INDICE
1. INTRODUZIONE
1.1 L’autofagia ...4 1.1.1 Generalità...4 1.1.2 Tipi di autofagia ...5 1.1.2.1 Autofagia chaperone-mediata ...5 1.1.2.2 Microautofagia...6 1.1.2.3 Macroautofagia ...7 1.1.3 Regolazione dell’autofagia. ...11 1.1.3.1 Regolazione metabolica ...11 1.1.3.2 Regolazione endocrina...131.1.3.3 Trasduzione dei segnali regolatori dell’autofagia ...14
1.1.4 Meccanismi molecolari ...15
1.1.5 Autofagia In Vivo durante il Digiuno ...18
1.1.6 Selettività dell’autofagia...21
1.1.7 Autofagia e invecchiamento ...23
1.2 Ruolo dei mitocondri nella patogenesi dell’invecchiamento...27
1.2.1 Invecchiamento e stress ossidativo...27
1.2.2 Teoria mitocondriale dello stress ossidativo ...31
1.3 Autofagia e mitocondri ...35
1.3.1 Mitofagia...35
1.3.1.1 Generalità...35
1.3.1.2 Selettività della mitofagia...36
1.3.1.3 Transizione di permeabilità di membrana (MPT): induttore della mitofagia ...37
2. MATERIALI E METODI
2.1 Valutazione degli effetti della somministrazione di un farmaco
antilipolitico ...42
2.1.1 Animali...42
2.1.2 Dosaggio dei livelli plasmatici di glucosio e insulina...42
2.1.3 Dosaggio dei livelli di valina e altri aminoacidi...43
2.1.3.1 Derivatizzazione ...43
2.1.3.2 Procedura cromatografica ...43
2.1.4 Procedura di microscopia elettronica ...44
2.2 Valutazione della stimolazione e dell’inibizione dell’autofagia sul danno ossidativo del mtDNA ...45
2.2.1 Animali e trattamenti...45
2.2.2 Dosaggio delle proteine...45
2.2.3 Isolamento del mtDNA...45
2.2.4 Dissociazione del mtDNA in singole basi ...47
2.2.5 Procedura cromatografica ...47
2.2.6 Dosaggio dell’attività della citocromo C ossidasi ...48
2.3 Analisi statistica...48
3. RISULTATI
3.1 Effetto della somministrazione del farmaco antilipolitico 3,5-dimetilpirazolo (DMP) sui livelli plasmatici di glucosio, insulina e valina in animali giovani (3 mesi d’età) sottoposti a digiuno di 18 ore...493.2 Effetto della somministrazione del farmaco antilipolitico 3,5-dimetilpirazolo (DMP) a ratti giovani digiuni da 18 ore sul compartimento autofagico epatico...55
3.3 Effetto della somministrazione di glucosio sui livelli plasmatici di glucosio e insulina in animali giovani trattati con farmaco antilipolitico
3,5-dimetilpirazolo (DMP) ...57
3.4 Effetto della somministrazione di glucosio al 42, 57, 87 minuto dopo la somministrazione di DMP sui livelli plasmatici di valina, leucina e glutamina…….. ...61
3.5 Effetto della somministrazione di glucosio a ratti giovani trattati con DMP sul compartimento autofagico epatico ...66
3.6 Effetti dell’età e della intensificazione farmacologica dell’autofagia sui livelli del danno ossidativo al mtDNA e sul contenuto mitocondriale del fegato...68
3.7 Effetti della somministrazione di insulina in animali giovani (3 mesi d’età) sui livelli del danno ossidativo al mtDNA e sul contenuto di mitocondri del fegato...72
4. DISCUSSIONE
...765. CONCLUSIONI
...831. Introduzione
1.1 L’autofagia
1.1.1 Generalità
L’autofagia è un processo cellulare complesso, universale, altamente conservato e regolato, ATP-dipendente che avviene in tutte le cellule eucariote. Esso coinvolge il riarrangiamento di membrane subcellulari al fine di sequestrare citoplasma e organuli da esporre all’azione degradativa di organuli, come il vacuolo nel lievito e i lisosomi nelle cellule di mammifero, dove il materiale sequestrato è degradato e riciclato (Kanazawa et al., 2003;Wang and Klionsky, 2003).
L’autofagia presenta molteplici funzioni. Questo processo è coinvolto: (1) nel rimodellamento durante lo sviluppo e il differenziamento cellulare (Levine and Klionsky, 2004); (2) nella degradazione delle proteine intracellulari (Mortimore et al., 1989); (3) nella produzione di aminoacidi quando si presenta un calo di nutrienti; (4) nel meccanismo di ricambio di citomembrane e di organuli come i mitocondri e i perossisomi (Klionsky and Emr, 2000); (5) nel controllo del meccanismo della morte cellulare programmata di tipo II (non-apoptotica) (Shintani and Klionsky, 2004). L’autofagia può avere anche un ruolo nel controllo della crescita cellulare, persino in quella tumorale (Klionsky and Emr, 2000).
L’autofagia è anche stata proposta come un meccanismo anti–invecchiamento in quanto è coinvolta nell’eliminazione di organuli e citomembrane danneggiati a
causa della perossidazione età-dipendente di diverse molecole (Bergamini et al., 2003).
L’autofagia viene stimolata in risposta a diverse situazioni di stress come il digiuno, i cambiamenti nel volume cellulare, lo stress ossidativo, l’accumulo di proteine danneggiate, segnali ormonali, l’irradiazione e il trattamento con xenobiotici (Meijer and Codogno, 2004).
1.1.2 Tipi di autofagia
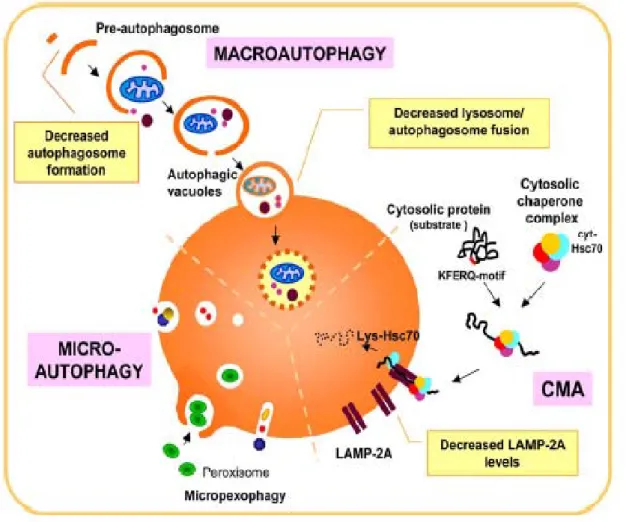
Le vie autofagiche meglio caratterizzate sono la macroautofagia, la microautofagia e l’autofagia chaperone-mediata (Cuervo, 2004; Klionsky, 2005). Esse si differenziano per: il modo con cui il materiale da degradare viene trasportato all’interno del lume lisosomiale, per il tipo di materiale trasportato e per la loro regolazione (fig. 1).
1.1.2.1 Autofagia chaperone-mediata
Molti tipi di cellule possono degradare proteine citosoliche attraverso la forma di autofagia chaperone-mediata (CMA) (Cuervo, 2004; Majeski and Dice, 2004; Massey et al., 2004). Fatto distintivo della CMA è la sua selettività per un particolare gruppo di proteine citoplasmatiche e per il fatto che questi substrati sono direttamente traslocati attraverso la membrana lisosomiale senza richiedere la formazione di vacuoli o la deformazione di membrana. Tutti i substrati di CMA contengono una sequenza biochimicamente legata al pentapeptide KFERQ (Majeski and Dice, 2004). Questo motivo è riconosciuto dalle chaperonine, tra cui la proteina heat shock di 70 kDa (hsc70) (Chiang et al., 1989), alle quali le proteine “target” si legano. Il complesso
chaperonina-proteina si lega sulla membrana lisosomiale interagendo con il recettore LAMP-2a ed il trasporto all’interno dell’organulo è assistito dalla hsc70 lisosomiale (lys-hsc70) (Majeski and Dice, 2004; Salvador et al., 2000).
I livelli di LAMP-2a, che sono in parte regolati da variazioni della sua degradazione (Yang et al., 2005), possono essere importanti nella modulazione di CMA (Majeski and Dice, 2004; Massey et al., 2004).
Il carattere selettivo di CMA fa sì che questa forma d’autofagia sia attivata quando la discriminazione tra le proteine da degradare è importante.
Durante condizioni di stress, come per esempio la mancanza di nutrienti, l’attività di CMA aumenta dopo che la macroautofagia, anch’essa attivata, comincia a calare (Cuervo, 2004). Questa attivazione sequenziale prima di una via autofagica non-selettiva poi di quella selettiva può essere finalizzata ad evitare la degradazione di componenti cellulari essenziali in caso di un digiuno prolungato (Martinez-Vicente et al., 2005).
1.1.2.2 Microautofagia
La microautofagia è coinvolta nell’inglobamento e nella degradazione di regioni complete del citosol, inclusi proteine e organuli citoplasmatici, direttamente da parte dei lisosomi, senza richiedere la formazione di vacuoli autofagici intermedi (Farre and Subramani, 2004; Klionsky, 2005; Mortimore et al., 1988). Il lisosoma, infatti, per invaginamento o protusione, avvolge il citoplasma e in seguito si chiude a formare una vescicola interna che contiene il materiale da degradare (Ahlberg and Glaumann, 1986).
La microautofagia è stata tradizionalmente considerata come una forma di autofagia attiva per garantire il turnover delle proteine a lunga vita in condizioni
basali (Mortimore et al., 1988), ma studi recenti (Farre and Subramani, 2004) la ritengono anche responsabile, in parte, della degradazione dei perossisomi.
