CAPITOLO 3
:
MATERIALI E METODI
Biologia molecolare
3.1 Produzione del costrutto di
packaging
A partire dal p34TF10, un clone molecolare FIV contenente l’intero genoma provirale dell’isolato FIV Petaluma (Talbott et al. 1989), è stato prodotto il costrutto di packaging utilizzato nei diversi esperimenti, p∆env2, nel quale l’LTR al 5’ è stata sostituita dal promotore del citomegalovirus (pCMV), mentre quella al 3’ con il segnale di poliadenilazione dell’ormone della crescita bovino (BGHpolyA) (Figura 3.1). L’LTR al 5’ del p34TF10 è stato rimosso utilizzando gli enzimi di restrizione PshAI (New England Biolabs, Milano, Italia) (posizione 105 sulla flankling al 5’) e SacI (New England Biolabs, Milano, Italia) (507) generando un frammento di 620 bp. Il plasmide in seguito è stato purificato dai sali di reazione mediante precipitazione con 1/10 di volume di sodio acetato 3M pH 5.2 e 2 volumi di etanolo assoluto freddo. La soluzione è stata incubata per 2 ore a –80°C, dopodiché è stata centrifugata a 12000 rpm per 30 minuti a 4°C, il pellet è stato lavato con etanolo al 70% e risospeso in acqua. Dopo la precipitazione, il plasmide è stato successivamente defosforilato aggiungendo una unità di Shrimp Alcaline Phosphatase (SAP) (Promega, Milano, Italia) per ogni µg di DNA
.
L’enzima viene lasciato agire 30 minuti a 37°C in presenza di Buffer SAP 1x (Promega, Milano, Italia) e poi disattivato per 15 minuti a 65°C. Il pCMV è stato amplificato dal plasmide pcDNA3 (Invitrogen), con una coppia di primer pCMV-S tccgacaccggtcgtacgggccagatatacg-3’) e CMV-long-AS(5’-cgagctcaatttcgataagccagtaagcagtgggttctctagttagccagagtgctctgc-3’), che contengono alle estremità 5’ i siti di taglio per gli enzimi PshAI e SacI. L’amplificato
è stato controllato mediante corsa elettroforetica su gel di agarosio, dopodiché è stato precipitato per eliminare i sali di reazione e digerito con i due enzimi PshAI e
rev
LTR gag pol env LTR FIV p34TF10 Ψ
vif
ORF-A RRE
PshAI
ORF-A
pCMV gag pol ∆env (2Kb)
∆Ψ vif rev RRE BGH polyA p∆env2
SacI BlpI SalI
Figura 3.1: Costrutto di packaging utilizzato nelle prove di trasfezione; in alto è rappresentato il virus wild type.
SacI. Il frammento digerito è stato poi caricato su gel di agarosio allo 0,8%, dal quale è stata estratta la banda di interesse (660 bp) con il Jet quick gel extraction spin kit (Genomed, Bad Oeynhausen, Germany). A questo punto il plasmide e l’inserto così preparati sono stati ligati usando l’enzima T4 DNA ligasi (MBI Fermentas, Milano, Italia) partendo da 100-200 ng di plasmide e una quantità di inserto che può variare da un rapporto molare di 1 a 5 a uno di 1 a 30. Sul plasmide ricombinante così ottenuto è stata effettuata la sostituzione dell’LTR al 3’ mediante digestione con gli enzimi BlpI (MBI Fermentas, Milano, Italia) (9203) e SalI (MBI Fermentas) (che riconosce un sito di taglio sul polylinker del pUC119). Al termine della digestione, che stacca una banda di 742 bp, il DNA è stato precipitato e defosforilato secondo il protocollo precedentemente descritto. Allo stesso modo la BGHpolyA è stata amplificata da pcDNA3 usando primer senso, BGH-S acgcgctaagctagtgctcgctgatcagcctcgactgtg-3’), e antisenso BGH-AS (5’-acgcgtcgactccccagcatgcctgctattgct-3’) opportunamente disegnati in modo tale da inserire i siti di taglio per gli enzimi BlpI e SalI. La reazione di PCR effettuata ha dato luogo ad un amplificato di 240 bp che è stato digerito, purificato da gel
utilizzando la metodica descritta in precedenza e ligato al plasmide. Il costrutto finale risultante con entrambe le LTR sostituite è stato denominato p∆LTR.
Il costrutto p∆LTR è stato ulteriormente modificato al fine di rimuovere un’ estesa regione di env senza intaccare la sequenza codificante di rev e dell’RRE, producendo un costrutto contenente una delezione in env di circa 2 Kb. A questo scopo è stata disegnata una strategia basata su due PCR, con le quali vengono amplificate separatamente la parte iniziale e quella finale di env con l’esclusione della regione centrale da deletare (Figura 3.2).
Figura 3.2: Strategia di PCR utilizzata per la produzione del costrutto p∆env2.
