16
Lo stress metabolico nelle cellule target dell’insulina provoca un
accumulo di specie reattive dell’ossigeno (ROS)
(2)Un ipotetico meccanismo comune per la resistenza all’insulina
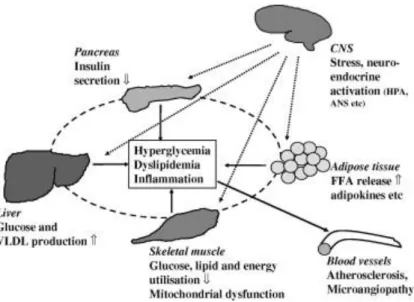
La sindrome metabolica è un insieme di fattori di rischio cardiovascolari, e l’adiposità viscerale è una componente centrale fortemente associata con la resistenza all’insulina. Sia l’obesità viscerale, sia la resistenza all’insulina rappresentano due fattori di rischio importanti per lo sviluppo del diabete di tipo 2. E’ probabile che il tessuto adiposo, ed in particolare quello localizzato nella regione intra-addominale, sia parte di un meccanismo complesso che coinvolge diversi tessuti, e che disordini ormonali, metabolici e neuronali tra diversi organi possano provocare l’insorgenza della malattia metabolica. Secondo un’ipotesi interessante, la maggior parte dei fattori di rischio che portano alla resistenza all’insulina sono dovuti all’accumulo anormale di specie reattive dell’ossigeno (ROS). Ci sono diverse prove che suggeriscono che gli effetti dannosi di glucosio, acidi grassi, ormoni e citochine, che provocano la resistenza all’insulina, possano seguire un percorso comune.
17
Lo stress e la resistenza all’insulina nella sindrome metabolica e nel diabete
di tipo 2
Con il termine stress viene definito un ampio concetto che racchiude un insieme di fattori capaci di alterare i processi omeostatici dell’organismo. Esistono attualmente diverse prove sottese a dimostrare che vari fattori di stress possano provocare l’insorgenza della sindrome metabolica e del diabete di tipo 2. Tali fattori di stress si possono trovare a vari livelli che vanno dall’ambiente socio-economico, alle condizioni psicologiche, neuroendocrine e metaboliche. Lo stress ossidativo causato dall’accumulo intracellulare di specie reattive dell’ossigeno (ROS, Reactive Oxigen Species) è stato implicato nell’aterosclerosi, nelle complicazioni microvascolari del diabete, ma anche nella degenerazione delle cellule beta nel diabete di tipo 2. Recenti evidenze suggeriscono, inoltre, che le specie reattive dell’ossigeno abbiano un ruolo importante in varie forme di resistenza all’insulina.
Nella maggior parte dei casi il diabete di tipo 2 è provocato da una disfunzione delle cellule beta e dalla resistenza all’insulina. L’inattività fisica, l’adiposità dovuta all’eccessiva alimentazione, lo stress ed il fumo sono fattori di rischio che, in sinergismo con la predisposizione genetica, provocano l’insorgenza della malattia. Il termine “sindrome metabolica” è spesso usato per definire un insieme di markers per la diagnosi di patologie cardiovascolari, ma anche del diabete di tipo 2. Un numero sempre maggiore di evidenze suggerisce che l’obesità addominale è una componente centrale di questa sindrome, che include anche ipertensione, dislipidemia e disordini nel metabolismo del glucosio. La resistenza all’insulina è stata indicata come uno dei principali responsabili della maggior parte dei sintomi che caratterizzano la sindrome metabolica.
La resistenza all’insulina sembra essere un componente importante dei complessi processi patofisiologici che sottendono allo sviluppo del diabete di tipo 2 e, possibilmente, anche all’insorgenza di altre patologie correlate come la dislipidemia, l’ipertensione e l’aterosclerosi. La resistenza all’insulina può essere definita come un effetto attenuato dell’insulina sulle cellule bersaglio, che sono principalmente il muscolo, il tessuto adiposo ed il fegato. Nel muscolo, l’uptake del glucosio transmembrana, stimolato dall’insulina, sembra essere il principale difetto che limita la velocità di assorbimento. Nel tessuto adiposo la resistenza all’insulina si manifesta, invece, come un difetto sia nell’uptake, sia nell’utilizzo del glucosio, ma in molti casi anche attraverso una mancata soppressione della lipolisi e del rilascio degli acidi grassi liberi (FFA, Free Fatty Acids). Nel fegato l’azione dell’insulina può essere attenuata riguardo all’uptake del glucosio ed al suo accumulo, ma anche per la riduzione nella produzione di glucosio stesso e della proteina VLDL (Very Low Density Lipoprotein). E’ interessante notare che può esserci insulinoresistenza anche nelle cellule beta del pancreas endocrino e questo può essere rilevante per il diabete di tipo 2, dal momento che provoca riduzione della sintesi della pre-proinsulina e,
18
conseguentemente, della produzione dell’ormone. Lo stress si può tradurre nel rischio di insorgenza del diabete di tipo 2 e della sindrome metabolica in parte attraverso il pathway di resistenza all’insulina, ma ci possono essere ovviamente anche altre vie attraverso le quali si possono alterare le funzioni delle cellule beta pancreatiche e delle cellule endoteliali dei vasi.