1.1.2.3 Macroautofagia
La macroautofagia è la principale forma di autofagia responsabile della degradazione sia di proteine solubili a lunga vita che di organuli in condizioni di stress (Klionsky, 2005; Shintani and Klionsky, 2004). Essa comporta la formazione di vacuoli a doppia membrana che sequestrano porzioni di citoplasma e le trasportano ai lisosomi (Seglen and Bohley, 1992). Per fare questo la macroautofagia impiega diversi organuli attraverso differenti passaggi. Un organulo membranoso, inizialmente, avvolge una porzione di citoplasma che poi chiudendosi forma un vacuolo, solitamente delimitato da due o più membrane, chiamato autofagosoma (Ohsumi and Mizushima, 2004).
Numerose membrane sono state proposte essere coinvolte nell’origine di questo vacuolo, in particolare le membrane del reticolo endoplasmatico (ER) e le cisterne del Golgi. Benché una parte dei vacuoli autofagici iniziali contenga delle proteine reticolari, una restante parte di essi è di diversa origine (Dunn, 1990). E’ stato proposto che in qualche caso gli autofagosomi possano avere origine da una struttura preesistente denominata fagoforo (Seglen, 1987): esso rappresenterebbe una struttura membranosa condensata, posizionata nel citosol e ricca di materiale osmolipidico.
In un secondo passaggio l’autofagosoma acquisisce proteine della membrana lisosomiale come LGP10 e la pompa protonica ATPasica, responsabile dell’acidificazione del vacuolo (Dunn, 1994). Con l’impiego di nocodazolo è possibile provocare l’accumulo d’autofagosomi acidi, privi però d’attività
idrolitica, morfologicamente indistinguibili dagli autofagosomi (Aplin et al., 1992). Si ritiene che questi vacuoli, chiamati autofagosomi tardivi, si formino dalla fusione degli autofagosomi con vescicole derivate sia dall’apparato del Golgi sia da endosomi tardivi. Altri studi hanno identificato come amfisomi, i vacuoli autofagici successivi agli autofagosomi iniziali (Seglen and Bohley, 1992).
L’amfisoma è una struttura capace di ricevere materiale proveniente sia dal processo d’endocitosi che da quello d’autofagia: ciò suggerisce che esso sia un vacuolo autofagico con proprietà anfofunzionali (Seglen and Bohley, 1992). La scoperta dell’amfisoma si è basata sulla capacità dell’asparagina di inibire la fusione tra lisosoma e autofagosoma senza interferire con l’endocitosi (Gordon et al., 1985). L’autofagia di lattosio elettroiniettato, infatti, può normalmente causare la degradazione del lattosio nei lisosomi (Hoyvik et al., 1986) ma in presenza di asparagina il lattosio si accumula nel compartimento prelisosomiale, dove è accessibile alla degradazione da parte di enzimi attivi nell’endocitosi (Gordon and Seglen, 1988). Questo dimostra che l’accumulo di materiale può avvenire in un compartimento prelisosomiale specifico capace di ricevere materiale dal processo d’autofagia e da quello d’endocitosi (Gordon and Seglen, 1988).
Anche se il meccanismo con cui avviene il trasferimento di materiale tra i due compartimenti non è ben noto, si ritiene che esso possa avvenire per un processo di fusione anche di più autofagosomi con lo stesso amfisoma (Seglen and Bohley, 1992).
In un terzo passaggio autofagico l’amfisoma si fonde con i lisosomi. La fusione porta il materiale autofagocitato in contatto con gli enzimi lisosomiali,
l’aspetto finale è quello di una struttura elettro-densa composta da materiale amorfo; in questa fase il vacuolo viene definito corpo denso (Pfeifer, 1981; Kovacs and Rez, 1989; Mortimore et al., 1989; Dunn, 1990).
Una volta degradate le sostanze molecolari semplici contenute nel vacuolo sono riassorbite o utilizzate dalle cellule o messe a disposizione dell’organismo. Il materiale non digerito viene di solito scaricato dalle cellule tramite esocitosi, oppure rimane all’interno del vacuolo andando ad acquisire con il tempo le caratteristiche biochimiche dei pigmenti da usura o lipofuscine (Glaumann et al., 1981).
Il termine d’autofagia è spesso sinonimo di macroautofagia, per questo, in seguito verranno utilizzati i due termini indifferentemente.
FIG 1. MODELLO SCHEMATICO DELLE PRINCIPALI FORME DI AUTOFAGIA NELLE CELLULE DI MAMMIFERO E LORO CAMBIAMENTI NELL’INVECCHIAMENTO.
Internalizzazione di regioni di citosol o per fusione dell’autofagosoma con il lisosoma (macroautofagia) o direttamente per invaginazione della membrana lisosomiale (microautofagia). Cattura selettiva di proteine citosoliche per via autofagica mediata da chaperonine(CMA). È stata anche mostrata un tipo di microautofagia che rimuove selettivamente i perossisomi dal citosol.
Sono anche mostrati i difetti età-dipendenti identificati nei diversi tipi di autofagia . Abbreviazioni: hsc70, heat-shock cognate protein of 70 KDa; LAMP-2A, lysosome associate membrane protein type 2A.
1.1.3 Regolazione dell’autofagia
Considerazioni funzionali suggeriscono che la regolazione biologica dell’autofagia possa essere esercitata a livello del sequestro del materiale da degradare (Seglen and Bohley,1992). In termini fisiologici, l’autofagia viene inibita dall’abbondanza di nutrienti: nello specifico il processo viene modulato dall’aumento a livello plasmatico delle concentrazioni aminoacidiche e dal rapporto insulina /glucagone.
1.1.3.1 Regolazione metabolica
Studi condotti da Mortimore e Schworer (1977) mostrano che gli aminoacidi, prodotto finale della degradazione autofagica, hanno un effetto inibitorio sul processo d’autofagia nelle cellule di fegato di ratto (Mortimore and Schworer, 1977). Solo pochi aminoacidi sono richiesti per la regolazione ed il tipo e il numero dipendono dalla natura del tessuto interessato. Ad esempio l’inibizione autofagica nel fegato (Woodside and Mortimore, 1972; Seglen et al., 1980; Poso et al., 1982) richiede l’azione concertata di più aminoacidi. Esistono evidenze sperimentali sul contributo di almeno 8 aminoacidi alla modulazione dell’autofagia negli epatociti di ratto (Woodside and Mortimore, 1972; Hopgood et al., 1980; Seglen et al, 1980; Sommercon and Swick, 1981; Poso et al., 1982). Questi aminoacidi sono leucina, fenilalanina, tiroxina, glutammina, prolina, istidina, triptofano, metionina insieme all’alanina, che da sola non ha effetti sull’autofagia ma agisce da co-regolatore (Poso et al., 1982). È stato osservato che nel fegato perfuso, leucina e glutammina insieme a tirosina o fenilalanina sono in grado di inibire quasi completamente l’autofagia (Kadowaki
et al., 1992) e che la glutammina è efficace nell’inibire l’autofagia epatica del ratto in vivo (Bergamini et al., 1994). Ulteriori studi su epatociti in coltura hanno stabilito che leucina, fenilalanina e tiroxina sono tra i più importanti regolatori del processo in vivo (Blommaart et al., 1993). In seguito a studi sul fegato, è stato suggerito che la leucina, la fenilalanina e la tiroxina sono in combinazione con altri aminoacidi, come la glutammina e l’alanina, i più importanti aminoacidi coinvolti nel controllo dell’autofagia (Bloommart et al., 1997).
Nel fegato ci sono due meccanismi di regolazione: (1) modo L, che richiede l’alanina affinché si verifichi l’inibizione a concentrazioni fisiologiche di aminoacidi; (2) modo H, che ha effetto a dosi alte di aminoacidi e l’alanina non svolge la funzione di co-regolatore (Venerando et al., 1994). Studi svolti tenendo conto dell’alimentazione dei ratti, hanno indicato che dopo l’alimentazione si ha una regolazione di tipo H, in seguito la regolazione è casuale e con il proseguire del digiuno è prevalentemente quella di tipo L (Mortimore et al., 1991).
Sebbene il controllo del processo avvenga principalmente a livello del sequestro di materiale da parte dell’autofagosoma si è visto che elevate concentrazioni di aminoacidi non bloccano completamente tale sequestro pur inibendo l’autofagia (Seglen and Gordon, 1984), ciò indica che gli aminoacidi possono inibire anche la fase successiva al sequestro. Ad esempio l’asparagina ad elevate concentrazioni inibisce la fusione tra autofagosoma e lisosoma (Hoyvik et al., 1991). È stato anche suggerito che gli aminoacidi potrebbero agire sui lisosomi alzando il loro pH e inibendo l’azione degli enzimi lisosomiali (Volk et al., 1993; Luiken et al., 1996).
1.1.3.2 Regolazione endocrina
È dimostrato che gli ormoni pancreatici sono coinvolti nella regolazione endocrina del processo autofagico. L’insulina inibisce l’autofagia, mentre il glucagone la stimola (Mortimore et al. 1989; Seglen and Bohley, 1992).
Nel fegato perfuso, il glucagone agisce come un potente induttore dell’autofagia sia in un mezzo privo di aminoacidi (Ashford and Porter, 1962; Schworer and Mortimore, 1979) che in presenza di aminoacidi a concentrazioni fisiologiche. Questo ormone, comunque, contrasta l’azione inibente degli aminoacidi, quando questi sono a concentrazioni superiori a quella fisiologica (Mortimore et al., 1987;Poli et al., 1981).
Il glucagone sembra abbassare i livelli intracellulari di glutamina (Schworer and Mortimore, 1979), uno dei più attivi aminoacidi inibitori della degradazione (Seglen et al., 1980). Benché l’esatto meccanismo biochimico non sia conosciuto, è noto che il glucagone stimola la gluconeogenesi, per la quale la glutamina è un importante substrato, e attiva la glutaminasi epatica (Joseph and McGivan, 1978).
