30
3. METODI DI ANALISI NELLA DIAGNOSI GENETICA
PRE-CONCEPIMENTO
Per mettere a punto il protocollo che consenta l’attuazione della Diagnosi Genetica Pre-Concepimento si è cercato di riprodurre e perfezionare nel nostro laboratorio un protocollo di minisequenziamento presente nell’articolo “The minisequencing method: an alternative strategy for preimplantation genetic diagnosis of single gene disorders” di F. Fiorentino et al del 2003, concentrandosi sulla mutazione maggiormente diffusa in Italia e in Etiopia responsabile della fibrosi cistica, la ΔF508 del gene CFTR (Cystic Fibrosis Transmembrane Conductance Regulator).
Il gene responsabile della patogenesi della fibrosi cistica è stato individuato da ormai 15 anni (Riordan JR et al, 1989; Rommens JM et al, 1989). Da allora, la crescente disponibilità di metodiche per l’analisi genetica ha cercato di soddisfare le richieste di test genetici per la fibrosi cistica e in parte ne ha anche indotto un costante aumento.
Oggi il test viene proposto a familiari di persone affette – che hanno un rischio di eterozigosi aumentato – e in alcune realtà locali anche a coppie della popolazione generale, per lo più durante la gravidanza.
La fibrosi cistica è la malattia autosomica recessiva grave più comune nella popolazione italiana. E’ verosimile che i dati raccolti nel registro italiano della fibrosi cistica sottostimino il numero degli affetti poiché il potenziale diagnostico della malattia è soggetto a un’ampia variabilità regionale. Sembra perciò più affidabile fare riferimento a programmi di screening neonatale che riportano un’incidenza compresa tra 1/2.730 e 1/3.170 nati (Castellani C et al, 1997)(Corbetta C et al, 2002).
Da questi dati si può desumere una frequenza di portatori compresa tra 1/26 e 1/30. Il gene responsabile della malattia identificato nel 1989 si trova sul braccio lungo del cromosoma 7, si estende per oltre 250.000 basi e contiene 27 esoni. La proteina codificata è chiamata Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), è composta da 1.480 aminoacidi, e la sua funzione principale riguarda il trasporto transmembrana del cloro. Chiunque possieda nel proprio corredo genetico sia una copia mutata che una normalmente funzionante di questo stesso gene è detto portatore. La copia funzionante del gene è ampiamente sufficiente a compensare il mancato funzionamento del gene mutato e pertanto chi è portatore non ha e non avrà
31
mai nessun sintomo di FC. Chi invece è malato ha nel proprio corredo genetico due geni mutati, avendone ereditato uno dalla madre e uno dal padre.
Ad ogni gravidanza, a seconda della diversa combinazione dei geni che essi trasmettono, una coppia di portatori ha 1 probabilità su 4 che il figlio sia malato, 1 probabilità su 4 che non sia né malato né portatore, 2 probabilità su 4 che sia portatore. Le mutazioni del gene CFTR sono molto numerose: a oggi ne sono state individuate più di 1.300 ( Cystic Fibrosis Genetic Consortium) .
La frequenza relativa delle mutazioni è quanto mai variabile in relazione all’area geografica: alcune sono molto più rappresentate in particolari popolazioni, altre sono estremamente rare. La più frequente la ΔF508 si concentra nella popolazione dell’Europa settentrionale, fino a costituire in Danimarca ed in Gran Bretagna l'85% degli alleli mutati; nell'Europa meridionale la frequenza è molto più bassa e varia tra il 35 ed il 55%, con un'incidenza media in Italia intorno al 50%. Alcune mutazioni sono molto più frequenti in particolari popolazioni, come W1282X negli ebrei Ashkenazi e R1162X e 2183AA/EG in Veneto e Trentino-Alto Adige, T338I in Sardegna.
Nell’insieme le 12 mutazioni più diffuse caratterizzano il 73% degli alleli responsabili di malattia, con differenze di rilievo tra regioni geografiche limitrofe e addirittura all’interno della stessa regione ( Rendine S et al , 1997)( Bonizzato A et al , 1995).
Conoscere il gene e le sue principali mutazioni ha reso in molti casi possibile l'identificazione della condizione di portatore con un semplice prelievo di sangue.
In commercio per questa patologia esistono vari kit utili per l’identificazione delle mutazioni, quello viene usato nell’articolo precedentemente citato e che abbiamo utilizzato anche noi per riprodurre e perfezionare il protocollo si basa su una metodica di estensione di una singola base (nota anche come mini-sequenziamento) è l’ABI PRISM® SNaPshot™ Multiplex Kit della Applied Biosystems.