Sebbene siano stati compiuti notevoli sforzi per delucidare i meccanismi primari responsabili dell’insulinoresistenza nella sindrome metabolica e nel diabete di tipo 2 (schema Fig.1), attualmente non vi è un largo consenso di opinioni. Naturalmente vi sono diverse prove che sottolineano l’importanza dei fattori genetici in questa patologia.
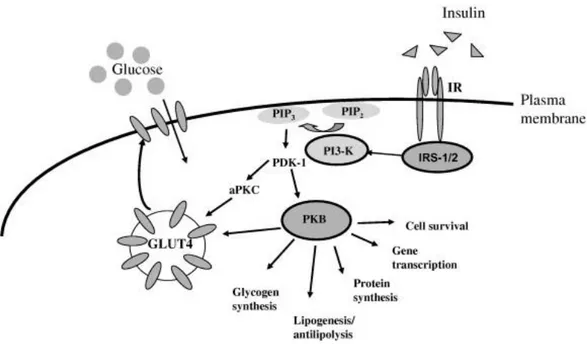
Un’ipotesi ovvia circa la causa della resistenza all’insulina riguarda l’esistenza di un possibile difetto primario, ereditato o acquisito, nel sistema di trasduzione del segnale delle cellule bersaglio dell’insulina (Fig.2). Mutazioni gravi a carico dei componenti del meccanismo cellulare di segnale dell’insulina potranno causare certamente una marcata resistenza all’ormone. Tuttavia questi difetti sono molto rari e non si riscontrano nella maggior parte dei pazienti affetti da sindrome metabolica o da diabete di tipo 2; al contrario ci sono molte prove che suggeriscono che la resistenza cellulare all’insulina non sia un disturbo primario nello sviluppo della resistenza esteso a tutti i tessuti dell’organismo. Per esempio, soggetti con predisposizione al diabete possono mostrare una responsività cellulare all’insulina regolare, e in cellule bersaglio ottenute da pazienti con diabete di tipo 2 sembra che l’insulinoresistenza sia largamente reversibile. Pertanto è possibile che si possano manifestare dapprima alterazioni nell’ambiente extracellulare che possono portare, in un secondo momento, ad una resistenza all’insulina a livello cellulare nel muscolo o nel tessuto adiposo. Questi fattori che riguardano l’ambiente tissutale possono coinvolgere segnali metabolici, neurali ed ormonali. Riguardo ai fattori metabolici è ben noto che alti livelli di glucosio e di acidi grassi liberi, che sono caratteristici del diabete di tipo 2, possano avere effetti deleteri su alcuni tessuti come il muscolo ed il fegato. Inoltre l’iperinsulinemia, che si verifica come meccanismo compensatorio durante lo sviluppo del diabete di tipo 2, può contribuire essa stessa allo sviluppo della resistenza all’insulina. Un concetto popolare nell’eziologia del diabete di tipo 2 e nella sindrome metabolica riguarda la vita intrauterina: la malnutrizione del feto sembrerebbe provocare, infatti, un incremento nel rischio di malattie metaboliche in età adulta. Anche questo può essere considerato come un fattore di stress ambientale che può condurre, presumibilmente, ad una iperattivià neuroendocrina attraverso l’imprinting nell’utero.
19
Fig. 11. Interplay between CNS and other tissues in the pathophysiology of type 2 diabetes and the metabolic syndrome. ANS, autonomic nervous system; FFA, free fatty acids; HPA, hypothalamo–pituitary–adrenal system; VLDL, very low density lipoprotein.
Nonostante numerosi sforzi per comprendere in meccanismi sottesi alla resistenza all’insulina, non si e ancora giunti ad un consenso circa i difetti che possono esservi con esattezza a livello molecolare e cellulare. Ci sono numerose vie che possono contribuire allo sviluppo della resistenza all’insulina e del diabete di tipo 2. Queste vie includono fattori metabolici, come il glucosio e gli acidi grassi, che in concentrazioni elevate possono provocare effetti dannosi. Anche i meccanismi neuro-ormonali possono essere coinvolti, infatti i glucocorticoidi, l’ormone della crescita, gli steroidi sessuali, le catecolamine, ed anche l’insulina stessa possono avere tutti degli effetti sulla responsività all’insulina in diversi tessuti.