L’inibizione insulinica è massima a una concentrazione di 10-7, invece a concentrazioni più alte l’azione sembra essere meno efficace (Poli et al., 1981). Contrariamente al glucagone, che ha un azione tessuto specifica, l’insulina esercita la sua azione su diversi tessuti e organi, come il fegato, cuore, muscolo scheletrico, rene, tessuto adiposo e sulle cellule in coltura (Mortimore and Poso, 1987).
E’ stato dimostrato, inoltre, che l’insulina è in grado di potenziare l’effetto di tutte le combinazioni degli aminoacidi, ma non quando è raggiunta l’inibizione ottimale massima (Poli et al., 1981).
Il fatto che quest’ormone abbia un’azione inibitoria più marcata in presenza di concentrazioni fisiologiche di aminoacidi (Blommaart et al., 1997), suggerisce che l’ormone agisca modulando l’azione degli aminoacidi.
1.1.3.3 Trasduzione dei segnali regolatori dell’autofagia
Si ritiene che la regolazione aminoacidica e insulinica possano agire in maniera sinergica nel controllo dell’autofagia. Sembra che il segnale, sia dell’insulina che degli aminoacidi possa convergere a livello dell’enzima Mammalian Target of Rapamycin (mTOR, omologa della TOR presente nel lievito), una chinasi che attiva la sintesi proteica e nel contempo inibisce l’autofagia (Schmelzleand Hall, 2000).
Le prime evidenze del ruolo del mTOR nel controllo dell’autofagia sono state ottenute da studi su epatociti di ratto, dove l’effetto inibitorio degli aminoacidi può essere diminuito dalla rapamicina (Blommaart et al., 1995), un inibitore dell’enzima. Al momento sembra evidente che gli aminoacidi determinino l’attivazione della mTOR: l’enzima è invece inibito da condizioni di carenza energetica come l’attivazione dell’enzima AMP chinasi dovuta ad una diminuzione del rapporto ATP/AMP (Hardie, 2003). Rimane da svelare, però, come gli aminoacidi segnalino la loro presenza alla cellula: alcuni ricercatori avrebbero identificato un complesso localizzato sulla superficie cellulare che si legherebbe con la leucina e hanno determinato l’inibizione dell’autofagia con un peptide globulare che lega 8 residui di leucina e non permea la membrana plasmatica (Miotto et al., 1994). Tali risultati a favore di un recettore sulla membrana plasmatica, non sono stati però confermati da altri laboratori usando la stessa molecola (Blommaart et al., 1997). In un altro ipotetico meccanismo, le
cellule risponderebbero all’aminoacilazione del tRNA ed in effetti è stato mostrato, nel lievito, che il tRNA libero, che non lega cioè aminoacidi, previene la fosforilazione della p70s6k mediata dalla Tor attivata (Iiboshi et al., 1999). Se anche non fosse questo il meccanismo, il fatto che l’inibizione del sistema di trasporto degli aminoacidi inibisca marcatamente la fosforilazione della p70s6k depone a favore del fatto che gli aminoacidi esercitino i loro effetti una volta entrati nella cellula (Iiboshi et al., 1999).
Come noto invece per l’insulina esiste un recettore specifico sulla membrana plasmatica (IR): il legame ormone-recettore determina una autofosforilazione dell’IR sul versante citoplasmatico che a sua volta catalizza la fosforilazione del substrato dell’IR (IRS): l’IRS così attivato probabilmente agisce attivando la fosfatidil inositolo 3-chinasi (PI3K) (Fulop et al., 2003). Studi condotti da Brazil e Hemmings (2001) hanno mostrato che l’inibizione del processo autofagico mediata dall’insulina (Pfeifer, 1978; Mortimore et al., 1987; Poli et al., 1981) potrebbe dipendere, dall’attivazione della classe I della fosfatidil inositolo 3-fosfato (PI3K) che converte il PI4P e PI(4,5)P2 in PI(3,4)P2 e PI(3,4,5)P3 che sono in grado di legarsi a dei domini aminoacidici dell’enzima PKB e del suo attivatore PDK1. PDK1 fosforila altre chinasi tra cui mTOR e p70s6k (Brazil and Hemmings, 2001).
1.1.4 Meccanismi molecolari
Il meccanismo molecolare alla base dell’autofagia è ad oggi ancora poco conosciuto. Alcuni anni fa studi genetici sul lievito Saccharomyces Cerevisiae hanno portato all’identificazione di geni, in parte conservati dal lievito all’uomo, coinvolti nel processo autofagico (Tsukada and Ohsumi, 1993; Thumm et al.,
1994; Khalfan and Klionsky, 2002); questi geni sono stati denominati geni ATG (Autophagy-related) (Klionsky et al., 2003).
In condizioni di ricchezza di nutrienti la degradazione autofagica nel lievito è inibita con un meccanismo che coinvolge l’attivazione della proteina chinasi Tor (Schmelzle and Hall, 2000). La Tor attivata sembra causare direttamente o indirettamente l’iperfosforilazione dell’Atg13. L’Atg13 iperfosforilata presenta una minore affinità per la chinasi Atg1 e la ridotta interazione tra Atg13 e Atg1 può inibire l’autofagia (Kamada et al., 2000). Atg1 e Atg13 infatti, fanno parte di un complesso più grande composto da altre molecole; la funzione di tale complesso potrebbe essere richiesta per aumentare la dimensione dell’autofagosoma (Abeliovich et al., 2003). È stato, inoltre, osservato che le chinasi mTor e Atg1 giocano un ruolo simile nell’autofagia negli eucarioti più evoluti (Abeliovich, 2004; Codogno and Meijer, 2004; Petiot et al., 2002).
Nelle cellule di lievito l’autofagosoma sembra essere generato a partire da una struttura membranosa di natura ignota chiamata struttura pre-autofagosomiale (PAS) (Suzuki et al., 2001; Noda et al., 2002) per aggiunta di altre componenti membranose. Nella formazione e nell’allungamento del PAS sembrano intervenire molti dei prodotti dei geni Atg.
Nel lievito un complesso denominato I formato da alcuni prodotti genici Atg e la PI3K, è pensato avere un ruolo nella formazione iniziale della membrana autofagosomiale. Nelle cellule di mammifero a differenza del lievito esistono 3 classi di PI3K. La classe I PI3K è un inibitore dell’autofagia (Arico et al., 2001) che coinvolge l’attivazione della mTOR (omologo della TOR). La classe III PI3K, un omologo funzionale della Vsp34 di lievito, è invece un enzima costitutivo
dell’autofagia e gioca un ruolo cruciale nei primi passaggi della formazione dell’autofagosoma nelle cellule di mammifero (Tassa et al., 2003).
Il complesso I può generare fosfatidilinositolo-3 fosfato a livello delle membrane dei PAS e facilitare il reclutamento di componenti proteiche utili nella formazione della membrana del vacuolo (Yorimitsu and Klionsky, 2005).
Nelle cellule di mammifero la classe III PI3K forma un complesso con la proteina Beclin-1 (Kihara et al., 2001), un omologo funzionale dell’Atg6 di lievito (Liang et al., 1999; Kihara et al., 2001). È stato ipotizzato che questo complesso possa svolgere un ruolo nel reclutamento di membrane da varie strutture (ER, trans-Golgi, mitocondri) per l’allungamento della membrana dell’autofagosoma (Liang et al., 1998).
Nella fase di allungamento della membrana che andrà a circoscrivere l’autofagosoma, il complesso Atg12-Atg5-Atg16 e il complesso Atg8- fosfolipide fosfatidiletanolamina (PE), due sistema di coniugazione simili a quelli dell’ubiquitina che funzionano in maniera coordinata (Nemoto et al., 2003; Suzuki et al., 2001; Tanida et al., 2002), sono attivati nel lievito dalla fosfatidilinositolo-3-chinasi.
Il complesso Atg12-Atg5-Atg16 è il primo ad entrare in funzione durante la fase d’espansione del vacuolo, formando una sorta di rivestimento intorno al vacuolo in formazione e controllando la curvatura di esso. Questo sistema di coniugazione è conservato nelle cellule di mammifero (Mizushima et al., 1998) dove svolge un ruolo nella formazione delle membrane autofagiche (Komatsu et al., 2001; Mizushima et al., 2001).
Il complesso Atg8-PE, invece, è coinvolto nell’ingrandimento del vacuolo agendo come una componente strutturale (Yorimitsu and Klionsky, 2005) . Nelle
cellule di mammifero diversi omologhi dell’Atg8 possono essere coinvolti nella reazione di coniugazione (Tanida et al., 2001). Uno di questi omologhi, la Microtubule Asssociated Protein 1 Light Chain 3 (MAP1-LC3), è coinvolta nella formazione dell’autofagosoma dopo un processo simile a quello che avviene nell’Atg8 di lievito (Kabeya et al., 2000).
LC3 interagisce con la membrana dell’autofagosoma in maniera Atg5-dipendente e rimane sulla membrana dopo la dissociazione del complesso Atg12-Atg5. Per questo motivo LC3 può essere usato come marker dall’autofagosoma nelle cellule di mammifero (Yoshimori, 2004).
Perché i due complessi possano localizzarsi sui PAS è richiesta la partecipazione dell’Atg9 (Suzuki et al., 2001).
Nel lievito Atg9 è scambiato continuamente tra il PAS e il mitocondrio, è stato ipotizzato che il ricircolo di Atg9 possa fornire i lipidi utili per l’espansione del vacuolo(Yorimitsu and Klionsky,2005) .
Un volta completata la formazione del vacuolo le proteine di rivestimento, eccetto i complessi Atg8-PE, orientati verso il lume, si dissociano dal vacuolo.