Per l’esecuzione dell’estensione di una singola base viene generato un prodotto di PCR standard, che potrebbe contenere più di un sito polimorfico da interrogare. Il prodotto di PCR viene purificato per rimuovere l’eccesso di primers e deossinucleotidi trifosfato (dNTP). Un’aliquota del prodotto di PCR purificato viene aggiunta alla reazione di estensione di una singola base nucleotidica insieme al primer di estensione specifico per lo SNP, una DNA polimerasi per l’estensione e dideossinucleotidi marcati (ddNTP).
32
3.1. LA PCR ( Reazione a Catena della Polimerasi)
Una delle tecniche più utilizzate nella genetica molecolare è la PCR (Saiki et al., 1985; Liet al., 1988), che molto rapidamente consente l'amplificazione in vitro di sequenze specifiche di DNA delle cellule prelevate,in modo da ottenere la quantità di materiale genetico necessaria per individuare, in tempi ragionevolmente brevi, gli embrioni che hanno ereditato la mutazione genetica dalla coppia e/o del singolo partner eterozigote .
3.1.1. Meccanismo di funzionamento
La PCR sfrutta alcune peculiarità della duplicazione del DNA ad opera della DNA polimerasi. Questa tecnica avviene in una serie di cicli composti da tre fasi( figura 7):
1. Denaturazione (separazione): il ds-DNA bersaglio (DNA a doppia catena) è denaturato alla temperatura di circa 95°C ed è convertito in DNA a singola catena.
2. Appaiamento ("annealing"): i "primers" oligonucleotidici complementari alle due estremità 3' della sequenza da amplificare ibridano con i due filamenti denaturati ad una temperatura che è orientativamente 5° C più bassa della Tm dei
"primers" stessi; la loro sequenza è orientata in modo da poter guidare la polimerizzazione del DNA (senso 5'- 3') nel tratto compreso tra le due regioni a cui essi si associano.
3. Estensione: i "primers" oligonucleotidici, in presenza dei quattro deossinucleotidi trifosfati e di una DNA polimerasi, vengono estesi ognuno in direzione dell'altro ma su due diverse catene complementari portando alla sintesi di due molecole di ds-DNA copie della regione bersaglio delimitata dagli inneschi.
33
Figura 7 : varie fasi della PCR (modificata da http://scienceblogs.com)
Il ciclo descritto viene ripetuto generalmente per circa 30-40 volte. In genere non si superano i 50 cicli in quanto ad un certo punto la quota di DNA ottenuto raggiunge un plateau. Ciò avviene, ad esempio, per carenza degli
oligonucleotidi usati come inneschi o per diminuzione dei dNTP. Bisogna inoltre considerare che si potrebbe amplificare in maniera eccessiva anche eventuale materiale genomico contaminante. Inoltre dopo l’ultimo ciclo si lascia il campione per 10 min. a 72°C. In questa maniera l’enzima ha il tempo di riempire ogni lacuna che dovesse essere eventualmente presente all’estremo 3’ di qualche filamento. Al termine degli n cicli, il miscuglio di reazione contiene un numero massimo teorico di molecole di DNA a doppia elica pari a 2n.
34
Tali molecole sono le copie della sequenza di DNA compresa tra i due primers.
Affinché tale processo avvenga si ha bisogno di:
- Sequenza da amplificare
- dNTP (nucleosidi trifosfati, come dATP, dCTP, dGTP, dTTP)
- Primers (Sequenze complementari agli estremi della sequenza da amplificare)
- Altri elementi di supporto (ad es. ioni magnesio), necessari per costituire l'ambiente adatto alla reazione
- Taq polimerasi (enzima necessario per l’estensione del DNA)
- Buffer (i tamponi necessari per la reazione di elongazione)
Il volume totale della reazione è generalmente di circa 50-100 µl
3.1.2. La quantità di materiale da amplificare
Per effettuare una PCR si può benissimo utilizzare una piccola quantità di bersaglio in quanto la sensibilità della reazione è molto alta. Si è visto che una quantità di DNA genomico di 80-90 ng/µl è sufficiente per identificare un gene bersaglio che è presente in una singola copia. La presenza di un basso quantitativo di bersaglio, comunque, aumenta la probabilità che vengano amplificate sequenze non specifiche.
Una quantità troppo elevata di DNA, al contrario, può diminuire l'efficienza dell'amplificazione a causa della presenza di troppi elementi contaminanti e può rendere complessa la valutazione della resa della reazione durante i processi di ottimizzazione dei singoli parametri per cercare di allestire tutta la PCR.
