INDICE
INTRODUZIONE GENERALE
………. 1 Canali al potassio………... 1
Canali al potassio ATP-dipendenti……… 1
Struttura proteica dei canali KATP………. 2
Diversità dei canali KATP……… 3
I canali KATP nella muscolatura liscia vascolare……… 4
I canali KATP nel Sistema Nervoso Centrale……….. 5
I canali KATP nella muscolatura liscia non-vascolare……… 6
Ischemia………. 8
Danno da Ischemia/Riperfusione……… 9
Precondizionamento Ischemico………. 10
Canali sarc-KATP e mito-KATP……….. 12
Potenziale terapeutico……… 17
Attivatori dei canali KATP………. 20
INTRODUZIONE ALLA PARTE SPERIMENTALE
31PARTE SPERIMENTALE
38BIBLIOGRAFIA
47I canali al potassio sono un gruppo ubiquitario di canali ionici coinvolti in svariate reazioni fisiologiche. Sono proteine integrali di membrana multimeriche in grado di formare pori acquosi transmembrana attraverso i quali permea il K+. Sono conosciute molte sottofamiglie molecolari dei canali al K+ classificati in base ai segnali fisiologici che controllano l’apertura del poro (voltaggio, Ca2+, ATP, proteine G o poliammine).1
CANALI AL POTASSIO ATP-DIPENDENTI
Tra le varie tipologie di canali del potassio, una molto importante è rappresentata dai canali al potassio ATP-dipendenti (KATP) scoperti nei primi anni ’80 nel muscolo cardiaco e nelle cellule
pancreatiche2. La loro apertura è regolata dalle variazioni della concentrazione intracellulare di ATP, quindi associano l’energia della cellula con il suo stato metabolico. I canali KATP sono
predisposti per funzionare come sensori del metabolismo intracellulare, modulando la permeabilità al potassio, e quindi l’attività elettrica, della cellula per il suo bilancio energetico. L’apertura dei canali KATP iperpolarizza la membrana plasmatica e riduce l’eccitabilità elettrica della cellula.
Questo ruolo metabolico prende parte in molti processi fisiologici, per esempio, svolge un ruolo importante nel modulare la glicemia nella cascata connessa alla secrezione insulinica a livello delle cellule del pancreas e partecipa al pre-condizionamento ischemico e perciò alla cardioprotezione durante un insulto ischemico del miocardio.3,4,5
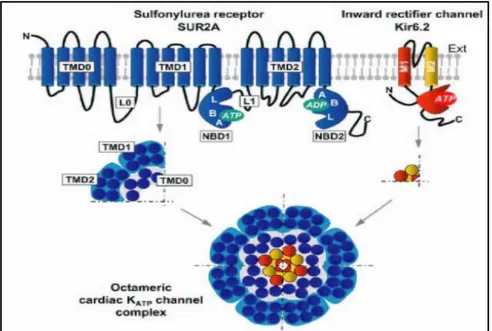
STRUTTURA PROTEICA DEI CANALI K
ATPI canali KATP sono formati dalla combinazione di due tipi di subunità proteiche6: una subunità
Kir6.x che costituisce il poro del canale e una subunità SUR che costituisce il sito recettoriale delle sulfaniluree. Il canale KATP è un ottamero formato da quattro
subunità Kir6 associate a quattro subunità SUR.7,8 Kir6 è costituito da sei eliche transmembrana (M1 e M2) unite a ponte da una regione extracellulare che genera la porzione ristretta del poro (H5) e controlla la selettività ionica. Si conoscono due membri della famiglia dei Kir6: Kir6.1 e Kir6.2, codificati rispettivamente dai geni KNCJ8 e KNCJ11, che hanno il 70% di identità aminoacidica.
SUR possiede omologia con i membri della sottofamiglia C delle proteine ABC ed è codificato da due geni, SUR1 e SUR2 con il 70% di omologia. Il gene SUR2 possiede due principali varianti, SUR2A e SUR2B, che differiscono di 42 aminoacidi nella porzione C-terminale. Le proteine SUR possiedono 17 segmenti transmembrana (TM) arrangiati in un dominio di cinque segmenti (TMD0) e due domini di sei segmenti ciascuno (TMD1 e TMD2), uniti tra loro da due larghi loops intracellulari detti NBF1 e NBF2.8
Figura 1. Struttura dei canali al potassio.
DIVERSITA’ DEI CANALI K
ATPI canali KATP furono descritti per la prima volta nei miocardiociti ventricolari, ma sono anche
espressi in vari tipi di tessuto tra cui reni, cervello, muscolatura scheletrica, cuore, cellule del pancreas, muscolatura liscia9,10 e sono stati identificati con certezza sia nel sarcolemma che nelle membrane mitocondriali (rispettivamente, sarc- e mito-KATP). Il loro ruolo funzionale in molti
secrezione di insulina conseguente ad un aumento ematico di glucosio; nel cuore esercitano una funzione protettiva in risposta a condizioni di ipossia o ischemia. Nel cervello i canali KATP
possiedono analoga funzione protettiva e sono inoltre coinvolti nel rilevamento dei livelli di glucosio nel sangue. L’apertura dei canali KATP determina il rilascio della muscolatura liscia, e in
particolare la loro attivazione a livello vascolare suggerisce un loro coinvolgimento nell’aumento del flusso ematico in risposta ad un aumentata richiesta metabolica. Nella muscolatura scheletrica, i canali KATP possono giocare un ruolo importante nell’affaticamento e nel recupero di glucosio.11
I canali KATP nei differenti tessuti sono costituiti da differenti combinazioni delle subunità Kir e
SUR, spiegando le peculiarità di ciascun canale tessuto-specifico. Per esempio, le cellule del pancreas sono costituite da canali KATP formati dalle subunità SUR1 e Kir6.212,13, mentre SUR2A è
associata a Kir6.2 nel canale sarcolemmatico cardiaco14,15. Nella muscolatura liscia vascolare Kir6.1 e SUR2B probabilmente costituiscono il principale tipo di canale KATP16, mentre i canali a livello
della muscolatura liscia non vascolare sono formati da SUR2B e Kir6.1 o Kir6.2. Diversi sottotipi di SUR corrispondono in modo diverso a nucleotidi intracellulari, sulfoniluree, e agonisti dei canali al potassio (KCOs), mentre Kir6.1 e Kir6.2 possiedono analoga sensibilità all’ATP. Perciò, la sensibilità metabolica e le proprietà farmacologiche dei canali KATP sono ampiamente determinati
dai costituenti SUR.