1.1.5 Autofagia In Vivo durante il Digiuno
Se studi in vitro hanno identificato specificamente negli aminoacidi e negli ormoni pancreatici i più importanti modulatori del processo autofagico, studi in vivo hanno dimostrato che in generale è la carenza energetica, specie il digiuno, ad essere un potente induttore del processo autofagico. Le proteine intracellulari sono, infatti, una delle fonti di aminoacidi più importanti quando l’assorbimento intestinale diminuisce o è assente (Mortimore et al., 1989).
bensì è rappresentata dalle normali proteine costituenti la cellula (Mortimore and Poso, 1987). La loro degradazione, che sappiamo essere finemente regolata da ormoni, aminoacidi e altre molecole, varia ampiamente a seconda del tipo di tessuto interessato (Mortimore and Poso, 1987). Durante il digiuno le perdite proteiche più consistenti si verificano nel fegato, cali minori si presentano nel rene, nell’intestino, nel cuore e nel muscolo scheletrico (Addis et al., 1936). Nel ratto è stato mostrato che durante le prime 24 ore di digiuno, si assiste alla perdita del 25-40% del contenuto proteico nel fegato senza che si verifichi un calo di proteine apprezzabile nel muscolo scheletrico (Addis et al., 1936; Hutson and Mortimore, 1982); dopo 48 ore di digiuno si verifica un incremento di proteolisi a livello del muscolo il cui contenuto proteico diminuisce (Millward and Waterlow, 1978). Si può concludere che i tessuti rispondono diversamente alla necessità di aminoacidi e che il rapido catabolismo proteico osservato nel fegato si instaura principalmente nell’intervallo tra i pasti mentre le risposte più ritardate come quella del muscolo si attivano nel digiuno prolungato (Mortimore and Poso, 1987).
L’entità della degradazione proteica del fegato rende possibile stabilire una correlazione quantitativa tra velocità di proteolisi e alterazioni del sistema vacuolare-lisosomiale durante il digiuno e nella successiva rialimentazione: la perdita netta delle proteine pùo essere attribuita all’autofagia (Hutson and Mortimore, 1982) e alla conseguente accelerazione della proteolisi che risulta consistentemente più veloce della sintesi proteica (Hutson and Mortimore, 1982; Pfeifer, 1973). Con la rialimentazione l’autofagia e l’incremento proteolitico sono repentinamente soppressi (Hutson and Mortimore, 1982; Khairallah, 1978; Pfeifer and Bertling, 1977).
Sebbene il fegato sia l’organo maggiormente interessato dall’incremento autofagico-proteolitico durante il digiuno è importante sottolineare che anche altri organi presentano un aumento dell’autofagia: nel muscolo le proteine miofibrillari, che rappresentano la maggior parte del contenuto proteico, sono degradate da meccanismi non-lisosomiali. Nonostante ciò è stato mostrato che anche la proteolisi lisosomiale è elevata durante il digiuno (Wing and Goldberg, 1993): inoltre, in un recente lavoro su linee di cellule muscolari, è stato evidenziato come la carenza di leucina attivi specificamente il processo di autofagia (Mordier et al., 2000).
La scoperta e lo studio dei meccanismi molecolari del processo di autofagia ha permesso di osservare che i geni coinvolti in questo processo nel lievito, hanno omologie con geni dei mammiferi che sono espressi in molti tessuti; ciò potrebbe indicare che il processo potrebbe essere attivo in maniera continua ubiquitariamente (Kim and Klionsky, 2000); in aggiunta la scoperta del segnale di trasduzione aminoacidica, in comune con quello della sintesi proteica e la progressione del ciclo cellulare suggerisce l’ipotesi che l’autofagia sia un meccanismo ubiquitario nell’organismo che si contrappone alla crescita cellulare durante la carenza di nutrienti (van Sluijters et al., 2000).
In studi condotti da Mizushima e coll. sull’autofagia in vivo in condizioni di digiuno, è stato utilizzato, come proteina marker per l’autofagosoma, LC3, un omologo del Atg 8 di lievito presente nei mammiferi (Mizushima et al., 2004). Questi studi hanno messo in evidenza che l’autofagia è indotta in quasi tutti i tessuti, benché il modello di risposta al digiuno è differente tra i diversi organi (Mizushima et al., 2004).
È stato, infatti, visto che l’autofagia può essere fortemente indotta durante le 48 ore di digiuno come avviene nel cuore, oppure essere indotta nelle prime 24 ore di digiuno ma ritornare a livelli quasi basali durante le restanti 24 ore di digiuno come avviene per il fegato, oppure non essere indotta nelle 48 ore di digiuno come avviene per il cervello.
Questi diversi modelli di risposta al digiuno suggeriscono che la regolazione dell’autofagia non sia uniforme, ma organo-dipendente (Mizushima et al., 2004).
1.1.6 Selettività dell’autofagia
Benché l’autofagia sia considerata un processo non selettivo, è stato più volte messo in evidenza che può esistere una degradazione autofagica preferenziale per alcuni tipi di proteine. Sebbene non sia possibile attribuire un ruolo esclusivo all’autofagia nella proteolisi cellulare, è stato però evidenziato come il processo di autofagia chaperone-mediata degradi specificamente proteine con una sequenza KFERQ comune sia nei fibroblasti in coltura (Chiang and Dice, 1988) sia negli epatociti isolati (Wing et al., 1991; Cuervo et al., 1995).
Per quanto riguarda gli organuli un sequestro selettivo può essere osservato in diverse situazioni patologiche e in condizioni di stress, sebbene ciò sia tuttora motivo di dibattito (Dunn, 1994).
Alcuni studi hanno mostrato che l’autofagia è responsabile della diminuzione della quantità di reticolo endoplasmatico liscio in epatociti di ratto dopo aver indotto la sua proliferazione con fenobarbital (Bolender and Weibel, 1973). L’autofagia, inoltre, potrebbe essere stimolata selettivamente dal reticolo endoplasmatico dopo che in esso si accumulano proteine strutturalmente alterate (Davies and Murphy, 2002; Teckman and Perlmutter, 2000).
In qualche caso, nella selettività del processo, potrebbe essere importante il segnale responsabile dell’attivazione dell’autofagia: ad esempio il sequestro autofagico di ribosomi, che è indotto dall’assenza d’aminoacidi, è soppresso dall’aggiunta di glucagone, un attivatore del processo autofagico (Shelburne et al., 1973).
Il termine di pexofagia è stato introdotto per descrivere l’eliminazione selettiva di perossisomi in diversi tipi di lievito (Scott and Klionsky, 1998); tale processo è reso evidente durante il passaggio di cellule o lieviti in coltura da un ambiente ricco di acidi grassi, dove i perossisomi proliferano, ad un ambiente prevalentemente ricco di carboidrati, come il glucosio (Kim and Klionsky, 2000). Nelle cellule di mammifero, l’autofagia è responsabile del rapido turnover dei perossisomi alterati nei fibroblasti di pazienti con sindrome di Zellweger, una malattia perossisomiale (Heikoop et al., 1992). I perossisomi sono degradati anche in epatociti di ratto in seguito alla stimolazione dell’autofagia (Bergamini et al., 1987; Locci-Cubeddu et al., 1985).
Il sequestro selettivo dei mitocondri sembra essere fondamentale durante i primi stadi dell’apoptosi (Elmore et al., 2001), ma le basi molecolari di questo processo selettivo cominciano ad essere conosciute. Infatti i mitocondri con potenziale di membrana diminuito e attivi nella produzione di specie reattive dell’ossigeno (ROS), sembrano più sensibili al sequestro autofagico (de Grey, 1997; Lemasters et al., 1998). Una migliore conoscenza dei meccanismi responsabili del sequestro selettivo dei mitocondri danneggiati può essere importante allo scopo di studiare come le cellule mantengano il proprio pool di mitocondri attivi. Questo può dare alcune indicazioni sul ruolo dell’autofagia
un ruolo come ad esempio l’aumento dello stress ossidativo caratteristico dell’invecchiamento (Cuervo and Dice, 2000; Terman et al., 2003).
1.1.7 Autofagia e invecchiamento
Normalmente, macromolecole e organuli danneggiati sono soggetti ad un efficace ricambio da parte della cellula, tuttavia alcune strutture alterate rimangono e quindi lentamente e inesorabilmente può essere accumulato del materiale “di scarto”(Brunk and Terman, 2002; Terman, 2001). Dato che questo materiale inizia ad accumularsi nelle cellule nei primi stadi della vita e poi aumenta gradualmente, è verosimile credere che la rimozione imperfetta di strutture alterate sia una caratteristica propria delle cellule, non necessariamente acquisita con l’età. Durante l’invecchiamento, inoltre, i sistemi cellulari in grado di garantire il turnover delle strutture appaiono progressivamente compromessi (Terman and Brunk, 2004).
Risultati ottenuti nei nostri laboratori e confermati da numerosi studi, mostano che l’autofagia, uno dei processi degradativi maggiormente coinvolti nel ricambio di macromolecole, organuli e citomembrane, declina con l’aumentare dell’età (Donati et al., 2001).
Studi condotti in vitro con fegato perfuso e con cellule isolate dal fegato di ratti alimentati ad libitum o sotto condizioni di restrizione calorica (Ward, 2002; Donati et al., 2001; Cavallini et al., 2001), mostrano che la funzione dell’autofagia e della degradazione lisosomiale raggiungono un picco a sei mesi d’età (età giovane adulta) (Ward, 1988; Donati et al., 2001), per poi diminuire progressivamente con l’invecchiamento.
Allo stesso modo, studi di microscopia elettronica hanno messo in evidenza che la velocità di formazione ed eliminazione di vacuoli autofagici diminuisce nelle cellule epatiche con l’invecchiamento (Pollera et al, 1990; Bergamini and Kovacs, 1990; Terman, 1995).
È stato proposto che l’alimentazione libera, sopprimendo l’autofagia a causa dell’abbondanza di nutrienti, porterebbe ad un minor ricambio delle strutture cellulari con aumento del danno a carico dei mitocondri e delle citomembrane (Bergamini et al., 2004). Ci sono indicazioni che il declino età-dipendente dell’autofagia sia collegato ad un progressivo deterioramento delle membrane come mostrato dall’accumulo età-dipendente del dolicolo, un lipide di membrana (Marino et al., 1997). L’alterazione delle citomembrane è probabilmente responsabile dell’ulteriore declino delle funzioni autofagiche per effetto dell’alterazione della regolazione del processo stesso.
