Regio- and stereoselective behaviour of
carbapyranose 1,2-epoxides
6. INTRODUCTION
6.1 Importance of carbapyranose 1.2-epoxides for the synthesis
of pseudosugars
Carbapyranose 1,2-epoxides have been extensively studied from Ogawa, as useful “glycosyl-donor mimics”, in that epoxide opening can be used to form ether-linked pseudosaccharides or pseudodisaccharides.[1-3] Carbasugar pseudooligosaccharides are important structures, because some of them have been found to have an interesting biological activity, in particular glucosidase inhibitory activity, but they may also act as substrates for glycosyltransferases or bind to lectins.
The bringing bond between the carba-pyranose unit and the O-functionality of the pseudosugar may be disconnected retrosynthetically to give a carbasugar C(1) electrophile and a carbohydrate (or a generic) O-nucleophile (Figure 2.1).[1]
The most used carbasugars C(1) electrophiles are carbapyranose 1,2-epoxides.[4] The epoxides are opened by amines in uncatalysed reactions, or by alcohols under Lewis acidic, but more usually basic, conditions. An important general feature of epoxide opening in 6-membered rings is the tendency for trans-diaxial ring opening.[5] The existing substituents on the cyclohexane ring favour one of the two possible half-chair conformations (4H
5). The principle of microscopic reversibility requires an antiperiplanar relationship between the nucleophile and the epoxide oxygen in the product immediately
Figure 2.1 RO RO OR OR O O OR OR RO RO carbohydrate nucleophile carbasugar electrophile
after reaction.
Carbapyranose 1,2-epoxides with the β-manno configuration 2.1β are opened efficiently with excellent regioselectivity by O- and N-nucleophiles; these epoxides are subjected by attack at C(1), that results in trans-diaxial opening as favoured by the Fürst-Plattner guidelines. Thus, epoxides ring-opening reaction on these systems gives 1,2-trans-diaxial carba-α-mannose derivatives and it is stereoelectronically favourable: indeed, C(1) is flanked by an electron-rich methylene group whereas C(2) is flanked by a carbon bearing an electron-withdrawing group [the C(3) benzyl ether], so that, from an electronic point of view, SN2 reaction should be more favourable at C(1) than at C(2). Besides, the group flanking C(1) are smaller than those flanking C(2), so attack at C(1) may be favoured also from a steric point of view (Scheme 2.1).
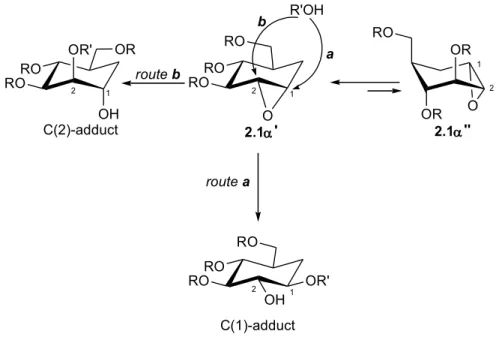
On the contrary, carbapyranose 1,2-epoxides with α-gluco configuration 2.1α do not give such good results: the opening process, extremely more difficult, is not regioselective and occurs only with some N-nucleophiles as azido ion and amines. The generally slower reactions and lower yields in this series can be put down to the conflict explained below: actually, in 2.1α the attack at C(1) carbon, that would be sterically and electronically favoured [both due to C(1) being flanked by the small and relatively electron-rich methylene group], leads to an unfavourable trans-diequatorial ring-opening (route a), whereas the attack at C(2) oxirane carbon, that would lead to a trans-diaxial opening (route b) is sterically hindered and electronically unfavourable (Scheme 2.2).
Scheme 2.1: Sterically and electronically favoured
nucleophilic attack to epoxide 2.1β.
RO RO RO O 1 2 RO RO RO 2 1 OH Nu Nu 2.1 -pseudomanno glycoside
As a result, epoxide 2.1α, which could reasonably be an effective β-carbaglucosyl donor for the synthesis of glycoconjugates bearing a β-carbaglucosidic bond, has been so far practically and unfortunately (2.1α is often the main product by oxidation of the corresponding olefin) not useful for this purpose.
Despite of this, carbapyranose 1,2-epoxides with α-gluco configuration, such as 2.2, theoretically turn out to be very useful for the synthesis of pseudosaccharides and pseudodisaccharides, such as 2.3, with pseudo-gluco configuration on carbapyranose unit (Figure 2.2).
In consideration of this, in the second part of my thesis, I have synthesized different
Scheme 2.2: The two regioisomeric routes
for nucleophilic attack to epoxide 2.1α.
Figure 2.2: Carbapyranose 1,2-epoxide precursor of
pseudosaccharide 2.3. HO OH OH ONu HO HO OH HO O 2.2 2.3 OR OH OR' RO RO RORO O RO 1 2 RO OR OR O 2 1 RO RO RO 2 1 OR' OH route a route b R'OH b a C(2)-adduct 2.1' 2.1'' C(1)-adduct 2 1
carbapyranose 1,2-epoxides, and I have studied the regio- and stereochemical behaviour of these substrates, under different reaction conditions and with different nucleophiles in order to direct and to make the ring opening process synthetically useful.
7. RESULTS AND DISCUSSION
We started our studies with the synthesis of 1,2 carbapyranoses 2.14α and 2.14β in order to better investigate nucleophilic addition reactions to these systems.
Among the various approaches available in the literature for their construction, we chose the one that allows the transformation of the glycal system 2.4 into a carbaglycal system 2.5, as shown in Figure 2.3.
In this transformation the key step is the glycal's Claisen thermal rearrangment of Nagarajan and Sudha,[6] which is particularly advantageous because the enantiomeric purity of the target carbasugar system is guaranteed from the starting glycal system.
The synthesis of the key intermediate olefin 2.12 has been achieved as reported in Scheme 2.3.
Figure 2.3: Conversion of the glycal system 2.4 into the carbaglycal system 2.5.
O RO OR HO 2.4 RO OR HO 2.5
Saponification of the tri-O-acetyl-D-glucal (+) 1.6 with MeONa/MeOH gave D-glucal
1.7, which was then protected at the primary hydroxy functionality with the sterically hindered TBSCl, in a 1:12 mixture of DMF/THF to give the -O-TBS derivative 2.6.[6,7] The -OBn protection of the remaining secondary hydroxy groups was obtained by reaction of trans diol 2.6 with NaH/BnBr/tetrabutylammonium iodide (TBAI) protocol in THF at 0 °C with the formation of the all-protected glycal 2.7, without observing any migration of the silyl group. Deprotection (TBAF/THF) of the primary -O-TBS led to primary alcohol 2.8 which was oxidized to aldehyde 2.9 by freshly prepared 2-iodoxy benzoic acid (IBX),[8] in anhydrous ethyl acetate at 80 °C for 3 h. Aldehyde 2.9 is not stable and should be used immediately or stored at low temperature (-78 °C) before subsequent treatment. Aldehyde 2.9 was transformed into allyl vinyl ether 2.10 by means of Wittig reaction, using the phosphorus ylide obtained in situ by reaction between methyl-triphenyl phosphonium iodide (MeI-Ph
3P+) and potassium hexamethyldisilazide (KHMDS) in THF.
Scheme 2.3: Synthesis of carbacyclic system 2.12.
O OAc AcO O OH HO MeONa MeOH AcO HO TBSCl Imidazole THF/DMF O OH HO TBSO BnBr NaH O OBn BnO TBSO TBAF THF O OBn BnO HO IBX AcOEt O OBn BnO O Ph3PCH3I KHMDS THF O OBn BnO 240 °C OBn BnO O NaBH4 OBn BnO HO 1.6 1.7 2.6 2.7 2.8 2.9 2.10 2.11 2.12 THF/ EtOH THF, TBAI 1,3-DCB
Thermal Claisen rearrangement of allyl vinyl ether 2.10 in 1,3 dichlorobenzene afforded aldehyde 2.11 which was not isolated but directly reduced by NaBH4 in THF/EtOH to the stable carba primary alcohol 2.12, which is the key precursor for the synthesis of carba 1,2-epoxides.
Thus, diastereoisomeric epoxides 2.14α and 2.14β[9] were syntesized starting from a common precursor, the endocyclic olefin 2.13, obtained by simple O-benzylation (NaH/BnBr in DMF) of the primary alcohol 2.12. The treatment of olefin 2.13 with MCPBA in CH2Cl2 afforded an 80:20 mixture of the diastereoisomeric epoxides 2.14α and 2.14β (1H NMR), which were separated by flash chromatography (Scheme 2.4):
Following this synthetic approach we obtained both diastereoisomeric 1,2-epoxides 2.14α and 2.14β and we started our studies towards their regio- and stereochemical behaviour in nucleophilic addition reactions under typical acid conditions. Data from the literature indicated that epoxide 2.14β has an usual behaviour, as for reactivity, regio- and stereoselectivity, whereas epoxide 2.14α turned out particularly not reactive, at least in the addition of nucleophiles of interest for the authors.[1,5,10] We started our studies with epoxide 2.14β.[11] This epoxide reacts with MeOH under different acid reaction conditions [0.2 N H2SO4/MeOH, MeOH (6 equiv)/CH2Cl2/Amberlyst 15, MeOH/Cu(OTf)2/CH2Cl2] affording only the product deriving from the above-mentioned trans diaxial opening with the exclusive obtainment of the corresponding C(1)-adduct 2.15, as shown in Table 2.1, entries 1,2,3. When TsOH is used as the acid catalyst [10-3 N TsOH/MeOH (6 equiv)/CH
2Cl2], we observed the formation of a 73:27 mixture of products deriving from attack at C(1) of MeOH (trans methoxy alcohol 2.15) and TsOH (trans hydroxy tosylate 2.16), as indicated in Table 2.1, entry 4. This was a surprising result, because TsOH is a weak nucleophile, and
Scheme 2.4: Synthesis of diastereoisomeric epoxides 2.14α and 2.14β.
BnO OBn HO 2.12 BnO OBn BnO 2.13 NaH BnBr DMF BnO OBn BnO 2.14 O BnO OBn BnO 2.14 O MCPBA CH2Cl2
it was unexpected that it was able to compete with the more nuclephilic MeOH, in order to do the already mentioned composition of the mixture. Probably, the oxirane oxygen is protonate by TsOH and this promotes in part the dissociation of TsOH to generate the species TsO-, a nucleophile competitive respect to MeOH.
The formation of trans hydroxy tosylate 2.16 becomes the only observed addition product when epoxide 2.14β is treated with 0.7 N TsOH/CH2Cl2 solution (entry 5, Table 2.1). The trans relative configuration of hydroxy tosylate 2.16 has been demonstrated by its rapid cyclization to epoxide 2.14β by treatment with t-BuOK.