La prima PCR è stata effettuata con i primer ORFA-FII tggcgaggatgctgtaatca-3’) e ∆env-AS (5’-ctggaccaagcggccgccagtacgtagtcaagatcatatgaacgccatgaaccagggac-3’), mentre l’altra con ∆env-S (5’-cttgactacgtactggcggccgcttggtccagatccacaagatactaggatacacag-3’) e LPCR-AS (5’-cgacttctacaacgggagacagc-3’). I due primer ∆env-S e ∆env-AS hanno la particolarità di contenere le sequenze FIV-specifiche alle rispettive estremità 3’ e due sequenze aspecifiche perfettamente complementari tra loro al 5’. Quando gli
amplificati delle due PCR vengono mescolati, previa estrazione da gel per eliminare il templato originale della miscela di reazione, si possono verificare due casi: 1) i filamenti di DNA delle due reazioni di amplificazione possono unirsi mediante le estremità 5’; 2) sono i filamenti complementari ad appaiarsi con le estremità 3’. Solo in quest’ultimo caso la Taq polimerasi può procedere con la polimerizzazione ed in presenza dei due primer ORFA-FII e LPCR-AS estendere i filamenti fino ad ottenere un amplificato che presenta l’estremità iniziale e quella finale di env, ma dal quale è stata tolta tutta la regione centrale (circa 2 Kb). A questo punto l’amplificato è stato digerito con KpnI (New England Biolabs, Milano, Italia) (6396) e AsuII (New England Biolabs) (8917) e, dopo estrazione da gel, il frammento di interesse di 420 bp è stato ligato al vettore p∆LTR precedentemente digerito con gli stessi enzimi e defosforilato.
3.2 Produzione dei costrutti vettore
Il costrutto vettore utilizzato nei diversi esperimenti di trasduzione è stato prodotto sulla base di uno precedentemente costruito a partire dal p34TF10 (KKS), un clone molecolare FIV contenente il gene env clonato direttamente da un isolato primario linfotropico. La precedente versione di costrutto vettore, denominata vCMV-GFP, contiene entrambe le LTR e una delezione nei geni gag-pol, al cui posto era stata inserita una cassetta di espressione contenente il pCMV e la green fluorescent protein (GFP), utilizzata come gene reporter (Figura 3.3).
vhLTR/CMV-GFP
vhLTR/PGK
vCMV-GFP
FIV p34TF10(KKS)
LTR gag pol env LTR
Ψ vif ORF-A RRE env LTR Ψ ∆vif RRE env LTR Ψ vif ORF-A RRE LTR env LTR Ψ vif ORF-A RRE pCMV GFP pPGK ∆gag ∆gag ∆gag ∆pol ∆pol ∆pol pCMV pCMV pCMV pCMV GFP GFP rev rev rev R U5 RU5 rev
Figura 3.3: Costrutti vettore utilizzati negli esperimenti.
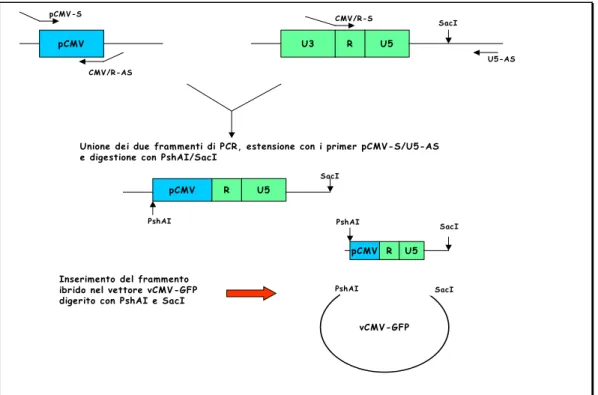
A partire dal vCMV-GFP è stato prodotto un nuovo vettore con LTR ibrida contenente il pCMV al posto dell’U3 al 5’. Il pCMV fuso con le regioni R/U5 dell’LTR al 5’, è stato ottenuto utilizzando la strategia illustrata nella Figura 3.4.
pCMV U3 R U5 CMV/R-AS pCMV-S SacI U5-AS CMV/R-S
Unione dei due frammenti di PCR, estensione con i primer pCMV-S/U5-AS e digestione con PshAI/SacI
pCMV R U5
SacI
PshAI
Inserimento del frammento ibrido nel vettore vCMV-GFP digerito con PshAI e SacI
vCMV-GFP pCMV R U5
PshAI SacI PshAI SacI
Figura 3.4: strategia usata per la produzione del vettore hLTR/CMV-GFP.