35
Durante le fasi d'allestimento di una PCR sarebbe bene, per evitare le problematiche appena riportate, cercare d'ottimizzare la quantità di DNA utilizzata (anche se non sempre ciò è possibile) effettuando una serie di reazioni d'amplificazione in cui tutti i parametri siano fissi tranne il quantitativo di DNA che viene impiegato in dosi scalari.
Per poter far ciò, comunque, è necessario poter valutare la quantità di DNA ottenuta durante il processo di estrazione e ciò può essere ottenuto tramite una lettura spettrofotometrica di una aliquota dell'estratto in cui viene misurata l'assorbanza a 260 nm.
3.1.3. I controlli
L'allestimento di opportuni controlli di qualità permette di valutare la sensibilità e specificità della metodica, nonché di evidenziare la presenza di falsi positivi o falsi negativi.
I controlli da utilizzare sono:
il controllo positivo, il controllo negativo.
Il controllo positivo consiste in un campione in cui la sequenza bersaglio è contenuta. Il controllo negativo consiste in un campione in cui la sequenza bersaglio manca. Esso serve per evidenziare eventuali contaminazioni che potrebbero riferirsi sia all'estrazione del materiale genomico, sia al momento di preparazione della PCR.
3.1.4. I primers
La scelta dei primers da utilizzare costituisce un aspetto essenziale per la buona riuscita della PCR. Essi, infatti, devono potersi ibridare in maniera specifica ed efficiente alla sequenza d'interesse, tralasciando quelle aspecifiche.
La lunghezza di un primer è, in genere, compresa tra le 20 e le 30 paia di basi e non dovrebbe essere inferiore alle 16 (al fine di non pregiudicare la
36 specificità del processo).
Anche la concentrazione risulta essere importante, infatti la concentrazione ottimale dei "primers" varia da 0,1 a 0,5 mM : concentrazioni più elevate possono promuovere l’accumulo di prodotti aspecifici, che determinano un notevole calo in resa dei prodotti desiderati (Steffan e Atlas, 1991).
Grazie alle banche dati ed alle pubblicazioni scientifiche stanno diventando sempre più disponibili le sequenze di DNA o di RNA necessarie per poter disegnare i primers da utilizzare nelle PCR. Durante la costruzione dei primers bisogna prestare molta attenzione alla temperatura di melting (la temperatura alla quale il 50% delle molecole si trova in forma di doppia elica stabile ed il restante 50% in forma di singola elica). Tale temperatura è strettamente correlata al contenuto nucleotidico in AT (o in CG).
Nella costruzione dei primers è necessario che essi presentino una temperatura di melting e di annealing molto vicine.
Per calcolare la Tm (Temperatura di melting) dei primers di lunghezza
inferiore a 20 basi, si usa la regola di Wallace (Wallace et al., 1979
)
:Tm = 2°C (A + T) + 4°C (G + C)
La temperatura di annealing di un esperimento di PCR si determina calcolando la Tm di ciascun primer e usando una temperatura più bassa 2-4°C.
E’ importante quindi che i due primers abbiano Tm simili, con uno scarto massimo di 2-3 °C.Una considerazione generale da tenere in considerazione è che i "primers" devono essere sufficientemente complessi affinché la probabilità di ibridare sequenze diverse da quella voluta sia estremamente bassa (Coyne et al., 1992).
3.1.5. Il magnesio
La concentrazione di magnesio è senza dubbio il fattore più critico di tutta la PCR. Questo parametro deve essere fatto oggetto d'una attenta procedura d'ottimizzazione in quanto può variare, anche se si utilizzano diversi primers per
37
amplificare una medesima regione di DNA. La presenza di magnesio condiziona l'attività della polimerasi, l'ibridizzazione dei primers ed aumenta la temperatura cui il DNA stampo si denatura.
Per allestire una PCR, di conseguenza, è bene allestire diverse miscele di reazione contenenti quantità progressivamente scalari di magnesio che varino da un minimo di 0.05 mM ad un massimo di 5 mM (il più delle volte si utilizza magnesio 1,5 mM).
3.1.6. I nucleotidi
Generalmente i nucleotidi vengono utilizzati alla concentrazione di 200 μM ciascuno. Un aumento di questa concentrazione non porta ad un aumento dell'efficienza della reazione in quanto i gruppi fosfato carichi negativamente possono legarsi al magnesio della miscela rendendolo meno disponibile.
E’ consigliabile utilizzare per i quattro nucleotidi la stessa concentrazione, questo infatti minimizza gli errori di incorporazione (Innis et al., 1988).