È noto che i mitocondri sono degradati grazie al processo d’autofagia garantendone il ricambio (Lemasters, 2005).
Il declino dipendente dell’autofagia potrebbe determinare un accumulo età-dipendente di mitocondri alterati che presentano un declino funzionale e una produzione eccessiva di ROS (Miquel et al., 1980). L’aumentata produzione dei ROS, oltre a danneggiare direttamente i mitocondri, in special modo il mtDNA, può avere effetti sul compartimento lisosomiale e indirettamente sull’autofagia (Lee and Wei, 2001). L’aumento dell’ossidazione di lipidi e proteine della membrana lisosomiale, infatti, può contribuire: (1) ad un aumento nella fragilità dei lisosomi, (2) ad una ridotta fusione dei lisosomi con i vacuoli autofagici durante l’autofagia, (3) una diminuita capacità del substrato proteico di legare
ed entrare nel lisosoma nel caso dell’attivazione della via autofagica chaperone-mediata (Cuervo and Dice, 2000).
La funzione lisosomiale può giocare un ruolo importante nel declino età-dipendente dell’autofagia. I lisosomi gradualmente accumulano un materiale polimerico non degradabile chiamato lipofuscine o pigmento dell’età, il quale probabilmente rappresenta un prodotto dell’attacco ossidativo a proteine e lipidi (Brunk and Terman, 2002). È stato ipotizzato che l’accumulo di lipofuscine possa fortemente diminuire la capacità degradativa lisosomiale impedendo il funzionamento degli enzimi lisosomiali.
Così come il declino dell’autofagia gioca un ruolo nel processo d’invecchiamento, il mantenimento della sua funzionalità potrebbe essere anche uno dei meccanismi anti-invecchiamento della restrizione calorica. È stato dimostrato che la restrizione calorica ha un effetto positivo sulla media e sulla massima lunghezza di vita in roditori e in diversi invertebrati e vertebrati (Masoro, 2002).Si ritiene che la restrizione calorica eserciti questo effetto sulla longevità rallentando il processo d’invecchiamento, sulla base di numerose e comprovate osservazioni sperimentali: (1) ritarda l’età di insorgenza e l’andamento di sviluppo di molte malattie associate all’età (Masoro, 1999; Masoro, 2002); (2) riduce il “rate metabolico” e lo stress ossidativo (Heilbronn and Ravussin, 2003); (3) diminuisce i livelli di zuccheri e d’insulina nel sangue (Masoro, 2002; Yamaza et al., 2002). È noto che la restrizione calorica, attraverso una prolungata condizione di carenza energetica, stimola l’autofagia e il turnover di membrane,organuli e proteine (Hagopian et al., 2003; Spindler, 2001).
L’autofagia così attivata potrebbe garantire un miglior mantenimento cellulare con l’avanzare dell’età e per questo essere il migliore candidato come mediatore dell’effetto anti-invecchiamento della restrizione calorica (Bergamini and Gori, 1995; Bergamini et al., 1998).
È stato, infatti, osservato che restrizioni caloriche di diversa intensità e durata prevengono il declino età-dipendente dell’autofagia in modo proporzionale al loro effetto sulla longevità (Cavallini et al., 2001).
1.2 Ruolo dei mitocondri nella patogenesi
dell’invecchiamento
1.2.1 Invecchiamento e stress ossidativo
Tutti gli organismi viventi, con l’avanzare dell’età, vanno incontro ad una progressiva diminuzione funzionale che si associa ad una ridotta capacità di rispondere agli stress ambientali e quindi ad un aumento della probabilità di andare incontro a malattie e a morte. Questi fenomeni sono le conseguenze di un processo biologico multifattoriale noto con il nome d’invecchiamento. L’insieme d’alterazioni, associate all’invecchiamento, identificabili a livello molecolare, cellulare, tissutale e fisiologico sono frutto di uno sbilanciamento tra le azioni di fattori endogeni ed esogeni, causa di danno, e le capacità di riparazione e di mantenimento proprie dei sistemi biologici. L’invecchiamento è accompagnato da cambiamenti e alterazioni biologiche che lo caratterizzano. Variazioni età –dipendenti sul piano molecolare sono state viste a carico del genoma cellulare e mitocondriale a causa dell’accumulo, progressivo nel tempo, di mutazioni provocate da danno ossidativo, da radiazioni ecc (Strehler, 1986). Con l’invecchiamento si hanno cambiamenti nel metabolismo delle proteine, si verifica infatti sia un rallentamento del loro ricambio (Van Remmen et al., 1995) sia un declino della loro sintesi (Makrides, 1983; Rattan and Clark, 1996). Si verifica, inoltre, l’accumulo di proteine abnormi recanti alterazioni post-traduzionali dovute a ossidazione,glicazione ecc (Rothstein, 1989).
In seguito a perossidazione lipidica è stato visto anche un accumulo di lipidi strutturali come colesterolo e dolicolo, che fanno ipotizzare un’alterazione della struttura e della funzione delle membrane cellulari (Marino et al., 1998).
Si sono osservate anche modificazioni a livello cellulare come la ridotta capacità delle normali cellule diploidi di dividersi in vitro e forse in vivo. Si riduce il numero delle replicazione in coltura (limite di Hayflick), e questo può influire sulla riparazione e sulla rigenerazione dei tessuti e sulla risposta immunitaria (Hayflick, 1980).
Diverse funzioni fisiologiche tra cui l’andamento metabolico basale, la capacità vitale, la velocità di conduzione neuronale, la capacità massima di respirazione e l’indice cardiaco subiscono un progressivo calo con l’avanzare dell’età (Masoro, 1999).
Si riduce, inoltre, la capacità di rispondere in maniera adattativa agli stimoli ambientali. Durante l’invecchiamento, l’organismo perde progressivamente la capacità di mantenere costante le condizioni chimico-fisiche interne anche al variare delle condizioni ambientali esterne. Si ha ad esempio, in un organismo anziano, una cattiva risposta sul piano adattativo alle escursioni termiche, all’esercizio fisico e alla malnutrizione (Masoro, 1999).
Aumenta la suscettibilità e vulnerabilità alle malattie. Ci sono molte malattie che sono più prevalenti nell’anziano o che avvengono solo ad età avanzata. Tali malattie sono chiamate malattie età-associate e comprendono patologie cardiache, neoplastiche ecc (Masoro, 1999).
Molte teorie che discutono le cause dell’invecchiamento sono basate sul concetto che il danno sia dovuto oltre che ai prodotti tossici in parte derivanti dal
metabolismo, alla progressiva inefficienza dei sistemi di difesa e riparazione (Masoro, 1999).
Nel 1956, Harman propose la Teoria dei radicali liberi, la cui versione più recente suggerisce che l’invecchiamento sia conseguenza del danno provocato da sostanze tossiche conosciute come ROS (Reactive Oxygen Species) (Harman, 1956; Harman, 1981). I ROS sarebbero prevalentemente il sottoprodotto del normale metabolismo ossidativo cellulare che per la maggior parte ha sede nei mitocondri (Finkel and Holbrook, 2000): si calcola che circa il 2% dell’ossigeno consumato a fine metabolico dia origine a radicali liberi che, successivamente, danneggiano lipidi, proteine e DNA (Harman, 1956, Harman, 1981). Molti siti cellulari sono sede di danno da parte delle specie reattive dell’ossigeno e dei radicali liberi, tuttavia tre reazioni risultano particolarmente importanti: (1) Perossidazione dei lipidi di membrana. Il danno ossidativo inizia, quando i doppi legami degli acidi grassi insaturi dei lipidi di membrana vengono attaccati dai ROS (Robbins, 1999). L’interazione ROS-lipidi porta alla formazione di perossidi, che a loro volta sono instabili e reattivi, e fanno sì che s’instauri una reazione a catena autocatalitica, portando in ultimo ad alterazioni nella funzione e nella struttura delle membrane stesse (Robbins, 1999). (2) Modificazioni ossidative delle proteine. I ROS promuovono l’ossidazione delle catene laterali dei residui aminoacidici, la formazione di legami crociati fra proteine e ossidazione della catena proteica (Robbins, 1999). Una delle maggiori modificazioni, causata dallo stress ossidativo, è l’introduzione di gruppi carbonilici nella catena proteica, che causa la perdita della funzione catalitica (Levine and Stadtman, 2001). (3) Lesioni al DNA (Robbins, 1999). La struttura che risente maggiormente dello stress ossidativo è il mtDNA. Adachi e coll.
dimostrarono che i ROS causavano un gran numero di delezioni nel mtDNA (Adachi et al., 1993). Oltre alle delezioni un altro tipo di danno al DNA è la modificazione delle basi. Una delle modificazioni più studiate è l’ossidazione della 2-desossiguanosina (2 dG) a 8-idrossi-2 desossiguanosina (8-OHdG). Numerosi studi riportano un accumulo età-dipendente di questa base alterata (Fraga et al., 1990).
In risposta all’azione dannosa dei ROS le cellule hanno sviluppato numerosi meccanismi. Esistono diversi sistemi enzimatici e non, che contribuiscono all’inattivazione delle reazioni da radicali liberi (Robbins, 1999). Di questi fanno parte: (1) antiossidanti che bloccano la formazione dei radicali liberi dall’inizio o li inattivano, come per esempio le vitamine E e A, l’acido ascorbico e il glutatione (Robbins, 1999); (2) diversi enzimi, come la superossido dismutasi dipendente da Mn2+ (MnSOD), la glutatione perossidasi (GPx), la glutatione reduttasi (GR) e la catalasi (CAT), agiscono come sistemi di disinnesco dei radicali liberi inattivando i ROS. MnSOD converte l’anione superossido a perossido d’idrogeno, il quale a sua volta è trasformato in acqua dalla GPx o dalla CAT (Robbins, 1999).