Il pCMV è stato amplificato dal pcDNA3 con i primer pCMV-S (contenente il
sito di restrizione PshAI all’etremità 5’) e CMV/R-AS. Quest’ultimo possiede, oltre ad una sequenza specifica per la produzione terminale del pCMV, una sequenza aggiuntiva al 5’ corrispondente al frammento iniziale della regione R dell’LTR di FIV. Una seconda PCR è stata effettuata a partire dal p34TF10 con i primer CMV/R-S e U5-AS, i quali amplificano un frammento di 526 bp che si estende dall’inizio di R fino ai primi 320 nucleotidi di gag. Al contrario del primer CMV/R-AS, il primer CMV/R-S, oltre alla sequenza specifica per R, contiene una sequenza aggiuntiva al 5’ specifica per la porzione terminale di pCMV. Come nel caso precedentemente descritto (Figura 3.4), il mescolamento dei due prodotti di PCR estratti da gel e ulteriormente amplificati con pCMV-S e U5-AS, porta alla formazione dell’LTR ibrida con il pCMV al posto dell’ U3. L’amplificato è stato successivamente digerito con gli enzimi PshAI e SacI (New England BioLabs, Milano, Italia) e, dopo estrazione da gel, è stato ligato al vettore vCMV-GFP precedentemente digerito con gli stessi enzimi e defosforilato. Il costrutto finale ottenuto, denominato hybrid LTR/CMV-GFP (vhLTR/CMV-GFP) (Figura 3.3), è stato sottoposto a successive modifiche per l’inserimento del promotore dell’enzima fosfoglicerato chinasi murino (pPGK) immediatamente a monte
del gene env. I due geni accessori vif ed ORF-A sono stati rimossi ed al loro posto inserito un polilinker mediante una strategia di PCR simile a quelle usate per la costruzione del costrutto di packaging e dell’ LTR ibrida. La prima PCR è stata effettuata con i primer IPe-S (5’-acatggtcctgctggagttcgtg-3’) disegnato sulla GFP contenuta nel vettore vhLTR/CMV-GFP, ed IPi-AS (5’-cggtccgcccgggctcgagttaattaagatatcatcatatgtaatatactctagtatgaaagc-3’) che contiene una parte della sequenza specifica di vif ed una parte contenente la sequenza di restrizione degli enzimi scelti per il polilinker: EcoRV, PacI, XhoI, SmaI, RsrII. Per la seconda PCR sono stati utilizzati i primer IPi-S (5’-gatatcttaattaactcgagcccgggcggaccgattttcatttgcaacaataagaatggc-3’) con la sequenza del polilinker complementare a quella del primer IPi-AS, ed una parte specifica all’estremità finale di ORF-A, ed IPe-AS (5’-gatcctaacttatttcgattacc-3’) disegnato sulla parte iniziale di env. I due frammenti ottenuti dall’amplificazione, rispettivamente di 641 e 408 bp, sono stati successivamente estratti da gel, mescolati e nuovamente amplificati con i due primer più esterni, IPe-S ed IPe-AS. Il frammento finale di 1016 bp contenente il polylinker, è stato precipitato e digerito con gli enzimi SacII e KpnI (New England BioLabs, Milano, Italia). Dopo estrazione da gel è stato legato al vettore vhLTR/CMV-GFP precedentemente digerito con gli stessi ezimi e defosforilato. Il pPGK è stato amplificato a partire da un plasmide già presente in laboratorio, con due primer contenenti uno la sequenza di restrizione di EcoRV e l’altro di SmaI, due degli enzimi di restrizione codificati dal polilinker di vhLTR/CMV-GFP. Il frammento, digerito con questi enzimi (Amersham Pharmacia Bioscience, Milano, Italia), è stato quindi inserito nel vettore linearizzato e defosforilato. Il prodotto è stato denominato vhLTR/PGK (Figura 3.3).
3.3 Produzione del plasmide esprimente Env ed il fGM-CSF
Il GM-CSF felino (fGM-CSF), precedentemente ricavato da macrofagi alveolari di un gatto FIV-infetto e clonato all’interno del plasmide pCR2.1-TOPO (InvitroGen, Milano, Italia), è stato amplificato a partire da quest’ultimo utilizzando i 2 primer GM-BspHI-S (5’-gctcatgagccaccatgtggctgcagaacctgc-3’), contenente il sito
di taglio per l’enzima BspHI, e GM-XbaI-AS (5’-gctctagattacttctggtctggtccccagc-3’), sul quale è stato inserito il sito di restrizione dell’enzima XbaI. La reazione di amplificazione è stata condotta a partire da 10 ng di templato iniziale utilizzando la DNA polimerasi proof reading PfuUltra (Stratagene, Milano, Italia) con un massimo di 30 cicli di amplificazione, seguendo le indicazioni fornite dal protocollo, per evitare l’inserimento di mutazioni nella sequenza codificante. L’amplificato è stato precipitato con sodio acetato ed etanolo assoluto, digerito con gli enzimi BspHI (New England BioLabs, Milano, Italia) e XbaI (MBI Fermentas, Milano, Italia) ed estratto da gel con il JET quick Gel Extration Spin kit (Genomed, Bad Oeynhausen, Germany). Il frammento è stato quindi clonato all’interno del plasmide di espressione eucariotica pVIVO-2 mcs (InvivoGen). Il plasmide è stato digerito con gli stessi enzimi utilizzati per il fGM-CSF, BspHI e XbaI, precipitato e defosforilato utilizzando il protocollo precedentemente descritto. A questo punto il plasmide e l’inserto così preparati sono stati legati utilizzando l’enzima T4 DNA Ligasi (MBI Fermentas, Milano, Italia). Il prodotto ottenuto è stato chiamato pVIVO/fGM-CSF.
L’intera regione comprendente env, rev e la sequenza RRE di FIV è stata amplificata a partire da p34TF10(KKS). Per effettuare l’amplificazione sono stati disegnati 2 primer, uno immediatamente a monte dell’ATG comune ai due geni env e rev, env-Bam-S (5’-cgcggatccgccaccatggcagaaggatttgcagccaatagac-3’) contenente il sito di taglio dell’enzima BamHI, e l’altro a valle del codone di stop di rev, rev-Eco-AS (5’-cggaattccagtccctagtccataagcattc-3’), sul quale è stato inserito il sito di restrizione dell’enzima EcoRI. La reazione di amplificazione è stata condotta a partire da 50-100 ng di templato iniziale, utilizzando la DNA polimerasi proof-reading PfuUltra (Stratagene) come precedentemente descritto. È stato ottenuto un frammento di 2600 bp che è stato digerito direttamente nel buffer di amplificazione con gli enzimi BamHI (New England BioLabs, Milano, Italia) e EcoRI (New England BioLabs), ed estratto da gel con il JET quick Gel Extration Spin kit (Genomed).