1
20
Dal momento che l’adiposità viscerale sembra essere una componente molto importante della sindrome metabolica ed anche uno dei maggiori fattori di rischio per lo sviluppo del diabete di tipo 2 e delle malattie cardiovascolari, anche i meccanismi legati al tessuto adiposo sono di interesse. Nella disfunzione del tessuto adiposo, mediatori dell’infiammazione come le citochine, le chemochine e le cellule infiammatorie, come linfociti, neutrofili e macrofagi, possono giocare un ruolo determinante.
Fig. 22. Insulin signalling via the PI3K-dependent pathway in adipocytes and skeletal muscle. Insulin binding to its receptor will lead to glucose transport activation and other metabolic effects and this is exerted via a cascade of signaling proteins. GLUT4, glucose transporter 4; IR, insulin receptor; IRS1/2, insulin receptor substrates 1 and 2; PDK1, phosphatidylinositol dependent protein kinase 1, PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol-3,4-phosphate; PIP3, phosphatidylinositol-3,4,5-phosphate; aPKC, atypical protein kinase C; PKB, protein kinase B.
2
21
Stress metabolico- glucosio, acidi, grassi ed insulina
A prescindere dalla causa primaria, quando la resistenza all’insulina si è instaurata, i livelli dell’ormone nel sangue aumentano per cercare di compensare l’incremento nei livelli di glucosio e degli acidi grassi in circolo. Normalmente esistono dei circuiti di feed-back finemente regolati a livello delle cellule beta secernenti insulina, in modo da mantenere non solo i livelli di glucosio, ma anche quelli degli acidi grassi nel range fisiologico. A seguito dell’instaurarsi del diabete di tipo 2, le alterazioni metaboliche peggiorano aggravando la resistenza all’insulina in una sorta di circolo vizioso. Nel diabete i valori di glucosio sono elevati cronicamente e si rilevano alti livelli di insulina nelle prime fasi della malattia, che si abbasseranno gradatamente man mano che procede la disfunzione delle cellule beta. Evidenze sperimentali suggeriscono che l’iperinsulinemia può causare la resistenza all’insulina. Inoltre, l’iperglicemia da sola può avere effetti deleteri sulla produzione di insulina e sull’azione stessa dell’ormone; un fenomeno questo noto con il termine di glucotossicità. Negli adipociti isolati, umani e di ratto, l’esposizione prolungata (di diverse ore) ad alti livelli di glucosio, particolarmente in presenza di alte concentrazioni di insulina, può alterare la capacità di trasporto degli zuccheri. Un comune denominatore per la perdita di funzione dell’insulina sull’uptake del glucosio negli studi sugli adipociti di ratto, ma non umani, sembra essere un ridotto contenuto di IRS-1 (Insulin Receptor Substrate-1). E’ interessante notare che bassi livelli di espressione del gene IRS-1, e della relativa proteina, sono stati considerati fattori di rischio per lo sviluppo dell’insulinoresistenza e del diabete di tipo 2; inoltre individui con diabete di tipo 2 conclamato mostrano una marcata riduzione dei livelli di espressione di IRS-1 e della sua funzione, mentre i livelli di IRS-2 sono inalterati. Sia nell’obesità che nel diabete di tipo 2 ci sono alti livelli di acidi grassi in circolo. Questi hanno effetti negativi sulla responsività all’insulina nel muscolo e nel fegato, ma non negli adipociti. L’aumento cronico degli acidi grassi può condurre anche ad una alterata secrezione di insulina nelle cellule beta a seguito dello stimolo del glucosio. Dati recenti suggeriscono che per quanto riguarda IRS-1 l’alterata funzionalità, piuttosto che la ridotta espressione, sia responsabile della resistenza all’insulina nello stress metabolico provocato da alti livelli di insulina, glucosio e di acidi grassi. E’ possibile che questo sia mediato da un alterato stato di fosforilazione di IRS-1: ciò cambia l’interazione con altre proteine di segnale e sembra anche veicolare IRS-1 verso la degradazione nei proteosomi.