Oltre a tali sistemi atti a neutralizzare i radicali liberi e ROS, la cellula ha sviluppato dei sistemi degradativi che costantemente rimuovono gli organuli e le macromolecole danneggiate.
Il proteasoma è un complesso multicatalitico che può essere considerato come un piccolo organulo. Esso è in grado di degradare una grande varietà di proteine danneggiate sia citosoliche che nucleari, dopo che queste si sono legate con l’ubiquitina (Myung et al., 2001; Wojcik and DeMartino, 2003).
I mitocondri possiedono, invece, dei propri sistemi proteolitici, come proteasi Lon (Bakala et al., 2003) e membrane-embedded AAA proteases (Arnold and Langer, 2002). Per rimuovere il danno ossidativo al DNA, inoltre, la cellula utilizza sistemi di riparazione tra cui la base excision repair (BER) e la nucleotide excision repair (NER).Il primo è presente nel nucleo e nei mitocondri, il secondo solo nel nucleo.
In risposta allo stress ossidativo la cellula può proteggersi usando l’autofagia che è in grado di rimuovere più o meno selettivamente macromolecole, organuli e citomembrane danneggiate.
1.2.2 Teoria mitocondriale dello stress ossidativo
Utilizzando come base comune la Teoria dei radicali liberi il mondo scientifico propone diverse ipotesi sulle cause che determinano il processo d’invecchiamento. Una di queste ipotesi argomenta che i mitocondri sono il principale bersaglio dell’attacco dei radicali liberi e che mutazioni nel DNA mitocondriale (mtDNA) accelerano il danno da radicali liberi mediante l’introduzione di componenti enzimatiche alterate nella catena di trasporto di elettroni (Miquel, 1980; Mandavilli et al., 2002). I radicali liberi possono essere generati in diversi siti cellulari, ma nei tessuti sani la principale risorsa di radicali liberi è la catena respiratoria mitocondriale (Barja, 2004). Durante questo processo è prodotta energia sotto forma d’ATP grazie ad un gradiente di protoni che porta alla riduzione finale di una molecola d’O2 a H2O. Il trasferimento di 4
elettroni porta alla formazione di un prodotto non tossico, mentre la riduzione parziale genera composti molto pericolosi per la vita della cellula, tra cui l’anione superossido (O2-.) e il perossido d’idrogeno (H2O2) (Stryer, 1996).
Questi ROS sono tossici per gli enzimi mitocondriale e possono portare anche alla formazione di una molecola più reattiva, il radicale idrossile (OH°), attraverso l’interazione con ioni metallici e la reazione di Fenton (Van Remmen et al., 2003). È stato visto che i siti di maggior interesse per la formazione di ROS all’interno della catena di trasporto di elettroni sono il complesso I (NADH-Q reduttasi) e il complesso III (citocromo reduttasi) (Wei et al., 1981).
Il danno ossidativo al DNA è il più importante per l’invecchiamento (Barja, 2004), poiché esso può portare alla perdita o all’alterazione irreversibile delle informazioni genetiche delle cellule. Per la sua vicinanza al sito di formazione dei ROS e per la mancanza di introni, il mtDNA è ritenuto il principale bersaglio del danno ossidativo (Alexeyev et al., 2004; Richter, 1995). Il tipo di danno ossidativo più conosciuto è 8OHdG. 8OHdG si forma per l’attacco al carbonio8 dell’anello purinico di un radicale idrossile che va a rimpiazzare un atomo d’idrogeno (Nakae et al., 1995). In accordo con quanto detto precedentemente, è stato visto che i livelli dell’8OHdG sono più alti nel mtDNA che nel nDNA sia nel cuore che nel cervello di molte specie di mammifero (Mecocci et al., 1993; Sohal et al.,1994; Richter et al., 1988; Chung et al., 1992). La concentrazione normale dell’8OHdG è più alta nelle specie che invecchiano rapidamente rispetto a quelle che invecchiano lentamente. Infine i livelli di 8OHdG nel mtDNA sono inversamente correlati con la massima lunghezza di vita delle specie, contrariamente a quello che avviene per il nDNA (Barja and Herrero, 2000).
Le mutazioni ossidative nel mtDNA si accumulano progressivamente con l’invecchiamento e raggiungono alti livelli negli organismi anziani, specialmente
a livello delle regioni di controllo responsabili della replicazione e trascrizione del mtDNA (Kraytsberg et al., 2003; Chomyn and Attardi, 2003).
Il danno ossidativo al mtDNA potrebbe essere conseguenza del grado d’insaturazione delle membrane mitocondriali come mostrano recenti studi (Pamplona et al., 2002). Questo è dovuto al fatto che un incremento dell’insaturazione, e quindi dei doppi legami, porta a un aumento della suscettibilità alla perossidazione lipidica con la produzione di sostanze, come la malondialdeide (MDA), che causano danno al DNA (Marnett, 2002).
Malgrado il mtDNA delle cellule animali contenga solo 1-3% del materiale genetico, diverse osservazioni suggeriscono che il suo contributo nella fisiologia cellulare può essere molto più grande.
È infatti noto che: (1) mtDNA muta a un andamento più elevato rispetto al nDNA e ciò può essere una conseguenza della sua vicinanza alla catena di trasporto degli elettroni (ETC); (2) mtDNA codifica per polipeptidi di ETC o componenti richieste per la loro sintesi, quindi, mutazioni nel mtDNA possono influenzare il ETC; (3) difetti nel ETC possono danneggiare la respirazione mitocondriale. Tutto questo ha portato ad ipotizzare che mutazioni al mtDNA, accumulate progressivamente durante la vita, siano direttamente responsabili del declino dell’attività della fosforilazione ossidativa cellulare, conducendo all’aumento della produzione di ROS (Wei, 1998).Questo aumento della produzione di ROS può comportare un ulteriore aumento del danno al mtDNA, causando, in questo modo, un “circolo vizioso”, che in ultimo può portare alla morte (Alexeyev et al., 2004) (fig. 2).
1.3 Autofagia e mitocondri
1.3.1 Mitofagia
1.3.1.1 Ruolo dell’autofagia nel ricambio dei mitocondri
Il danno ossidativo del mtDNA può causare, come precedentemente detto, un ulteriore intensificazione dello stress ossidativo durante l’invecchiamento (Wei, 1998). La degradazione autofagica dei mitocondri alterati può rappresentare un importante meccanismo usato dalla cellula per eliminare mtDNA danneggiato, dato che la riparazione del DNA nei mitocondri è meno efficace rispetto a quella del nucleo (Bohr, 2002). Numerosi studi hanno evidenziato il ruolo dell’autofagia nella degradazione dei mitocondri: studi morfologici hanno mostrato, innanzi tutto, che i mitocondri si possono trovare nei vacuoli autofagici (Klionsky and Emr, 2000). Altri studi condotti su lievito dimostrano che mutanti con deficit autofagico presentano un fenotipo mitocondriale alterato, con un ridotto consumo d’ossigeno (Jin, 2006). Ulteriori studi genetici su mammiferi, inoltre, rivelano che tessuti di fegato di ratto, con delezioni di geni coinvolti nell’autofagia (atg7) accumulano mitocondri con morfologia anormale (Komatsu et al., 2005). Studi sul lievito hanno mostrato che i mitocondri sono identificati e sequestrati come tali dalla funzione autofagica: ceppi mutanti per la proteina della membrana mitocondriale esterna Uth1p hanno una normale attività autofagica che però non è in grado di degradare i mitocondri (Kissova et al., 2004). Questa forma di autofagia, per mezzo della quale i mitocondri sono degradati è conosciuta come mitofagia.
1.3.1.2 Selettività della mitofagia
Una questione aperta è se la degradazione autofagica dei mitocondri sia un processo selettivo, il quale etichetta e rimuove selettivamente i mitocondri danneggiati, o un processo non-selettivo.
È stato visto che linee cellulari tumorali sia con mtDNA normale sia con delezioni del mtDNA presentano un basso livello di autofagia mitocondriale in presenza di nutrienti. Tuttavia la riduzione di nutrienti provoca un aumento dell’autofagia nelle cellule con delezioni al mtDNA ma non in quelle con mtDNA normale (Gu et al., 2004).
L’incremento della mitofagia nelle linee cellulari con delezione nel mtDNA è correlata, anche, con una marcata riduzione nella produzione di ROS (Gu et al., 2004).
Ulteriori studi svolti utilizzando queste linee cellulari con mtDNA normale, incubate con H2O2, hanno messo in evidenza che a basse concentrazioni di
nutrienti essi vanno incontro a un aumento della mitofagia (Gu et al., 2004). Tutto questo ha fatto ipotizzare che l’autofagia possa essere capace di riconoscere e eliminare i mitocondri non funzionali che presentano un danno al mtDNA.
In questo senso, la compromissione dell’autofagia, che sappiamo avvenire progressivamente durante l’invecchiamento, potrebbe essere una causa dell’accumulo di mitocondri alterati (Jin, 2006).
Studi eseguiti da Barja e coll. su ratti, inoltre, hanno messo in evidenza che la restrizione calorica, un trattamento anti-invecchiamento che mantiene alti i livelli di autofagia, provoca una diminuzione del danno ossidativo nel mtDNA
(Lopez-descritti nel cuore (Gredilla et al., 2001b) e anche nel fegato di ratti a restrizione calorica (Kang et al., 1998; Gredilla et al., 2001a).
Al contrario, studi eseguiti da Sanz et al. sul fegato di ratto trattato con insulina, un inibitore del processo autofagico, hanno mostrato che l’ormone determina un aumento del danno ossidativo al mtDNA (Sanz et al., 2005).