Questa regione quindi è stata clonata all’interno del plasmide pVIVO-2 mcs (InVivoGen, Milano, Italia). Il plasmide è stato precedentemente linearizzato attraverso la digestione effettuata con gli stessi enzimi utilizzati per il frammento, defosforilato, seguendo il protocollo descritto in precedenza, e legato con la regione
contenente env-rev per azione dell’enzima T4 DNA ligasi (MBI Fermentas, Milano, Italia), ottenendo il prodotto denominato pVIVO/env-rev.
Allo stesso modo il frammento amplificato è stato clonato all’interno del costrutto CSF, ottenendo il plasmide ricombinante pVIVO/fGM-CSF/env-rev.
3.4 Propagazione dei plasmidi
I prodotti di ciascuna ligazione vengono usati per trasformare il ceppo JM109 di E.coli reso competente mediante trattamento con calcio cloruro (CaCl2 100
mM). Il protocollo di trasformazione prevede l'aggiunta del DNA plasmidico alla soluzione contenente i batteri competenti seguita da una incubazione per 30 minuti in ghiaccio. Viene poi effettuato lo shock termico a 42°C per 1 minuto dopodichè le cellule vengono subito trasferite in ghiaccio per 3 minuti. A questo punto viene aggiunto ad ogni provetta di cellule 1 ml di SOC (2% bacto-triptone, 0.5% estratto di lievito, 10mM NaCl, 25mM KCl, 50 mM MgCl2, 20mM glucosio) e le cellule vengono
lasciate a crescere per 1 ora a 37°C in agitazione. Al termine, le cellule vengono piastrate su piastre Petri contenenti LB agar (1% bacto-triptone, 0.5% estratto di lievito, 1% NaCl, 1.5% agar batteriologico) più ampicillina 50 µg/ml. Nel caso del plasmide pVIVO-2, contenente il gene di resistenza all’igromicina, il terreno selettivo di crescita è costituito da HigroAgar (InvivoGen). Dopo un'incubazione overnight a 37°C le singole colonie sono inoculate in terreno liquido LB (1% bacto-triptone, 0,5% di estratto di lievito, 1% NaCl) più ampicillina (oppure HygroTB nel caso del pVIVO-2) e cresciute in agitazione a 37°C per 16 h. Quindi viene prelevato 1.5 ml di crescita, centrifugato 5 minuti a 12000 rpm e dal pellet di batteri viene estratto il DNA plasmidico mediante lisi alcalina. Brevemente, il pellet è risospeso in 100 µl di Soluzione I (50mM glucosio, 25mM tris a pH 8, 10mM EDTA a pH 8) a cui sono aggiunti 200 µl di Soluzione II di lisi (0.2N NaOH, 1% SDS) e 150 µl di Soluzione III di neutralizzazione (3M potassio acetato e 5M acido acetico glaciale). Il lisato batterico ottenuto viene centrifugato per 5 minuti a 4°C a 12000 rpm ed al surnatante, contenente il DNA plasmidico, viene aggiunto un uguale volume di fenolo-cloroformio. Una centrifugazione a 4°C a 12000 rpm per 5 minuti permette la
formazione di due fasi, quella inferiore contenente fenolo-cloroformio e proteine, che ha una densità maggiore, e quella superiore contenente il DNA. Viene prelevata la fase superiore alla quale sono aggiunti 0.7 volumi di isopropanolo ed il DNA è lasciato precipitare centrifugando a 4° a 12000 rpm per 30 minuti. Al termine, il pellet di DNA, evidente sul fondo della provetta, è lavato con 200 µl di etanolo al 70%, asciugato e risospeso in 20-25 µl di H2O più RNasi (20µg/ml). Dopo aver fatto agire
l’RNasiA (Sigma Aldrich) a 37°C per 30 minuti viene controllato 1 µl della soluzione contenente il DNA plasmidico su un gel di agarosio all'1%.
Per ottenere preparazioni pulite e su più larga scala di DNA plasmidico da impiegare nei saggi di trasfezione, è stato invece utilizzato il kit di estrazione Plasmid Midi Kit (Qiagen, Hilden, Germany) seguendo le istruzioni del protocollo e partendo da 50 ml di crescita batterica. Il DNA estratto è stato infine quantificato mediante lettura spettrofotometrica ed un'aliquota è stata corsa su gel di agarosio allo 0.8% per verificarne l'integrità. Per appurare l’integrità delle sequenze dei frammenti clonati e valutare l’eventuale presenza di mutazioni, i plasmidi estratti sono stati di volta in volta sequenziati mediante reazione basata sul metodo di terminazione a catena di Sanger, con l’utilizzo di primer marcati con un fluorocromo (Cy5’). I frammenti ottenuti con questa reazione sono stati caricati su gel di poliacrilammide (Amersham Pharmacia Bioscence) e la corsa elettroforetica è stata analizzata mediante sequenziatore automatico, Automatic Laser Fluorescence (A.L.F.) DNA Sequencer (Amersham Pharmacia Biosciences).