I CANALI KATP NELLA MUSCOLATURA LISCIA VASCOLARE
I canali KATP a livello della muscolatura liscia, modulano la contrattilità attraverso variazioni del
potenziale di membrana. La loro attivazione, fisiologica o indotta da ligandi farmacologici, porta all’iperpolarizzazione della membrana e al rilasciamento muscolare. I canali KATP della muscolatura
liscia vascolare sono quelli maggiormente studiati, e attualmente costituiscono il principale target terapeutico degli attivatori dei canali KATP (KCOs).11
La natura molecolare dei canali KATP vascolari è meno definita di quella dei canali KATP presenti
nelle cellule pancreatiche o cardiache. I canali KATP vascolari mostrano una diversa sensibilità ai
KCOs rispetto a quelli pancreatici o cardiaci, i quali sono attivati da pinacidil, levcromakalim e diazossido (KCOs noti in letteratura), e mostrano anche una forte sensibilità per i nucleotidi difosfato.
Figura 2: Muscolatura liscia e canali al potassio
Molti vasodilatatori attivano i canali KATP e causano iperpolarizzazione di membrana; ne risulta una
diminuzione nella probabilità di apertura dei canali al calcio voltaggio-dipendenti, che contribuisce all’azione vasorilasciante.17Analogamente questo avviene per i canali presenti in altri tessuti.
In conclusione, sembra che tutte questi processi servono a mantenere un’attività basale dei canali per ridurre le resistenze vascolari e contribuire al mantenimento del flusso sanguigno, e che un’ulteriore attivazione dei canali KATP contribuisca ad aumentare il flusso sanguigno in risposta ad
un aumentata esigenza metabolica tissutale.11
I CANALI KATP NEL SISTEMA NERVOSO CENTRALE
I canali al potassio giocano un ruolo centrale nel controllo dell’eccitabilità neuronale, del potenziale d’azione e del rilascio di neurotrasmettitori nel SNC.18,19
La loro localizzazione a livello della corteccia cerebrale e dell’ippocampo ne suggerisce un loro coinvolgimento nel meccanismo della memoria. Inoltre, l’iperpolarizzazione delle cellule eccitabili attraverso l’apertura di canali al potassio può mostrare una certa utilità terapeutica soprattutto in patologie, come l’epilessia, in cui la genesi e la propagazione dell’impulso elettrico non-fisiologico sono causa di disfunzione.
Può essere ipotizzato anche un ruolo potenziale per i KCOs come analgesici, dato che il meccanismo antinocicettivo degli oppioidi prevede l’apertura dei canali K+
e l’iperpolarizzazione neuronale.20
Figura 3:Canali al potassio e mediatori centrali.
CANALI KATP DELLA MUSCOLATURA LISCIA NON-VASCOLARE
Vescica urinaria
I canali KATP sono espressi nelle cellule muscolari lisce della vescica, e possono essere attivati da
diversi KCOs. L’attivazione farmacologica di una piccolissima porzione della popolazione totale di canali KATP porta ad una significativa inibizione dell’eccitabilità elettrica, ed a contrazioni fasiche
della vescica.21 I canali K
ATP giocano quindi un ruolo chiave nella regolazione del potenziale di
KATP rappresenta perciò un potente mezzo per controllare la funzionalità contrattile della vescica.
Grazie al potenziale terapeutico dei canali KATP nel trattamento dell’incontinenza urinaria, la ricerca
si è rivolta allo sviluppo di KCOs selettivi per i canali KATP della vescica.
Figura 4: Funzionamento e localizzazione dei canali al potassio nel rene
Tratto gastrointestinale
Studi sulla muscolatura liscia intestinale hanno mostrato la capacità rilasciante dei KCOs anche in questo tessuto; è stato inoltre osservato che la glibenclamide, farmaco bloccante non selettivo dei canali KATP, riduce la conduttanza basale del potassio e provoca depolarizzazione e aumento della durata del potenziale d’azione. Questo suggerisce che i canali KATP contribuiscono al ripristino del
potenziale di membrana delle cellule intestinali, e quindi alla regolazione della loro eccitabilità e contrattilità.22
Vie aeree
Esistono pochi studi sul ruolo fisiologico dei canali KATP nella muscolatura liscia bronchiale, ma è
noto che la stimolazione muscarinica inibisce la corrente di K+, mediante l’attivazione della PKC nelle cellule della trachea, e ciò è in accordo con il ruolo dei canali KATP nella modulazione del
potenziale di membrana e della contrattilità delle vie aeree.23 Un’ampia varietà di KCOs risulta capace di rilasciare la muscolatura bronchiale sia in vitro che in vivo, indicando la presenza di una significativa popolazione di canali KATP. Gli attivatori dei canali KATP rappresentano potenziali
farmaci per il trattamento dell’ipercontrattilità delle vie aeree.
Muscolo cardiaco
I canali KATP, individuati per la prima volta da Norma, nel 1983, nelle membrane dei miociti
ventricolari del maiale della Guinea24, giocano un ruolo centrale nella protezione del cuore da danni ischemici. In condizioni di normossia, i canali KATP si trovano in uno stato chiuso e inattivo, mentre
durante l’ischemia miocardica o, più in generale, in condizioni di stress cellulare metabolico, la loro apertura viene stimolata da una riduzione della concentrazione di ATP intracellulare e/o da un accumulo di metaboliti ischemici. I canali KATP cardiaci sono coinvolti in un meccanismo
fisiologico per cui brevi episodi di ischemia/riperfusione rendono il cuore più resistente nei confronti di un successivo evento ischemico prolungato: tale fenomeno è noto come precondizionamento ischemico (IPC)25
ISCHEMIA
Le malattie cardio-cerebrovascolari (infarto miocardico, ictus, scompenso cardiaco)
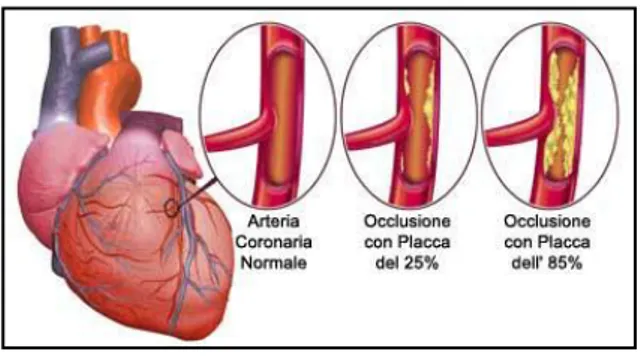
rappresentano la principale causa di morte nei Paesi del mondo industrializzato. Queste malattie, sono tutte legate alla parziale o totale occlusione delle arterie; l’occlusione è dovuta alla formazione di placche aterosclerotiche che ispessiscono la parete del vaso (aterosclerosi), così da rallentare il flusso sanguigno.