Entry Reaction Conditions C(1)-adduct 2.15 C(1)-adduct 2.16
1 0.2 N H2SO4/MeOH >99% -2 Amberlyst 15/MeOH/CH2Cl2 >99% -3 MeOH/Cu(OTf)2/CH2Cl2 >99% -4 10-3 N TsOH/MeOH/CH 2Cl2 73.0% 27.0% 5 0.7 N TsOH/CH2Cl2 - >99%
Then we switched to study the behaviour of epoxide 2.14α in nucleophilic addition reactions. As already explained in Chapter 6 and applied to our epoxide 2.14α,[4] the different stereoelectronic factors determining the different behaviour of epoxide 2.14α can be resumed in the following way: epoxide 2.14α exists in two chair conformations which have got different energies; conformer 2.14α' is reasonably the more stable because it is characterized by the triequatorial arrangement of the bulky -OBn groups. In this conformer 2.14α', nucleophilic attack can occurr at the sterically and electronically favoured C(1)
Table 2.1: Acid methanolysis of 1,2 epoxide 2.14β.
BnO OBn BnO 2.14 O MeOH H+ BnO OBn BnO 2.15 OMe OH BnO OBn BnO 2.16 OTs OH
oxirane carbon through an unfavoured trans-diequatorial opening (route a) and/or at the sterically and electronically unfavourable C(2) oxirane carbon [because of the presence of the adjacent C(3)-OBn substitution and its -I electron-withdrawing effect] through a favoured trans-diaxial ring opening (route b). The other conformer 2.14α'' is less stable, because of the triaxial arrangement of the hindered -OBn groups, but only in this conformer 2.14'', a completely favoured (sterically and electronically) trans-diaxial opening at C(1), as previously found in diastereoisomeric epoxide 2.14β, can occur (route c).
Besides requiring the occurrence of a triaxial conformer, route d shows the theoretical interference of an undesired intramolecular trans-diaxial addition by an internal nucleophile, that is the oxygen of the ether functionality present in the axial and appropriately disposed C(5)-CH2OBn group. Following this addition, with supposed elimination of benzyl methyl ether, the formation of the bicyclic compound 2.19 can occur (route d, Scheme 2.5).
In this framework, it is clear that bicyclic compound 2.19 can arise from the less stable triaxial conformer 2.14α'' (R = Bn), in which the attack of an internal nucleophile, that is the appropriately disposed oxygen of the -OBn group, determines the above described
Scheme 2.5: Possible routes for nucleophilic attack in the ring opening of 1,2-epoxide 2.14α.
OBn OH OMe BnO BnO 2 1 1 2 OBn O 2.14'' BnO BnO O BnO 2 1 MeOH b a 2.14' H route b H O Ph BnO MeOH d c route d OH 2 1 OBn OBn O BnOMe BnO BnO 2 1 BnO OH OMe route c route a 2.17 2.18 2.19
secondary addition reaction. All this can occur through the independent route d in which only small competition exists with any external O-nucleophile (for example, MeOH in the methanolysis).
With this background, we tried to study the behaviour of epoxide 2.14α in ring opening reactions with different O-nucleophiles and different reaction conditions, in order to observe if the stereo- and regioselectivity of nucleophilic attack could be directed.
Firstly, we studied reactivity of epoxide 2.14α using the same procedures already described for epoxide 2.14β (Table 2.1). The typical methanolysis reaction of epoxide 2.14α under protocol B conditions [MeOH (6 equiv)/CH2Cl2/10-3 N TsOH] (entry 3, Table 2.2), experimentally idoneous (low amount of the nucleophile) for every type of O-nucleophile, did not lead to any addition products by MeOH, but only to the bicyclic compound 2.19 (85%) and, also in this case, to a C(1)-addition product deriving from nucleophilic attack by the acid catalyst (TsOH) on C(1), the trans hydroxy tosylate 2.20 (15%). Only when MeOH was used under the less generally applicable protocol A reaction conditions (0.2 N H2SO4/MeOH), (entry 1 Table 2.2), that is when the nucleophile MeOH is also the solvent, addition by MeOH occurs and the corresponding two regioisomeric trans methoxy alcohol 2.17 [attack on C(1)] and 2.18 [attack on C(2)] were obtained in an almost 1:1 ratio (80%), accompanied by a still consistent amount of the bicyclic compound 2.19 (20%). A similar result was obtained when Cu(OTf)2 was used as the acid catalyst under protocol B conditions [MeOH (6 equiv)/CH2Cl2/Cu(OTf)2], as shown in Table 2.2, entry 4, whereas the use of an acidic resin, such as Amberlyst 15, under protocol B [MeOH (6 equiv)/CH2Cl2/Amberlyst 15] (entry 2, Table 2.2) led only to the bicyclic compound 2.19. Probably the formation of the bicyclic compound 2.19 under all the reaction conditions reported, is the consequence of the fact that, under acid condition and after protonation of the oxirane oxygen, the internal nucleophile immediately reacts by attacking C(1) oxirane carbon, through the small amount of the traxial conformer present in the reaction mixture (see Scheme 2.6). In this way the triaxial conformer 2.14α'' is made available for the reaction (Scheme 2.6) and the bicyclic compound 2.19 is formed. The internal cyclization through the triaxial conformer 2.14α'' proceeds with higher rate compared to the attack of an external nucleophile on C(1)- or on C(2)-position of the more stable conformer 2.14α' and this fact is caused by various factors: indeed, an internal
addition reaction is always favoured in comparison with an external one for entropic factors; besides, this internal cyclization occurs through a trans-diaxial transition state and the nucleophilic attack is at C(1), that is on the sterically and electronically favoured oxirane carbon. The higher rate of the intramolecular secondary reaction is probably responsible of the consistent amount of bicyclic byproduct 2.19 present in all the crude reaction mixture (from 20 to >99%, entries 1-4, Table 2.2) also in the presence of external nucleophiles (MeOH) and in spite of the small amount of triaxial conformer 2.14α'' present at the ground-state equilibrium of the epoxide 2.14α.
Interestingly, the treatment of the epoxide 2.14α with a 0.7 N TsOH in CH2Cl2 solution afforded a 75:25 mixture of the bicyclic compound 2.19 and the trans hydroxy tosylate 2.20, derived by an attack of the acid catalyst TsOH at C(1). This results was particurarly relevant because it represented a simple procedure, in which the regioselective attack of a weak nucleophile as TsOH had occurred at C(1). The structure and configuration of trans hydroxy tosylate 2.20 has been firmly established by accurate examination of the 1H NMR spectrum of the corresponding O-acetyl derivative. In spite of its certain relative trans configuration, trans hydroxy tosylate did not cyclized to epoxide 2.14α by treatment with a base (t-BuOK) also after for two days stirring at room temperature (entry 5, Table 2.2).
Entry Reaction conditions C(1)-adduct 2.17 C(2)-adduct 2.18 C(1)-adduct 2.20 Bicyclic compound 2.19 1 0.2 N H2SO4/MeOH 40.0% 40.0% - 20.0% 2 Amberlyst 15/ MeOH/CH2Cl2 - - - >99% 3 10-3 N TsOH/ MeOH/CH2Cl2 - - 15.0% 85.0% 4 MeOH/Cu(OTf)2/CH2Cl2 35.0% 25.0% - 40.0% 5 0.7 N TsOH/CH2Cl2 - - 25.0% 75.0%
These preliminary results clearly indicated that the undesired competitive intramolecular addition, leading to bicyclic compound 2.19, prevented the complete evaluation of the regiochemical behaviour of epoxide 2.14α under acid condition. Besides, this secondary undesired reaction leading to the bicyclic byproduct 2.19 influenced negatively the yield of the typical ring-opening process.
For these reasons, we tried to introduce some structural modifications to epoxide 2.14α, in order to eliminate, or at least drastically reduce, the interference of the intramolecular reaction.
Therefore, our first attempt was oriented to switch the C(6)-OBn group with an O-TIPS protective group, characterized by the presence of a less basic, potentially less or
non-Table 2.2: Acid methanolysis of 1,2 epoxide 2.14α.
BnO OBn BnO 2.14 O MeOH H+ BnO OBn BnO 2.17 OMe OH BnO OBn BnO 2.18 OH OMe BnO OBn BnO 2.20 OTs OH OH OBn OBn O 2.19
nucleophilic oxygen.[10]
The new epoxide (+)-2.22α was still prepared starting from the primary alcohol 2.12, which was treated with TIPSCl to give the corresponding C(6)-O-TIPS derivative (+)-2.21. Subsequent oxidation of (+)-2.21 by MCPBA/CH2Cl2 protocol afforded an 80:20 mixture of the diastereoisomeric epoxides (+)-2.22α and (+)-2.22β, which were separated and obtained as pure diastereoisomers by flash chromatography (Scheme 2.6).
Then we decided to better investigate the regio- and stereochemical behaviour in nucleophilic addition reactions of epoxide (+)-2.22α. Epoxide (+)-2.22α was subjected to methanolysis reaction by the simple protocol MeOH/CH2Cl2/Cu(OTf)2 protocol B reaction conditions. Among the several ring-opening reaction conditions we tested (and that we have previously described), we chose the protocol which uses MeOH/CH2Cl2/Cu(OTf)2 (with low amount of nucleophile) because it is easily applicable to different O-nucleophiles; besides, these conditions are quite simple and determine the obtainment of the products with elevate yields. The crude reaction mixture showed the presence of both the regioisomeric trans methoxy alcohol 2.23 and 2.24 (almost 1:1 ratio, 80% yield), characterized as acetates (2.23-Ac and 2.24-Ac) accompanied also in this case by the bicyclic compound 2.19 (20%), even if in a reduced amount with respect to previously obtained results on epoxide 2.14α (Entry 5, Table 2.2). This experimental evidence demonstrated that the new C(6)-O-protective group was only able to reduce the amount of the intramolecular pathway that allowed to obtain the bicyclic compound 2.19 but not to completely eliminate its formation (Scheme 2.7).
Scheme 2.6: Synthesis of C(6)-O-TIPS protected epoxides (+)-2.22α and (+)-2.22β.
OBn BnO HO 2.12 TIPSCl Imidazole DMF OBn BnO TIPSO (+)-2.21 MCPBA OBn BnO TIPSO (+)-2.22 O OBn BnO TIPSO (+)-2.22 O CH2Cl2
This result appeared promising, so that we decided to continue following this approach in order to reduce (or better to eliminate) the intramolecular undesired secondary reaction. Therefore, we tried to substitute the C(6)-OBn group with a C(6)-O-TBDPS group, that is a protective group which is not only characterized by the presence of a less basic and non-nucleophilic oxygen, but also by a consistent steric hindrance. Using this protective group we wanted to observe if bulky substitutes on C(6)-position could prevent the formation of the bicyclic byproduct 2.19.