3.5 Lettura spettrofotometrica
Per valutare la purezza e la concentrazione del DNA estratto viene effettuata una lettura allo spettrofotometro UV-VIS (Beckman D.U. 640, Fullerton, CA, USA). La densità ottica (O.D.) è stata misurata alla lunghezza d’onda di 260 nm, alla quale gli acidi nucleici presentano un massimo di assorbimento, e a 280 nm, a cui assorbono diversi aminoacidi. Per definire la presenza di eventuali contaminanti nel campione, è stato considerato il rapporto O.D.260/O.D.280 accettando i valori vicini a
1,8 considerati con un buon grado di purezza. La concentrazione di DNA viene calcolata assumendo che un O.D.260 corrisponde ad una concentrazione di DNA a
doppia elica di 50 ng/µl ed utilizzando quindi la seguente relazione: DNA (ng/µl)= O.D.260 x 50 x fattore di diluizione.
3.6 Elettroforesi su gel di agarosio
L’agarosio è stato sciolto in tampone TAE (Tris-acido acetico, EDTA) 0,5X (242 g di Tris base, 57,1 ml di EDTA 0,5 M PH 8), scegliendo la concentrazione in base alla risoluzione desiderata, dopodiché è stato aggiunto bromuro di etidio (50 ng/ml) e la miscela è stata fatta solidificare in una vasca elettroforetica. I campioni di DNA da analizzare sono stati mescolati con loading buffer 10X (0,25% di blu di bromofenolo, 0,25% xilene cianolo, 30% glicerolo in H2O) in rapporto 10:1, e quindi
caricati sul gel a cui è stata applicata una differenza di potenziale di 60-90 V. A corsa terminata i gel sono stati fotografati mediante transilluminatore ad UV (Ultra Violet Products).
Biologia cellulare
3.7 Linee cellulari utilizzate
• La linea cellulare di fibroblasti renali felini Crandell (CrFk) viene mantenuta a 37°C, in atmosfera umidificata, con il 5% di CO2 in terreno DMEM (Dulbecco’s
Modified Eagle’s Medium, Sigma Aldrich, S.Louis, USA) addizionato con il 10% (vol/vol) di FBS (siero fetale bovino, Sigma Aldrich) inattivato al calore, 2mM di L-Glutammina, 1000U/ml di penicillina (Eurobio, Francia), 100µg/ml di streptomicina (Eurobio, Francia), 1% di aminoacidi non essenziali (Sigma Aldrich) (DMEM completo). Per l’espansione delle cellule, al raggiungimento della confluenza viene rimosso il terreno e le cellule sono lavate con PBS (NaCl 8 g/l, KCl 0,2 g/l, KH2PO4 0,2 g/l, Na2HPO4 (H2O)12 2,89 g/l)e staccate
con tripsina (Tripsina/EDTA, Eurobio, Francia). Le fiasche con tripsina vengono incubate per alcuni minuti a 37°C per permettere all’enzima di agire,
per bloccare l’azione della tripsina. Le cellule staccate vengono raccolte e centrifugate a 1200 rpm per 5 minuti dopodiché sono risospese in terreno fresco, contate nella camera di Burker, poi seminate di nuovo, in diluizioni opportune, nelle fiasche o nelle piastre da trasfezione.
• La linea cellulare 293T, fibroblasti renali embrionali umani trasformati con l’antigene T del simian virus 40 (SV40), viene mantenuta a 37°C in atmosfera umidificata, con il 5% di CO2 , in terreno DMEM completo al 10% di FBS. Per
l’espansione si procede nello stesso modo delle CrFk.
• La linea cellulare eritroblastoide umana TF-1 viene mantenuta in terreno RPMI 1640 (Sigma Aldrich) addizionato con il 10% di FBS, 2mM di L-Glutammina, 1,5 g/l di sodio bicarbonato, 4,5 g/l di D-glucosio, 10mM di HEPES, 1,0 mM di sodio piruvato, 1000 U/ml di penicillina e 100 µg/ml di streptomicina. Al terreno vengono aggiunti 2 ng/ml di hGM-CSF (Mielogen, Schering-Plough, Milano, Italia), componente fondamentale per la crescita delle TF-1. Le cellule vengono cresciute in fiasca ad una concentrazione compresa tra 1x10^5 e 1x10^6 cellule per ml di terreno, a 37°C in atmosfera umidificata con il 5% di CO2. Le cellule sono state diluite ogni 2 giorni: sono
state raccolte e centrifugate a 1200 rpm per 5 minuti, contate in Tripan Blu nella camera di Burker e risospese in terreno fresco alla concentrazione consigliata.
• La linea cellulare linfoblastoide T felina MBM (Matteucci et al. 1995) CD4-, CD8-, IL-2 e Con-A dipendenti, è stata mantenuta in terreno RPMI 1640 (Sigma Aldrich), addizionato con 10% di FBS, 2mM di L-Glutammina, 1% di aminoacidi non essenziali (Sigma Aldrich), 5 µg/ml di ConA, 20 U/ml di IL-2 ricombinante umana (Roche, Basel, Svizzera). Due volte alla settimana le colture sono state passate e addizionate di terreno fresco e IL-2, mentre una volta alla settimana è stata aggiunta ConA.
• La linea cellulare FL4, linfociti T felini cronicamente infettati con FIV-Petaluma, è stata mantenuta in terreno RPMI 1640 (Sigma Aldrich) addizionato con 10% di FBS, 2mM di L-Glutammina, 1% di aminoacidi essenziali (Sigma Aldrich), 1000U/ml di penicillina, 100 µg/ml di streptomicina. Le colture sono state cresciute in fiasca in atmosfera umidificata a 37°C con il 5% di CO2.