Questi risultati sono compatibili con l’ipotesi che meccanismi che inibiscono o stimolano l’autofagia possano avere un influenza sulla rimozione di mitocondri con un elevato grado di danno ossidativo al mtDNA.
1.3.1.3 Transizione di permeabilità di membrana (MPT): induttore della mitofagia
Se davvero la mitofagia avviene selezionando mitocondri danneggiati rimane ancora da comprendere in quale modo tale danno rappresenti un segnale per l’attivazione del processo. Basandosi su osservazioni riguardanti colture cellulari di mammifero, Lemasters propose che il processo mitofagico fosse altamente selettivo e l’evento scatenante la mitofagia fosse l’inizio della transizione di permeabilità di membrana (MPT) (Lemasters, 2005; Lemasters et al., 1998) (fig. 3).
La transizione di permeabilità di membrana è un processo che provoca un improvviso aumento della permeabilità della membrana interna mitocondriale a tutte le molecole di dimensioni minori di 1.5 kDa (Rodriguez-Enriquez et al., 2004). Il rapido cambiamento di permeabilità associato con MPT causa rigonfiamento mitocondriale, depolarizzazione di membrana e danneggiamento della fosforilazione ossidativa. Questo ultimo fenomeno conduce all’inibizione della produzione di ATP e conseguente abbassamento dei livelli di ATP che
culmina nel mal funzionamento della membrana plasmatici e conseguente rilascio di enzimi intracellulari (Bernardi et al., 1999; Lemasters et al., 1998). Da un punto di vista funzionale, la MPT è quindi indice di un danno mitocondriale che di solito culmina con l’apoptosi. Secondo un recente modello, quando pochi mitocondri vanno incontro a MPT, viene attivata la mitofagia che consente di eliminare i mitocondri danneggiati; quando molti mitocondri sono coinvolti invece la MPT promuove l’apoptosi (Rodriguez-Enriquez et al., 2004). A sostegno di tale ipotesi, grazie a studi di microscopia confocale che hanno reso possibile visualizzare la depolarizzazione mitocondriale, è stato mostrato che almeno i due terzi dei mitocondri che vanno incontro a depolarizzazione spontanea entrano nella via di degradazione auto-lisosomiale (Elmore et al., 2004). In aggiunta l’inibizione della MPT con ciclosporina A blocca la depolarizzazione mitocondriale e la proliferazione del compartimento autofagico (Rodriguez-Enriquez and Lemasters, 2003).
Per quanto riguarda gli eventi che conducono alla MPT, sembra che siano coinvolti diversi fattori (Bernardi, 1996; Zoratti and Szabo, 1995). Fattori come Ca2+, fosfato inorganico, acidi grassi liberi, numerosi ossidanti chimici (ROS), ossidazione del glutatione e di pirimidine e la depolarizzazione del potenziale di membrana mitocondriale promuovono un aumento della permeabilità. Al contrario il pH della matrice al di sotto di 7, Mg2+, ADP e potenziale mitocondriale negativo ritardano l’inizio di MPT.
Il processo di MPT inizia con l’apertura di pori di transizione di permeabilità (PT) nella membrana interna mitocondriale, ma la struttura molecolare dei pori PT rimane sconosciuta.
Molti modelli sono stati proposti per spiegarne la struttura, ma due sono di principale interesse: (1) secondo il modello più conosciuto i pori PT sono visti come un complesso strutturale composto da diverse molecole associate tra loro (Crompton, 1999; Marzo et al., 1998; Zoratti and Szabo, 1995). (2) secondo il nuovo modello la formazione di pori è dovuta da proteine della membrana mitocondriale mal ripiegate (He and Lemasters, 2002).
In accordo con questo modello è possibile che il danno ossidativo che si accumula all’interno del mitocondrio e quindi anche a livello proteico, conduca ad un’alterazione tridimensionale delle proteine, causando infine la MPT (Rodriguez-Enriquez et al., 2004).
FIG 3. SCHEMA DEL RUOLO DELLA TRANSIZIONE DI PERMEABILITÀ DI MEMBRANA NELL’AUTOFAGIA E NELLA MORTE CELLULARE.
1.4 Scopo della ricerca
È stato visto che con l’aumento dell’età si ha un accumulo di mitocondri danneggiati che potrebbero aumentare lo stress ossidativo durante l’invecchiamento. In aggiunta a questo è noto che l’autofagia, un processo cellulare che svolge un ruolo nel ricambio di macromolecole, organuli e membrane, diminuisce con l’avanzare dell’età.
Lo scopo di questa ricerca è stato, quindi, valutare il ruolo dell’autofagia nella rimozione dei mitocondri che mostravano un danno ossidativo al mtDNA.
A tale scopo abbiamo cercato di intensificare l’autofagia nell’animale adulto quando sia il processo autofagico che i mitocondri hanno subito un declino funzionale: a tale fine abbiamo quindi validato l’uso di un farmaco che bloccando la lipolisi durante il digiuno costringesse il metabolismo a sfruttare più intensamente la proteolisi autofagica.
Con un approccio inverso al precedente abbiamo, poi, valutato in animali giovani, dove la funzionalità mitocondriale e autofagica sono ottimali, l’effetto di una prolungata inibizione dell’autofagia tramite la somministrazione di insulina a lento rilascio.
2. Materiali e metodi
2.1 Valutazione degli effetti della somministrazione di un
farmaco antilipolitico
2.1.1 Animali
Sono stati usati ratti maschi di ceppo Sprague-Dawley alimentati ad libitum. All’età di 3 mesi, ad un gruppo di 5 ratti tenuto a digiuno per 18 ore è stato iniettato intraperitonealmente DMP (12 mg/Kg p.c. in fisiologica).
Gli animali sono stati sacrificati sotto anestesia con nembutal (50 mg/Kg p.c. intraperitonealmente) 15, 30, 60, 90 e 120 minuti dopo l’iniezione di DMP.
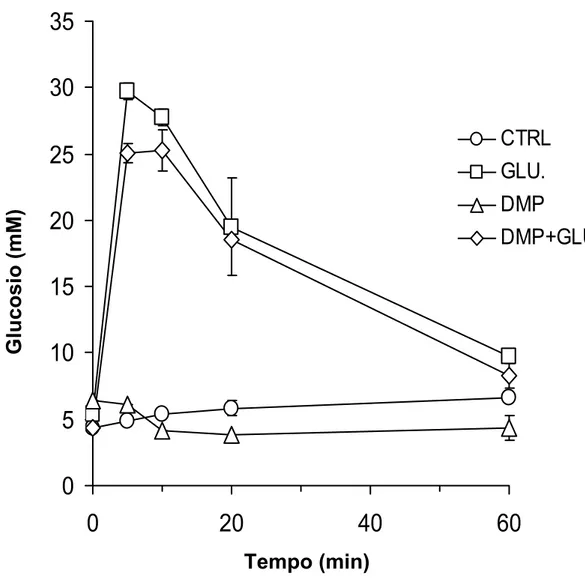
Ad un altro gruppo d’animali giovani tenuti a digiuno per 18 ore, è stato iniettato DMP e 42, 57 e 87 minuti dopo l’iniezione di DMP è stato dato loro glucosio in fisiologica intraperitonealmente (2 g/Kg p.c.). Questo gruppo di animali è stato sacrificato 2 ore dopo la somministrazione di DMP.
2.1.2 Dosaggio dei livelli plasmatici di glucosio e insulina
Ai tempi indicati, sono stati prelevati campioni di sangue periferico e su questi sono stati saggiati i livelli di glucosio, usando un kit disponibile in commercio (Glucosio: Glucinet Sclavo ISVT), che utilizza l’enzima glucosio ossidasi insieme ad una perossidasi.
L’insulina plasmatica è stata misurata grazie a dosaggio RIA (Radio Immuno Assay), usando insulina di ratto come standard.
2.1.3 Dosaggio dei livelli di valina e altri aminoacidi
I livelli plasmatici di valina e altri aminoacidi sono stati saggiati nel sangue periferico e di fegato al fine di valutare l’effetto stimolatore del farmaco antilipolitico sull’andamento della proteolisi autofagica del fegato.
Dopo aver separato per centrifugazione il plasma dal sangue intero, i campioni sono stati diluiti 1:10 con fisiologica e ad essi è stato aggiunto acido perclorico (PCA) a concentrazione finale 6%. Sono stati poi messi in ghiaccio per 30 min e centrifugati per 10 min ed il sovranatante è stato prelevato e utilizzato per la derivatizzazione degli aminoacidi.
2.1.3.1 Derivatizzazione
I campioni, dopo l’aggiunta di norvalina, utilizzata come standard interno dei campioni, sono portati a pH alcalino con KOH 3 M e Na2CO3 0.5 M pH 9.5 e
lasciati in ghiaccio per circa 30 min.
Il pH viene poi portato, grazie all’utilizzo di PCA 6%, PCA 12% e KOH 3 M, ad un valore compreso tra 9.68 e 9.80. Dopo centrifugazione, i sovranatanti sono stati fatti reagire con l’agente derivatizzante, il dansil cloruro e dopo 40 min con metilammina 2%. Dopo centrifugazione i sovranatanti vengono filtrati (filtro da 0.46 μm) e letti per mezzo dell’HPLC.
2.1.3.2 Procedura cromatografica
I dansil-derivati degli aminoacidi sono stati separati con una colonna a fase inversa Ultrasphere OSD Beckman (lunghezza 25 cm, diametro interno 4,6 mm, dimensione delle particelle 5Å) e rilevati tramite fluorimetria (λ eccitazione 340nm; λ emissione 525 nm). I solventi di eluizione (A e B) sono stati miscelati
secondo un gradiente lineare che porta la percentuale del tampone A dal 60% al 50% in 10 minuti. Il tampone A è costituito dal 12% da acetonitrile, 0,3% di acido acetico, 0,035% di trietilammina in H2O milliQ (millipore) e il tampone B da
metanolo 100%.
