AR antagonists can be used to improve renal function without adversely affecting patients with heart failure.102
Action on heart. A1 AR antagonists are tested in the treatment of bradyarrhythmias associated with inferior myocardial infarction, cardiac arrest and cardiac transplant rejection and could be useful in the treatment of chronic heart diseases, suppressing cardiac fibrosis and decreasing albuminuria, without effects on blood pressure.102
Antiasthma activity. As previously reported, adenosine plays a significant
role in lung diseases and is produced endogenously by many cells during hypoxia, stress, allergic stimulation, and exercise. Agonists and antagonists of the different receptor subtypes have been tested in many preclinical in vitro and in vivo studies and have progressed to clinical studies in order to evaluate their potential as novel antiasthma drugs. In particular A1 AR antagonists could represent useful drugs for
108
the treatment of chronic lung diseases such as asthma, Chronic Obstructive Pulmonary Disease (COPD) and pulmonary fibrosis.102
Actions on CNS. The observation of the effects of caffeine, a classical non
selective adenosine antagonist, on the CNS, including an improvement of awareness and learning, encouraged the search of selective antagonists endowed with central activity. It has been reported that A1 AR blockade is involved in the discriminative-stimulus effects of behaviorally relevant doses of caffeine.
Nevertheless other authors showed that the A2A AR is the main mediator of the behavioral stimulatory effect of caffeine. Selective A1 AR antagonists induce cognition enhancement, leading to a general improvement in memory performance. This is potentially useful in the treatment of dementia and anxiety disorders. In a recent article, Trevitt and colleagues reported that treatment with the A1 AR antagonist CPT (8-cyclopentyl-1,3-dimethylxanthine) in a model of Parkinson’s disease (PD) produced a dose dependent improvement in locomotion, suggesting that, although the role of A1 AR in Parkinson’s disease (PD) is still unclear, the A1 AR antagonism may produce beneficial therapeutic effects, particularly at the beginning of treatment.102
Other activities. Adenosine is involved in the regulation of bone function
and recently it has been reported that A1 AR antagonists reduced bone loss, indicating their potential use in the treatment of osteopoenia, osteoporosis and other bone diseases (e.g. Paget disease). Other potential therapeutic applications of A1 AR antagonists are represented by the treatment of sepsis, in association with antibiotics and the treatment of hepatic ischemia-reperfusion injury.
The prototypic A1 AR antagonists were the xanthines, teophylline and caffeine, and a multitude of xanthine derivatives has been synthesized and studied for this activity. However also a high number of non-xanthine derivatives has been reported as potent and selective A1 antagonists, for examples: adenine derivatives, pyrazolo[1,5-a]pyridines, tricyclic imidazolines. Both xanthine and non-xanthine derivatives are devoid of the sugar moiety that characterizes the majority of A1 AR agonists. In general A1 AR antagonists are bicyclic or tricyclic compounds, flat, aromatic or π-electron rich, nitrogen-containing heterocycles; hydrophobic substituents may enhance affinity, whereas hydrophilic substituents,
109 which render many of the high-affinity antagonists quite insoluble in water, are usually not well tolerated.102
2.1.3.1 Xanthines derivatives
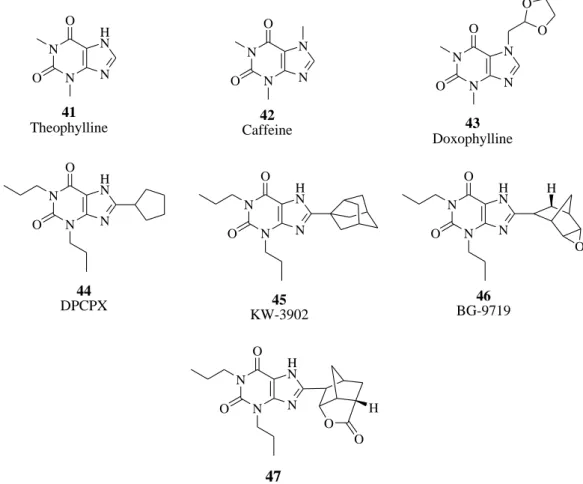
Alkylxanthines are the best-known class of compounds characterized as adenosine antagonists. There have been a large number of these derivatives prepared in an effort to increase potency and selectivity. Compounds of much higher potency than the prototypical caffeine have been identified, usually resulting from 1,3-dialkyl and 8-aryl or 8-cycloalkyl substitution. However, this class of compounds shows additional activities unrelated to adenosine receptor blockade, and these has been only limited success in preparing adenosine A1 -selective agents as therapeutic agents.103 Theophylline (41, 1,3-dimethylxanthine) and caffeine (42, 1,3,7-trimethylxanthine) are the classical nonselective xanthine antagonists of ARs that display micromolar affinity at all AR subtypes. Figure 47 Theophylline was first extracted from tea leaves around 1888. The drug was chemically identified and synthesized in 1896. There are many examples of potent A1 antagonists that contain bulky lipophilic substitution at the 8-position of 1,3-dipropylxanthines. Doxofylline (43, 7-(1,3-dioxalan-2-ylmethyl)theophylline) is a xanthine bronchodilator which differs from theophylline in that it contains a dioxalane group in position 7. A large number of modifications on the xanthine core at the 1-, 3-, and 8-positions led to the discovery of 8-cyclopentyl-1,3-dipropyl xanthine (44, DPCPX, Figure 47), which was selective for rat A1 AR compared with the A2A AR and less selective at the human (h) A1 compared with hA2A and hA2B ARs. Other substituted xanthines have been proposed as A1 AR antagonists, in particular, 1,3-dipropyl-8-(3-noradamantyl)xanthine (45, KW-3902) and 1,3-dipropyl-8-[2-(5,6-epoxynorbornyl)xanthine (46, BG-9719). In this compound (also named Naxifylline), the xanthine ring and the epoxide are, respectively, situated endo and exo to the norbornane moiety, and the latter has an asymmetric center at C-2. Figure 47 While a small stereochemical effect on the affinity was present between the enantiomers at guinea pig and hA1 ARs, the R-isomer appeared to be less potent than the S-R-isomer in the rat. Very recently, a series of xanthines substituted with norbornyl-lactones structurally related to
BG-110
9719 (47, Figure 47) was investigated. These derivatives, in which the xanthine occupies the exo position on the norbornyl ring system, showed high A1 binding affinity and selectivity over the closely related A2A AR. The lactones possessed similar if not better in vivo activity to BG-9719 in the rat diuresis models.103
N N N H N O O 41 Theophylline N N N N O O 42 Caffeine N N N N O O O O 43 Doxophylline N N N H N O O 44 DPCPX N N N H N O O 45 KW-3902 N N N H N O O H O 46 BG-9719 N N N H N O O 47 O H O
Figure 47. A1 AR antagonists (Xanthines).