Figura 5:principali cause di insorgenza di ischemia cardiaca
Quando il flusso si riduce o si interrompe per un tempo limitato si verifica l’ischemia. Se l’ischemia colpisce le coronarie (le arterie che irrorano il cuore), si verifica l’angina pectoris, caratterizzata da un forte dolore al torace e al braccio sinistro. Le conseguenze possono essere gravi, perché la mancanza di ossigeno causa la necrosi dei tessuti.
Nel caso dell’infarto miocardico questa occlusione impedisce al sangue di alimentare parti del cuore, nel caso dell’ictus cerebrale invece l’organo in cui è carente l’apporto di ossigeno è il cervello. In entrambi i casi i tessuti si necrotizzano, con conseguenze invalidanti o decesso. La causa più frequente di arresto cardiaco è la fibrillazione ventricolare, indotta da eventi ischemici acuti; i soggetti che hanno già subito un infarto del miocardio rappresentano la categoria più a rischio.
Quando l’apporto di sangue al cuore viene interrotto per 30-40 min, la contrattilità dei miociti diminuisce, con conseguente morte cellulare. Nelle fibre miocardiche ischemiche la concentrazione di ATP è ridotta, poiché in assenza di ossigeno solo una piccola percentuale di ADP viene trasformato in ATP dalla glicolisi anaerobica. In oltre, la quantità di ADP che non è convertita in ATP dà origine a composti, come adenosina, inosina e xantine, responsabili dell’aumento di osmolarià intracellulare e del rigonfiamento delle fibre. A causa della ridotta attività della pompa ionica ATP-dipendente, si ha anche un innalzamento della concentrazione intracellulare di ioni Ca++; in seguito a tutti questi processi l’integrità della membrana cellulare è quindi seriamente compromessa dall’ischemia. La riperfusione che segue un lungo periodo di ischemia può aumentare il danno tissutale, a causa dell’aumento del rigonfiamento
cellulare, e della possibile distruzione del sarcolemma. Questi fenomeni portano a morte cellulare e all’ampliamento dell’area necrotica. Nelle fibre cardiache si verifica un ulteriore aumento della concentrazione di ioni Ca++, con conseguente attivazione di enzimi degradativi come le proteasi, le endonucleasi e le fosfolipasi. Tra questi enzimi, le fosfolipasi contribuiscono alla degradazione del sarcolemma idrolizzando i fosfolipidi di membrana. Infine, anche la produzione di specie radicaliche dell’ossigeno (ROS) riveste un ruolo fondamentale nell’induzione del danno tissutale.26
PRECONDIZIONAMENTO ISCHEMICO
Il precondizionamento ischemico (IPC) può essere considerato una potente forma di cardioprotezione endogena. L’IPC fu inizialmente scoperta da Murry e colleghi27
, che hanno dimostrato che 4 cicli di 5 minuti di ischemia, intervallati da riperfusione, limitavano del 75% l’entità dell’infarto nelle cellule cardiache. Questo meccanismo di “auto-difesa” del cuore, inoltre, migliora il recupero post-ischemico e la protezione dell’endotelio coronarico.
L’IPC si articola in due fasi: una prima fase, detta IPC classico, che dura per 1 o 3 ore dopo lo stimolo di precondizionamento e una ritardata, seconda finestra dell’IPC, che dura da 24 a 96 ore. I meccanismi esatti coinvolti nel precondizionamento ischemico sono ancora oggetto di dibattito, ma è comunque chiaro che sono implicati svariati processi.
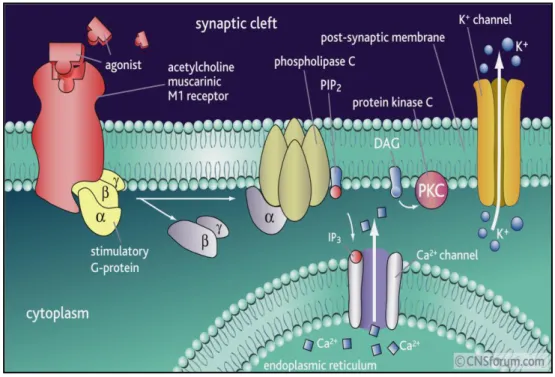
Alcune sostanze, dette “inneschi” (o triggers), come per esempio, adenosina, bradichinina, norepinefrina e oppioidi, che hanno mostrato effetti cardioprotettivi, si accumulano nell’interstizio durante l’ischemia ed interagiscono con i propri recettori di membrana. Questi recettori sono accoppiati alla fosforilasi C (PLC), la cui attivazione catalizza l’idrolisi di fosfatidil-inositolo 4,5-bifosfato (PIP2), generando il messaggero secondario inositolo 1,2,4-trifosfato (IP3) e diacilglicerolo (DAG), che stimola a sua volta la protein chinasi C (PKC), che è, ritenuto il
mediatore centrale dell’IPC.28-33
Attraverso un processo di fosforilazione, la PKC provoca l’attivazione di varie proteine e, tra queste, anche i canali KATP.
I triggers dell’IPC possono essere classificati come dipendenti o come recettori-indipendenti; i primi esercitano la loro azione attraverso interazioni con recettori specifici (adenosina, oppioidi, bradichinina, etc) mentre i secondi sono sostanze endogene, così come l’ossido nitrico (NO), radicali liberi e calcio i quali non necessitano di un legame con il recettore. L’adenosina, un trigger recettore-dipendente, costituisce il primo attivatore ad essere stato identificato nel processo IPC. Gli effetti cardiaci dell’adenosina vengono mediati principalmente dai recettori A1 e A3.34,35 È stato osservato anche un importante coinvolgimento della bradichinina, un altro trigger recettore-dipendente, nell’IPC breve. Sembra infatti che adenosina e bradichinina agiscono in sinergia.28
L’NO, un trigger recettore-indipendente, è stato identificato come un fattore endogeno che viene coinvolto nella cardioprotezione. Attualmente, il principale ruolo dell’NO consiste nel ritardare la fase di IPC, tuttavia, il suo coinvolgimento nella prima fase di IPC è ancora controverso.36 Esiste, però, una chiara evidenza che la somministrazione di NO esogeno dà luogo ad una significativa diminuzione del danno al miocardio durante l’ischemia.