3.1.7. L’enzima
La Taq polimerasi (DNA polimerasi termostabile) è una polimerasi, proveniente dall'organismo termofilo Thermus aquaticus. L'enzima è costituito da una singola catena polipeptidica, con massa molecolare di 94 KDa. Presenta una attività principale DNA-polimerasi DNA-dipendente in direzione 5'->3', per la quale è richiesta una temperatura ottimale di 75° e la presenza di ioni Mg2+ (ad es. MgCl2); in tali condizioni la reazione avviene con elevata processività e alto numero di turn over.
La ricerca, comunque, si è volta a ricercare altre polimerasi con frequenza d'errore minore e con una più elevata resistenza alle alte temperature. Ciò ha fatto sì che venissero messe in commercio altri enzimi come quelli ottenuti per purificazione da Thermococcus litoralis, Pyrococcus furiosus o Thermotoga maritima. La prima, infatti, associa un'elevata termoresistenza ad una maggior
38
fedeltà nella sintesi del filamento complementare mentre le altre presentano un'interessante attività di correzione di bozze.
Il frammento di Stoffel è una DNA polimerasi ottenuta eliminando i 289 aminoacidi della porzione N-terminale della Taq polimerasi. Ciò fa sì che l'enzima risultante sia privo dell'attività esonucleasica 5'-3' e che abbia una maggior resistenza alle alte temperature (ha infatti un'emivita di circa 20 minuti a 97,5 °C). Una tale caratteristica permette l'utilizzo di temperature di denaturazione più elevate del solito e facilità nella sintesi di frammenti ricchi in guanine e citosine che presentano una struttura secondaria alquanto elaborata.
In commercio si trova anche la AmpliTaq Gold DNA polimerasi che è in grado di attivarsi gradualmente a seguito d'una esposizione a 95 °C per 10 minuti. Tale attivazione, che rientra nel concetto delle PCR "hot start", permetta un aumento della sensibilità e specificità della reazione. Risulta, infine, molto utile nelle PCR multiplex in quanto diminuisce l'aggancio aspecifico dei primers e la formazione di dimeri.
Anche la quantità d'enzima da utilizzare può essere un fattore limitante l'accuratezza della PCR in quanto se la concentrazione è troppo bassa la resa dell'amplificato è scarsa mentre se è troppo alta si possono generare dei prodotti aspecifici.
3.1.8. Le contaminazioni
Paradossalmente, il più grande problema della PCR deriva proprio dalla sua elevata sensibilità ed efficienza. In effetti la reazione risulta molto sensibile alla presenza di materiale genetico contaminante che si può trovare in differenti posti: strumentazione, operatore, ambiente esterno. Una delle maggiori fonti di contaminazione consiste nell'apertura di provette contenenti materiale amplificato (contaminazione da carry over) il quale, a seguito dell'apertura del recipiente, può disperdersi nell'aria sotto forma di aerosol che potrebbe contaminare successive PCR. Il problema delle contaminazioni è tanto maggiore quanto la sensibilità della PCR è elevata. Una PCR meno sensibile risulterà, ovviamente, meno soggetta a contaminazioni ma necessiterà d'una maggior presenza del proprio bersaglio per poterlo amplificare.
39
Un altro aspetto che deve essere considerato è la presenza di materiale contaminante di origine ambientale o cellulare. Volendo, ad esempio, amplificare materiale genomico umano, vi sarà la possibilità che lo stesso operatore sia una fonte di contaminazione (per esempio per perdita di frammenti di cute che si desquamano o per il rilascio di goccioline di saliva).
È anche possibile la contaminazione crociata a partire dal materiale utilizzato come controllo positivo il quale potrebbe andarsi a depositare nelle provette dei campioni da testare. Esiste, infine un'altra possibilità di contaminazione che si può avere durante le procedure di rilevazione del prodotto della PCR (ad esempio su gel d'agarosio per la corsa elettroforetica). In questo caso è possibile che materiale di un campione possa aggiungersi in piccola quantità ad un altro con la possibilità d'un risultato falsato.
Di fronte ad un problema così importante come quello delle contaminazioni (si pensi soprattutto al campo della diagnostica) è opportuno che vengano intrapresi degli accorgimenti idonei a minimizzare tale rischio.
È assolutamente indispensabile che l'area di preparazione della miscela della reazione sia ben distinta da quella in cui i campioni vengono inoculati e da quella in cui vengono analizzati. Ciò vale anche per tutta la strumentazione da utilizzarsi. Il fine di ciò consiste nell'evitare che eventuale materiale genomico possa contaminare la soluzione mentre viene preparata.