2.1.3.2 Water-Soluble Xanthine Derivatives as A1 AR Antagonists
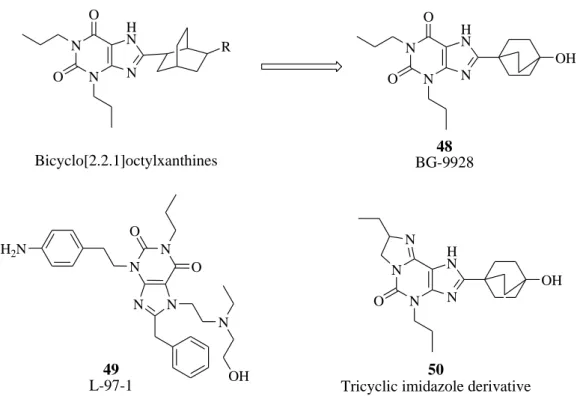
The highest affinity xanthine-based molecules pictured in Figure 47 lack appreciably polar substituents. The utility of most of these compounds for intravenous administration in the treatment of acutely decompensate congestive heart failure patients in the clinic may be limited because of their low water solubility. In the search for a selective A1 AR antagonist with greater aqueous solubility, a series of 1,3-substituted-8-cyclohexyl- and 8-bicyclo-[2.2.2]octylxanthines that contain linear substitution patterns was investigated by Kiesman et al.104 Figure 48 These authors have initially drawn their attention to
111 the 8-cyclohexyl-trans-4-carboxylic acid xanthine derivatives published by Katsushima and co-workers,105 that showed low binding affinity at A1 AR. They expanded this series of compounds and prepared the bicyclo-[2.2.2]octane derivatives by addition of a two-carbon bridge linking the 1- and 4-positions across the cyclohexane ring. Figure 48 Optimization of the bridgehead substituent led to propionic acid (48, BG-9928, Figure 48),106 which retained high potency (dog A1, Ki 29 nM) and selectivity for the A1 AR (163-fold vs A2A AR; 24-fold vs A2B AR; 1452 vs A3 AR) with improved oral efficacy in a rat diuresis model as well as high oral bioavailability in rat, dog, and cynomolgus monkey. Another water-soluble molecule that has been described as a potential new oral drug for asthma is 3-[2-(4-aminophenyl)ethyl]-8-benzyl-7-{2-ethyl-(2-hydroxyethyl)amino]ethyl}-1-propyl-3,7-dihydropurine-2,6-dione] 49 labeled L-97-1. Figure 48 This compound is an adenosine A1 antagonist (Ki 580 nM) with at least 100-fold selectivity over A2A and A2B ARs.
N N N H N O O R Bicyclo[2.2.1]octylxanthines N N N H N O O OH 48 BG-9928 N N N N N OH O O H2N 49 L-97-1 N N N H N O OH 50
Tricyclic imidazole derivative N
112
2.1.3.3 Tricyclic imidazoline derivatives
These compounds are essentially derivatives of xanthenes in which the additional basic site significantly increased their water solubility relative to xanthines without substantial loss in A1 binding affinity. In connection with the discovery of BG-9928 and the tricyclic imidazoline derivatives reported in the literature, Vu et al.107 reported the synthesis of compound 50, (Figure 48) the R-isomer of 7,8-dihydro-8-ethyl-2-(4-bicyclo[2.2.2]octan-1-ol)-4-propyl-1H-imidazo-[2,1-i]purin-5(4H)-one, a potent A1 AR antagonist with good selectivity over the other three ARs. Imidazoline 50 is a potent competitive A1 AR antagonist, highly soluble in water (>100 mg/mL). In addition, it has an oral bioavailability of 84% and an oral half-life of 3.8 h in rats. When orally administered in a rat diuresis model, compound 50 promoted sodium excretion (ED50 0.01 mg/kg). This level of efficacy is comparable to that of BG-9928. Additional modifications of 50 also showed that the bridgehead hydroxyl group could be replaced with a propionic acid without a significant loss in binding affinity or in vivo activity.