Epoxide (-)-2.40α was still prepared starting from the common precursor primary alcohol 2.12, by treatment with TBDPS-Cl to give the corresponding C(6)-O-TBDPS derivative (-)-2.25. Subsequent oxidation of (-)-2.25 by means of MCPBA/CH2Cl2 protocol afforded a 80:20 mixture (1H NMR) of the two diastereoisomeric epoxides (-)-2.26α and (+)-2.26β, which were separated and obtained pure by flash chromatography (Scheme 2.8).
Scheme 2.7: Cu(OTf)2-catalyzed methanolysis of C(6)-O-TIPS protected epoxide (+)-2.22α.
OBn BnO TIPSO (+)-2.22 O MeOH/Cu(OTf)2 CH2Cl2 OBn BnO TIPSO 2.23 OH OMe OBn BnO TIPSO 2.24 OMe OH OH OBn OBn O 2.19 Ac2O Py AcPy2O OBn BnO TIPSO 2.23-Ac OAc OMe OBn BnO TIPSO 2.24-Ac OMe OAc
Scheme 2.8: Synthesis of C(6)-O-TBDPS protected epoxides (-)-2.26α and (+)-2.26β.
OBn BnO HO 2.12 TBDPSCl Imidazole DMF OBn BnO TBDPSO (-)-2.25 MCPBA OBn BnO TBDPSO (-)-2.26 O OBn BnO TBDPSO (+)-2.26 O CH2Cl2
Epoxide (-)-2.26α was subjected to methanolysis reaction by MeOH/CH2Cl2/Cu(OTf)2 protocol B and the crude reaction mixture showed the presence of both the regioisomeric trans methoxy alcohols 2.27 and 2.28 (about in a 2:1 ratio), which were completely characterized by means of the corresponding acetates 2.27-Ac and 2.28-Ac, but also in this case 1H NMR spectrum indicated the presence of bicyclic compound 2.19 (10%). Thus, also in this case we did not are able to completely eliminate the undesired addition of the internal nucleophile, however, we could observe an interesting trend, because the formation of byciclic 2.19 was reduced by introduction of bulky protecting group, in which oxygen acquired less basic character. Besides, methanolysis reaction on epoxide (-)-2.26α was even more notable, because 1H NMR analysis of the crude mixture of acetates, derived from the crude mixture of inseparable trans methoxy alcohols 2.27 and 2.28, showed that 2.27-Ac and 2.28-Ac were present in a 2:1 ratio, indicating that the attack of MeOH had occurred preferentially on C(1)-position (Scheme 2.9).
This result was the first case in which we observed a regioselective attack of the nucleophile on this system under acid conditions and, in my opinion, the reason of this regioselectivity is based on the ground-state equilibrium of epoxide (-)-2.26α. Similarly to the other α-epoxide examined so far, epoxide (-)-2.26α exists in two conformers, the more stable 2.26α' and the less stable 2.26α''. This second conformer, which is characterized by
Scheme 2.9: Cu(OTf)2-catalyzed methanolysis of C(6)-O-TBDPS protected epoxide (-)-2.26α.
OBn BnO TBDPSO (-)-2.26 O MeOH/Cu(OTf)2 CH2Cl2 OBn BnO TBDPSO 2.27 OH OMe OBn BnO TBDPSO 2.28 OMe OH OH OBn OBn O 2.19 Ac2O Py Ac2O Py OBn BnO TBDPSO (+)-2.27-Ac OAc OMe OBn BnO TBDPSO (+)-2.28-Ac OMe OAc
the triaxial arrangement of the -OR groups, is, as already explained, the conformer in which the intramolecular undesired reaction occurs, because of the right disposition of the internal nucleophile. In the case of epoxide (-)-2.26α, the bulky protective group on C(6) partially inhibits the internal addition of the nucleophilic oxygen (only 10% of bicyclic compound 2.19), so that the small amount of conformer 2.26α'' can undergo the competitive attack of an external nucleophile (MeOH); the external nucleophile attacks regioselectively at C(1), because it is the favoured position for steric and electronic reasons, and this fact probably determines the formation of a higher amount of methoxy alcohol 2.27 (Scheme 2.10).
Neverthless, the obtained results indicated that the secondary reaction could not completely be eliminated, but only drastically reduced. In order to eliminate the formation of bicyclic compound 2.19 starting from 1,2-carbaepoxide with α-gluco configuration, we thought to prevent completely the possibility of an internal cyclization by eliminating the internal nucleophile, that is by transforming the -CH2OBn substituent into a methyl group.
The 6-deoxy epoxide (+)-2.31α was prepared starting from the usually primary alcohol 2.12, which was transformed into the corresponding tosylate (-)-2.29. The LiAlH4 reduction of tosylate (-)-2.29 afforded the methyl-substituted olefin (+)-2.30 which was subjected to MCPBA/CH2Cl2 oxidation protocol. A 80:20 mixture of the diastereoisomeric
Scheme 2.10: Possible ring opening pathways for nucleophilic attack to 1,2 epoxide (-)-2.26α.
OTBDPS OH OMe BnO BnO 2 1 1 2 OBn O 2.26'' BnO BnO O TBDPSO 2 1 MeOH b a 2.26' H route b H O TBDPS BnO MeOH d c route d OH 2 1 OBn OBn O BnO BnO 2 1 TBDPSO OH OMe route c route a 2.27 2.28 2.19
epoxides (+)-2.31α e (+)-2.31β was obtained and the products were separated pure by flash chromatography (Scheme 2.11).
The treatment of epoxide (+)-2.31α under MeOH/CH2Cl2/Cu(OTf)2 protocol B reaction conditions afforded an almost 1:1 mixture of the corresponding regioisomeric not separable methoxy alcohols 2.32 and 2.33 which were completely characterized by means of the corresponding acetates (+)-2.32-Ac and (+)-2.33-Ac and separated pure by preparative TLC (Scheme 2.12).
Scheme 2.11: Synthesis of methyl-substituted epoxides (+)-2.31α and (+)-2.31β.
OBn BnO HO 2.12 TsCl Py OBn BnO TsO (-)-2.29 LAH OBn BnO (+)-2.30 MCPBA OBn BnO (+)-2.31 O OBn BnO O (+)-2.31 Et2O CH2Cl2
Therefore, even if in this case, as desired, the formation of bicyclic 2.19 was not observed, the reaction still was not regioselective (the methoxy alcohols 2.32 and 2.33 were obtained in a 1:1 ratio). This could indicate that methyl-substituted epoxide (+)-2.31α commonly reacts through three independents routes by means of the corresponding triequatorial (+)-2.31α' and triaxial (+)-2.31α'' conformers: both C(2)- (2.33) and C(1)-addition (2.32) products arise from triequatorial conformer (+)-2.31α' by trans-diaxial (route b) and trans-diequatorial (route a) opening process, respectively, and routes a and b are at the same time so equally favoured or unfavourable by steric or electronic considerations that a non-regioselective results is appropriately obtained. Moreover, in this case, in absence of an internal nucleophile on C(6) position, the small amount of triaxial conformer (+)-2.31α'' can undergo only the attack of the external nucleophile MeOH to produce, by a trans-diaxial ring opening process (route c), the methoxy alcohols 2.32 (Scheme 2.13).
Scheme 2.12: Cu(OTf)2-catalyzed methanolysis of methyl-substituted
epoxide (+)-2.31α. OBn BnO (+)-2.31 O MeOH/Cu(OTf)2 CH2Cl2 OBn BnO 2.32 OH OMe OBn BnO 2.33 OMe OH Ac2O Py AcPy2O OBn BnO (+)-2.32-Ac OAc OMe OBn BnO (+)-2.33-Ac OMe OAc
The introduction of a methyl substituent on C(5) position allowed to obtain the first goal of this thesis project, which was to completely avoid the formation of bicyclic compound 2.19. The following step was to better direct the regiochemistry of the ring opening process with nucleophiles. In particular, we hoped that the use of strongly coordinating reaction conditions, by the presence of an ion such as Li+ (from LiClO
4), could modify, at least partially, the conformers population toward the triaxial conformer (+)-2.31α'', through bidentate chelation by the metal cation (2.31α''-Li) (Scheme 2.14). Subsequent nucleophilic attack on 2.31α''-Li would necessary occur at C(1), following a trans diaxial opening process, with the formation of the desired pseudo-β-O-glicoside 2.32. In order to verify if our hypothesis could be correct we treated epoxide (+)-2.31α with a suspension of LiClO4 in MeOH, working under protocol A conditions, that is using MeOH at the same time as the nucleophile and as the solvent. The resulting reaction mixture was stirred for 7 days at 80 °C affording, as the only reaction product, the methoxy alcohol 2.32 (1H NMR), derived from C(1) attack of the nucleophile. The exact regiochemistry of the methoxy alcohol was confirmed by its transformation into the corresponding acetate 2.32-Ac. Therefore, with much of our delight, this result confirmed our initial idea: in presence of an ion with chelation property, such as Li+, the ground-state equilibrium of epoxide (+)-2.31α could be shifted toward the less stable triaxial conformer (+)-(+)-2.31α'', in which oxirane oxygen and -OBn group on C(3) position were appropriately disposed to form a
Scheme 2.13: Possible ring opening routes of nucleophilic attack to
1,2-epoxides (+)-2.31α. OH OMe BnO BnO 2 1 1 2 Me OBn O 2.31'' BnO BnO O 2 1 MeOH b a 2.31'H route b H BnO MeOH c BnO BnO 2 1 OH OMe route c route a 2.32 2.33 Me Me Me
bidentate chelate system with Li+. As already demonstrated, conformer (+)-2.31α'' reacts with nucleophile only at C(1) oxirane carbon, because it is the sterically and electronically favoured position and, furthermore, ring opening process occurrs through a trans-diaxial transition state, according to Fürst-Plattner rule.
At this point of my thesis work, we had sufficient information about reactivity of carba 1,2-epoxides with α-gluco configuration with a small and quite good O-nucleophile such as MeOH. Then it was interesting to study the regio- and sereoselectivity of nucleophilic addition reactions with more hindered alcohols, in order to investigate if the nature of the O-nucleophile could, in some way, influence the regioselectivity of the attack.
Therefore, epoxide (+)-2.31α was treated under t-BuOH (10 equiv)/CH2Cl2/Cu(OTf)2 protocol B conditions; 1H NMR of the crude reaction mixture showed, as we expected, that bicyclic compound 2.19 was not present, but that both regioisomeric t-buthoxy alcohols 2.34 and 2.35 was formed in a 2:1 ratio; the two t-buthoxy alcohols were separated pure by preparative TLC and completely characterized. Thus, the reaction of epoxide (+)-2.31α with t-BuOH presented an interesting, even if incomplete, regioselectivity in favour of C(1)-adduct.