Naturalmente, gli alti livelli di insulina, glucosio o acidi grassi non sono considerati cause primarie della insulinoresistenza, tuttavia come fenomeni secondari possono indubbiamente contribuire alla permanenza del disturbo a livello cellulare e tissutale. Un
meccanismo importante attraverso il quale l’iperglicemia può condurre
all’insulinoresistenza è attraverso il rilascio di citochine. E’ interessante notare che alti livelli di acidi grassi, in particolare nella vena porta, possono condurre all’attivazione dell’asse ipotalamo-ipofisi-surrene, determinando una risposta simpatoadrenergica. Non a caso nei pazienti affetti da diabete di tipo 2 è stato riscontrato uno scarso controllo metabolico associato con alti livelli di citochine e cortisolo nel torrente ematico.
22
Stress neuroendocrino
Ci sono diversi ormoni con azione antagonista sull’insulina che svolgono ruoli importanti nel trasporto immediato di nutrienti ai tessuti, nel digiuno prolungato e per contrastare l’ipoglicemia indotta da farmaci. Questi ormoni includono il glucagone, l’adrenalina, la noradrenalina, l’ormone della crescita ed il cortisolo. La capacità di aumentare i livelli ematici di glucosio rappresenta comunque uno svantaggio quando c’è una iperproduzione di tali ormoni. In questo contesto l’asse ipotalamo-ipofisi-surrene, che controlla la secrezione di cortisolo ed il sistema simpatico, è stato studiato attentamente come una delle possibili vie attraverso le quali possono instaurarsi l’insulinoresistenza ed il diabete di tipo 2. Queste due vie originano dai “centri dello stress” nell’ipotalamo dove interagiscono l’un l’altra modulandosi vicendevolmente.
La sindrome di Cushing, dovuta ad un eccesso di glucocorticoidi, è associata con la resistenza all’insulina, l’intolleranza al glucosio, l’obesità e l’ipertensione. Anche trattamenti farmacologici con alte dosi di glucocorticoidi possono condurre ad una alterazione nella responsività all’insulina. Nell’obesità, la conversione del cortisone a cortisolo nel tessuto adiposo sembra essere elevata e questo può contribuire ad uno squilibrio nel metabolismo del glucosio e dei lipidi, ma anche all’accumulo stesso dell’adipe. L’effetto antagonista sull’insulina mediato dai glucocorticoidi include sia un’alterazione nell’uptake del glucosio nei tessuti periferici, sia una stimolazione della gluconeogenesi nel fegato. Sembra che ci siano degli effetti diretti a livello cellulare che possono condurre ad uno squilibrio nel trasporto del glucosio e, infatti, l’insulinoresistenza indotta dal desametasone nelle cellule adipose coinvolge probabilmente il processo di traslocazione del trasportatore del glucosio GLUT4. Parte dell’effetto antagonista mediato dai glucocorticoidi sembra essere secondario ad un effetto lipolitico che comporta l’innalzamento dei livelli ematici di acidi grassi. Inoltre i glucocorticoidi possono anche inibire la secrezione di insulina dalle cellule beta del pancreas e questo può ovviamente contribuire alle loro proprietà diabetogene.
L’attività del sistema simpatico e dei suoi mediatori esercita profonde interazioni con gli effetti metabolici dell’insulina. In questo modo, attraverso il rilascio delle catecolamine, il sistema nervoso autonomo può condurre direttamente alla resistenza all’insulina e, da qui, promuovere l’insorgenza del diabete di tipo 2. Inoltre, ci sono anche degli effetti secondari poiché l’attivazione del sistema simpatico induce la lipolisi, ed il conseguente aumento degli acidi grassi circolanti contrasta, a sua volta, l’azione dell’insulina nel muscolo e nel fegato.
Anche il sistema renina-angiotensina può rappresentare un’importante via neuroendocrina per lo sviluppo dell’insulinoresistenza. La renina è un enzima che catalizza la formazione dell’angiotensina I dall’angiotensinogeno e, successivamente, l’angiotensina II è formata per azione dell’enzima ACE (Angiotensin Converting Enzyme). L’angiotensina esercita effetti rilevanti a livello vascolare, ma promuove anche la produzione di aldosterone dalla
23
corteccia surrenale. Complessivamente gli effetti del sistema renina-angiotensina includono vasocostrizione, aumento della pressione sanguigna, ritenzione di liquidi e sodio ed escrezione del potassio a livello renale. I farmaci antiipertensivi che attivano il sistema renina-angiotensina sembrano causare l’insulinoresistenza, mentre i farmaci che riducono l’attività del sistema, come gli ACE inibitori o i bloccanti del recettore dell’angiotensina (ARB, Angiotensin Receptor Blockers) hanno un effetto positivo o neutro sulla responsività all’insulina.