2.1.3.4 Pyrazolo[1,5-a]pyridines
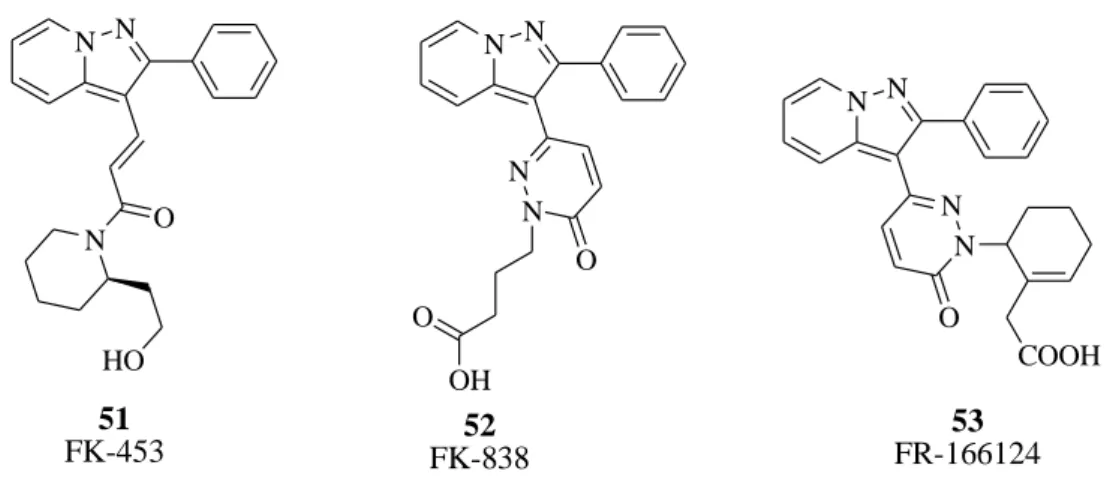
Another class of analogs structurally related to the xanthine core consists of derivatives the pyrazolo[1,5-a]pyridines nucleus. One compound in this series, FK-453, (51, Figure 49), synthesized by Akahane et al.,108 showed potent and selective adenosine A1 antagonist activity and was selected for further evaluation. Various modifications have been performed on this nucleus such as constraining of acryloyl amide into a pyridazinone nucleus (compound 52, FK-838, Figure 49) that produced a significant increase of potency and selectivity.109 FK-838 is an A1 -selective adenosine antagonist, as demonstrated in radioligand binding (IC50 A1 120 nM; IC50 A2 5900 nM) and functional assays, whose diuretic and natriuretic effects appear to be due to both its renal hemodynamic effects and a direct inhibition of proximal tubular Na+ reabsorption. The design process leading to the discovery of FR-166124 (53, 2-[2-[6-oxo-3-(2-phenylpyrazolo[1,5-a]pyridin-3-yl)-1,6-dihydropyridazin-1-yl]-1-cyclohexenyl]acetic acid, Figure 49) involved introduction of various cyclic acid groups to the N2 of the pyridazinone ring, in
113 place of the butyric acid group of FK-838, as substituents to induce rigidity and mimic the postulated conformation of FK-453. A cyclohexenyl acetic acid group was found to be especially effective. Compound 53 is reported as a more potent derivative with higher A2A/A1 selectivity and very high water solubility as the sodium salt (>200 mg/mL).110-112 N N N O HO 51 FK-453 N N 52 FK-838 N N O OH O N N 53 FR-166124 N N O COOH
Figure 49. A1 AR antagonists (pyrazolopyridines).
2.1.3.5 Adenine derivatives
The initial report of adenine derivatives showing adenosine antagonist activity only covered 1-methyl- and 9-methyladenine.113 The observation was made that adenine itself and 9-methyladenine showed specific competitive antagonism at low concentrations but exhibited nonspecific inhibitory activity at higher concentrations. Since N6-substitution of adenosine had been used to confer A1 AR agonist selectivity, it was clear that a selective antagonist could potentially be obtained by replacing the ribose sugar with a methyl group to generate N6 -substituted 9-methyladenines.114-115 Further structure-activity work has identified ((±)-N6-(endo-2-norbornyl)-9-methyladenine N-0861 (54, Figure 50) as a lead compound, which has been undergoing development as a cardiovascular agent for treatment of adenosine-related ARs, having negligible affinity (Ki > 10 000 nM) for alpha 1, alpha 2, and beta adrenoceptors, D1 and D2 dopamine receptors, and 5-HT2 serotonin receptors.117 Radioligand binding studies have shown that N-0861 (54, Figure 50) binds with high affinity to A1 ARs in bovine caudate membranes, where it is 600-fold selective for the A1 vs the A2 subtype of ARs.117
114
N-0861 (54, Figure 50) binds with lower affinity to A1 ARs in rat and guinea pig cerebral cortex and to A1 ARs in atrial tissue from guinea pig and human heart.118 Substitution in the 8-position of adenine with an isopropylmethyl-amine moiety gave the best results as observed from compound 55 (WRC-0571, Figure 50), which is a highly potent and selective A1 AR antagonist with superior potency and aqueous solubility relative to N-0861 (54, Figure 50). In radioligand binding studies, it displayed high affinity for cloned hA1 (Ki 1.7 nM) and much lower affinity for cloned hA2A and hA3 ARs (Ki 105 and 7940 nM, respectively). In functional studies, it potently inhibited the A1-mediated negative inotropic response to 5’-(N-ethyl-carboxamido)adenosine (NECA) in isolated guinea pig atria (pKb 8.41), whereas it was much less active against NECA-induced, A 2B-mediated, relaxation in guinea pig aorta (pKb < 5).119
N N N N NH 54 N-0861 N N N N NH 55 WRC-0571 HO N
Figure 50. A1 AR antagonists (adenines).