Scheme 2.14: Ring opening at C(1) in nucleophilic addition by MeOH to
epoxide 2.31α in the presence of LiClO4 . 2 1 1 2 O Me BnO BnO O Me OBn Li BnO MeOH LiClO4 MeOH 2 1 Me OBn BnO OMe OH 1 2 Me BnO BnO OMe OH (+)-2.31''-Li (+)-2.31' 2.32 Ac2O Py 1 2 Me BnO BnO OMe OAc 2.32-Ac
This experimental result complied with our hypothesis, because we thought that a hindered nucleophile would have attacked preferentially the sterically favoured position, that is C(1) oxirane carbon, which is not flanked by the bulky -OBn group. The fact that t-buthoxy alcohol (+)-2.34 was the principle product of this reaction confirmed our idea.
Nevertheless, we did not obtain a complete regioselectivity, so that we decided to use a different O-nucleophile, less reactive and more hindered, that is p-methoxyphenol. We treated epoxide (+)-2.31α with p-CH3OPhOH (3 equiv)/CH2Cl2/Cu(OTf)2 under protocol B reaction conditions. This nucleophile gave an exciting result, because the 1H NMR of the crude of reaction showed alcohol 2.36 as the only reaction product, indicating that nucleophile attack occurred exclusively at C(1). This fact is, in my opinion, due to the steric hindrance and to the less nucleophilic character of p-methoxyphenol, that impedes its attack on the sterically and electronically unfavourable C(2)-position. In other words, these experimental evidences underlines that, using a hindered O-nucleophile, steric and electronic factors weight more on determining the process regioselectivity, compared to Fürst-Plattner guide-lines.
With this important consideration the second part of my thesis work ended; next
Scheme 2.15: Cu(OTf)2-catalyzed ring opening by t-BuOH to epoxide 2.31α.
Scheme 2.16: Cu(OTf)2-catalyzed ring opening by p-methoxyphenol to epoxide 2.31α.
OBn BnO (+)-2.31 O p-OCH3PhOH/Cu(OTf)2 CH2Cl2 OBn BnO 2.36 OH O OCH3 OBn BnO 2.36-Ac OAc O OCH3 Ac2O Py OBn BnO (+)-2.31 O t-BuOH/Cu(OTf)2 CH2Cl2 OBn BnO (+)-2.34 O-tBu OH OBn BnO (+)-2.35 OH O-tBu
researches will aimed to find and optimize the coordinating reaction conditions in order to apply them in a more general and synthetically useful protocol, appropriate for every kind of nucleophiles. Indeed, the coordinating reaction conditions we successfully used are not easily applicable in reactions with alcohol different from MeOH, because they require the use of an alcohol which acts at the same time as the nucleophile and as the solvent. On the contrary, a protocol which allows the use of low amount of nucleophile would be more interesting. For this purpose, the choice of an adequate solvent represents a crucial point.
8. EXPERIMENTAL
General procedures: see Part I, Chapter 3.
Materials: MeONa, 0.5 M KHMDS in THF, TBSCl, 1.0 M TBAF in THF, MeOH, t-BuOH, tri-O-acetyl-D-glucal, Ph3PMeI, BnBr, TBAI, 1,3-dichlorobenzene, AP 100 Silicone oil, LiAlH4, Cu(OTf)2, 70% MCPBA, 60% mineral oil dispersion NaH, NaBH4, anhydrous CH2Cl2 over molecular sieves, anhydrous pyridina over molecular sieves, anhydrous DMF over molecular sieves were purchased from Aldrich and used without purification. Imidazole, TIPSCl, TBDPSCl, TsCl were purchased from Fluka and used without purification. 2-Iodoxybenzoic acid (IBX) was synthesized according to the literature methods.[8] Toluene, Et
2O and THF were distilled from sodium/benzophenone.
Instrumentation: see Part I, Chapter 3.
6-O-tert-buthyldimethylsilyl-D-glucal[11]
Imidazole (5.3g, 77.96 mmol, 2.0 equiv) was added to a solution of D
-glucal 1.2 (5.70 g, 38.98 mmol, 1.0 equiv) in anhydrous DMF (20.0 mL) and anhydrous THF (240.0 mL), cooled at 0 °C. Then TBDMSCl (8.77g, 58.47 mmol, 1.5 equiv) was added and the resulting reaction mixture was stirred for 30 min allowing the temperature to rise to r.t. Dilution with Et2O and evaporation of the washed (distilled water, saturated aqueous NaCl) organic solution afforded TBS-O-derivative (11.37 g, >99% yield), practically pure as an oil, which was directly utilized in the next step without any further purification: Rf = 0.51 (1:1 CH2Cl2/MeOH); 1H NMR (CDCl3) δ 6.31 (dd, 1H, J = 6.0, 1.8 Hz), 4.73 (dd, 1H, J = 6.0, 2.2 Hz), 4.30-4.23 (m, 1H), 4.05-3.87 (m, 2H), 3.86-3.69 (m, 2H), 0.9 (s, 9H), 0.1 (s, 6H). 6-(O- tert-buthyldimethylsilyl)-3,4-di-O-benzyl-D-glucal[11]
A solution of trans-diol TBS-O-derivative 2.6 (6.0 g, 23.08 mmol, 1.0 equiv) in anhydrous THF (20.0 mL) was added dropwise at 0°C to a suspension of 60% NaH in mineral oil (3.5 g, 85.40 mmol, 3.7 equiv) in
O OBn BnO TBSO O OH HO TBSO 2.6
anhydrous THF (80.0 mL) and the resulting reaction mixture was stirred for 30 min at room temperature. Subsequently BnBr (6.0 mL, 50.78 mmol, 2.2 equiv) and TBAI (430.0 mg, 1.15 mmol, 0.05 equiv) were added to the solution and the resulting mixture was stirred overnight at room temperature. After dilution with Et2O and ice, evaporation of the washed (distilled water, saturated aqueous NaCl) organic solution afforded a crude product consisting of compound 2.7 (9.83 g, 96.8% yield), practically pure as a liquid: Rf = 0.45 (9:1 hexane/AcOEt); 1H NMR (CDCl 3) δ 7.42-7.26 (m, 10H), 6.38 (dd, 1H, J = 6.1, 1.2 Hz), 4.86 (d, 1H, J = 11.2 Hz), 4.85-4.79 (m, 1H), 4.73 (d, 1H, J = 11.2 Hz), 4.64 (d, 1H, J = 11.2 Hz) 4.57 (d, 1H, J = 11.2 Hz), 4.27-4.14 (m, 1H), 4.03-3.83 (m, 4H), 0.9 (s, 9H), 0.06 (s, 6H). 6-hydroxy-3,4-di-O-benzyl-D-glucal[11]
A 1.0 M solution of TBAF in THF (25.0 mL, 25.0 mmol, 1.1 equiv) was added dropwise at 0 °C to a solution of compound 2.7 (10.0 g, 22.73 mmol, 1.0 equiv) in anhydrous THF (230.0 mL). The reaction mixture was stirred overnight at 0 °C. After dilution with Et2O, evaporation of the washed (saturated aqueous NaCl) and dried (MgSO4) organic solution afforded a crude product (9.505 g), which was subjected to flash chromatografy (8:2 hexane/AcOEt) to afford alcohol 2.8 (5.0, 67.6% yield), pure as a white solid: mp = 32-35 °C; Rf = 0.18 (8:2 hexane/AcOEt); 1H NMR (CDCl 3) δ 7.41-7.27 (m, 10H), 6.41 (dd, 1H, J = 6.1, 1.3 Hz), 4.94-4.88 (m, 1H), 4.87 (d, 1H, J = 11.6 Hz), 4.73 (d, 1H, J = 11.6 Hz), 4.68 (d, 1H, J = 11.6 Hz), 4.57 (d, 1H, J = 11.6 Hz), 4.28-4.17 (m, 1H), 3.99-3.91 (m, 1H), 3.90-3.75 (m, 3H). 2-Formyl-3,4- di-(benzyl)-3,4-dihydro-2H-pyrane
IBX (3.66 g, 13.06 mmol, 3.0 equiv)[8] was added to a solution of primary alcohol 2.8 (1.42 g, 4.35 mmol, 1.0 equiv) in anhydrous AcOEt (100.0 mL) and the reaction mixture was stirred at 75 °C for 4 h, affording pure aldehyde 2.9 (1.41 g, >99% yield), as an oil, which was used in the next step without any further purification. Rf : 0.29 (7:3 hexane/AcOEt); [α]20D +10.20 (c 1.12, CHCl3); 1H NMR (CDCl3) δ 9.55 (s, 1H), 7.42-7.12 (m, 10H), 6.68 (d, 1H, J = 6.3 Hz), 5.16-4.98 (m, 1H), O OBn BnO O 2.9 O OBn BnO HO 2.8
4.81-4.49 (m, 3H), 4.38 (s, 2H), 4.19-3.98 (m, 1H), 3.88-3.69 (m, 1H). 1,5-Anhydro-di-O-(benzyl)-2,6,7-trideoxy-D-arabino-Hept-1,6-Dienitol(-)
A solution 0.5 M KHMDS in THF (13.06 mL, 6.53 mmol, 1.5 equiv) was added dropwise to a solution of Ph3PMe+I- (2.81g, 6.96mmol, 1.6 equiv) in anhydrous THF (25.0 mL) at -78 °C and the reaction mixture was stirred at the same temperature for 30 min and at 0 °C for 1 h. After cooling at -78 °C, a solution of aldehyde 2.9 (1.41 g, 4.35 mmol, 1.0 equiv) in anhydrous THF (20.0 mL) was added dropwise and the reaction mixture was stirred at room temperature for 3 h, affording a crude product (2.689 g) yielding olefin (-)-2.10, practically pure as a solid (0.943 g, 67.3% yield): Rf = 0.22 (9:1 hexane/AcOEt); [α]20D -34.7 (c 1.1, CHCl3); 1H NMR (CDCl3) δ 7.45-7.28 (m, 10H), 6.42 (dd, 1H, J = 6.1, 1.2 Hz), 6.1 (ddd, 1H, J = 17.1, 10.6, 6.4 Hz), 5.44 (dt, 1H, J = 17.1, 2.9, 1.4 Hz), 5.31 (dt, 1H, J = 10.6, 2.9, 1.4 Hz), 4.89 (dd, 1H, J = 6.1, 2.7 Hz), 4.79 (d, 1H, J = 11.2 Hz), 4.69 (d, 1H, J = 11.2 Hz), 4.65 (d, 1H, J = 11.7 Hz), 4.58 (d, 1H, J = 11.7 Hz), 4.34 (dd, 1H, J = 8.1, 6.7 Hz), 4.21 (ddd, 1H, J = 6.0, 2.7, 1.6 Hz), 3.61 (dd, 1H, J = 8.4, 6.0 Hz); 13C NMR (CDCl 3) δ 144.7, 138.6, 138.3, 134.5, 128.6, 128.2, 127.9, 127.8, 118.5, 100.5, 78.5, 78.2, 75.6, 74.0, 70.9. 3,4-Di-O-(benzyl)-5a-carba-D-glucal