Stress infiammatorio
Studi recenti hanno dimostrato che livelli elevati della proteina C reattiva (CRP, C Reactive Protein) e di diverse citochine sono associati con l’obesità, la resistenza all’insulina e con il diabete di tipo 2. Nel tessuto adiposo c’è una produzione locale di TNF-α e di interleuchina-6 (IL-6), ciascuna delle quali mostra proprietà antagoniste sull’insulina dovute ad un’interferenza nella trasduzione del segnale dell’ormone. Queste citochine hanno anche la capacità di attivare la lipolisi, ed è stato rilevato anche che il TNF-α può inibire la produzione di adiponectina. E’ interessante notare che è stata implicata anche un’interazione tra i livelli di cortisolo e di TNF-α, poiché quest’ultimo è in grado di promuovere la conversione del cortisolo in cortisone negli adipociti. Sia TNF-α che IL-6 possono stimolare la produzione di cortisolo in modo diretto, o attraverso l’asse ipotalamo-ipofisi-surrene. Dati recenti indicano che alti livelli di TNF-α, ma non di IL-6, sono correlati con l’insulinoresistenza nei pazienti affetti da diabete di tipo 2, mentre l’iperproduzione di entrambe le citochine è associato con l’iperglicemia. Sembrerebbe, pertanto, che nello stato iperglicemico TNF-α sia un fattore che contribuisce all’insulinoresistenza e che questo potrebbe verificarsi in parte a causa dell’aumento dei livelli di cortisolo. Anche i livelli di CRP sono fortemente correlati con la glicemia, ma sorprendentemente non con l’insulinoresistenza. Questi risultati suggeriscono che l’iperglicemia è un fattore importante che contribuisce all’aumento dei livelli di CRP nel
diabete conclamato e nel pre-diabete; perciò l’iperglicemia può condurre
24
Il tessuto adiposo come bersaglio per i fattori di stress
Attualmente ci sono numerosi studi che correlano i fattori psicosociali all’adiposità viscerale, alla sindrome metabolica ed al diabete di tipo 2. Gli eventi stressanti della vita, ma anche bassi livelli di educazione sono considerati fattori di rischio per il diabete di tipo 2 nella popolazione generale. Nelle donne, lo stress dovuto al lavoro ed uno scarso sostegno emotivo sono stati associati con lo sviluppo del diabete, mentre negli uomini il principale fattore di rischio è rappresentato dai disturbi del sonno. Le situazioni stressanti inducono risposte neuroendocrine che in una prospettiva a lungo termine possono portare allo sviluppo dell’adiposità viscerale ed al diabete di tipo 2. L’asse ipotalamo-ipofisi surrene ed il sistema nervoso simpatico mediano gli effetti dello stress in diversi organi: gli ormoni secreti da questi due sistemi, come l’adrenalina, la noradrenalina ed il cortisolo, contrastano gli effetti dell’insulina. Tuttavia, non ci sono ancora molti dati per dimostrare che i livelli di questi ormoni sono alterati in individui esposti ad un marcato stress psicosociale.
C’è una chiara relazione tra l’obesità viscerale e le caratteristiche della sindrome metabolica. Sebbene il rapporto causale non sia stato ancora stabilito, l’esistenza della relazione tra l’accumulo di adipe in sede addominale e l’insorgenza della resistenza all’insulina, così come del diabete di tipo 2, è stata generalmente accettata. E’ stato anche proposto un legame tra l’obesità viscerale ed i disordini dell’asse ipotalamo-ipofisi-surrene; sembra, infatti, che alti livelli di glucocorticoidi promuovano l’accumulo dei lipidi nelle viscere piuttosto che nel tessuto adiposo subcutaneo. Inoltre nell’uomo, il tessuto adiposo viscerale mostra una maggiore propensione all’infiammazione rispetto al tessuto adiposo subcutaneo, come evidenziato dagli alti livelli di citochine e di macrofagi. Gli adipociti viscerali sono molto più sensibili all’effetto lipolitico delle catecolamine e all’azione antagonista dell’insulina mediata dai glucocorticoidi, piuttosto che all’azione lipogenica ed antilipolitica dell’insulina. Questo potrebbe provocare un direzionamento degli acidi grassi verso altri tessuti come il muscolo ed il fegato.