Tutti i reagenti della PCR dovrebbero venir suddivisi in aliquote piuttosto piccole in maniera tale da evitare che una provetta venga aperta e chiusa un numero troppo elevato di volte. In caso di presenza di materiale contaminante, poi, non sarà necessario buttare tutto quanto il reagente considerato inquinato ma solo l'aliquota di esso che è stata utilizzata.
I reagenti, inoltre, dovrebbero essere conservati in aree dove non sono presenti prodotti di altre PCR od eventuale DNA estratto.
Le pipette utilizzate nei laboratori costituiscono una delle maggiori fonti di contaminazione in quanto, durante la fase d'aspirazione d'una soluzione contenente DNA, possono creare degli aerosol che si vanno a depositare sulla punta e che possono successivamente andare ad inquinare altri campioni (specie i controlli negativi). Per ovviare a questa problematica è bene utilizzare puntali dotati di filtro o pipette ad espulsione positiva. Un altro accorgimento utile da usare consiste nell'utilizzo di pipette differenti per la preparazione della miscela di
40 reazione e per l'inoculo del DNA.
Tutte queste misure vanno, ovviamente, inserite in un contesto generale di buona pratica laboratoristica che dovrebbe prevedere, tra l'altro: il cambio frequente dei guanti, la pulizia accurata di tutte le superfici e strumentazioni e la chiusura di tutte le provette subito dopo il loro utilizzo.
3.1.9. Varianti
Attualmente esistono delle varianti della PCR classica tra cui:
Real time PCR RT-PCR PCR-RFLP Nested PCR Touchdown PCR Ecc..
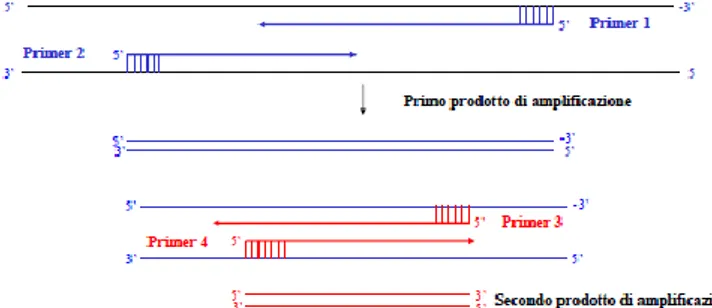
La metodica di PCR che viene principalmente utilizzata per effettuare le analisi per la Diagnosi Genetica Pre-Concepimento è la Nested PCR.
La nested PCR è una variante della tecnica di PCR che consiste nello utilizzo di due coppie di primers, una esterna che genera un normale prodotto di PCR ed una coppia con primers all’interno del prodotto amplificato: Se il prodotto di amplificazione fosse aspecifico la seconda PCR non andrebbe a buon fine. In questo modo si aumenta la specificità e il tasso di amplificazione(figura 8).
41 3.1.10. Rilevazione degli amplificati
La PCR consente di ottenere migliaia di copie di una specifica sequenza di DNA a partire da una o poche copie. I prodotti amplificati sono frammenti di DNA della stessa lunghezza; la lunghezza dei frammenti è nota poiché è determinata dal modo in cui sono stati costruiti i primers.
La rivelazione dei prodotti di reazione è facilmente eseguita mediante elettroforesi su gel di agarosio, poliacrilammide o elettroforesi capillare. L’elettroforesi è una delle tecniche per la visualizzazione e la purificazione di molecole di interesse biologico quali acidi nucleici e proteine. Tale separazione è resa possibile dal peso molecolare e dalla carica elettrica di cui sono dotate le particelle, ioni o macromolecole. Tali particelle poste in un campo elettrico generatosi dalla d.d.p.(differenza di potenziale) applicata a due elettrodi, si muoveranno in direzione dell'elettrodo di carica opposta. Per poter effettuare l’elettroforesi è necessario prima preparare il gel di agarosio.
L’agarosio è un polisaccaride purificato dall’agar-agar, una sostanza gelatinosa isolata a sua volta dalle alghe. L’agarosio si presenta in polvere solubile in acqua alla temperatura di ebollizione, mentre diventa solido man mano che si raffredda formando un gel grazie alla formazione di una matrice tridimensionale costituitasi attraverso dei legami a idrogeno tra le catene lineari.
Un gel si prepara sciogliendo una quantità nota di polvere di agarosio in un tampone salino come il TAE o il TBE.
Maggiore è la concentrazione di agarosio (peso/volume),maggiore sarà la resistenza opposta alla migrazione delle molecole di DNA; sarà maggiore però anche la risoluzione delle bande,cioè la separazione tra due bande molto simili.