A solution of olefin (-)-2.10 (0.878 g, 2.73 mmol) in 1,3-dichlorobenzene (4.0 mL) was stirred at 240°C for 20 min in AP 100 Silicone oil (Aldrich) warming bath. After cooling, the reaction mixture was added to a solution of NaBH4 (0.155 g, 4.01 mmol, 6.0 equiv) in 2:1 THF/EtOH (4.0 mL) and the resulting mixture was stirred at room temperature for 20 min. After dilution with CH2Cl2, evaporation of the washed (saturated aqueous NaCl) and dried organic solution, afforded a crude reaction product (0.950 g), which was subjected to flash chromatography. Elution with an 8:2 hexane/AcOEt mixture yielded primary alcohol 2.12 (0.500 g, 60% yield), pure as a liquid. Rf = 0.25 (7:3 hexane/AcOEt); 1H NMR (CDCl3) δ 7.50-7.27 (m, 10H), 5.85-5.63 (m, 2H), 4.99 (d, 1H, J = 11.3 Hz), 4.74 (d, 1H, J = 11.3 Hz), 4.64 (d, 1H, J = 11.2 Hz), 4.24 (ddd, 1H, J = 7.1, 2.7, 1.4 Hz), 3.71-3.51 (m, 3H), 2.61 (dd, J = 8.0, 4.0 Hz), 2.13-1.74 (m, 2H). 13C NMR (CDCl 3) δ 138.4, 131.0, 129.5, 127.8, 127.5, 125.9, 82.0, BnO OBn HO 2.12 O OBn BnO 2.10
81.2, 74.4, 71.3, 65.4, 40.5, 29.8, 28.1. 3,4,6-Tri-O-benzyl-5a-carba-D-glucal
A solution of primary alcohol 2.12 (0.788 g, 2.42 mmol) in anhydrous DMF (6.0 mL) was added dropwise at 0 °C to a suspension of 60% NaH in mineral oil (0.15 g, 3.78 mmol, 3.0 equiv) in anhydrous DMF (5.0 mL) and the resulting reaction mixture was stirred at room temperature for 40 min. After cooling at 0°C, BnBr (0.31 mL, 2.66 mmol, 1.1 equiv) was added dropwise, and the reaction mixture was stirred at the same temperature for 24 h. Evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude reaction mixture (0.631 g) which was subjected to flash chromatography. Elution with an 8:2 hexane/AcOEt mixture afforded tri-O-benzyl-derivative 2.13,[10] pure as a liquid (1.10 g, 99% yield). R
f= 0.52 (8:2 hexane/AcOEt). 1H NMR (CDCl
3) δ 7.45-7.28 (m, 10H), 5.83-5.63 (m, 2H), 4.90 (d, 1H, J = 11.0 Hz), 4.72-4.46 (m, 5H), 4.20 (ddd, 1H, J = 9.05, 4.02, 2.01 Hz), 3.77-3.54 (m, 3H), 2.31-2.22 (m, 2H), 2.17-2.01 (m, 1H).
Reaction of tri-O-benzyl-protected olefin 2.13 under MCPBA/CH2Cl2 protocol:[7]
70% MCPBA (1.313 g, 5.33 mmol, 2.0 equiv) was added to a solution of tri-O-benzyl-protected olefin 2.13 (1.102 g, 2.663 mmol, 1.0 equiv) in anhydrous CH2Cl2 (92.0 mL) at 0 °C and the reaction mixture was stirred overnight at room temperature. After dilution with CH2Cl2, evaporation of the washed (10% Na2S2O3, saturated aqueous NaHCO3, saturated aqueous NaCl) and dried (MgSO4) organic solution afforded a crude mixture (1.186 g), consisting of a 80:20 (1H NMR) mixture of diastereoisomeric epoxides 2.14α and 2.14β, which was subjected to flash chromatography. Elution with a 8:2 hexane/AcOEt mixture afforded the two diastereosomeric epoxides 2.14α (0.456 g, 40% yield) and 2.14β (0.123 g, 11% yield).
3,4,6-tri-O-benzyl-1,2-anhydro-5a-carba-α-D,L-glucopyranose:[7] a
colorless liquid; Rf α: 0.29 (8:2 hexane/AcOEt); 1H NMR (CDCl3) δ 7.45-7.23 (m, 15H), 4.86 (d, 1H, J = 11.1 Hz), 4.84 (d, 1H, J = 11.1 Hz), 4.72 (d, 1H, J = 11.1 Hz), 4.58 (d, 1H, J = 11.1 Hz), 4.48 (s, 2H), 3.80 (dd, 1H, J = 8.1, 0.6 Hz), 3.75 (dd, 1H, J = 9.0, 4.5 Hz), 3.53-3.38 (m, 2H), 3.27 (d, BnO OBn BnO 2.13 BnO OBn BnO 2.14 O
1H, J = 3.5 Hz), 3.18 (d, 1H, J = 3.5 Hz), 2.26 (ddd, 1H, J = 15.1, 4.8, 2.1 Hz), 2.12 (dd, 1H, J = 11.7, 1.7 Hz), 1.86-1.69 (m, 1H).
3,4,6-tri-O-benzyl-1,2-anhydro-5a-carba-β-D,L-glucopyranose:[7] a
colorless liquid; Rf β: 0.18 (8:2 hexane/AcOEt); 1H NMR (CDCl3) δ 7.46-7.28 (m, 15 H), 4.84 (d, 1H, J = 10.6 Hz), 4.82 (s, 2H), 4.52 (d, 1H, J = 10.6 Hz), 4.46 (s, 2H), 3.84 (dd, 1H, J = 8.3, 1.7 Hz), 3.66 (dd, 1H, J = 10.9, 8.3 Hz), 3.53-3.45 (m, 2H), 3.34 (dd, 1H, J = 4.04, 1.7 Hz), 3.26 (t, 1H, J = 4.04 Hz), 2.21-1.97 (m, 2H), 1.91-1.74 (m, 1H).
Reaction of epoxide 2.14β with a 10-3 N TsOH MeOH/CH
2Cl2 solution:
Epoxide 2.14β (0.030 g, 0.069 mmol) was added to a CH2Cl2 solution (7.0 mL) containing MeOH (17.0 L, 0.42 mmoli, 6.0 equiv), and TsOH.H
2O (0.001 g, 0.007 mmol, 0.1 equiv) (epoxide: TsOH: MeOH = 1: 0.1: 6) and the resulting mixture was stirred 2 h at room temperature. Dilution with CH2Cl2 and evaporation of the washed (saturated aqueous NaHCO3 and saturated aqueous NaCl) organic solution afforded a crude reaction product (0.042 g) consisting of a 73:27 mixture of trans methoxy alcohol 2.15 and trans hydroxy tosylate 2.16 (1H NMR) which was subjected to preparative TLC, using a 1:1 hexane/AcOEt mixture as the eluant. Extraction of the two most intense bands (the slower moving band contained 2.15) afforded trans methoxy alcohol 2.15 (0.022 g, 69% yield) and trans hydroxy tosylate 2.16 (0.010 g, 24% yield):
Methyl 3,4,6-tri-O-benzyl-α-D-mannopyranoside: a colorless
liquid, Rf = 0.58 (1:1 hexane/AcOEt); 1H NMR (CDCl3) 7.37-7.26 (m, 15H), 4.83 (d, 1H, J = 10.8 Hz), 4.71 (d, 1H, J = 11.5 Hz), 4.66 (d, 1H, J = 11.5 Hz), 4.51 (d, 1H, J = 10.8 Hz), 4.47 (s, 2H), 4.12 (t, 1H, J = 3.2 Hz), 3.76 (dd, 1H, J = 8.8, 3.9 Hz), 3.64 (dd, 1H, J = 8.8, 5.2 Hz), 3.60-3.54 (m, 1H), 3.49 (dd, 1H, J = 8.8, 2.8 Hz), 3.31 (s, 3H), 2.50 (bs, 1H, -OH), 2.08-1.94 (m, 1H), 1.93-1.83 (m, 2H). 13C NMR (CDCl 3) 139.2, 138.9, 138.5, 128.7, 128.5, 128.0, 127.7, 82.6, 78.3, 75.0, 73.2, 72.8, 70.6, 69.5, 56.7, 37.3, 29.9, 26.4, 22.9.
Tosyl 3,4,6-tri-O-benzyl-α-D-mannopyranoside: a colorless liquid,
Rf = 0.65 (1:1 hexane/AcOEt); 1H NMR (CDCl3) 7.76 (dt, 2H, J = 8.5, 1.8 Hz), 7.45-7.19 (m, 17H), 4.82 (d, 1H, J = 11.2 Hz), 4.77-BnO OBn BnO 2.15 OMe OH BnO OBn BnO 2.14 O BnO OBn BnO OTs OH
4.72 (m, 1H), 4.66 (d, 1H, J = 11.5 Hz), 4.57 (d, 1H, J = 11.5 Hz), 4.48 (d, 1H, J = 11.2 Hz), 4.41 (s, 2H), 4.06 (bs, 1H), 3.77-3.69 (m, 2H), 3.63 (dd, 1H, J = 9.2, 3.9 Hz), 3.35 (dd, 1H, J = 9.2, 2.3 Hz), 2.53 (s, 3H), 2.52 (bs, 1H, -OH), 2.06-1.95 (m, 2H), 1.73 (dd, 1H, J = 10.5, 3.2 Hz). 13C NMR (CDCl 3) 145.1, 138.8, 138.6, 137.9, 133.9, 130.1, 128.5, 128.0, 127.9, 127.7, 81.6, 78.9, 77.4, 76.9, 75.2, 73.1, 72.9, 69.6, 69.0, 37.3, 27.9, 21.8. Reaction of epoxide 2.14β with 0.2 N H2SO4/MeOH
Epoxide 2.14β(0.010 g, 0.023 mmol) was added to a 0.2 N H2SO4/MeOH (1.0 mL) and the resulting reaction mixture was stirred 30 minutes at room temperature. After dilution with CH2Cl2, solid NaHCO3 was added. Evaporation of the washed (saturated aqueous NaHCO3 and saturated aqueous NaCl) organic solution, afforded a crude product (0.011 g, 99% yield) consisting of trans methoxy alcohol 2.15, as the only reaction product (1H NMR). Reaction of epoxide 2.14β with Amberlyst 15/MeOH/CH2Cl2
A solution of epoxide 2.14β(0.020 g, 0.046 mmol) in anhydrous CH2Cl2 (0.5 mL) was treated with MeOH (11.1 L, 0.276 mmol, 6.0 equiv) and Amberlyst 15 (4.7 meq/g, 0.001 g, 0.005 mmol, 0.11 equiv) and the resulting reaction mixture was stirred 6 h at room temperature. After dilution with CH2Cl2, evaporation of the filtered solution, afforded a crude product (0.024 g, 99% yield) consisting of trans methoxy alcohol 2.15 (1H NMR). Reaction of epoxide 2.14β with a solution of TsOH in CH2Cl2
A solution of epoxide 2.14β (0.020 g, 0.046 mmol) in anhydrous CH2Cl2 (0.2 mL) was treated with TsOH.H
2O (0.026 g, 0.138 mmol, 3.0 equiv) and the resulting reaction mixture was stirred 18 h at room temperature. After dilution with CH2Cl2, evaporation of the filtered solution, afforded a crude product (0.032 g, >99% yield) consisting of pure trans hydroxy tosylate 2.16, as the only reaction product (1H NMR).