2.1.4 A2A Adenosine Receptor Antagonists
Within the brain A2A ARs are richly expressed in the striatum, nucleus accumbens, and olfactory tubercle. A coexpression of A2A with D2 dopamine receptors has been reported in the GABAergic striatopallidal neurons where adenosine and dopamine agonists exert antagonistic effects in the regulation of locomotor activity. Activation of A2A ARs in striatopallidal neurons decreases the affinity of D2 receptors for dopamine, antagonizing the effects of D2 receptors. The negative interaction between A2A and D2 receptors is at the basis of the use of A2A antagonists as a novel therapeutic approach in the treatment of Parkinson’s disease.121 In addition, A2A ARs may have an important role in the neurodegenerative process. Accordingly, a neuroprotective effect was
115 demonstrated after caffeine intake or A2A AR inactivation against dopaminergic neurodegeneration in a neurotoxin model of Parkinson’s disease.122 Concomitantly, two large prospective epidemiological studies have strongly associated caffeine consumption to a reduced risk of developing Parkinson’s disease.123,124 Last, the recent discovery that the A2A can form functional heteromeric receptor complexes with other G protein-coupled receptors such as D2 and the mGlu5 receptors has also suggested new opportunities for the potential of A2A antagonists in PD.125 In the future development of bivalent ligands, able to activate D2 and block A2A ARs or antagonize both A2A and mGlu5 subtypes, would be a promising strategy for the treatment of this neurodegenerative disease.126-128 In addition to the protection against striatal and nigral neuron loss by A2A antagonists, there are data also supporting their protective role outside the basal ganglia.129 Local injection of an A2A antagonist prevents glutamate-dependent death of neurons in hippocampal cortex130 and also reduced cortical damage in a variety of ischemic stroke models. In A2A knockout (KO) mice transient focal ischemia caused less neuronal damage in comparison to their wild-type (WT) littermates.131 Therefore, it seems that tonic activation of A2A ARs may be responsible for dangerous signal during injury, in contrast to the neuroprotective effects induced by endogenous A1 activation. Recently, selective inactivation or reconstitution of A2A ARs in bone-marrow cells revealed their contribution to the development of ischemic brain injury.132 The involvement of A2A ARs in neuroprotection is likely to be complex as stimulation of this subtype also diminishes brain damage after excitotoxic and traumatic injury.133,134 A2A -mediated protection has been reported against ischemia in the myocardia, kidney, and liver and in ischemia reperfusion injury in the spinal cord.135-138 High expression of A2A ARs has been found in platelets, leukocytes, vascular smooth muscle, and endothelial cells with important implications in the regulation of inflammatory responses. It is now well established that stimulation of the A2A AR in immune cells induces anti-inflammatory effects, mostly due to its ability to increase cAMP levels, which has strong immunosuppressive effects.139 Stimulation of A2A ARs inhibits neutrophil adherence to the endothelium, degranulation of activated neutrophils and monocytes, plus superoxide anion
116
generation. A2A ARs have been recently defined as sensors and terminators of proinflammatory activities. The strongest evidence for the key role of A2A in inflammation derived by the elegant study of Ohta et al.140 using mice deficient in A2A ARs. In this model the lack of A2A subtype leads to increased tissue inflammation and damage, thus suggesting a negative and nonredundant regulatory role for the A2A AR. This model permits one to appreciate that adenosinergic regulation of immune cells is fundamental in normal physiological control of inflammation in vivo in spite of the fact that other Gs-protein-coupled receptors and cAMP elevating ligands are present such as cathecolamines, prostaglandins, dopamine, and histamine.139 Interestingly, the A2A AR has been demonstrated to be involved in promotion of wound healing and angiogenesis in healing wounds.141,142 Moreover, it plays an active role in the pathogenesis of dermal fibrosis, suggesting a role for antagonists as novel therapeutic approach in the treatment and prevention of dermal fibrosis in diseases such as scleroderma.143
2.1.4.1 Styrylxanthines
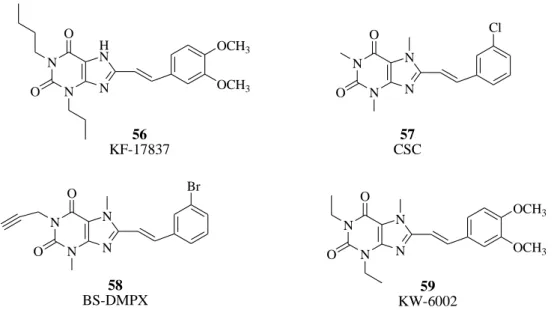
1,3-Dipropyl-7-methyl-8-(3,4-dimethoxystyryl)xanthine (56, KF17837,
Figure 51) was the first A2A AR antagonist in this chemical class of compounds.144 The 3-chlorostyrylcaffeine 57 (CSC, Figure 51) was identified as being less potent than KF17837 but with an increased selectivity vs A1 AR subtype.145 Introduction of a propargyl at the 1-position in combination with the 8-styryl group in compound 58 (BS-DMPX, Figure 51) increased affinity to the A2A AR with retention of selectivity.146 1,3-Diethyl-7-methyl-8-(3,4-dimethoxystyryl)- xanthine 59 (KW-6002, also named istradefylline, Figure 51) is an 8-styrylxanthine with high affinity for the rat striatal A2A AR. Due to its high affinity and selectivity, a radiolabeled derivative, [11C]-KW-6002 labeled at the aromatic O-methyl position, was developed to be used in pharmacological testing to trace the A2A ARs in vivo.147,148 Recently, KW-6002 (compound 59, Figure 51) has been selected for phase III clinical trials for treatment of Parkinson’s disease. However, this compound present metabolic issues and poor photostability in both solid form and therefore its approval was declined by the FDA in 2008.120
117 N N N H N O O OCH3 OCH3 56 KF-17837 N N N N O O 57 CSC Cl N N N N O O 58 BS-DMPX Br N N N N O O 59 KW-6002 OCH3 OCH3
Figure 51. A2A AR antagonists (styrylxanthines).
2.1.4.2 9H-Purine derivatives
Minetti et al., on the basis of the molecular modeling of a number of potent AR antagonists, designed and synthesized a number of 2-alkyl-substituted purine derivatives as A2A AR antagonists.120 From them ST-1535 (2-n-butyl-9-methyl-8-[1,2,3]triazol-2-yl-9H-purin-6-ylamine 60, Figure 52), was the most interesting.
N N N N N N N NH2 60 ST-1535
Figure 52. 9H-purine derivative.