Sebbene il fenomeno terminale dell’IPC non sia stato ancora completamente identificato, diversi studi riportano che nell’IPC, c’è una diminuzione di accumulo di cataboliti, come il lattato37
, una riduzione dell’acidità intracellulare, e della glicolisi anaerobica. Inoltre si ha un cambiamento della concentrazione Na+/H+, down-regolazione di TNFalfa e attivazione dei canali KATP mitocondriali
.
Figura 6:Meccanismo di induzione del precondizionamento endogeno
CANALI SARC-K
ATPe MITO-K
ATPI canali KATP cardiaci sono stati chiaramente identificate sia nelle membrane sarcolemmali che
mitocondriali (rispettivamente sarc- e mito- KATP).
Il canale cardiaco sarc-KATP è costituito da una molecola ottomerica composta da quattro subunità
Kir6.2 e da quattro subunità SUR2A. La struttura del canale cardiaco mito-KATP non è stata ancora
chiarita, sebbene una recente pubblicazione ha evidenziato la presenza delle subunità Kir6.1, Kir6.2 e SUR2, mentre la subunità SUR1 non risulta presente.
Figura 7: Struttura molecolare del canale sarc-KATP
L’apertura del canale sarc-KATP, indotta da ipossia, ischemia, o da agenti farmacologici accelera la
ripolarizzazione delle membrane dei cardiomiociti, riducendo la durata del potenziale di azione e inibendo lo scambio Na+/K+, con conseguente diminuzione dell’ingresso di ioni Ca++ nella cellula. Queste azioni prevengono la saturazione di Ca++ nella cellula e migliorano la conducibilità della cellula stessa, riaggiustando il bilanciamento tra consumo energetico e domanda energetica.
Queste osservazioni farebbero ipotizzare una correlazione tra l’aumento della resistenza al danno ischemico e l’attivazione dei canali sarc-KATP, ma pubblicazioni recenti hanno indicato nei canali
mito-KATP, anziché nei canali sarc-KATP, i maggiori responsabili della protezione cardiaca
bimakalim e cromakalim, erano in grado di indurre un effetto cardioprotettivo anche a dosi prive di efficacia sul potenziale di membrana del sarcolemma.
I mitocondri sono responsabili della sintesi di ATP nelle cellule. In caso di crisi energetica, la funzionalità mitocondriale è compromessa ma l’apertura dei canali mito-KATP ripristina
parzialmente il potenziale di membrana,e previene la deplezione dei fosfati ad alta energia, creando un gradiente elettrochimico più favorevole per la sintesi dell’ATP. Nella cellula cardiaca questi canali hanno la funzione di proteggere la cellula da un sovraccarico di calcio durante ipossia prolungata, e pertanto rappresentano potenziali target per agenti terapeutici in grado di proteggere il cuore dal danno ischemico.
CANALI SARC-KATP E LORO RUOLO NELL’IPC
L’aprikalim, attivatore dei canali sarc-KATP, mima l’effetto dell’IPC nei cani trattati con barbital,
producendo una significativa riduzione delle dimensioni dell’infarto. La glibenclamide, inibitore dei canali sarc-KATP, invece abolisce questo effetto protettivo.40 Questi risultati sono stati confermati da
altri studi, come quelli condotti da Cole & altri, che hanno dimostrato che la glibenclamide
determina un danneggiamento delle funzioni ventricolari durante il periodo di riperfusione.41
E’ stato, anche, verificato il coinvolgimento dei canali sarc-KATP nell’IPC attraverso l’uso di un
inibitore dei canali sarc-KATP, HMR 1883, che ritarda la caduta del potenziale di azione e riduce la
protezione indotta dal diazossido42.
Cascata dei segnali coinvolta nella fase acuta dell’IPC indotto attraverso i canali
sarc-KATP
Nei miociti ventricolari del coniglio, l’attivazione di adenosina e di PKC determina un aumento del flusso attraverso i canali sarc-KATP durante un episodio ischemico transitorio. Questo effetto è
inibito dalla somministrazione di un antagonista selettivo dell’adenosina.43
Inoltre, è stato dimostrato che NO attiva i canali sarc-KATP sia in condizioni di normossia che in di
ipossia.44 Il rilascio di NO endogeno sembra non essere coinvolto nella fase iniziale dell’IPC.45
Esiste un’interazione tra l’attivazione dei canali sarc-KATP e quella dei mito-KATP, infatti
l’iperpolarizzazione dovuta all’attivazione del primo può portare all’attivazione del secondo e viceversa.