Reaction of epoxide 2.14α with a 10-3 N TsOH MeOH/CH
2Cl2 solution
Following the procedure used for epoxide 2.14β, epoxide 2.14α (0.030 g, 0.069 mmol) was added to a CH2Cl2 solution (7.0 mL) containing MeOH (17.0 L, 0.42 mmoli, 6.0 equiv) and TsOH.H
resulting reaction mixture was stirred 2 h at room temperature. Dilution with CH2Cl2 and evaporation of the washed (saturated aqueous NaHCO3 and saturated aqueous NaCl) organic solution afforded a crude reaction product (0.024 g) consisting of a 15:85 mixture of trans hydroxy tosylate 2.20 and bicyclic compound 2.19 (1H NMR) which was subjected to preparative TLC, using a 1:1 hexane/AcOEt mixture as the eluant. Extraction of the two most intense bands (the slower moving band contained 2.20) afforded trans hydroxy tosylate 2.20 (0.005 g, 21% yield), then acetylated (Ac2O/Py in 1:2 ratio) to give the corresponding acetyl derivative 2.20-Ac, and bicyclic compound 2.19 (0.003 g, 7% yield).
Tosyl 3,4,6-tri-O-benzyl-β-D-glucopyranoside: a colorless liquid,
Rf = 0.38 (1:1 hexane/AcOEt). 1H NMR (CDCl3) 7.76 (d, 2H, J = 8.1 Hz), 7.41-7.26 (m, 17H), 7.15-7.03 (m, 1H), 4.89 (d, 1H, J = 10.9 Hz), 4.83 (d, 1H, J = 10.9 Hz), 4.82 (d, 1H, J = 10.9 Hz), 4.67 (d, 1H, J = 11.3 Hz), 4.53 (d, 1H, J = 11.3 Hz), 4.34 (d, 1H, J = 10.9 Hz), 4.14 (dd, 1H, J = 9.3, 5.3 Hz), 4.07 (dd, 1H, J = 9.3, 2.8 Hz), 3.57 (t, 1H, J = 8.5 Hz), 3.45-3.24 (m, 2H), 2.65 (bs 1H, -OH), 2.40 (s, 3H), 2.15-1.98 (m, 1H), 1.84-1.65 (m, 1H), 1.42-1.30 (m, 1H).
Tosyl 2-O-acetyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside: a
liquid, 1H NMR (CDCl 3) 7.76 (d, 2H, J = 8.3 Hz), 7.39-7.26 (m, 15H), 7.16-7.10 (m, 2H), 5.07 (t, 1H, J = 8.5 Hz), 4.76 (d, 1H, J = 10.3 Hz), 4.74 (d, 1H, J = 11.2 Hz), 4.63 (d, 1H, J = 11.2 Hz), 4.61 (d, 1H, J = 11.8 Hz), 4.42 (d, 1H, J =11.8 Hz), 4.35 (d, 1H, J =10.3 Hz), 4.11-4.06 (m, 2H), 3.48-3.29 (m, 3H), 2.41 (s, 3H), 2.15-1.95 (m, 1H), 1.93 (s, 3H), 1.80-1.72 (m, 1H), 1.40-1.33 (m, 1H). 13C NMR (CDCl 3) δ 170.2, 145.2, 138.3, 137.9, 132.8, 130.1, 128.6, 128.3, 128.1, 127.8, 127.5, 84.2, 79.6, 75.6, 75.3, 71.6, 69.8, 38.2, 29.9, 29.4, 21.8, 21.2. (1S,2S,3R,4R,5S)-3,4-diacetoxy-7-oxabicyclo[3.2.1]octan-2-ol: a colorless liquid, Rf = 0.07 (1:1 hexane/AcOEt): 1H NMR (CDCl3) 7.40-7.27 (m, 10H) 4.65 (d, 1H, J = 12.1 Hz), 4.58 (d, 1H, J = 12.1 Hz), 4.56 (d, 1H, J = 12.0 Hz), 4.48 (d, 1H, J = 12.0 Hz), 4.26 (dd, 1H, J = 5.7, 3.7 Hz), 3.97 (d, 1H, J = 8.1 Hz), 3.86-3.80 (m, 1H), 3.76 (dd, 1H, J = 8.1, 4.9 Hz), 3.59 (bs, 1H), 3.49 (bs, 1H), 2.67-2.59 (m, 1H), 2.40 (d, 1H, J = 12.3 Hz), 1.84 (d, 1H, J = 6.5 Hz), 1.69-1.54 (m, 1H). 13C NMR (CDCl 3) δ 138.5, 138.2, 128.7, 128.0, 127.9, 127.8, 81.0, 79.8, 72.4, 72.2, BnO OBn BnO 2.20-Ac OTs OAc BnO OBn BnO 2.20 OTs OH OH OBn OBn O 2.19
71.1, 69.6, 38.4, 25.6.
Reaction of epoxide 2.14α with 0.2 N H2SO4/MeOH
As previously described for epoxide 2.14β, epoxide 2.14α(0.010 g, 0.023 mmol) was added to 0.2 N H2SO4/MeOH (1.0 mL) and the resulting reaction mixture was stirred 12 h at room temperature. After dilution with CH2Cl2, solid NaHCO3 was added. Evaporation of the washed (saturated aqueous NaHCO3 and saturated aqueous NaCl) organic solution, afforded a crude reaction product (0.009 g) consisting of a 50:30:20 mixture of the two regioisomeric trans methoxy alcohols 2.17 and 2.18 and bicyclic compound 2.19 (1H NMR), which was subjected to preparative TLC, using a 9:1 CH2Cl2/(i-Pr)2O mixture as the eluant (3 runs). Extraction of the most intense bands (the faster and the slower moving band contained 2.19 and 2.18, respectively) afforded bicyclic compound 2.19 (0.002 g, 25% yield), the trans methoxy alcohol 2.17 (0.004 g, 36% yield) and the trans methoxy alcohol 2.18 (0.003 g, 27% yield).
Methyl 3,4,6-tri-O-benzyl-β-D-glucopyranoside: a colorless liquid, Rf = 0.12 [9:1 CH2Cl2/(i-Pr)2O]. 1H NMR (CDCl3) 7.43-7.27 (m, 15H), 4.88 (s, 2H), 4.86 (d, 1H, J = 11.8 Hz), 4.51 (d, 1H, J = 11.8 Hz), 4.46 (s, 2H), 3.61-3.46 (m, 5H), 3.43 (s, 3H), 3.18-3.05 (m, 1H), 2.66 (bs, 1H, -OH), 2.34-2.12 (m, 1H), 1.77-1.65 (m, 1H), 1.39-1.29 (m, 1H). 13C NMR (CDCl 3) 138.9, 138.7, 138.5, 128.6, 128.2, 128.1, 127.9, 127.8, 86.1, 81.1, 80.7, 77.2, 75.5, 75.4, 73.4, 70.3, 57.4, 39.4, 29.1.
Methyl 2-acetyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside: a
liquid, 1H NMR (CDCl 3) 7.32-7.27 (m, 15H), 5.04 (t, 1H, J = 9.1 Hz), 4.82 (d, 1H, J = 11.2 Hz), 4.81 (d, 1H, J = 10.6 Hz), 4.66 (d, 1H, J = 11.2 Hz), 4.48 (d, 1H, J = 10.6 Hz), 4.46 (s, 2H), 3.57-3.45 (m, 4H), 3.34 (s, 3H), 3.27-3.14 (m, 1H), 2.27-2.14 (m, 1H), 1.98 (s, 3H), 1.77-1.65 (m, 1H), 1.47-1.36 (m, 1H). 13C NMR (CDCl 3) 170.5, 138.7, 138.5, 128.6, 128.3, 127.8, 84.5, 80.9, 79.2, 75.5, 73.3, 70.1, 57.3, 39.2, 29.9, 29.6, 21.3.
3,4,6-tri-O-benzyl-2-O-methyl-α-D-mannopyranose: a colorless
liquid, Rf = 0.09 [9:1 CH2Cl2/(i-Pr)2O]. 1H NMR (CDCl3) 7.36-7.27 (m, 15H), 4.74 (d, 1H, J = 11.5 Hz), 4.68 (d, 1H, J = 12.4 Hz), BnO BnO OH OMe BnO OBn BnO 2.17-Ac OMe OAc BnO OBn BnO 2.17 OMe OH
4.62 (d, 1H, J = 12.4 Hz), 4.51 (d, 1H, J = 11.5 Hz), 4.45 (s, 2H), 4.11-4.02 (m, 1H), 3.86 (dd, 1H, J = 7.5, 2.9 Hz), 3.79 (d, 1H, J = 7.5 Hz), 3.60-3.54 (m, 2H), 3.49-3.43 (m, 1H), 3.40 (s, 3H), 2.24-2.14 (m, 1H), 1.99-1.84 (m, 1H), 1.79-1.69 (m, 1H). 13C NMR (CDCl 3) 139.0, 138.8, 128.5, 128.1, 127.8, 81.9, 79.4, 74.1, 73.2, 72.9, 71.0, 66.9, 58.6, 37.7, 30.4. 1-O-Acetyl-3,4,6-tri-O-benzyl-2-O-methyl-α-D-mannopyranose: a liquid, 1H NMR (CDCl 3) 7.39-7.27 (m, 15H), 5.15-5.09 (m, 1H), 4.92 (d, 1H, J = 10.7 Hz), 4.75 (d, 1H, J = 12.2 Hz), 4.67 (d, 1H, J = 12.2 Hz), 4.50 (d, 1H, J = 10.7 Hz), 4.46 (s, 2H), 3.76 (t, 1H, J = 9.6 Hz), 3.66 (dd, 1H, J = 9.6, 3.1 Hz), 3.59 (dd, 1H, J = 8.9, 4.3 Hz), 3.53-3.49 (m, 2H), 3.48 (s, 3H), 1.95 (s, 3H), 1.99-1.88 (m, 2H), 1.83-1.74 (m, 1H). 13C NMR (CDCl3) 170.2, 139.1, 138.8, 138.7, 128.5, 128.3, 127.8, 81.6, 78.2, 75.8, 75.7, 73.4, 73.1, 70.5, 69.5, 59.5, 59.3, 38.5, 27.9, 21.4.