Un’ipotesi interessante è quella secondo cui il tessuto adiposo possa “saturarsi”. In situazioni di eccessiva introduzione di cibo, l’eccesso di calorie sarà conservato dapprima nel tessuto adiposo. Il posto migliore a questo scopo è rappresentato dal tessuto adiposo subcutaneo. Quando il sovraccarico calorico diventa cronico, tuttavia, il tessuto adiposo subcutaneo raggiunge il limite della sua capacità per l’accumulo delle riserve di trigliceridi, e questo può innescare sia l’infiammazione del tessuto adiposo sia la fuoriuscita dei lipidi. Questo significa che le riserve energetiche saranno frazionate negli accumuli di grasso viscerali e successivamente in depositi ectopici come i lipidi intraepatocellulari ed intramiocellulari, ciascuno dei quali ha delle conseguenze dirette negative per gli effetti dell’insulina in questi tessuti. La capacità di accumulo dei grassi nel tessuto adiposo subcutaneo potrebbe essere determinata da fattori genetici che regolano il reclutamento di nuovi adipociti, ma anche da interazioni neuroendocrine o dall’infiammazione del tessuto adiposo. Pertanto un tessuto adiposo sottocutaneo
25
disfunzionale può rappresentare una causa importante per una deposizione errata dei lipidi nei compartimenti intra-addominali ed intramiocellulari.
E’ stato dimostrato che l’aumento delle dimensioni degli adipociti è associato con l’insulinoresistenza, inoltre la presenza di adipociti subcutanei ingranditi può predire lo sviluppo del diabete di tipo 2. La leptina e l’adiponectina sono due prodotti specifici secreti dagli adipociti che eventualmente possono comparire nella circolazione. L’iperleptinemia sembra essere un marker indipendente dell’ipertrofia degli adipociti, inoltre i livelli di leptina sono anche associati alla quantità di grasso del corpo. Perciò alti livelli di leptina possono essere considerati come il segnale che indica il sovraccarico delle riserve di lipidi subcutanee. La leptina ha effetti ipotalamici che portano al senso di sazietà ed all’aumento dell’impiego delle risorse energetiche. A differenza della leptina, i livelli di adiponectina circolanti hanno rapporti inversi con l’insulinoresistenza ed il grasso corporeo, e dati sperimentali suggeriscono che l’adiponectina può ridurre l’iperglicemia, la resistenza all’insulina, l’infiammazione, l’aterosclerosi e, potenzialmente, l’adiposità.
L’aumentata secrezione delle adipochine, cioè delle molecole simili alle citochine prodotte nel tessuto adiposo, può spiegare in parte in che modo l’incremento delle dimensioni degli adipociti possa promuovere l’insulinoresistenza. Nel complesso quindi ci sono diverse prove sottese a dimostrare che le dimensioni degli adipociti, ma anche i fattori prodotti dagli stessi (TNF-α; IL-6 e acidi grassi) sono associati con l’insulinoresistenza.
26
Stress Ossidativo - un pathway comune per l’insulinoresistenza?
Diversi tipi di insulinoresistenza sembrano essere legati allo stress ossidativo cellulare. Questo termine sta ad indicare l’accumulo, nella cellula, di composti reattivi dell’ossigeno e dell’azoto, i cosiddetti ROS (Reactive Oxigen Species). I principali esempi
di ROS sono il perossido di idrogeno (H2O2), l’acido ipocloroso (HClO), il superossido
(.O2-), l’idrossile (.OH), il perossile (.RO2-) e l’idroperossile (.HO2-). Nella respirazione
mitocondriale i ROS sono generati nella catena di trasporto degli elettroni, come co-prodotto del processo di generazione dell’ATP. Questo si verifica quando c’è una marcata ossidazione dei substrati energetici come il glucosio e gli acidi grassi, a meno che l’uncoupling non ne prevenga la formazione. L’enzima NAD(P)H ossidasi svolge un ruolo importante per stimolare la produzione di ROS, ed è attivato da varie citochine. I meccanismi mitocondriali coinvolgono numerosi donatori di elettroni, principalmente
FADH2 e NADH, che sono generati dal ciclo degli acidi tricarbossilici e spingono gli
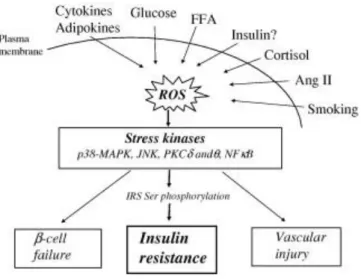