In queste condizioni, l’iperpolarizzazione aumenta l’attivazione della fosforilasi D, che a sua volta determina l’attivazione e la traslocazione della PKC, responsabile a sua volta dell’apertura di entrambi i canali sarc- e mito-KATP.46Questa teoria è stata provata anche attraverso l’utilizzo di 5-HD
l’azione di uno dei due antagonisti singolarmente non riesce a produrre effetto protettivo mentre la combinazione dei due antagonisti inibisce completamente tale effetto.47 Inoltre, studi recenti hanno confermato che l’IPC era parzialmente inibita dal 5-HD, ma completamente inibita da antagonisti non-selettivi dei canali KATP come glibenclamide.48
CANALI MITO-KATP E LORO RUOLO NELL’IPC
Già nel 1980 è stato scoperto l’effetto protettivo dei canali mito-KATP contro danni da ischemia,
sebbene a quel tempo non fosse ancora noto il meccanismo.49
Successivamente, tale ipotesi fu confermata mediante l’utilizzo di farmaci agonisti non selettivi (bimakalim) che, a basse dosi, non avevano effetto sui canali KATP del sarcolemma e sulla
concentrazione di APD.50
La prima prova diretta a supporto del coinvolgimento dei canali mito-KATP nella cardioprotezione
fu fornita dall’osservazione che nei mitocondri del tessuto cardiaco bovino, il diazossido induce l’apertura dei canali mito-KATP a concentrazione più bassa di quella necessaria ad aprire i canali
sarc-KATP. Anche nel cuore isolato del ratto, diazossido e cromakalim, a concentrazioni che non
inducono l’apertura dei canali sarc-KATP producono effetto cardioprotettivo. Questi effetti
cardioprotettivi vengono annullati non solo dalla glibenclamide, ma anche dal 5-HD, che è un bloccante selettivo dei mito-KATP, dimostrando così il maggior coinvolgimento di questo tipo di
canale KATP.51
Cascata dei segnali nella fase acuta dell’IPC indotta dai canali mito-KATP
L’attivazione e la traslocazione di una specifica isoforma di PKC sembra essere il nodo centrale nell’apertura dei canali mito-KATP. Infatti, la protezione indotta dall’apertura dei canali mito-KATP
può essere abolita dalla somministrazione di antagonisti della PKC e l’effetto protettivo mediato dall’attivazione di PKC stessa può essere inibito da antagonisti mito-KATP; perciò, PKC e canali
mito-KATP sono entrambi necessari e interconnessi nell’effetto cardioprotettivo.52 Studi successivi
hanno rilevato che la PKC è “a monte” rispetto all’attivazione dei canali mito-KATP infatti, il
pretrattamento con diazossido porta alla riduzione della dimensione dell’infarto, senza aumentare il processo di traslocazione della PKC, mentre, 5-HD inibisce l’effetto del precondizionamento senza però bloccare la traslocazione della PKC.53
Esistono inoltre evidenze sperimentali che dimostrano che l’NO incentiva l’apertura dei canali mito-KATP: l’inibizione dell’ossido nitrico sintasi (NOS) ostacola l’azione protettiva del diazossido,
il quale dà origine ad entrambe le fasi, precoce e tardiva, dell’IPC attraverso la cascata di attivazione NO-dipendente.54 Studi condotti su modelli in vivo di cuore di coniglio hanno
dimostrato che la protezione diazossido-mediata risulta bloccata per trattamento con 5-HD, suggerendo così il ruolo dei canali mito-KATP e, per trattamento con un inibitore della NOS,
confermando così il ruolo svolto dall’NO in questo fenomeno.55
Ad oggi, esistono risultati controversi sulla funzione delle specie reattive dell’ossigeno (ROS). Da un lato, vi sono dati sperimentali che dimostrano come i ROS siano essenziali nella cardioprotezione, poiché determinano l’attivazione della cascata della PKC aumentando la probabilità di avere l’apertura dei canali mito- KATP. D’altra parte, il rilascio di ROS favorisce
l’apertura dei pori di transizione di permeabilità mitocondriale (MitoPTP) durante la riperfusione. Sebbene l’apertura del poro durante la riperfusione sembra essere una delle principali cause di morte cellulare, il rilascio di ROS e l’apertura transitoria del MitoPTP durante la fase di innesco dell’IPC sono stati considerati come effetti scatenanti nella cardioprotezione.57
Infatti, l’apertura momentanea dei MitoPTP in uno stato di bassa conduttanza, eviterebbe un pre-carico di ioni Ca++, così da prevenire un ulteriore attivazione calcio-indotta che comporta una più ampia e dannosa apertura di MitoPTP.58
Meccanismi di protezione attraverso i canali mito-KATP
Il flusso di ioni K+ verso la matrice mitocondriale è regolato dal potenziale della membrana interna e la sua fuoriuscita è compensata dall’antiporto K+
/H+, che rimuove K+ sfruttando il gradiente protonico.
I canali mito-KATP si trovano nella membrana interna di mitocondri e la loro apertura consente
l’ingresso di ioni K+
dentro la matrice. In condizioni fisiologiche l’ingresso di K+ è molto scarso e perciò l’effetto sul potenziale di membrana mitocondriale è marginale, causa però un significativo aumento del volume della matrice.
La permeabilità della membrana all’ADP e all’ATP è bassa, in condizioni di normossia, mentre in condizioni di ipossia la membrana esterna aumenta la conduttanza, determinando di conseguenza la contrazione della matrice e dunque l’espansione dello spazio intermembrana. L’apertura dei canali mito-KATP provvede a mantenere il volume della matrice a valori omeostatici, preservando la
distanza normale tra la porzione interna e quella esterna della membrana, coinvolta nella regolazione della permeabilità ai nucleotidi (ADP e ATP). Si ha un aumento della conduttanza al K+, per compensare l’abbassamento del gradiente elettrico, mantenendo così il volume della matrice praticamente costante e riducendo quindi l’idrolisi di ATP durante l’evento ischemico. Dato che il trasporto totale di ioni deve essere elettricamente neutro, l’influsso di K+
viene esattamente bilanciato da un efflusso di H+ tramite una pompa scambiatrice K+/H+. Se questa pompa fosse l’unico sistema a funzionare, lo scambio K+
intracellulare. In realtà la perdita di protoni è compensata da un ingresso di acido fosforico elettricamente neutro, anche se tale compensazione è solo parziale, e ne risulta comunque una alcalinizzazione della matrice.
L’apertura di canali mito-KATP causa inoltre una depolarizzazione della membrana interna che
riduce l’aumento della concentrazione di Ca++
intra-mitocondriale.59,60 Quando il metabolismo energetico cellulare risulta compromesso, come in caso di ischemia, i livelli cellulari di Ca++ aumentano a causa di una massiccia entrata di Ca++ dal compartimento extracellulare. Questo innalzamento della concentrazione citosolica di ioni Ca++ liberi viene in parte contrastata dal passaggio di questi ioni all’interno dei mitocondri, attraverso dei canali Ca++
selettivi. Ad alti livelli di Ca++ mitocondriale, il MitoPTP favorisce la fuoriuscita di Ca++ dalla matrice nel corso della riperfusione, ma non durante la fase ischemica, durante la quale il MitoPTP potrebbe non essere aperto. Questa apertura del MitoPTP rappresenta l’innesco all’apoptosi cellulare, poiché conduce al rilascio di proteine (dallo spazio mitocondriale intermembrana al citosol) pro-apoptotiche, come il citocromo c. Perciò l’apertura del MitoPTP durante la riperfusione rappresenta il fattore più importante nel determinare le lesioni irreversibili riperfusione-indotte a livello dei cardiomiociti. Il legame tra l’attivazione dei canali mito-KATP e del MitoPTP non è ancora del tutto chiaro. E’
ampiamente accettato che una porzione significativa della cardioprotezione sia dovuta all’inibizione dell’attivazione di MitoPTP durante la riperfusione e, di conseguenza, la prevenzione del danno cellulare indotto dalla riperfusione. Pertanto, l’attivazione dei canali mito-KATP durante la fase
ischemica può determinare la riduzione dell’accumulo di Ca++
nella matrice mitocondriale. Tale riduzione a livello della matrice stessa può prevenire l’apertura protratta del MitoPTP nella fase di riperfusione. Quindi, l’inibizione del MitoPTP è probabilmente il meccanismo d’azione più rilevante, sfruttato da agonisti mito-KATP per ottenere effetti cardioprotettivi.