Reaction of epoxide 2.14α with Amberlyst 15/MeOH/CH2Cl2
As previously described for epoxide 2.14β, a solution of epoxide 2.14α(0.020 g, 0.046 mmol) in anhydrous CH2Cl2 (0.5 mL) was treated with MeOH (11.1 L, 0.276 mmol, 6.0 equiv) and Amberlyst 15 (4.7 meq/g, 0.001 g, 0.005 mmol, 0.11 equiv) and the resulting reaction mixture was stirred 18 h at room temperature. After dilution with CH2Cl2, evaporation of the filtered solution afforded a crude product (0.017 g, >99% yield) consisting of bicyclic compound 2.19 (1H NMR).
Reaction of epoxide 2.14α with Cu(OTf)2/MeOH/CH2Cl2
A solution of epoxide 2.14α (0.020 g, 0.046 mmol) in anhydrous CH2Cl2 (0.2 mL) was treated with MeOH (7.5 L, 0.186 mmol, 4.0 equiv) and Cu(OTf)2 (0.005 g, 0.014 mmol, 0.3 equiv) and the resulting reaction mixture was stirred 18 h at room temperature. After dilution with CH2Cl2, evaporation of the washed (saturated aqueous NaHCO3 and saturated aqueous NaCl) organic solution afforded a crude reaction product (0.022 g, >99% yield) consisting of a 35:25:40 mixture the two regioisomeric trans methoxy alcohols 2.17 and 2.18 and bicyclic compound 2.19 (1H NMR).
BnO OBn BnO 2.18-Ac OAc OMe
Reaction of epoxide 2.14α with a solution of TsOH in CH2Cl2
A solution of epoxide 2.14α (0.020 g, 0.046 mmol) in anhydrous CH2Cl2 (0.2 mL) was treated with TsOH.H
2O (0.026 g, 0.138 mmol, 3.0 equiv) and the resulting reaction mixture was stirred 18 h at room temperature. After dilution with CH2Cl2, evaporation of the filtered solution, afforded a crude product (0.034 g, 99% yield) consisting of a 25:75 mixture of trans hydroxy tosylate 2.20 and bicyclic compound 2.19 (1H NMR).
6-O-Triisopropylsilyl-3,4-di-O-benzyl-5a-carba-D-glucal
A solution of primary alcohol 2.12 (0.250 g, 0.77 mmol) in anhydrous DMF (2.0 mL) was treated with imidazole (0.157 g, 2.31 mmol, 3.0 equiv) and TIPSCl (0.25 mL, 1.15 mmol, 1.5 equiv) at 0°C and the reaction mixture was stirred 18 h at room temperature. Dilution with Et2O and evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude product (0.444 g) consisting of -O-TIPS derivative (+)-2.21 which was subjected to flash chromathography. Elution with a 9:1 hexane/AcOEt mixture afforded -O-TIPS derivative (+)-2.21 (0.411 g, 90% yield), pure as a yellow liquid: Rf = 0.57 (8:2 hexane/AcOEt); []20 D +15.1 (c 0.42, CHCl3). 1H NMR (CDCl3) δ 7.44-7.25 (m, 10H), 5.88-5.77 (m, 1H), 5.75-5.67 (m, 1H), 4.97 (d, 1H, J = 11.3 Hz), 4.88 (s, 2H), 4.74 (d, 1H, J = 11.3 Hz), 4.30-4.18 (m, 1H), 3.95 (dd, 1H, J = 9.6, 5.4 Hz), 3.86 (dd, 1H, J = 9.6, 3.0 Hz), 3.71 (dd, 1H, J = 10.7, 7.4 Hz), 2.34-2.22 (m, 2H), 2.10-1.92 (m, 1H), 1.14 (s, 9H), 1.09 (s, 9H), 1.08 (s, 3H). 13C NMR (CDCl 3) δ 139.3, 138.6, 128.9, 128.6, 128.3, 128.0, 127.8, 126.9, 126.1, 125.9, 81.8, 79.6, 74.6, 71.6, 65.1, 41.6, 28.7, 18.3, 12.2.
Reaction of -O-TIPS protected olefin (+)-2.21 under MCPBA/CH2Cl2 protocol.
Following the typical procedure described for the synthesis of diastereoisomeric epoxides 2.14α and 2.14β, a solution of -O-TIPS derivative (+)-2.21 (0.030 g, 0.063 mmol) in anhydrous CH2Cl2 (0.6 mL) was treated with MCPBA (0.031 g, 0.125 mmol, 2.0 equiv). After stirring for 18 h, typical workup afforded a crude (0.030 g) consisting of a 80:20 mixture (1H NMR) of the diasteroisomeric epoxides (+)-2.22α and (+)-2.22β which was purified by preparative TLC, by using a 9:1 hexane/AcOEt mixture as the eluant. Extraction of the more intense bands [the faster band contains (+)-2.22α] afforded
OBn BnO
TIPSO
epoxides (+)-2.22α (0.021 g, 68% yield) and (+)-2.22β (0.005 g, 15% yield). 6-O-Triisopropylsilyl-3,4-di-O-benzyl-5a-carba-α-D
-glucopyranose: a liquid, Rf = 0.32 (9:1 hexane/AcOEt); []20
D +6.26 (c 0.82, CHCl3). 1H NMR (CDCl3) δ 7.41-7.24 (m, 10H), 4.87 (d, 1H, J = 11.0 Hz), 4.81 (d, 1H, J = 11.4 Hz), 4.70 (d, 1H, J = 11.4 Hz), 4.66 (d, 1H, J = 11.0 Hz), 3.97 (dd, 1H, J = 9.7, 4.3 Hz), 3.80 (dd, J = 8.0, 0.5 Hz), 3.67 (dd, 1H, J = 9.7, 2.7 Hz), 3.41 (d, 1H, J = 11.3, 8.0 Hz), 3.30-3.25 (m, 1H), 3.18-3.15 (m, 1H), 2.28-2.16 (m, 1H), 2.11-1.95 (m, 1H), 1.74-1.58 (m, 1H), 1.06 (s, 18H), 1.04 (s, 3H). 13C NMR (CDCl 3) δ 139.2, 138.1, 128.7, 128.5, 128.2, 128.1, 127.8, 127.6, 81.3, 79.4, 77.2, 74.6, 72.9, 63.0, 54.4, 53.4, 35.2, 27.7, 18.3, 12.2. 6-O-Triisopropylsilyl-3,4-di-O-benzyl-5a-carba-β-D
-mannopyranose: a liquid, Rf = 0.21 (9:1 hexane/AcOEt); []20 D +38.2 (c 0.33, CHCl3). 1H NMR (CDCl3) δ 7.45-7.26 (m, 10H), 4.87 (d, 1H, J = 11.0 Hz), 4.81 (s, 2H), 4.60 (d, 1H, J = 11.0 Hz), 3.82 (dd, 1H, J = 11.0, 1.7 Hz), 3.75-3.70 (m, 2H), 3.60 (dd, 1H, J = 11.0, 8.2 Hz), 3.32 (dd, 1H, J = 4.0, 1.7 Hz), 3.30-3.24 (m, 1H), 2.18-1.93 (m, 2H), 1.69-1.63 (m, 1H), 1.06 (s, 6H), 1.04 (s, 12H), 1.03 (s, 3H). 13C NMR (CDCl 3) δ 139.0, 138.7, 128.6, 128.5, 128.2, 127.9, 127.7, 82.1, 78.9, 75.3, 72.6, 63.5, 55.5, 53.7, 42.2, 26.8, 18.5, 12.4.
Reaction of epoxide 2.22α with Cu(OTf)2/MeOH/CH2Cl2
As described for epoxide 2.14α, a solution of epoxide (+)- 2.22α (0.023 g, 0.046 mmol) in anhydrous CH2Cl2 (0.2 mL) was treated with MeOH (7.5 L, 0.186 mmol, 4.0 equiv) and Cu(OTf)2 (0.005 g, 0.014 mmol, 0.3 equiv) and the resulting reaction mixture was stirred 48 h at room temperature. After dilution with CH2Cl2, evaporation of the washed (saturated aqueous NaHCO3 and saturated aqueous NaCl) organic solution afforded a crude product (0.022 g, 99% yield) which was treated with Ac2O (0.2 mL) in anhydrous pyridine (0.4 mL) at 0°C and the resulting mixture was stirred 18 h at room temperature. Co-evaporation with toluene afforded a crude product (0.030 g, 99% yield) consisting of a 40:40:20 mixture of the two regioisomeric trans methoxy acetates (+)-8.23-Ac and (+)-8.24-Ac and bicyclic compound 2.19-Ac (1H NMR), which was subjected to preparative TLC, by using a 3:6:1 hexane/CH2Cl2/(i-Pr)2O mixture. Extraction of the more intense bands (the faster
OBn BnO TIPSO (+)-2.22 O OBn BnO TIPSO (+)-2.22 O
band contains (+)-2.23-Ac and the slower band contains 2.19-Ac) afforded pure methoxy derivatives (+)-2.23-Ac (0.006 g, 23% yield) and (+)-2.24-Ac (0.007 g, 27% yield) and bicyclic compound 2.19-Ac (0.004 g, 15% yield).
