3. Risultati e discussione inerenti la sintesi dell’azadisaccaride
a struttura β-
D-GalNAc-(1
→4)-DNJ
Il secondo obiettivo di questo lavoro di Tesi è stata la sintesi del mimico disaccaridico a struttura β-D-GalNAc-(1→4)-DNJ (74, Schema 28). Fra le possibili strade sintetiche applicabili alla preparazione di disaccaridi contenenti unità DNJ, descritte nel paragrafo 1.5, è stata scelta quella basata sulla trasformazione del lattosio (18), un disaccaride disponibile da fonti naturali, che consente di evitare lo step di glicosidazione. Formalmente, la conversione del lattosio (18) nell’azadisaccaride 74 (Schema 28) comporta una reazione di amminazione con ritenzione di configurazione della posizione 2’ dell’unità β-D-galattopiranosidica, seguita da ossidazione selettiva
della posizione 5 dell’unità riducente in modo da ottenere l’esos-5-ulosio disaccaridico
94 a configurazione D-xilo su cui applicare la reazione di doppia amminazione riducente intramolecolare a dare il mimico disaccaridico 74. E’ da sottolineare che queste trasformazioni sono puramente ipotetiche e possono essere realizzate solo attraverso la preparazione di opportuni precursori protetti sugli ossidrili che non partecipano alle specifiche reazioni. Schema 28 O O OH O H OH OH O H O OH NHAc CHO OH O O H OH OH O O O H OH O H OH OH O O H OH NHAc O N H O H OH OH 74 94 18 β-GalNAc-(1>4)-DNJ
Nel Laboratorio dove ho svolto il mio lavoro di Tesi è stata acquisita nell’ultimo decennio una vasta esperienza sulla chimica del lattosio, e sono state messe a punto numerose sintesi di carboidrati, difficilmente accessibili da fonti naturali, basandosi sulla considerazione che il lattosio può essere trattato come un semplice glucopiranosio sostituito in posizione 4 con un gruppo β-D-galattopiranosidico oppure come un β-D -galattopiranoside avente come aglicone un residuo glucopiranosico. Nell’ipotesi che una delle due unità monosaccaridiche sia completamente protetta, sarà possibile applicare all’altra unità tutte le manipolazioni tipiche della chimica dei carboidrati (ossidazioni, epimerizzazioni, amminazioni, etc.). Ugualmente sarebbe possibile
applicare ad una sola delle due unità tutte quelle reazioni che, a causa delle differenze strutturali fra i due monosaccaridi, avvengono in maniera completamente chemoselettiva su uno solo di essi. Questa semplificazione richiede, per aver validità, una totale o, comunque molto avanzata, protezione con gruppi sufficientemente stabili, dell’una o dell’altra unità monosaccaridica, in modo da poter considerare, all’occorrenza, come sostituente la porzione non interessata. Proseguendo su questo ragionamento, i processi sintetici necessari per trasformare il lattosio (18) nel mimico azadisaccaridico 74 richiedono, basandoci sull’esperienza acquisita nella sintesi di β-D
-TalNAc-(1→4)-DNJ (29, Figura 22, pag. 16) e β-D-ManNAc-(1→4)-DNJ (30, Figura
22, pag. 16),10 quattro processi sintetici illustrati nello Schema 29:
1. realizzazione di un’efficiente differenziazione delle due unità monosaccaridiche del lattosio (18) che determini la completa protezione dell’unità riducente di D -glucosio e la protezione differenziata in 3’, 4’ e 6’ dell’unità β -D
-galattopiranosidica, fornendo così un precursore di tipo 95;
2. amminazione in posizione 2’ di 95 con formale ritenzione di configurazione per accedere ad un derivato β-GalNAc (96);
3. elaborazione dell’unità di D-glucosio di 96, sostituita in 4 con l’unità β-D -esosamminica, con l’obiettivo di realizzare un’ossidazione regioselettiva del gruppo alcolico in 5 ottenendo un derivato D-xilo-esos-5-ulosio (97);
4. doppia amminazione riducente intramolecolare (amminociclizzazione) del derivato 1,5-dicarbonilico 97 con formazione dell’azapiranoso a struttura DNJ, β-glicosilata in 4 con l’unità esosamminica, da cui, dopo deprotezione finale, si otterrà il target 74. Schema 29 OH O O O H NHAc OH O H N H OH OH OHO O OH OH O O H OH OH OH OH O O O O O (MeO)2CH OP2 O P1O OH OP3 O O O O O (MeO)2CH OP2 O P1O NP OP3 OP2 O O P1O NP OP3 O H OH OH O CHO 95 18 97 96 74
La presentazione e la discussione dei risultati di questa seconda parte della Tesi saranno, quindi, riportati seguendo l’ordine logico delle 4 sequenze sopra descritte.