3.8 Preparazione DNA per trasfezione
Il DNA da utilizzare nella trasfezione viene precipitato in 1/10 di volume di sodio acetato che viene aggiunto al volume di templato, contenente la quantità di DNA desiderata, insieme a 2 volumi di etanolo assoluto freddo. I campioni cosi preparati vengono lasciati a –20°C overnight, ed il giorno dopo, a precipitazione avvenuta, vengono centrifugati a 12000 rpm per 30 minuti a 4°C. Una volta eliminato il surnatante, sono stati aggiunti 200 µl di etanolo al 70% ed i campioni sono stati nuovamente centrifugati a 12000 rpm per 10 minuti a 4°C. Il pellet ottenuto è stato separato dal surnatante ed asciugato.
3.9 Trasfezione
Il vettore è stato prodotto sia su CrFk che su 293T, mediante cotrasfezione del costrutto di packaging, costrutto vettore e plasmide esprimente Env, in rapporto molare variabile. Ventiquattro ore prima di iniziare la trasfezione le cellule sono state lavate con PBS 1X, contate e seminate nei pozzetti (8x10^5 nelle piastre da sei pozzetti; 2x10^6 nelle piastre da 100 mm, la quantità di cellule è stata scelta in modo tale da avere al momento della trasfezione una confluenza del 60-80%), contenenti terreno DMEM completo al 10% di FBS. Un’ ora circa prima della trasfezione, il terreno è stato sostituito con terreno fresco, 4,5 ml nel caso delle piastre da sei pozzetti, 9 ml per le piastre da 100 mm.
Il protocollo di trasfezione usato è quello con calcio fosfato. Il DNA precipitato è stato risospeso in buffer TE 0,1x a pH 8.0 (1mM TRIS-HCl, 0,1 mM EDTA), dopodiché è stato aggiunto CaCl2 2,5 M. Questa miscela è stata aggiunta a
gocce all’ HEPES 2x a PH 7.0 (280 mM NaCl, 10 mM KCl, 1,5 mM Na2PO4 x 2H2O,
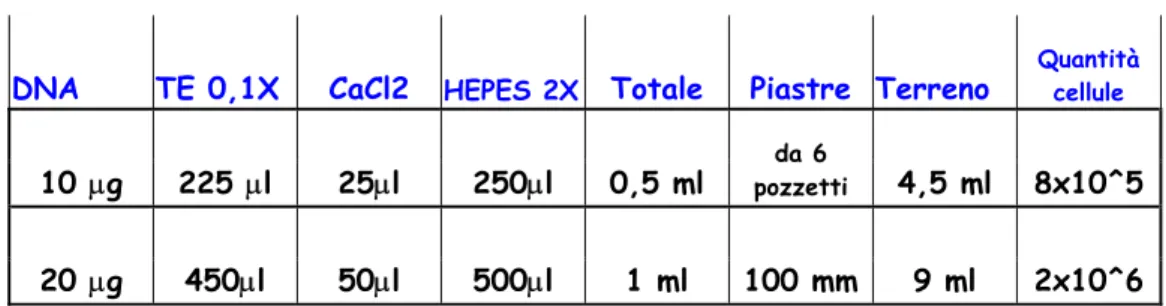
12mM destrosio, 50 mM Hepes). Le soluzioni sono state aggiunte nelle proporzioni indicate nella tabella 3.1.
DNA TE 0,1X CaCl2 HEPES 2X Totale Piastre Terreno Quantità cellule
10 µg 225 µl 25µl 250µl 0,5 ml pozzetti da 6 4,5 ml 8x10^5
20 µg 450µl 50µl 500µl 1 ml 100 mm 9 ml 2x10^6
Tabella 3.1: Protocollo trasfezione con calcio-fosfato.
I campioni vengono quindi incubati per 20 minuti a temperatura ambiente, dopodiché la soluzione viene distribuita goccia a goccia in modo uniforme sulla piastra contenente le cellule da trasfettare. La piastra è stata successivamente incubata per 6-8 ore a 37°C in presenza di CO2 al 5%, dopodiché è stato rimosso il terreno, è
stato effettuato un lavaggio con PBS 1X per eliminare DNA plasmidico non incorporato e quindi incubate in presenza di terreno fresco aggiunto in un volume di 3 ml e 10ml in piastre da 6 pozzetti e piastre da 100 mm rispettivamente.
3.10 Trasduzione
Per testare l’efficienza di trasduzione del vettore FIV sono state utilizzate diverse linee cellulari. Il vettore è stato testato sia su CrFk che su MBM. Per quanto riguarda le CrFk, 24 ore prima della trasduzione sono state seminate in piastre da 24 pozzetti contenenti 1X10^5 cellule/pozzetto con 1 ml di terreno DMEM completo. Le MBM sono state seminate nelle piastre da 24 pozzetti alla concentrazione di 1-5X10^5 cellule/pozzetto, in 1 ml di RPMI 1640 completo al 10% di FBS, addizionato di Con-A e IL-2.
Tre giorni dopo la trasfezione il surnatante delle cellule trasfettate è stato raccolto e chiarificato con centrifugazione a 1700 rpm per 10 minuti, per eliminare eventuali cellule morte presenti in sospensione, poi filtrato con filtri da 0.45 µm (Sarstedt, Numbrecht, Germania). Sono stati seguiti due diversi protocolli di trasduzione, testati in parallelo, consistenti in due diversi metodi di concentrazione del vettore prodotto.