2.1.4.3 Bicyclic derivatives
In the past years several researchers focused their attention on nitrogen bicyclic derivatives formed by 1,2,4-triazole nucleus fused to pyridine, pyrimidine ot thiazole ring in which the nitrogen atom is shared by the to rings. In particular ZM 241385 (61, Figure 53) is one the most potent A2A AR antagonists ever reported with favorable water solubility. Although different substituents can be introduced in the bicyclic system, the antagonistic activity increase when a
2-118
furanyl substituent is introduced at the carbon of the triazole nucleus and free or substituted aryl and amine groups are present on hexagonal ring.120 Modification of the pharmacokinetic features of this type of derivatives can be achieved by the introduction of hydroxyl or amino substituent on the aryl group or various substituents on the position 5. In particular, derivative 62 (Figure 53), showed great potency and selectivity for the A2A AR as compared with the A1 AR.
Several isoster of the triazolo-triazine nucleus have been synthesized; in particular some oxazolo-pyrimidines (63, Figure 53) and triazolo-pyrazine (64,
65, Figure 53) showed good potency at the A2A AR and good selectivity versus A1 AR.120 N N N N N O NH2 N H HO 61 ZM241385 N N N N N O NH2 N N O F 62 N N N N N O NH2 63 O H N HO N N N N N O NH2 64 N
Figure 53. A2A AR antagonists (bi-heterocyclic derivatives).
2.1.4.4 Pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines
9-Chloro-2-furan-2-yl-[1,2,4]triazolo[1,5-c]quinazolin-5-ylamine named CGS-15943 (65, Figure 54) represented the first potent but poorly selective antagonist for the A2A AR subtype.149 Bioisosteric replacement of the phenyl ring of CGS-15943 with an N7-substituted pyrazole led to the first example of an adenosine antagonist displaying the pyrazolotriazolo-pyrimidine core named 8FBPTP (66,
119 of this compound highlighted the essential requirements for the A2A affinity, i.e., the furyl moiety and the free amino group at the 5-position. Starting from these observations Baraldi et al.151,152 focused their interest on the pattern of substitution on the pyrazolo preserving the other structural elements. Several alkyl, aryl, and phenylalkyl substituents have been introduced at both the N7 and the N8 positions. The biological data derived from the molecules obtained indicated that the best radicals were phenylalkyl chains, and among these it was possible to discern the length of the spacer introduced between the phenyl ring and the pyrazolo nitrogen that was optimized in two or three carbon atoms. Two selected compounds of this family named SCH-58261 (67, 5-amino-7-(β-phenylethyl)2-(2-furyl)-pyrazolo[4,3
e][1,2,4]triazolo-[1,5-c]pyrimidine) and SCH-63390 (68, 5-amino-7-(3 phenylpropyl)2-(2-furyl)-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine)151,152 proved to be potent and selective A2A AR antagonists both in rat and human models. Figure 54 It was also noted that the N7 derivatives were more selective for the A2A AR than the corresponding N8 derivatives. From the family of SCH compounds, 5-amino-7-(3-(4-methoxyphenyl)propyl)-2-(2 furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (SCH-442416, 69, Figure 54) was selected for the development of a new positron emission tomography (PET) ligand, whose chemical structure allows an easy introduction of a methyl group by direct O-alkylation of the phenolic function with [11C]CH3I under alkaline conditions.153 The aim of this study was to use [11C]SCH-442416, 69, as a new ligand for the in vivo imaging of A2A ARs using PET. The in vitro binding in the brain and periphery, the good signal-to-noise ratio observed between 5 and 15 min after injection, and the low occurrence of radioactive metabolites all suggested that [11C]SCH-442416 was applicable as the first non-xanthine ligand suitable for the in vivo imaging of A2A ARs using PET. In addition, the data obtained from the binding experiments showed a higher affinity of the title compound for hA2A vs rat ARs (0.048 vs 0.5 nM).153
120 N N N N O NH2 Cl 65 CGS-15943 N N N N O NH2 66 8FBPTP N N F N N N N O NH2 67 SCH-58261 N N N N N N O NH2 68 SCH-63390 N N N N N N O NH2 69 SCH-442416 N N H3CO
Figure 54. A2A AR antagonists (Pyrazolo-triazolo-pyrimidines).
2.1.4.5 Water-Soluble A2A Adenosine Receptor Antagonists
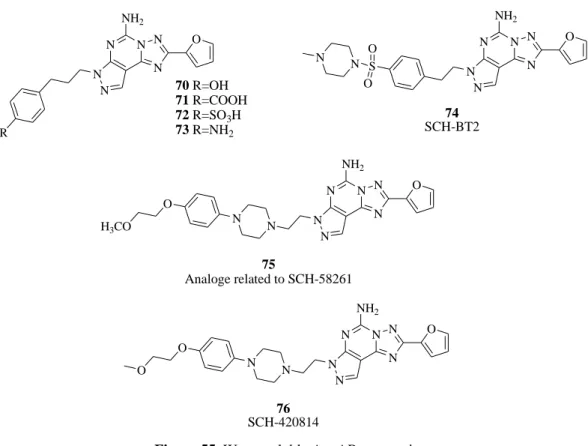
The major restriction of the tricyclic adenosine antagonists was the low solubility in aqueous media that limited the pharmacological screening. Starting from this limit Baraldi et al.152,154 reported a second generation of pyrazolo-triazolopyrimidines bearing oxygenated substituents on the phenylalkyl chains at the 7-position (compounds 70-73). The most interesting compounds are depicted in Figure 55. Compound 70 displayed the best value of A2A AR affinity indicating that the 4-hydroxy group positively influenced the receptor interaction but was not enough for reaching a good profile of water solubility. A water-soluble analogue of SCH-58261, named SCH-BT2 (74), was prepared by introduction of a 4-methyl-piperazine-1-sulfonyl moiety at the para position of the phenyl ring. SCH-BT2 altered neither motor behavior nor produced postural
121 asymmetry by itself. However, when infused concomitantly with levodopa (L-DOPA) (capable of inducing modest controlateral rotational behavior), SCH-BT2 significantly potentiated the number of contraversive rotations.155-157 Very recently, a novel series of 3-substituted 8-furyl-[1,2,4]-triazolo[1,5-i]purin-5-amine analogs related to SCH-58261 was reported as A2A AR antagonists.158 Most of the N3-substituted aryl piperazine and piperidine analogs demonstrated in vivo A2A receptor binding affinity and A1 receptor selectivity profiles superior to those of SCH-58261. In these series compound 75 displayed both superior in vitro and promising in vivo profiles. Figure 55 Neustadt et al.159 recently reported the arylpiperazine derivatives of pyrazolo[4,3-e]triazolo[1,5-c]pyrimidines with antagonist activity on the A2A AR. Among these derivatives, SCH-420814 (76,
Figure 55) demonstrated potent antagonist activity at the A2A AR. Structure-activity relationship studies revealed additional compounds incorporating an aryl-piperazine side chain that also showed potent oral activity in the haloperidol-induced catalepsy model in rats.