Infine recenti studi hanno mostrato che l’apertura dei canali mito-KATP comporta
un’alcalinizzazione della matrice della membrana e una conseguente diminuzione della produzione mitocondriale di specie reattive dell’ossigeno.60
Figura 8:Rappresentazione del meccanismo di attivazione correlato del canale sarc-KATP
e del mito-KATP
POTENZIALE TERAPEUTICO
I canali al potassio sono un gruppo di canali ionici eterogenei e ubiquitari, che mostrano un certo coinvolgimento nei processi di controllo cellulare, perciò i farmaci che riducono l’eccitabilità cellulare attraverso l’apertura di questi canali possono avere un ampio potenziale per l’uso clinico. Nello specifico, andando ad analizzare il solo ruolo dei canali KATP nel mantenimento
dell’omeostasi cellulare, gli agonisti dei canali al potassio (KCOs) possono trovare impiego nel trattamento di patologie come: asma, ipertensione, incontinenza urinaria, ischemia cardiaca, disfunzione del CNS.
L’utilità terapeutica dei KCOs dipende in definitiva dalla loro selettività tissutale.
ATTIVATORI DEI CANALI K
ATPI ligandi dei canali ionici in genere esibiscono una pronunciata diversità chimica, e questo vale anche per gli attivatori dei canali KATP, che comprendono differenti classi strutturali. I KCOs
(Kallium channel Openers) di “prima generazione”, comprendono i benzopirani, le cianoguanidine, le tioformammidi, le tiadiazine e i piridilnitrati. Successivamente sono stati sintetizzati nuovi composti, migliori dal punto di vista dell’attività e della selettività, definiti di seconda generazione, e includono i ciclobutenedioni, i derivati delle diidropiridine e i carbinoli terziari.
KCOs DI PRIMA GENERAZIONE
Benzopirani
Tra gli attivatori dei canali KATP di “prima generazione”, i benzopirani rappresentano la classe di
composti più conosciuta e studiata. Il cromakalim rappresenta il capostipite di questa classe, è stato sintetizzato nel 1980 ed è stato sottoposto a varie modifiche strutturali. Questo composto è frequentemente usato come farmaco cardioprotettivo per studi sperimentali, sebbene non abbia mostrato alcuna selettività verso i canali mito-KATP, essendo responsabile infatti di una marcata
Il cromakalim contiene due centri stereogenici, i carboni 3 e 4: l’attività di KCO risiede nell’enantiomero 3S,4R- detto levcromakalim; il gruppo 3-OH e l’anello 4pirrolidinone sono in posizione trans tra loro.
Modifiche strutturali sulla struttura benzopiranica
1. Variazioni in posizione 4:
Il gruppo carbonilico, in posizione 4 della pirrolidina del cromakalim, è stato considerato essenziale per l’attività biologica poiché i lattami sono vasodilatatori più potenti rispetto alle ammine cicliche originarie.
Le prime variazioni in posizione 4 comprendono lattami e sostituenti contenenti gruppi carbonilici in posizione α dal punto di attaccamento sul nucleo benzopiranico, mentre recenti variazioni includono sostituenti contenenti carbonili, ma non in posizione originale.
Le modifiche strutturali principali effettuate in posizione 4 sono: Sostituzione di natura ciclica;
Sostituzioni con eteroatomi a loro volta sostituiti con raggruppamenti ciclici (sostituenti “a ponte”);
Sostituzione di tipo aciclico.
2. Variazioni strutturali in posizione 3:
La maggior parte dei benzopirani risultano non sostituiti in posizione 3 oppure presentano un gruppo idrossilico. In una serie di derivati benzopiranici, diversamente sostituiti in posizione 3, è stato osservato che in presenza di gruppi come CHO o CH2OH si mantiene una moderata potenza
ipotensiva mentre con gruppi come Br o CH3 si ha una totale perdita di efficacia.Sono stati eseguiti
studi sugli effetti della trasposizione dei sostituenti dalla posizione 3 alla 4; questi hanno condotto ad una serie di composti, tipo BRL 49381, che risultano avere una potenza simile al cromakalim. L’importanza del gruppo OH è ambigua. Mentre la presenza del 3–OH nel levcromakalim aumenta la potenza di 15 volte rispetto all’analogo con 3–H, i cromeni come KC-399, che non hanno sostituenti in questa posizione, sono di gran lunga più potenti del levcromakalim. Di conseguenza, è
N
O
OH
N O
improbabile che il gruppo 3-OH interagisca con il sito di legame. E’ ancora più improbabile che questo gruppo stabilizzi la conformazione bioattiva.
3. Variazioni strutturali in posizione 2:
L’influenza sulla potenza dei derivati benzopiranici del sostituente nella posizione 2, dipende dalla natura del sostituente in posizione 4.
Per il cromakalim il sostituente ottimale in posizione 2 è il gruppo dimetilico, che risulta essere più attivo di quello monometilico e di quello etilico. Il composto di-idro è relativamente privo di
attività.
38
4. Sostituzioni aromatiche:
La posizione e la natura del sostituente aromatico, sono due fattori capaci di influenzare notevolmente la potenza dei derivati benzopiranici. L’attività è abolita da sostituzioni aromatiche in posizione 5 e 8, mentre aumenta passando alle posizioni 7 e 6. Nelle prime ricerche, la sostituzione ottimale è stata attribuita ai sostituenti elettronegativi. Successivamente, è stato scoperto che i sostituenti più ingombranti, come il fenilsulfonile in posizione 6 del rilmakalim, sono capaci di conferire un’elevata potenza.39Da studi di relazione struttura-attività e dall’analisi conformazionale, è possibile ritenere che i sostituenti in 6 accrescano l’interazione al sito di legame.
5. Trasformazioni del nucleo benzopiranico:
Il sito di legame per i benzopirani accoglie cambiamenti sia dell’anello aromatico che dell’anello pirano. La piridina può sostituire il cianofenile, come mostrato nel composto A. La posizione 6 dell’azoto piridinico è migliore di quello in posizione 7, le posizioni 5 e 8 danno composti inattivi.
Rispetto al cromakalim, l’introduzione di NH o CH2 riduce la potenza di circa 10 volte, mentre il carbonile di circa 30 volte. L’ossidazione a solfossido o solfone è dannosa. Le sostituzioni più vantaggiose dell’anello piranico sono state effettuate con sistemi eterociclici, come le 1,4-benzossazine, tra cui il YM 934 che è un potente vasodilatatore con particolari effetti sulle
coronarie
40
, e le 1,4-benzotiazine come B che mostra una buona attività sia in vitro che in vivo.