Nel primo caso il surnatante filtrato e chiarificato è stato ultracentrifugato a 25000 rpm per 2 ore a 4°C utilizzando la centrifuga Optima TL (Beckman, Fullerton, CA, USA) con rotore TLA 100.4, dopodiché il pellet è stato risospeso in terreno fresco ed aggiunto alle cellule da trasdurre. Nel secondo caso al surnatante è stata aggiunta poli-L-lisina 500 µg/ml (Poly-L-Lysine hydrobromide, Sigma Aldrich), la soluzione è stata incubata a 4°C per 30 minuti e infine centrifugata a 10000 rpm per 2 ore a 4°C nella centrifuga J2-MC (Beckman) con rotore JA-20. Il pellet è stato risospeso in terreno fresco ed aggiunto alle cellule. In tutte le linee cellulari è stato utilizzato in parallelo sia il surnatante non concentrato che quello concentrato con ultracentrifugazione e poli-L-lisina.
Dopo circa 6 ore il terreno è stato aspirato, le cellule sono state lavate con PBS 1X, ed è stato aggiunto terreno fresco.
Le colture sono state propagate fino ad un massimo di 30 giorni, andando a valutare a vari tempi l’espressione di GFP mediante analisi al FACS: l’efficienza di trasduzione è stata valutata come percentuale di cellule GFP-positive. Inoltre per valutare l’infettività residua del vettore, le cellule trasdotte sono state monitorate periodicamente per produzione virale misurando il rilascio dell’antigene p25 nel mezzo di coltura.
3.11 Analisi al FACS
Per effettuare la lettura al citofluorimetro (FACScan, Becton Dickinson), le cellule trasfettate e trasdotte sono state lavate, tripsinizzate e centrifugate a 1200 rpm per 5 minuti. Il pellet è stato risospeso in FACS Buffer (PBS 1X, 0,2% di BSA, 0,1% di NaN3), centrifugato nuovamente e fissato con FACS FIX (FACS Buffer, 1%
di paraformaldeide).
I dati sono stati interpretati andando ad osservare lo spostamento delle cellule esprimenti GFP verso valori più alti di fluorescenza rispetto a cellule di controllo (cellule non trattate o dopo trasduzione mock).
3.12 Dosaggio immunoenzimatico della proteina p25
Questo saggio è stato utilizzato per valutare la produzione della poliproteina Gag-Pol da parte dei costrutti di packaging in seguito a trasfezione delle cellule CrFK e delle 293T, e per valutare l'infettività residua del vettore dopo traduzione di CrFk ed MBM. A tale scopo è stato utilizzato un test immunoenzimatico (ELISA) impiegando 100 µl di surnatante delle cellule raccolti a vari tempi.
Questo saggio permette la rivelazione e la quantificazione della proteina capsidica p25 di FIV sfruttando due anticorpi monoclonali rivolti verso due epitopi diversi (Lombardi et al. 1994) per immobilizzare la proteina rilasciata nel surnatante di coltura.
Il coating viene effettuato su piastre ELISA lasciando incubare overnight l'anticorpo monoclonale DF3 (0.25 µg/pozzetto) in tampone carbonato a pH 9.5 (Na2CO3 1,59 g/l, NaHCO3 2,93 g/l, NaN3 0,2 g/l). Il giorno successivo, per saturare
completamente la superficie del pozzetto viene effettuato il post-coating aggiungendo ai pozzetti 100 µl di skim milk all’1% diluito in PBS. Dopo un’ora di incubazione a temperatura ambiente vengono seminati i campioni in presenza di tampone di lisi (Tween 20 0,5%, Triton 5%, PBS) e dopo altre due ore viene aggiunto l’anticorpo DF10 biotinilato (100 µl/pozzetto). La rivelazione viene effettuata mediante un anticorpo anti-biotina coniugato con l’enzima perossidasi (100 µl/pozzetto) ed aggiungendo il substrato di reazione. Nei campioni positivi viene osservata la comparsa di un colore azzurro di intensità proporzionale alla quantità di p25 presente. La lettura dell’assorbanza viene eseguita ad una lunghezza d’onda compresa tra i 450 e i 650 nm. Sono considerati positivi i campioni che mostrano valori di densità ottica (D.O.)maggiori di cinque volte la media dei valori dei controlli negativi o comunque maggiori di 0.050.
3.13 Valutazione dell’attività biologica del fGM-CSF
3.13.1 Valutazione della cinetica di crescita delle TF-1 in presenza di fGM-CSF
Al fine di valutare l’attività biologica del fGM-CSF clonato, è stata utilizzata la linea cellulare eritroblastoide TF-1, mantenuta e propagata come descritto nella sezione 3.7.
Il fGM-CSF è stato prodotto mediante trasfezione su CrFk di 10 µg del costrutto pVIVO/fGM-CSF. Sono state seminate 3 quantità diverse di CrFk in una piastra da sei pozzetti, rispettivamente 1X10^6, 8X10^5, 5X10^5, per raccogliere il surnatante a tre tempi diversi. In parallelo è stato utilizzato anche il solo vettore pVIVO-2, come controllo negativo. A 6 ore dalla trasfezione, le cellule sono state lavate in PBS 1X e coltivate in terreno completo utilizzato per le TF-1. Il surnatante delle cellule trasfettate è stato raccolto dopo 24 ore, 2 giorni e 3 giorni, ed è stato testato sulle TF-1, utilizzando diverse diluizioni (1:5, 1:10, non diluito).