N N N N N N NH2 R O 70 R=OH 71 R=COOH 72 R=SO3H 73 R=NH2 N N N N N N NH2 O S O O N N 74 SCH-BT2 N N N N N N N NH2 O N O H3CO 75 Analoge related to SCH-58261 N N N N N N N NH2 O N O O 76 SCH-420814
122
2.1.5 A2B Adenosine Receptor Antagonists
A2B receptor is mainly expressed in the gastrointestinal tract, bladder, lung, and on mast cells, but also in eye, adipose tissue, brain, kidney, liver, and other tissue. Apart from stimulation of adenylate cyclase through Gs proteins, A2B AR are associated to Ca2+ mobilization through Gq proteins and mitogen-activated protein kinase (MAPK) activation. Furthermore, in human mastocytoma-1 cells (HMC-1) adenosine can stimulate interleukin (IL)-8 secretio, in addition to IL-1b, IL-3, IL-4 and IL-13 secretion, all via the A2B AR.160 This enhances the release of inflammatory mediators in addition to pro-inflammatory effects on airway smooth muscle cells, epithelial cells, and fibroblasts, confirming that A2B AR plays a key role in the inflammatory response associated with asthma. First studies on the involvement of A2B AR in asthma-concerned selectivity of enprofylline, a methylxanthine structurally related to theophylline. This anti-asthmatic drug was the most selective, though not potent A2B antagonist. Further studies showed that A2B antagonists inhibited NECA (5'-N-ethylcarboxamidoadenosine)-induced interleukin-8 secretion in HMC-1 and attenuated the release of IL-19 from human bronchial epithelial cells.160 Recently, it has been reported that the selective A2B antagonist CVT-6883 attenuated the airway inflammation and fibrosis induced by inhaled AMP or allergens. In the human intestinal epithelial cells, adenosine was shown to stimulate via A2B AR activation an increase in cAMP levels that is responsible for Cl- secretion, which allows the natural movement of isotonic fluid into the lumen, but in abnormal conditions could cause a secretory diarrhea. Furthermore, a recent study demonstrated that the A2B AR blockade significantly down-regulates proinflammatory cytokines and reduces the symptoms of colitis, indicating that A2B antagonists could be an effective therapeutic strategy to treat colonic inflammation.
Studies on the antinociceptive effects of some A2B antagonists supported the role of A2B AR inperipheral pain signaling, with a synergistic effectsbetween A2B-selective compounds and Delta 9-tetrahydrocannabinol (THC), not due to a general increase in antinociceptive drug efficacy but specific for the opioid system. Specific A2B antagonists could serve as adjuvant drugs for opioid analgesiawith minimal side effects. Some A2B antagonists were evaluated also for
123 their inhibitory activity versus agonist-induced hepatic glucose production, with the aim of developing a novel type of antidiabetic agent. Their examination in a murine diabetic model showed an oral activity as hypoglycemic agents.160 Furthermore, some authors reported a prospective function of the A2B antagonism due to the stimulation of proliferation, differentiation, and migration of retinal endothelial cells. This effect could inhibit retinal angiogenesis and provide a novel therapeutic approach to the treatment of diseases associated with aberrant neovascularization, such as diabetic retinopathy and retinopathy of prematurity.