O N NH NH O BRL 49381 O CH 2F CH2F NH S N N+ O -O KC 399
Cianoguanidine
Pinacidil e i suoi analoghi cianoguanidinici sono stati sviluppati a partire da composti a struttura N-alchil-N’-piridin tioureica aventi proprietà antipertensive.
N N NH N H CN t-Bu Pinacidil N N NH N H CN N P-1075
TRASFORMAZIONE DELL’ANELLO AROMATICO
TRASFORMAZIONE DELL’ANELLO PIRANICO
N O N OH O A N S N+ O -O O B N O N+ O -O N O YM 934
Il pinacidil è il prototipo della serie e la sua attività risiede nell’enantiomero R-(-).
Il più potente composto di questa categoria risulta essere P-1075, le cui proprietà farmacologiche sono state ampiamente studiate. P-1075 si lega con elevata affinità alle subunità SUR2A e SUR2B e di conseguenza è stato proposto come attivatore selettivo dei canali sarc-KATP. Questa sua
caratteristica ha permesso di usarlo in studi farmacologici allo scopo di evidenziare il coinvolgimento dei sarc- e mito-KATP nell’IPC. P-1075 riduce la disfunzione contrattile e il
sovraccarico di calcio in seguito a inibizione metabolica nelle cellule miocitarie del ratto. Questo effetto è inibito in seguito a trattamento con un bloccante selettivo dei canali sarc-KATP, come
HMR1098.
Primi studi di SAR su derivati del pinacidil hanno dimostrato che i derivati 3-piridilici come P1075 sono più potenti dei corrispondenti 4-piridinici, ad eccezione del pinacidil stesso. Nella porzione centrale, la catena cianoguanidinica si è rivelata migliore della tiourea e molto superiore all’urea. Lo scambio del gruppo ciano con altri gruppi polari porta invariabilmente a composti privi di attività. Nella parte terminale della catena lipofila sono ottimali gruppi alchilici piccoli e ramificati.
Altre due derivati cianoguanidinici, PNU-99963 e PNU-94750, meritano di essere menzionate in quanto presentano un’attività di tipo bloccante sui canali KATP. La modificazione chimica principale
è la presenza di un anello fenilico nella catena laterale lipofila. L’attività antagonista di PNU-99963 ha permesso di ipotizzare che questo composto sia in grado di modulare il legame dell’ATP e l’attività ATPasica della subunità SUR in modo tale da determinarne il blocco.
Piridil nitrati
Nicorandil è il composto più rappresentativo della classe dei piridil-nitrati. Mostra attività vasorilasciante dovuta non soltanto all’attivazione dei canali KATP, ma anche
al fatto che il nicorandil stimola l’attività della guanilato-ciclasi nella muscolatura liscia vascolare e agisce come NO-donor.. I dati di SAR su questa classe di composti sono pochi, e il numero di analoghi di nicorandil presenti in letteratura è limitato. Tra questi si annovera una struttura
Cl N NH N H CN N cBut PNU-99963 Cl N C H3 NH N H CN N PNU-94750
presentante la sostituzione dell’anello piridinico con un piperazinico C, e altri due composti che presentano rispettivamente un anello pirazinico D o tiazolico E.
Benzotiazidi
Il diazossido, prototipo di questa classe di composti, determina l’apertura dei canali mito-KATP con
una buona selettività, senza l’attivazione dei canali cardiaci sarc-Katp. Infatti, è usato normalmente a basse dosi per rivelare il coinvolgimento dei canali mito-KATP nel precondizionamento; ad alte
dosi, il diazossido attiva i canali KATP dei muscoli dell’endotelio vasale e debolmente i canali
cardiaci sarc-KATP.
Il suo sito di legame è posizionato in regioni della proteina SUR diverse rispetto a quelle del sito di legame per i benzopirani e le cianoguanidine. Il diazossido è l’unico che lega con simile affinità sia la SUR1 che la SUR2B, pertanto rilascia la muscolatura vascolare liscia e stimola la secrezione insulinica in maniera equipotente. Ulteriori esperimenti hanno dimostrato che, nel trattamento con
N NH O ONO2 Nicorandil N N N NH O O NO2 C N H N NH O O NO2 D N S NH O O NO2 E N H S N O O DIAZOSSIDO
diazossido, si può avere una maggior probabilità di apertura dei canali sarc-KATP durante
l’inibizione metabolica, questo effetto, però, non è necessariamente dovuto ad un azione diretta sul canale, ma può essere attribuita al disaccoppiamento mitocondriale o all’inibizione della succinato deidrogenasi.
Le proprietà di questo composto tiadiazidico sono state determinate attraverso studi funzionali e di binding. Partendo dal diazossido come lead compound, sono stati sviluppati alcuni derivati del diazossido selettivi sia per il pancreas che per la muscolatura liscia. Queste nuove strutture sono ibridi tra diazossido e pinacidil. Da questi studi è derivato un modello farmacoforico per l’attività agonista di tali composti sui canali KATP delle cellule del pancreas. In particolare, la sostituzione
della porzione 7-clorobenzenica con il nucleo bioisosterico piridinico ha migliorato significativamente la selettività; un esempio è il prototipo BPDZ-44. Il cambiamento dell’azoto della piridina porta a
composti con una selettività tissutale opposta: risultano infatti più selettivi nel rilasciamento dell’aorta come nel caso dei composti BPDZ-79 e sia BPDZ-83. Questo conferma come anche sottili variazioni strutturali possano influenzare la selettività tissutale.
Altri studiosi hanno sviluppato composti ancora più potenti e selettivi sostituendo la piridina con un tiofene, come nel caso del composto F, che attiva i canali KATP pancreatici ed è almeno 1000 volte più potente del diazossido per quanto riguarda il rilascio d’insulina.
È stata anche progettata la sintesi di ibridi delle benzotiadiazine con le sulfoniluree (composto G), che hanno mostrano attività vasorilasciante comparabile con quella del cromakalim.
N S N N H NH O O BPDZ 44 N N S N H NH O O BPDZ 79 N N S N H NH O O Cl BPDZ 83 S N S N H Cl NH cBut O O F
Tioformammidi
Nella classe delle tioformammidi il prototipo è rappresentato da Aprikalim.