Le TF-1 sono state seminate in una piastra da 24 pozzetti in una concentrazione di 1X10^5 cellule/pozzetto in 1 ml di terreno. Ogni prova è stata fatta in triplicato, per effettuare la lettura dopo tempi diversi di incubazione con la citochina. Le cellule contenute in ogni pozzetto, infatti, sono state contate nella camera Burker dopo 24 , 48 e 72 ore di incubazione con il fGM-CSF.
3.13.2 Saggio di linfoproliferazione con timidina triziata
Il fGM-CSF è stato prodotto mediante trasfezione su CrFk come descritto nel paragrafo precedente. Il surnatante delle cellule trasfettate è stato raccolto a 48 ore dalla trasfezione, è stato chiarificato a 1700 rpm per 5 minuti per eliminare eventuali cellule morte e filtrato con filtri da 0,45 µm (Sarstedt, Numbrecht, Germania).
Le cellule della linea TF-1 sono state fatte crescere per 2 giorni in terreno privato di hGM-CSF, dopodiché sono state seminate in una piastra da 96 pozzetti in quadruplicato alla concentrazione di 5X10^4 cellule/pozzetto, a cui sono è stato aggiunto il surnatante (200µl) proveniente dalla trasfezione delle CrFk, adattate per questo scopo, alla crescita in RPMI 1640 completo, il terreno di crescita delle TF-1. Sono state testate diverse diliuizioni: 1:5, 1:10, oltre al surnatante non diluito. Oltre
al controllo costituito dal solo pVIVO-2 trasfettato sulle CrFk, sono stati utilizzati altri 3 controlli, due negativi costituiti dal surnatante delle CrFk non trasfettate e dal solo terreno RPMI 1640 fresco, ed uno positivo costituito dalle TF-1 coltivate in terreno completo addizionato di hGM-CSF (Mielogen). Le TF-1 sono state quindi incubate in presenza della citochina per 48 ore a 37°C, dopodiché ad ogni pozzetto sono stati aggiunti 0,75 µCi di timidina triziata (3H-timidina), con cui le cellule sono
state incubate per 5 ore. Alla fine dell’incubazione le cellule sono state raccolte su un filtro ed è stata analizzata la quantità di 3H-timidina incorporata nel DNA con un
contatore di particelle β (1450 Microbeta Trilux, Perkin Elmer) che misura l’emissione di radioattività in cpm. I risultati sono stati interpretati in funzione dell’indice di stimolazione (IS), ossia il rapporto tra la media del numero di cpm prodotto dalle cellule stimolate con la citochina, con la media del numero di cpm prodotto dalle cellule mock stimolate, il controllo negativo. Per convenzione solo quando IS è maggiore o uguale a 2 indica proliferazione specifica.
3.14 Valutazione dell’espressione di Env
Per valutare l’espressione di Env da parte del costrutto vettore e dei due plasmidi esprimenti Env (pVIVO/env-rev e pcDNA3 L-env/CTE), è stato effettuato
un western blot sulle cellule trasfettate e tradotte. Circa 1X10^5 cellule, precedentemente lavate in PBS, sono state risospese in 20 µl del buffer di lisi (Tris 10 mM, EDTA 2mM, NaCl 0,15 mM, Nonidet 0,5%), a cui sono stati poi aggiunti 20 µl del colorante SDS Sample Buffer 2X (Tris-HCl 0,5 M PH 6.8, glicerolo 10%, SDS 10%, β-mercaptoetanolo, 0.05% blu di bromofenolo). I campioni così preparati sono stati bolliti per 7 minuti e caricati su gel di acrilammide/bisacrilammide al 10%. Lo stacking gel è stato preparato con il 30% di mix acrilammide/bisacrilammide (Acrilamide/Bis Solution, 29:1. Bio-Rad Laboratories, Hercules, CA, USA), Tris-HCl 0.5 mM pH 6.8, 10% SDS, 10% APS, TEMED (Sigma Aldrich). Il resolving gel è stato preparato con il 30% di mix acrilammide/bisacrilammide (BioRad Laboratories), Tris-HCl 1.5 mM pH 8.8, 10% SDS, 10% APS, TEMED. La corsa elettroforetica è stata effettuata in tampone running buffer 1x (Tris base 25 mM, 0.1% SDS, 1.44% glicina, pH 9.0), ad una differenza di potenziale di 80-100 volt.
Il trasferimento delle bande sulla membrana di nitrocellulosa (Amersham Pharmacia Bioscience) è stato effettuato in presenza del buffer di trasferimento (0.3% Tris-base, 1.44% glicina, 20% metanolo) ad una differenza di potenziale di 100 volt, dopodichè è stato effettuato il bloccaggio con PBS-skim milk al 3%. La membrana è stata ibridata con l’anticorpo monoclonale αenv -71.2 diluito 1:500 in PBS Twin-skim milk all’1%. Dopo un’ora è stato aggiunto l’anticorpo secondario α-mouse perossidato (BioRad Laboratories) diluito 1:1000 in PBS Twin-skin milk allo 0.5%. La rivelazione delle bande è stata effettuata con un substrato contenente diaminobenzidina (DAB 40 mg/ml),Tris HCl 100 mM, perossido di idrogeno (H2O2),
NiCl2 80 mg/ml.