Il gel ancora liquido viene versato in un apposito stampo fornito di un pettine, per creare i pozzetti in cui depositare i campioni. Prima di versare l’agarosio nell’apposita formina si aggiungono 5 µl di bromuro di etidio che è una molecola intercalante gli acidi nucleici (DNA e RNA) comunemente utilizzata in tecniche di biologia molecolare come l'elettroforesi in gel d'agarosio.
Quando è esposto ai raggi ultravioletti, infatti, esso è in grado di emettere fluorescenza che si intensifica di quasi 20 volte se intercalato nel DNA. Inserendo
42
dunque nel gel il bromuro di etidio, è possibile evidenziare lo stato della migrazione del DNA all'interno del gel.
Una volta raffreddato e solidificato il gel viene immerso nel tampone salino all’interno della camera elettroforetica (figura 9).
Figura 9: camera elettroforetica (da http://web.tiscalinet.it/lauramorelli/protocols.htm)
Si procede al caricamento dei campioni da analizzare che vanno depositati, con una micropipetta, in apposite fenditure verticali, dette "pozzetti", praticate nel gel a poca distanza dal margine dalla parte del polo negativo e anche in posizione centrale del gel. Essendo il DNA un polianione i frammenti migreranno in avanti verso il polo positivo. All'atto del caricamento, al campione (si prendono all’incirca 10µl) viene solitamente aggiunta una "soluzione di caricamento" colorata ( all’incirca 5µl ) contenente glicerolo per agevolare la precipitazione del campione sul fondo del pozzetto. Inoltre in almeno un pozzetto viene caricato il DNA ladder un marcatore costituito da frammenti a peso molecolare noto così da poterlo utilizzare come standard di peso molecolare per l’approssimazione della massa delle bande ignote ( figura 10).
43 3.2. IL MINISEQUENZAMENTO
La tecnica di minisequenziamento si basa sull’estensione di una singola base nucleotidica; un Oligonucleotide (primer) non marcato si lega alla sequenza complementare in presenza di ddNTPs (dideossinucleotidi) marcati e dell’enzima polimerasi. La polimerasi estende il primer solo di una base, aggiungendo il ddNTP alla sua estremità 3’ libera. Il ddNTP incorporato è complementare alla singola base variante di interesse (Pastinen et al., 1997; Syvänen et al 1999).
Questo processo viene ripetuto in successivi cicli di estensione e terminazione per generare dei frammenti marcati con un fluorocromo per l’analisi. Dopo la fase di estensione, il prodotto di minisequenziamento viene purificato tramite delle colonnine con Sephadex G50 e successivamente 1 µl viene mischiato con 15 µl di Hi-Di Formamide e denaturato per 4 min a 90°C. Il campione viene poi analizzato tramite elettroforesi capillare eseguita su un sequenziatore automatico di DNA .
Per la tecnica del minisequenziamento, i colori assegnati per identificare i vari ddNTP sono: verde / A, nero / C, blu / G, rosso / T. La reazione di minisequenziamento produce uno (omozigoti) o due (eterozigote) picchi a seconda del genotipo di questo locus.
Lisi cellulare
La singola cellula viene posta in una eppendorf da 0,2 ml nella quale si aggiungono 5 µl di buffer di lisi alcalino (200 mmol/l KOH, 50 mmol/l DTT) e si copre con una goccia di olio minerale.
La lisi cellulare a questo punto viene effettuata tramite incubazione a 65°C per 10 min. Il buffer di lisi alcalino viene poi neutralizzato tramite aggiunta di 5 µl di buffer di neutralizzazione (900 mmol/l Tris–HCl, 200 mmol/l KCl, 200mmol/l HCl) prima di procedere alla PCR.
44 PCR
La strategia di PCR prevede un’iniziale amplificazione esterna seguita poi da una nested PCR, specifica per la regione coinvolta nella mutazione.
Fase di amplificazione esterna
L’amplificazione esterna è svolta dopo la lisi cellulare e la neutralizzazione aggiungendo alla eppendorf 1,5 mmol/l MgCl2 , 200µnol/l di ogni dNTP, 2,5 IU di
Amply TaqPolymerase(Applied Biosystems), 10 pmol di ogni primer esterno, per un volume totale di 50 µl come riportato nel protocollo. La PCR è svolta effettuando un iniziale denaturazione di 4 min a 94°C , poi vengono svolti 10 cicli con fase di denaturazione a 96°C così da ridurre l’ADO (Ray, P.F. and Handyside, 1996), seguiti poi da i restanti 25 cicli con fase di denaturazione a 94°C . Alla fine dei 35 cicli è svolta una fase finale di estensione a 72°C per 10 minuti.