Methyl 2-O-acetyl-6-O-triisopropylsilyl-3,4-di-O-benzyl-5a-carba-β-D-glucopyranoside: a liquid, Rf = 0.71 [3:6:1 hexane/CH2Cl2/(i-Pr)2O]; []20D +33.6 (c 0.17, CHCl3). 1H NMR (CDCl3) δ 7.33-7.26 (m, 10H), 5.02 (t, 1H, J = 9.6 Hz), 4.85 (d, 1H, J = 10.9 Hz), 4.82 (d, 1H, J = 10.1 Hz), 4.66 (d, 1H, J = 10.1 Hz), 3.62 (d, 1H, J = 10.9 Hz), 3.87 (dd, 1H, J = 9.6, 4.9 Hz), 3.78 (dd, 1H, J = 9.6, 2.6 Hz), 3.57-3.47 (m, 2H), 3.34 (s, 3H), 3.28 -3.16 (m, 1H), 2.36-2.24 (m, 1H), 2.19-2.01 (m, 2H), 1.98 (s, 3H), 1.05 (s, 21H). 13C NMR (CDCl 3) δ 171.8, 138.9, 128.6, 128.1, 127.9, 85.0, 81.2, 79.8, 76.1, 76.0, 63.3, 57.6, 41.2, 32.0, 22.9, 18.3, 12.4. 1-O-Acetyl-6-O-triisopropylsilyl-3,4-di-O-benzyl-2-O-methyl-5a-carba-α-D-mannopyranose: a liquid, Rf = 0.61 [3:6:1
hexane/CH2Cl2/(i-Pr)2O]; []20D +53.8 (c 0.13, CHCl3). 1H NMR (CDCl3) δ 7.39-7.27 (m, 10H), 5.17-5.09 (m, 1H), 4.97 (d, 1H, J = 10.6 Hz), 4.76 (d, 1H, J = 11.9 Hz), 4.67 (d, 1H, J = 11.9 Hz), 4.62 (d, 1H, J = 10.6 Hz), 3.92 (dd, 1H, J = 9.5, 4.6 Hz), 3.81 (t, 1H, J = 9.5 Hz), 3.73-3.62 (m, 2H), 3.51-3.48 (m, 1H), 3.47 (s, 3H), 2.36-2.24 (m, 1H), 2.11-1.98 (m, 2H), 1.96 (s, 3H), 1.04 (s, 18H), 1.03 (s, 3H). 13C NMR (CDCl 3) δ 170.9, 137.8, 128.6, 128.3, 128.2, 127.9, 81.7, 77.1, 75.5, 73.1, 69.3, 67.0, 63.4, 58.9, 40.3, 32.1, 22.9, 18.3, 12.2. 6-O-t-buthyldiphenylsilyl-3,4-di-O-benzyl-5a-carba-D-glucal
Following the typical procedure previously described for the synthesis of compound (+)-2.21, a solution of primary alcohol 2.12 (0.100 g, 0.31 mmol, 1.0 equiv) in anhydrous DMF (1.0 mL) was treated with imidazole (0.065 g, 0.93 mmol, 3.0 equiv) and TBDPSCl (0.12 mL, 0.47 mmol, 1.5 equiv) at 0 °C. After stirring for 18 h, typical workup afforded a crude product (0.325 g), consisting of -O-TBDPS derivative (-)-2.25, which was subjected to flash chromatography. Elution with a 9:1 hexane/AcOEt mixture afforded -O-TBDPS derivative (-)-2.25 (0.120 g, 70% yield), pure as a liquid: Rf = 0.44 (9:1 hexane/AcOEt);
OBn BnO TBDPSO (-)-2.25 OBn BnO TIPSO 2.24-Ac OMe OAc OBn BnO TIPSO 2.23-Ac OAc OMe
[]20 D -21.13 (c 1.08, CHCl3). 1H NMR (CDCl3) δ 7.66-7.62 (m, 4H); 7.44-7.23 (m, 14H); 7.18-7.14 (m, 2H), 5.86 – 5.74 (m, 1H), 5.71-5.67 (m, 1H), 4.92 (d, 1H, J = 11.0 Hz), 4.73 (d, 1H, J = 11.6 Hz), 4.67 (d, 1H, J = 11.6 Hz), 4.65 (d, 1H, J = 11.0 Hz), 4.22-4.18 (m, 1H), 3.97 (dd, 1H, J = 9.9, 5.0 Hz), 3.80-3.71 (m, 2H), 2.46-2.29 (m, 1H), 2.26-2.11 (m, 1H), 2.07-1.90 (m, 1H), 1.08 (s, 9H). 13C NMR (CDCl 3) δ 139.1, 138.8, 135.9, 135.8, 133.8, 133.7, 129.8, 129.8, 128.8, 128.6, 128.5, 128.0, 128.0, 127.8, 127.8, 127.7, 127.5, 126.2, 81.8, 79.5, 74.6, 71.8, 63.6, 53.6, 41.4, 28.7, 27.1, 19.5.
Reaction of -O-TBDPS protected olefin (-)-2.25 under MCPBA/CH2Cl2 protocol.
Following the typical procedure described for the synthesis of diastereoisomeric epoxides 2.14α and 2.14β, a solution of -O-TBDPS derivative (-)-2.25 (0.050 g, 0.078 mmol, 1.0 equiv) in anhydrous CH2Cl2 (4.0 mL) was treated with MCPBA (0.040 g, 0.22 mmol, 2.0 equiv). After stirring for 18 h, typical workup afforded a crude (0.060 g) consisting of a 80:20 mixture (1H NMR) of the diastereoisomeric epoxides (-)-2.26α and (+)-2.26β which was purified by preparative TLC, by using a 9:1 hexane/AcOEt mixture as the eluant. Extraction of the more intense bands [the faster band contains (-)-2.26α] afforded epoxides (-)-2.26α (0.033 g, 73% yield) and (+)-2.26β (0.010 g, 20% yield).
6-O-t-buthyldiphenylsilyl-3,4-di-O-benzyl-5a-carba-α-D -glucopyranose: a liquid, Rf = 0.44 (9:1 hexane/AcOEt); []20
D -7.15 (c 0.71, CHCl3). 1H NMR (CDCl3) δ 7.64-7.60 (m, 3H), 7.60-7.31 (m, 15H), 7.30-7.10 (m, 2H), 4.88 (d, 1H, J = 10.9 Hz), 4.82 (d, 1H, J = 11.3 Hz), 4.71 (d, 1H, J = 11.3 Hz), 4.61 (d, 1H, J = 10.9 Hz), 4.00 (dd, 1H, J = 9.9, 4.0 Hz), 3.81-3.78 (m, 1H), 3.58 (dd, 1H, J = 9.8, 2.4), 3.50 (dd, 1H, J = 11.3, 8.1 Hz), 3.30-3.29 (m. 1H), 3.18 (d, 1H, J = 3.7 Hz), 2.18-2.11 (m, 2H), 1.74-1.60 (m, 1H), 1.07 (s, 9H). 13C NMR (CDCl 3) δ 140.0, 138.1, 135.9, 135.9, 133.7, 133.6, 129.9, 129.8, 128.7, 128.5, 128.2, 128.1, 127.9, 127.8, 127.5, 81.2, 79.4, 74.6, 73.0, 63.1, 54.4, 53.4, 34.9, 27.7, 27.2, 19.5. 6-O-t-buthyldiphenylsilyl-3,4-di-O-benzyl-5a-carba-β-D
-glucopyranose: a liquid, Rf = 0.3 (9:1 hexane/AcOEt); []20 D +9.33 (c 0.63, CHCl3). 1H NMR (CDCl3) δ 7.65-7.60 (m, 4H), 7.45-7.28 (m, 10H), 7.26-7.22 (m, 4H), 7.13-7.09 (m, 2H), 4.85 (d, 1H, J OBn BnO TBDPSO (-)-2.26 O OBn BnO TBDPSO (+)-2.26 O
= 10.6 Hz), 4.82 (s, 2H), 4.54 (d, 1H, J = 10.6 Hz), 3.85-3.78 (m, 2H), 3.73-3.62 (m, 2H), 3.34-3.27 (m, 2H), 2.18-2.05 (m, 2H), 1.73-1.32 (m, 1H), 1.07 (s, 9H). 13C NMR (CDCl
3) δ 138.8, 138.7, 136.0, 135.9, 133.7, 133.6, 129.8, 129.8, 128.6, 128.5, 128.1, 128.1, 127.9, 127.8, 127.7, 82.0, 77.6, 75.3, 72.7, 63.3, 55.6, 53.8, 53.6, 41.9, 27.1, 26.9, 19.5.
Reaction of epoxide (-)-2.26α with Cu(OTf)2/MeOH/CH2Cl2
As described for epoxide 2.14α, a solution of epoxide (-)-2.26α (0.026 g, 0.046 mmol, 1.0 equiv) in anhydrous CH2Cl2 (0.2 mL) was treated with MeOH (20 μL, 0.46 mmol, 10 equiv) and Cu(OTf)2 (0.010 g, 0.028 mmol, 0.6 equiv) and the resulting reaction mixture was stirred 48 h at room temperature. Typical workup afforded a crude product (0.023 g), consisting in a 60:30:10 mixture of the two regioisomeric trans methoxy alcohols 2.27 and 2.28 and bicyclic product 2.19 (1H NMR), which was subjected to preparative TLC, by using a 7:3 hexane/AcOEt mixture as the eluant. Extraction of the most intense bands (the slower band contains 2.19) afforded pure bicyclic product 2.19 (0.001 g, 1%) and the mixture of the two methoxy alcohols 2.27 and 2.28 (0.015 g), which turned out to be not separable under all the chromatographic conditions tried.
A solution of the 2:1 mixture of trans methoxy alcohols 2.27 and 2.28 in anhydrous pyridine (0.2 mL) was treated at 0 °C with Ac2O (0.1 mL) and the reaction mixture was stirred 18 h at room temperature. Co-evaporation with toluene afforded a 2:1 mixture of the corresponding acetyl derivatives (+)-2.27-Ac and (+)-2.28-Ac, which was subjected to preparative TLC by using a 7:3 hexane/AcOEt mixture (two runs) as the eluant. Extraction of the most intense bands [the faster band contains (+)-2.28-Ac] afforded pure methoxy derivatives (+)-2.27-Ac (0.005 g, 20%) and (+)-2.28-Ac (0.003 g, 10%).
2-O-acetyl-6-O-t-buthyldiphenylsilyl-3,4-di-O-benzyl-2-O-methyl-5a-carba-β-D-glucopyranoside: a liquid, Rf = 0.46 (7:3 hexane/AcOEt); []20 D +40.8 (c 0.22, CHCl3). 1H NMR (CDCl3) δ 7.66-7.58 (m, 4H), 7.46-7.26 (m, 12H), 7.25-7.21 (m, 2H), 7.12-7.04 (m, 2H), 5.05 (t, 1H, J = 9.6 Hz),4.83 (d, 1H, J = 10.5 Hz), 4.83 (d, 1H, J = 11.2 Hz), 4.66 (d, 1H, J = 11.2 Hz), 4.54 (d, 1H, J = 10.5 Hz), 3.93 (dd, 1H, J = 9.8, 3.2 Hz), 3.72-3.57 (m, 2H), 3.53-3.40 (m, 1H), 3.34 (s, 3H), 3.28-3.15 (m, 1H), 2.10-2.00 (m, 1H), 1.99 (s, 3H), 1.66-1.59 (m, 1H), 1.54-1.46 (m, 1H),1.07 (s, 9H). 13C NMR (CDCl 3) δ 170.5, OBn BnO TBDPSO (+)-2.27-Ac OAc OMe