I precursori di questo composto sono derivati tioammidici di tetraidrotiofene, tetraidrofurano, 1,3-ossatiano o 1,3-ditiano con note proprietà anti-ulcera e anti-secretorie, e recanti una porzione piridilica in posizione 2.
La picartamide e una serie di suoi derivati sono stati sottoposti a vari test per valutarne il profilo biologico, tra questi composti il derivato H ha mostrato una marcata attività anti-ipertensiva, mentre è inattivo come anti-ulcera. L’effetto ipotensivo è stato attribuito al metabolita di ossidazione, cioè il solfossido. Sono stati effettuati studi di SAR principalmente considerando le tre porzioni più importanti: la funzione tioamidica, l’eterociclo aromatico e l’eterociclo saturo.
Funzione tioammidica: in questa regione della molecola sono permesse
solo poche modificazioni; la preferenza per i gruppi alchilici indica la presenza di una tasca idrofobica ristretta sul sito recettoriale, i gruppi migliori sono l’etile o il propile, ma butile o fenile sono di dimensioni ancora accettabili. L’enorme perdita di attività che si verifica quando si
R S NH O CH3 NH S N O O S NH NH O O O O O G S N NH S O APRIKALIM S N N H S S N N H S PICARTAMIDE H
sostituisce la tioammide con un ammide sottolinea i precisi requisiti strutturali necessari per l’interazione in questa parte della molecola.
Gruppo eterociclico aromatico: la posizione dell’azoto sull’anello piridinico è importante per l’attività di questi composti, infatti nei derivati 2 e 4 sostituiti si ha una drastica perdita di attività. Questo può essere compensato da un’adeguata sostituzione con gruppi elettrondonatori come si può osservare nel caso del derivato 3,4-dicloro sostituito che mostra l’attività migliore. Particolarmente interessante è il fatto che il gruppo 3-chinolil- sia migliore del 3-piridile, e il 2-naftil meglio del fenile. Questo dimostra una buona tolleranza nell’ingombro sterico in questa parte della molecola, e sostiene l’ipotesi di interazioni idrofobiche.
Eterociclo saturo: il gruppo tiopiranico è essenziale per l’attività dimostrano chiaramente che questa porzione corrisponde esattamente ai requisiti recettoriali. Analoghi contenenti un solfone, un gruppo
tetraidrotiofenico o tetraidrofuranico determinano, infatti, un drastico calo dell’attività. Solo con un cicloesano l’attività viene quasi mantenuta. Questa scoperta ha dato inizio allo sviluppo di una nuova serie di composti contenenti la porzione cicloesanica.
KCOs DI SECONDA GENERAZIONE
Per quanto riguarda i KCOs di prima generazione la loro utilità terapeutica risulta limitata a causa della loro scarsa selettività tissutale. Infatti solo quattro KCOs sono stati introdotti nella pratica clinica: il nicorandil per l’angina, il minoxidil per l’alopecia, il diazossido e il pinacidil per l’ipertensione.
Studi successivi sono stati quindi incentrati sulla progettazione di KCOs dotati di maggiore selettività, dando origine ai KCOs di seconda generazione, che presentano un migliore profilo soprattutto dal punto di vista della selettività,
KCOs cardioselettivi
I KCOs di prima generazione non sono stati utilizzati per il trattamento delle patologie del cuore ischemico a causa degli effetti secondari dovuti all’attivazione dei canali KATP presenti nella
muscolatura liscia vascolare. È quindi risultata evidente la necessità di sviluppare composti più
N
R
S R
selettivi nei confronti dei canali mito-KATP, considerati i principali responsabili del
precondizionamento ischemico
A questo scopo sono stati sviluppati molti ibridi tra i benzopirani, e le cianoguanidine. Il primo composto interessante di questa serie di composti è stato BMS 180448, che mostra una scarsa capacità vasorilasciante, ma conserva l’effetto cardioprotettivo.
BMS 180448 presenta un’attività antiischemica comparabile a quella del cromakalim, con un miglior recupero della funzionalità contrattile dopo riperfusione, aumentando il tempo di contrazione (definito come l’arco di tempo necessario durante l’ischemia globale ad aumentare la pressione diastolica finale di 5mmHg) e diminuendo il rilascio di lattato deidrogenasi (LDH). Inoltre, il cromakalim produce un sensibile aumento del flusso sanguigno coronarico, mentre BMS 180448 non ha mostrato tale effetto. In aggiunta, cromakalim riduce significativamente l’APD e l’intervallo QT, mentre BMS 180448 non altera questi parametri.
A partire da questo farmaco, i ricercatori hanno effettuato dettagliati studi di SAR
41
per ottimizzare le proprietà cardioselettive. Per quanto riguarda le modifiche sull’anello benzopiranico, la sostituzione dell’ossigeno in posizione 1 con un metilene è tollerata, invece la sostituzione con NH porta a composti inattivi. I gruppi metilici geminali sono essenziali, infatti gli analoghi demetilati sono privi di attività. La presenza del gruppo OH in posizione 3 non è necessaria ai fini dell’efficacia del farmaco, ma se è presente, il trans-OH risulta essere più attivo del cis-OH. L’introduzione di un doppio legame 3-4 abolisce l’attività antischemica suggerendo che un carbonio sp3è preferibile ad un C4.
Un altro composto interessante è BMS 191095, che appartiene alla serie dei benzopirani 4-(N-aril)-sostituiti. Questo derivato risulta essere almeno trenta volte più selettivo rispetto al BMS 18044842.
O N OH NC NH NH Cl NC BMS 180448
L’attività cardioprotettiva di questo farmaco è simile a quella del cromakalim e del BMS 180448, ed è antagonizzata dall’azione di farmaci come la glibenclamide e 5-HD. Inoltre BMS 191095, come il cromakalim e BMS 180448, ha mostrato un miglioramento della funzionalità cardiaca post-ischemica, un aumento del tempo di contrazione e un ridotto rilascio di LDH. In più, ha mostrato una ridotta influenza sull’APD, sull’intervallo QT e una minor azione vasodilatante. Sfortunatamente gli studi su BMS 191095 sono stati interrotti a causa della sua neurotossicità. Sono stati sviluppati altri derivati a struttura benzopiranil-indolinica o indolo-sostituita: in particolare, l’acido 6-nitro-benzopiranil-indolin-2-carbossilico ha mostrato, in studi sui ratti in vitro, una buona azione cardioprotettiva e un basso potere vasodilatante.43
O OH NH N Cl BMS 191095 N H O O2N COOH Acidi 6-nitro-benzopiranil-2-carbossilici