elettroni verso i complessi proteici della catena respiratoria mitocondriale. Questo porterà all’accumulo degli elettroni nella catena di trasporto mitocondriale, che eventualmente, saranno donati all’ossigeno molecolare generando il superossido, il quale, successivamente, sarà degradato in perossido di idrogeno dalla manganese superossido dismutasi. E’interessante notare che eccessive quantità del messaggero ubiquitario NO (monossido d’azoto) possono avere un effetto dannoso paragonabile a quello dei ROS. E’ stato riportato che la produzione di NO da parte dell’enzima NO sintasi inducibile (iNOS) è accresciuto nei modelli animali di obesità, e che la delezione del gene che codifica per iNOS (Nos2) può prevenire l’insulinoresistenza indotta dalla dieta nel muscolo scheletrico. Paradossalmente, alcuni ROS hanno effetti che mimano quelli dell’insulina, e per esempio, il perossido di idrogeno può aumentare il trasporto transmembrana del glucosio ed inibire la lipolisi. Tuttavia, l’accumulo dei ROS in generale è associato con la resistenza all’insulina e questo si può verificare attraverso l’attivazione di chinasi dello stress, danni alle membrane cellulari, al reticolo endoplasmatico ed al DNA nucleare. La modificazione ossidativa delle proteine e dei lipidi cellulari può avere conseguenze funzionali che contribuiscono all’insulinoresistenza. Gli effetti dannosi dei ROS sono mediati da meccanismi effettori a valle i cui dettagli sono scarsamente noti, e probabilmente, ci sono diversi meccanismi coinvolti in base al tipo ed alle funzioni cellulari. Tuttavia, riguardo alla riduzione della responsività all’insulina, si pensa che siano coinvolte diverse chinasi dello stress come JNK, p38-MAPK, NFκ ed alcune isoforme di PKC. Il comune denominatore che accomuna queste chinasi sembra essere la loro capacità di aumentare, direttamente o indirettamente, la fosforilazione delle proteine IRS sui residui di serina e treonina, attenuando in questo modo il segnale dell’insulina. Inoltre alcune di queste chinasi possono aumentare la produzione di citochine, ed anche questo altera le azioni dell’insulina. Ci sono ora diverse prove emergenti che suggeriscono che diversi fattori che provocano l’insulinoresistenza hanno una via comune nell’eccessiva formazione di ROS.
27
Questo sembra essere vero per l’infiammazione, la glucotossicità, la lipotosicità ed alcuni mediatori endocrini (Fig. 3).
Le citochine infiammatorie possono accrescere lo stress ossidativo, e ci sono risultati che suggeriscono che questo possa essere mediato attraverso la formazione della ceramide che stimola la produzione mitocondriale di ROS. Nelle cellule adipose, TNF-α può così aumentare i livelli di ROS che attiveranno la chinasi JNK, la quale può, a sua volta, aumentare la fosforilazione delle serine di IRS-1. Gli agenti antiossidanti dati in presenza di TNF-α in vitro, possono prevenire sia l’insulinoresistenza sia lo stress ossidativo. Il trattamento con antiossidanti in vivo, in topi insulinoresistenti, porta inoltre ad un miglioramento del controllo glicemico e dell’insulinoresistenza. Pertanto c’è un forte consenso per un ruolo fondamentale dei ROS nella resistenza all’insulina indotta dalle citochine.
Fig. 33. ROS accumulation as a unifying pathway leading to and insulin resistance. Several factors exerting cellular stress are proposed to cause insulin resistance via increased ROS production and oxidative stress.
3
28
L’aumento del flusso dei substrati energetici nelle vie ossidative comporta un aumento dell’attività della catena respiratoria mitocondriale e della generazione di ROS. L’accumulo delle specie reattive potrebbe essere visto come un sensore per il sovraccarico di energia nelle cellule, e può agire come sistema di sicurezza per prevenire l’ulteriore influsso ed accumulo di substrati. Perciò alti livelli di acidi grassi e di glucosio portano ad un incremento nell’ossidazione dei substrati ed ad un secondario aumento della formazione di specie reattive mitocondriali. Negli adipociti sovraccarichi è possibile che un aumento nei livelli di acidi grassi sia accompagnato da stress ossidativo. Questo potrebbe spiegare in parte la relazione tra l’ingrossamento delle cellule adipose e la resistenza all’insulina. Dati clinici suggeriscono inoltre che lo stress ossidativo è aumentato nei soggetti con adiposità viscerale.