Fase di nested PCR
Vengono prelevati 2µl del prodotto della PCR precedente e aggiunti ad una eppendorf da 0,2 ml contenente 5 µl di 10XPCR buffer (Applied Biosystems), 1,5 mmol/l MgCl2 , 200µnol/l di ogni dNTP, ,5 IU di Amply TaqPolymerase(Applied
Biosystems), 10 pmol di ogni primer interno, per un volume totale di 50 µl come riportato nel protocollo.
Per monitorare il successo dell’amplificazione , 10 µl del prodotto di PCR sono sottoposti ad elettroforesi per 5 min a 150V in agarosio al 2% in Tris-borate/EDTA buffer colorato con 0,5 µg/ml di bromuro di etidio.
Fase di purificazione
Per permettere la successiva reazione di minisequenziamento i primers e i dNTP non incorporati sono stati eliminati dal prodotto di PCR tramite purificazione con QIAquick PCR Purification Kit (Qiagen).
Il QIAquick PCR Purification Kit consente la purificazione rapida di amplificati di DNA direttamente dalla miscela di PCR. Il kit è in grado di
45
rimuovere con elevata efficienza dai prodotti di PCR olio minerale, enzimi, nucleotidi non incorporati, primers e dimeri dei primers, Sali. Il DNA purificato è di elevata qualità e può essere utilizzato in una vasta gamma di applicazioni.
La tecnologia di purificazione si basa sulla cromatografia mediante colonnine utilizzando una membrana in silice(figura 11).
Figura 11 colonnina per la purificazione (da http://www.qiagen.com)
La membrana in silicio lega il DNA in presenza di un’elevata concentrazione di sali e pH acido e rilascia il DNA legato in presenza di una bassa concentrazione di sali e di condizioni leggermente alcaline. Il prodotto di PCR viene prima mescolato con 200 µl di Binding Solution . Quindi, il campione viene applicato all’interno di una colonnina e centrifugato. La resina lega il DNA in modo dipendente dalla concentrazione ionica, mentre i primers, i dimeri e altri contaminanti passeranno attraverso la resina e verranno subito eluiti. Il DNA legato viene quindi lavato due volte utilizzando la Wash Solution per rimuovere le impurità residue e il prodotto di amplificazione è eluito con l’Eluition Buffer .
Fase del minisequenziamento
Il minisequenziamento è svolto usando 10 ng del prodotto purificato di PCR e il kit ABI Prism SnaPshot Multiplex Kit (Applied Biosystems).Il volume di reazione è di 10 µl, inclusi 5µl del Ready Reaction Premix e 10 pmol di ogni primer per il minisequenziamento.Le condizioni di reazione sono le seguenti: 25 cicli di PCR, step di denaturazione di 10 s a 96°C, annealing per 10 s a 50°C e estensione per 30 s a 60°C. Dopo i 25 cicli, si purifica la reazione di minisequenziamento per eliminare gli elementi in eccesso. Questa procedura è
46
stata fatta utilizzando il Sephadex G‐50 che è una matrice solida che viene sospesa in acqua. Dopo centrifugazione si ottiene una colonnina che serve da filtro.
Per effettuare la purificazione bisogna:
Mettere una colonnina da purificazione in un eppendorf tube da 1.5 ml Nella colonnina aggiungere 800 μl di Sephadex
Centrifugare 2 minuti a 3000 RPM.
Pipettare 15‐20 μl di reazione di minisequenziamento nella colonnina Centrifugare per 2 minuti a 3000 RPM.
Nella eppendorf si ottiene il risultato della reazione di minisequenziamento purificato.
Si preleva poi 1 µl del prodotto purificato che viene mischiato con 15 µl di Hi-Di Formamide e denaturato per 4 min a 90°C. Il campione è poi analizzato tramite l’uso del sequenziatore automatico ABI Prism 3130xl (figura 12).
47
3.3. SEQUENZIAMETO DEGLI ACIDI NUCLEICI
“Sequenziare” un frammento di DNA significa stabilire la successione dei nucleotidi che ne formano la molecola:
…ACGGGGGGTTTAGCGCGATTCCAT…
Il sequenziamento del DNA viene attualmente effettuato con un metodo descritto già alla metà degli anni settanta da un certo Sanger (Sanger F et al , 1975) (F. Sanger et al, 1977). Sono stati abbandonati i metodi chimici di Maxam e Gilbert (Maxam AM, Gilbert W, 1977). Oggi in molte applicazioni si effettua il sequenziamento diretto: che cosa vuol dire? Fino a non molto tempo fa una sequenza di DNA prevedeva il clonaggio del frammento da sequenziare, una procedura che richiedeva e richiede l’inserimento del frammento in un vettore, ad esempio un plasmide, che viene poi inserito in un batterio per poterlo replicare.