2.1.4 Procedura di microscopia elettronica
Le immagini di microscopia elettronica sono state ottenute e gentilmente fornite dal laboratorio di microscopia elettronica nella persona della Dott.ssa Matilde Masini.
2.2 Valutazione della stimolazione e dell’inibizione
dell’autofagia sul danno ossidativo del mtDNA
2.2.1 Animali e trattamenti
Sono stati utilizzati ratti maschi di ceppo Sprague-Dawley di 3 e 16 mesi di età alimentati ad libitum.
Per intensificare il processo di autofagia gli animali adulti sono stati messi a digiuno e dopo 18 ore è stato loro somministrato l’antilipolitico 3,5-dimetilpirazolo (12 mg/Kg p.c.), o fisiologica per i controlli, per due volte a distanza di 3 ore.
Per inibire in maniera prolungata l’autofagia, gli animali giovani sono stati sottoposti ad un periodo di 20 ore di alimentazione libera all’inizio del quale veniva somministrata insulina a lento rilascio (Insulina Glargine, 5 U) che veniva ripetuta dopo 10 ore. Gli animali di controllo sono stati sottoposti ad un periodo di digiuno di 20 ore.
2.2.2 Dosaggio delle proteine
Il dosaggio delle proteine presenti nel fegato è stato fatto dopo omogenizzazione del tessuto con il Bio-Rad protein assay che si basa sul metodo di Bradford (1976).
2.2.3 Isolamento del mtDNA
Per isolare il DNA mitocondriale è stato usato il metodo di Balansky e coll. (1995).
Il fegato è stato prelevato sotto anestesia con Nembutal (50mg/Kg p.c.) omogenizzato in Potter-Elvehjem e sottoposto ad una serie di centrifughe in saccarosio 0.25 M, EDTA 10 mM e TRIS-HCl pH 7.2-7.4 10mM, al fine di precipitare selettivamente dapprima i nuclei e frammenti cellulari e successivamente i mitocondri e altri organuli.
Il pellet mitocondriale risospeso in tampone TRIS 25 mM, glucosio 50 mM, EDTA 10 mM, pH 8.0 è stato trattato con una soluzione contenente SDS 1% e NaOH 0.2 M, e dopo 3 minuti neutralizzato con una soluzione acido acetico/acetato di sodio pH 4.8, lasciato per 35 min a 0° C e poi centrifugato a 12000 x g per 20 min a 4°C. Durante questa fase si ha la precipitazione selettiva del DNA nucleare e delle proteine. Al sovranatante recuperato viene aggiunto un ugual volume di isopropanolo, lasciato sedimentare a temperatura ambiente per 30 min e poi centrifugato a 14000 x g per 30 min a 15°C. Il pellet contenente il mtDNA è stato portato a secco in azoto, lavato con poche gocce di etanolo ice-cold 70% e riportato nuovamente a secco.
Il pellet viene risospeso in una soluzione tampone TRIS 25 mM, EDTA 10 mM, pH 8.0 e successivamente viene aggiunto un egual volume di LiCl 11 M, che consente un ulteriore precipitazione di DNA nucleare e di proteine.
Dopo 20 min a temperatura ambiente, la soluzione è stata centrifugata a 12000 x g per 20 min a 4°C e al sovranatante recuperato sono stati aggiunti due volumi di etanolo ice-cold 100%: dopo 30 min a -20°C la soluzione è stata centrifugata di nuovo a 12000 x g per 20 min a 4°C e il sovranatante è stato eliminato.
mM, pH 8.0. Il campione è stato congelato all’interno di Eppendorf e conservato a –80°C.
2.2.4 Dissociazione del mtDNA in singole basi
Alla soluzione contenente il mtDNA sono stati aggiunti due volumi di etanolo freddo: dopo un’incubazione a -20°C per 30 min, si è centrifugato per 10 min in microfuge e il pellet è stato portato a secco in azoto.
Il mtDNA (dai 15 ai 25 μg) viene risospeso in un tampone sodio acetato 20mM pH 4.8: ad esso viene aggiunta dapprima la nucleasi P1 (5 U in 20 μl di acetato di sodio 20 mM, cloruro di zinco 10 mM, glicerolo 15 %, pH 4.8) che è lasciata reagire 40 min a 37°C e dopo la fosfatasi alcalina (1 U in 20 μl di Tris-HCl 1 M, pH 7.0) per 1h a 37°C. A questo punto il campione viene filtrato in microfuge per mezzo di eppendorf con filtri a 10 kDa.
2.2.5 Procedura cromatografica
Il campione preparato con questa procedura è poi analizzato grazie all’utilizzo di un sistema HPLC con rivelatori spettrofotometrico ed elettrochimico in serie che permettono la determinazione rispettivamente della 2-desossiguanosina e della 8-idrossi-2-desossiguanosina. Viene utilizzata una corsa isocratica di 40 min a 1ml/min in tampone fosfato 45 mM, 2,5% acetonitrile, pH 5.0. Lo spettrofotometro legge ad una lunghezza d’onda di 254nm e il rivelatore elettrochimico ha i potenziali E1=0 mV, E2=200 mV e una sensibilità di 10nA.
2.2.6 Dosaggio dell’attività della citocromo C ossidasi
Per il dosaggio dell’attività della citocromo C ossidasi è stato utilizzato il Cytochrome C Oxidase Assay Kit fornito dalla SIGMA che utilizza la diminuzione dell’assorbanza del citocromo C a 550 nM quando passa dallo stato ridotto a quello ossidato per effetto dell’enzima.
2.3 Analisi statistica
Negli esperimenti per il modello d’induzione con DMP per valutare delle differenze tra le condizioni multiple è stato utilizzato il test ANOVA (One- or two-way analysis of variance). Per i confronti multipli dei trattati è stato utilizzato il test di Tukey- Kramer.
Negli esperimenti d’isolamento del mtDNA per l’analisi statistica dei risultati è stato utilizzato il test di Student.
Sono considerati significativi i valori di p<0.05 e altamente significativi quelli di p<0.01.
3. Risultati
3.1 Effetto della somministrazione del farmaco antilipolitico
3,5-dimetilpirazolo (DMP) sui livelli plasmatici di glucosio,
insulina e valina in animali giovani (3 mesi d’età) sottoposti
a digiuno di 18 ore.
Studi precedenti hanno mostrato che la somministrazione di DMP ad animali digiuni provoca un repentino e drastico abbassamento dei livelli plasmatici di acidi grassi liberi (FFA, -80%) che dura circa 2 ore prima di ritornare lentamente ai valori iniziali (figura 1): l’effetto primario del farmaco è infatti l’inibizione della lipolisi nel tessuto adiposo mediante azione sulla lipasi.
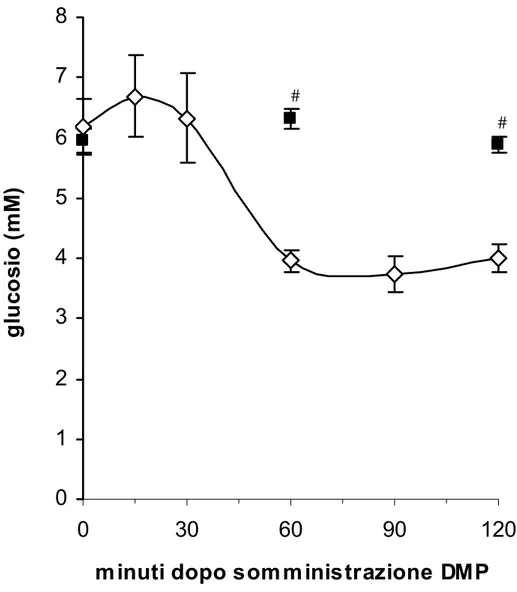
Nelle figure 2, 3 e 4 sono mostrati gli effetti della somministrazione del farmaco antilipolitico 3,5-dimetilpirazolo (DMP, 12 mg/Kg p.c.) sui livelli plasmatici di glucosio, insulina e valina in animali giovani (3 mesi d’età) sottoposti a digiuno di 18 ore prima della somministrazione.
In ogni grafico sono stati indicati anche i valori di glucosio, insulina e valina dei ratti di controllo ai quali veniva somministrata fisiologica.
Sull’asse delle ascisse sono indicati i minuti dalla somministrazione di DMP. Sull’asse delle ordinate sono riportati i valori delle concentrazioni di glucosio, insulina e valina espressi in mM, pM e μM rispettivamente.
Nelle figura 2 si osserva che il DMP determina una diminuzione dei livelli plasmatici di glucosio nelle 2 ore successive alla somministrazione. La glicemia
cala dopo 30 minuti dalla somministrazione del farmaco e rimane fino a 2 ore su dei livelli inferiori di circa il 35% rispetto ai controlli.
Nella figura 3 si osserva che in seguito a somministrazione di DMP avviene anche un calo dei livelli plasmatici di insulina. Osservando tale diminuzione che sembra iniziare precocemente, va specificato che i valori di insulina sono significativamente inferiori a quelli dei controlli solo dopo i 30 minuti dalla somministrazione del farmaco e che tali livelli permangono fino ai 120 minuti. Nella figura 4 sono riportati i livelli di valina che rappresentano una stima delle variazioni del processo di proteolisi autofagica. I valori plasmatici di valina sono stati saggiati sia su campioni di sangue provenienti dal fegato, dove l’autofagia è più attiva rispetto ad altri organi, sia in quelli di sangue periferico. Si osserva che in seguito alla somministrazione di DMP si ha un aumento dei valori di valina sia a livello epatico che a quello periferico e che tale aumento è già visibile a livello epatico dopo 1 ora dal trattamento mentre nel sangue periferico diviene evidente dopo altri 30 minuti. In entrambi i casi i valori della valinemia continuano ad aumentare fino ad arrivare ad un incremento di circa il 60% rispetto ai valori iniziali.