Another prospective field of application for A2B antagonists could be the potential to inhibit tumor vascularization. In fact, it was found that A2B AR up-regulates tumor tissue VEGF levels and increases intratumor vascular density, thus promoting tumor growth.160
2.1.5.1 Xanthines
Over the past several years the effort has been focused on studying the structure-activity relationships of xanthine derivatives to search for more selective and potent adenosine A2B ligands. Potent and selective xanthine antagonists stem from multiple substitutions of the parent heterocycle. C8-Substitution combined with N1- and N3- (and sometimes N7) substitution have led to the development of potent and selective antagonists.103 In the series of 8-phenyl xanthines, a large number of amide derivatives was shown to be selective for hA2B vs hA1, hA2A, and hA3, although less selective vs rat A1 and A2A ARs. As an example, the p-cyanoanilide derivative MRS-1754 (N-(4-cyanophenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]-acetamide, 77, Figure 56), was shown to be 204-, 255-, and 289-fold selective for hA2B vs hA1, hA2A, and hA3 ARs.103 Moreover, the tritium-labeled form of MRS-1754 has been prepared and utilized in radioligand binding assays. Baraldi et al.161,162 published a series of 8-heterocyclesubstituted xanthines as A2B adenosine receptor antagonists. Several heterocycles, such as pyrazole, isoxazole, pyridine, and pyridazine at the 8-position of the xanthine nucleus, were studied. The synthesized compounds showed A2B AR affinity in the nanomolar range and good levels of selectivity evaluated in radioligand binding assays at hA1, hA2A, hA2B, and hA3 ARs. These
124
studies allowed identification of the derivatives 2-(3,4-dimethoxy-phenyl)-N-[5- (2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)-1-methyl-1Hpyrazol-3-yl]-acetamide (78, MRE-2028-F20), N-benzo[1,3]-dioxol-5-yl-2-[5-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)-1-methyl-1H-pyrazol-3-yloxy]- acetamide (79, MRE-2029-F20), and N-(3,4-dimethoxy-phenyl)-2-[5-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)-1-methyl-1H-pyrazol-3-yloxy]- acetamide (80, MRE-2030-F20, Figure 56), which showed high affinity to the A2B AR subtype and very good selectivity vs the other AR subtypes. Substitution of the acetamide with an urea moiety afforded bioisosteric xanthines with good affinity and selectivity comparable to the acetamide derivatives. The derivatives with higher affinity to hA2B AR proved to be antagonists, in the cAMP assay, capable of inhibiting the stimulatory effect of NECA (100 nM) with IC50 values in the nanomolar range and a trend significantly related to that observed in the binding assay. Consequently, Baraldi’s group163
synthesized the N-benzo- [1,3]dioxol-5-yl-2-[5-(1,3-diallyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)-1-methyl-1H-pyrazol-3-yloxy]-acetamide that led to preparation of the tritium-labeled form [3H]MRE-2029-F20 (KD value of 1.65 ± 0.10 nM in Chinese Hamster Ovary (CHO) cells expressing hA2B receptors). This compound was found to be a selective, high-affinity radioligand useful for characterizing recombinant hA2B ARs. N N N H N O HN N O O O 77 MRS-1754 N N N H N O O 78 MRE-2028-F20 N N N H OCH3 OCH3 O N N N H N O O 79 MRE-2029-F20 N N O H N O O O N N N H N O O 80 MRE-2029-F20 N N O H N O OCH3 OCH3
125 In this field of research, very recently Zeng et al.164 patented a series of 8-pyrazole xanthine derivatives as A2B AR antagonists. The new 8-(1H-pyrazole-4-yl)xanthines displayed good affinity to A2B AR, confirming the relevance of a pyrazole ring at the 8-position of xanthine nucleus. The chemical structures of the most potent compounds are reported in Figure 57. In this report the authors presented compounds 81 and 82 (3-ethyl-1-propyl-8-[1-[3- (trifluoromethyl)benzyl]-1H-pyrazol-4-yl]-2,3,6,7-tetrahydro-1H-purine-2,6-dione, CVT-6883), which showed good affinity to the hA2B AR. The same authors synthesized a series of heterocyclic 5-membered rings by bioisosteric replacement of the amide bond with different heterocyclic 5-membered rings (1,2,4-oxadiazoles and isoxazoles).165 Derivative 83 was the most active and selective analogue among these classes of compounds, displaying high affinity (Ki 1 nM) and selectivity for the hA2B AR vs A1, A2A, and A3 AR subtypes (A1/A2B, A2A/A2B, and A3/A2B selectivity ratios of 370, 1100, and 480, respectively; Figure 57).
N N N H N O O 81 N N H N O Cl N N N H N O O 82 CVT-6883 N N F F F N N N H N O O 83 N N N O N
Figure 57. A2B AR antagonists (8-pyrazolo-4-yl-xanthines).
2.1.5.2 Pyrrolopyrimidines
Recently in a patent by OSI Pharmaceuticals Inc., a series of pyrrolopyrimidines is reported to be A2B AR antagonists.166 The most important compound of this family, coded OSIP-339391 (84, Figure 58), displays a 70-fold selectivity for A2B ARs over the other hAR subtypes. The radiolabeled form
126
[3H]OSIP-339391 was prepared and characterized in kinetic, saturation, and competition binding experiments at recombinant hA2B ARs. From the association and dissociation rate studies, the affinity was 0.41 nM, and that found in saturation binding experiments was 0.17 nM.
N N N H N N NH HN O O 84 OSIP-339391
Figure 58. A2B AR antagonist (pyrrolopyrimidine derivative).
2.1.6 A3 Adenosine Receptor Antagonists
The human A3 AR is widely distributed in peripheral organs with high levels in testis and lower levels in lung, kidney and heart; it is also expressed in low levels in regions of the central nervous system (CNS). The A3 AR is involved in a variety of important physiological processes, including modulation of cerebral and cardiac ischemic damage, inflammation, modulation of intraocular pressure, regulation of normal and tumor cell growth, and immunosuppression.167 Consequently, A3 AR selective ligands may represent important pharmacological tools in the treatment of a variety of diseases. Indeed, the development of potent and selective A3 AR ligands has been the subject of medicinal chemistry research for more than two decades. Although to date a considerable number of selective A3 AR antagonists have been discovered, much is still to be learned about the exact function of this subtype in different pathophysiological conditions. Among the ARs, the A3 is perhaps the most enigmatic, as in several therapeutic fields including ischemia, inflammation, and cancer, a double nature emerges for this receptor, rendering it as a single entity behaving in two different ways often in direct conflict between them. Actually, it is widely recognized that in ischemic and inflammatory conditions, as well as in cancer, the A3 AR may play a