Oggi è possibile evitare il clonaggio del frammento da sequenziale grazie alla possibilità di amplificare il DNA bersaglio con l’uso della PCR (clonaggio a-cellulare).
Il metodo di Sanger si basa sulla metodica di terminazione della catena con i di-deossi nucleotidi. E’ attualmente il metodo più usato per definire mutazioni specifiche. I vantaggi principali di questo metodo risiedono nella facilità d’uso, e questa è stata aumentata negli anni recenti con l’introduzione di marcature fluorescenti e sistemi automatizzati che hanno ovviato alla necessità di sostanze radioattive.
Il vantaggio principale di sequenziare direttamente il DNA usando la PCR anziché sequenziare col clonaggio è che solo una sequenza deve essere determinata.
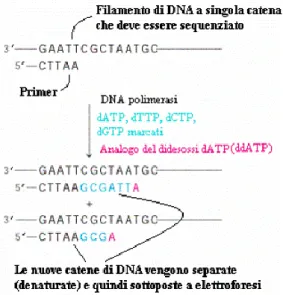
Il principio su cui si basa il sequenziamento del DNA consiste nel blocco della reazione di estensione della catena DNA mediante i cosiddetti analoghi delle basi. Queste sono molecole molto simili a deossiribonucleotidi ma mancano dell’ossidrile nel carbonio 3’ (di-deossiribonucleotidi) per cui possono essere incorporati in una catena di DNA in fase di sintesi ma ad essi non possono essere legati altri dNTP. Quindi se un “di-deossi” viene per caso incorporato nella catena
48
nascente di DNA l’ulteriore allungamento viene bloccato e la reazione termina. Quindi i di-deossi sono chiamati anche “terminatori di catena” (figura 13) .
Figura 13 Confronto tra dideoxynucleotide e deoxynucleotide (http://www.vialattea.net)
In pratica viene allestita una reazione contenente tutte le 4 basi in concentrazione normale e tutte le 4 basi sotto forma di di-deossi in concentrazione molto debole. Il frammento di DNA da sequenziare (stampo) viene incubato con una DNA polimerasi termostabile in presenza di deossi-nucleotidi-tri-fosfato (dNTPs) e di un primer oligonucleotidico, complementare alla parte iniziale del DNA da sequenziare. La polimerasi a partire dal primer genera un filamento complementare allo stampo, che si estende per una lunghezza indefinita a valle del primer. Ciò dà luogo a una amplificazione lineare dei prodotti di estensione (figura 14).
Figura 14 Amplificazione lineare prodotti di estensione (da www.dia.unisa.it)
Siccome la sintesi del DNA viene effettuata per esterificazione dell’ossidrile 3’ di un nucleotide, già incorporato mediante l’estremità 5’ fosfato, sul carbonio in alfa di un “deossi” nel momento in cui viene incorporato un
“di-49
deossi” la sintesi si arresta (poiché non possiede un 3’ ossidrile). Dal momento che questo nucleotide è presente in una concentrazione molto bassa esso verrà incorporato molto raramente e in modo casuale. Statisticamente si otterranno così tanti frammenti abortivi quante sono le volte in cui le basi corrispondenti sono rappresentate nel pezzo di DNA in questione.
La dimensione dei frammenti sintetizzati corrisponde alla distanza tra l’inizio del primer e la base a livello della quale si è fermata la replicazione.
La marcatura più usata attualmente è quella basata sui terminatori di catena chiamati “big-dyes derminator”, che consistono di didesossinucleotidi marcati con molecole con sistema di trasferimento di energia da un donatore a un accettore .
L’introduzione della marcatura fluorescente permette di passare dal sequenziamento manuale a quello automatico che prevede una corsa elettroforetica su supporto capillare. La migrazione dei vari frammenti è seguita rilevando le emissioni in fluorescenza a diverse lunghezze d’onda dei diversi fluorocromi dopo l’eccitazione provocata dal laser. Le emissioni vengono raccolte e analizzate da una camera CCD (charge coupled device) che elabora i diversi segnali di fluorescenza con elevata sensibilità. La sequenza delle bande di DNA marcato viene visualizzata in un unico grafico detto elettroferogramma, caratterizzato da una successione di picchi di quattro colori diversi, che corrispondono alle emissioni fluorescenti dei diversi fluorocromi, ogni volta che i vari frammenti di diversa lunghezza nucleotidica raggiungono, lungo la corsa elettroforetica, la posizione del rilevatore (figura 15) .