Lo stress ossidativo indotto dall’iperglicemia può essere di grande importanza nel diabete; infatti è importante notare che secondo alcuni studi l’insulinoresistenza indotta dall’iperglicemia può essere prevenuta attraverso il trattamento con gli antiossidanti. Inoltre ci sono dati che indicano che i picchi di glicemia postprandiale nel diabete di tipo due mostrano una particolare tendenza ad aumentare le specie reattive nelle pareti dei vasi. Questo può essere rilevante per lo sviluppo di complicazioni micro- e macrovascolari nel diabete. Nel complesso tutti questi dati dimostrano che lo stress ossidativo è un meccanismo critico sia nella glucotossicità che contribuisce all’insulinoresistenza, sia nel malfunzionamento delle cellule beta e nello sviluppo di danni agli organi che sono associati con l’iperglicemia cronica e la disfunzione vascolare. In realtà ci sono anche dati che suggeriscono che non c’è aumento dello stress ossidativo nel diabete di tipo 2, come, ad esempio, nel muscolo scheletrico. Negli organi bersaglio dell’insulina come il tessuto adiposo ed il muscolo scheletrico, la glucotossicità sembra essere mediata attraverso la cosiddetta via delle esosamine. In sinergismo con lo stress ossidativo questa potrebbe servire come segnale per un meccanismo compensatorio che indica che i livelli di energia sono sufficienti e, conseguentemente, per limitare l’uptake di glucosio mediato dall’insulina. D’altronde in altri tessuti come i nervi ed i vasi sanguigni, dove il flusso di glucosio non dipende dall’insulina, ci sarebbe un continuo sovraccarico di glucosio e danni irreversibili, come ad esempio le complicazioni croniche del diabete che interessano il sistema nervoso, i reni e la retina.
E’ interessante notare come anche alcuni pathway neuroendocrini che comportano resistenza all’insulina, coinvolgano anche la formazione di ROS nelle cellule bersaglio. Ad esempio, riguardo al sistema renina-angiotensina, sembra che le specie reattive dell’ossigeno siano coinvolte in alcuni effetti dell’angiotensina II che portano all’infiammazione ed all’insulinoresistenza; inoltre i farmaci antagonisti per il recettore dell’angiotensina sembrano attenuare lo stress ossidativo.
29
Il fumo è un altro fattore che notoriamente causa resistenza all’insulina ed è associato con il rischio di sviluppo del diabete di tipo 2. i meccanismi coinvolti non sono ben definiti, ma possono includere l’attivazione del sistema nervoso simpatico, alterazione dei livelli di citochine e disfunzioni endoteliali.
Ci sono alcuni studi clinici rivolti allo sviluppo di potenziali terapie per la riduzione dello stress ossidativo mirati principalmente al trattamento, o alla prevenzione, delle malattie cardiovascolari. In generale in questi questi programmi sono state testate vitamine antiossidanti ed altri potenziali “spazzini” delle specie reattive. Tuttavia i risultati sono stati deludenti. Lo stesso vale per alcuni trials indirizzati al controllo metabolico nel diabete di tipo 2.
In alcuni studi sono stati considerati anche alcuni biomarkers per lo stress ossidativo come lipidi, lipoproteine, derivati delle prostaglandine ed aminoacidi che possono essere modificati dai ROS. Tuttavia questi parametri devono essere ulteriormente validati prima che possano essere impiegati in studi su vasta scala o nella pratica clinica.
30
Conclusioni
I meccanismi che causano la resistenza all’insulina nel diabete di tipo 2 e la
sindrome metabolica non sono ancora compresi totalmente. In generale è stata accettata l’idea secondo cui esiste un complesso intreccio tra i fattori genetici e quelli acquisiti. E’ probabile che i fattori di stress in varie forme colpiscano l’organismo a diversi livelli, e che questo si aggiunga a processi patogeni diretti verso l’insuliniresistenza ed il diabete. Questi fattori di stress sociali, psicologici, neurali, endocrini, metabolici ed infiammatori, possono convergere in un’unica via a livello cellulare che va sotto il nome di stress ossidativo. E’ stato proposto che questo rappresenta un pathway critico per lo sviluppo della resistenza all’insulina, ma anche per la disfunzione delle cellule beta ed il danno vascolare (Fig. 4). La ricerca futura nel campo delle malattie metaboliche dovrebbe includere delucidazioni su nuovi potenziali meccanismi, biomarkers, bersagli farmacologici correlati alla produzione di ROS e sui segnali attivati dall’accumulo delle specie reattive. Questo migliorerebbe la nostra comprensione dei complessi meccanismi della malattia e faciliterebbe lo sviluppo di nuovi approcci terapeutici.
Fig. 44. A hypothesis on the development of insulin resistance and type 2 diabetes over time. Various stress mechanisms challenge the organism’s metabolic homeostasis. In a decompensated situation, where the total stressor load exceeds the capacity of compensatory mechanisms, oxidative stress occurs at the cellular level and this leads to insulin resistance and eventually beta cell failure and vascular damage.
4