127 beneficial or detrimental role in dependence on the system investigated (tissue, cell line, or species).167 In this view, both A3 agonists and antagonists could be potentially effective in cancer therapy. Actually, an intriguing challenge for the current research is to identify selective A3 ligands that allow to understand whether and in what pathological conditions agonists or antagonists represent the most efficacious choice. A3 AR selective antagonists have been proposed as novel anti-inflammatory drugs, as cerebroprotective agents for preventing ischemic brain damage and as new agents for the treatment of glaucoma. In the last two decades, numerous medicinal chemistry groups have made intense efforts in searching for ideal ligands for the A3 AR subtype. In particular, the search for selective A3 AR antagonists held greater appeal than selective agonists, not only for their potential therapeutic applications, but also because antagonists are preferred molecular probes for pharmacological characterization of receptors.167
2.1.6.1 Xanthines
Natural antagonists for ARs, such as caffeine and theophylline, show in general low affinity for the A3 AR subtype. Different positions of the xanthine core have been modified with the aim of improving A3 AR affinity. A series of tricyclic imidazo[2,1-i]purinones and ring-enlarged analogues derived from xanthine derivatives has been prepared as AR antagonists. In comparison with xanthines, the tricyclic compounds exhibit increased water solubility due to a basic nitrogen atom, which can be protonated under physiological conditions.103 Among this series PSB-10, 8(R)-ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (85, Figure 59), is a high-affinity ligand for A3 ARs (hA3, Ki 0.43 nM) with high selectivity over hA1 and hA2A ARs (Ki 1700 and 2700 nM, respectively). The compound showed inverse agonist activity in binding studies in CHO cells expressing recombinant hA3 ARs (IC50 4 nM).103 Another similar compound is 2-(4-bromophenyl)-7,8-dihydro-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one, also named KF-26777 (86, Figure 59), endowed with subnanomolar affinity to hA3 ARs (Ki 0.20 nM) and high selectivity over A1, A2A, and A2B ARs (9000-, 23 500-,and 31 000-fold, respectively). It concentration-dependently inhibited the 2-chloro-N6
-(3-iodobenzyl)-N-methyl-5’-128
carbamoyladenosine (Cl-IB-MECA)-induced [35 S]guanosine-5’-O-(3-thiotriphosphate) ([35S]-GTPγS) binding to human embryonic kidney 293 cells (HEK293) (IC50 270 nM) and enhanced intracellular Ca2+ concentration in human promyelocytic cells (KB 0.42 nM). This agent was indicated for potential interest for treatment of brain ischemia and inflammatory diseases such as asthma.103
N N N H N N Cl Cl Cl O C2H5 85 PSB-10 N N N H N N O 86 KF-26777 N N N N O 87 O NH
Figure 59. A3 AR antagonists (xanthines).
The discovery of 1-benzyl-3-propyl-1H,8H-imidazo[2,1-f]purine-2,4-diones by cyclization between the 7- and 8-positions of the xanthine core lead to
87 (Figure 59), a highly potent and selective A3 AR antagonist. This compound shows a subnanomolar affinity (hA3, Ki 0.8 nM) toward the desired receptor target with a noteworthy selectivity versus the other AR subtypes. In this field of research the triazolopurine derivatives in which the xanthine structure is extended, are also reported. One example is OT-7999 (88, Figure 60), which proved to be a potent and selective hA3 AR ligand. In receptor binding assays, OT-7999 displayed high affinity for the A3 AR (Ki 0.95 nM) and >10 500-fold selectivity relative to other AR subtypes. Significant reductions in intraocular pressure were obtained in cynomolgus monkeys at 2-4 h following topical application to the eye of OT-7999 (500 mcg).103
129 N N N N N N 88 OT-7999 F F F
Figure 60. A3 AR antagonist (triazolopurine derivative).
2.1.6.2 1,4-Dihydropyridines and piridines
Starting from the experimental observations that 1,4-dihydropyridines bind A1 AR in the rat brain,168,169 Jacobson et al. used the 1,4-dihydropyridine nucleus as a template for probing the SAR profile at the A3 AR subtype.170 SAR studies of AR antagonists indicated that sterically bulky groups are well tolerated at the 4-, 5-, and 6-positions. The combination of substitutions led to the discovery of MRS 1097 (2-methyl-6-phenyl-4-styryl-1,4-dihydro pyridine-3,5-dicarboxylic acid diethyl ester, 89, Figure 61), MRS 1191, (2-methyl-6-phenyl-4-phenylethynyl-1,4-dihydro pyridine-3,5-dicarboxylic acid 5-benzyl ester, 90, Figure 61), and MRS 1334 (2-methyl-6-phenyl-4-phenylethynyl-1,4-dihydro pyridine-3,5-dicarboxylic acid 3-ethyl ester 5-(4-nitro-benzyl) ester, 91, Figure 61) as the first A3 antagonists related to 1,4-dihyropyridines. In this study, they also synthesized pyridine derivatives170,171 through oxidation of the corresponding 1,4-dihydropyridine. In this class of compounds, small groups at the 4-position were found to be essential such as in MRS 1523 (6-ethyl-5-ethylsulfanylcarbonyl-2-phenyl-4-propyl-nicotinic acid propyl ester, 92, Figure 61), which showed favorable affinity at the hA3 AR subtype. Comparing the structural requirements for the two related classes of compounds indicated that bulky substituents at the 4-position and a 5-benzyl ester, which are affinity enhancing in dihydropyridines, are not well tolerated in the pyridine series for A3 receptor binding. At other positions, structural parallels occur between corresponding dihydropyridine and pyridine analogues.172
130 N H O O O O 89 MRS 1097 N H O O O O 90 MRS 1191 N H O O O O 91 MRS 1334 NO2 N H O O S O 92 MRS 1523
Figure 61. Dihydropyridine and Pyridine derivatives as A3 AR antagonists.
2.1.6.3 Pyrazolo-triazolo pyrimidines
The pyrazolo-triazolo-pyrimidine nucleus, due to its strong structural correlation with the nonselective antagonists 65, CGS-15943, and the adenine nucleus present in the endogenous modulator adenosine 40 (Figure 62), has been strongly investigated in the past decade as a prototypical template for adenosine antagonists. The triazolo-quinazoline derivative CGS-15943 represented the starting point in searching for new potent and selective hA3 AR antagonists. MRS-1220, a 5-N-phenylacetyl derivative of CGS-15943, in receptor binding studies displayed Ki values of 305 ± 51, 52.0 ± 8.8, and 0.65 ± 0.25 nM for rat A1, A2A, and hA3 ARs, respectively, being 470- and 80-fold selective for hA3 ARs vs rat A1 and A2A ARs, respectively. MRS-1220 also antagonized the effects of an A3 agonist in functional assays.173,174