3.1. Protezione differenziata delle unità monosaccaridiche del lattosio
Un metodo molto efficiente di differenziazione delle due unità monosaccaridiche costituenti il lattosio è stato sviluppato nel nostro Laboratorio, sottoponendo il lattosio ad una doppia reazione di acetonazione con 2,2-dimetossipropano (DMP) promossa da quantità catalitiche di acido p-toluensolfonico (TsOH).20 La prima acetonazione prevede un trattamento a caldo (80°C) di una soluzione di lattosio (18) in un largo eccesso di DMP, che funziona, quindi, da solvente e agente acetonante (Schema 30). Dopo 12 ore il prodotto di partenza risulta scomparso, mentre si nota la formazione di due prodotti principali, attribuibili al triacetonlattosio dimetilacetale (99, Rf 0.26) ed al suo
6’-O-metossiisopropil derivato (98, Rf 0.47). I due poliacetonuri derivano, oltre che dalla
formazione dei gruppi isopropilidenici e, nel caso di 98, anche di quello acetalico misto in posizione 6, da reazione di acetalazione fra il gruppo aldeidico dell’unità riducente del D-glucosio con il metanolo che si libera nella reazione con DMP. Il rapporto fra i due acetonuri 98 e 99 è, in questo primo step di acetonazione, a favore del secondo, per un rapporto di circa 2:3. Schema 30 OH O OH O O OH OH OH O H OH O H O O O O (MeO)2CH OGlc O O OH OC(OMe)Me2 O OGlc O O OH OH O 18 98 (65%) Glc = 1) TsOH, DMP, 80°C 2) TsOH, DMP, t.a. + 99 (22%)
Poiché il derivato del lattosio più utile per i nostri scopi è 98, che presenta solo la funzione ossidrilica al C-2’ deprotetta, dopo neutralizzazione del grezzo con NEt3,
seguita da evaporazione dell’eccesso di DMP e di MeOH e da rigoroso allontanamento dell’eccesso di base mediante ripetute coevaporazioni con toluene, si procede ad una seconda acetonazione riprendendo semplicemente il residuo con DMP. Dopo 3 ore a temperatura ambiente si osserva (TLC) che il rapporto fra le due macchie corrispondenti a 98 e 99 risulta invertito, grazie allo stabilirsi di una nuova posizione di equilibrio fra i due componenti come risultato dell’allontanamento del metanolo. Una flash-cromatografia su silice porta ad ottenere i due poliacetonuri 98 e 99 puri, in resa rispettivamente del 65 e 22%, con proprietà chimico-fisiche e caratteristiche NMR corrispondenti a quanto riportato in letteratura.20 Di particolare interesse è la possibilità
di eseguire questa reazione su larga scala (decine di grammi) consentendo, in una sola reazione, di ottenere quantità di precursore 98 sufficiente per le sequenze successive.
3.2. Trasformazione dell’unità
β-
D-Galp nell’unità
β-
D-GalNAc
Il tetracetonuro del lattosio 98 risulta un ottimo punto di partenza per la nostra sequenza sintetica per due motivi: presenta un’unica funzione ossidrilica libera (OH-2’) e, inoltre, le due tipologie di gruppi protettivi presenti sull’unità D-galatto sono ortogonali tra loro. Di fatti, il gruppo acetalico misto 2-metossiisopropilidenico (MIP) in 6’ è molto più labile in ambiente acido rispetto ai gruppi isopropilidenici, tanto che può essere semplicemente idrolizzato in condizioni (MeOH-H2O e tracce di AcOH) in cui i
tre acetali ciclici sono perfettamente stabili, ma allo stesso tempo è sufficientemente stabile da permettere eventuali funzionalizzazioni (alchilazioni, acilazioni) o elaborazioni della posizione 2’.20
La trasformazione dell’unità β-D-galattopiranosidica di derivati disaccaridici tipo 100 (Schema 31) in unità β-D-GalNAc 100c prevede un’amminazione in posizione 2’
con formale ritenzione di configurazione realizzabile, in linea di principio, attraverso due semplici metodologie. Secondo un primo approccio, è possibile ipotizzare la preparazione dei derivati 100c attraverso una sequenza che prevede la trasformazione di
100 nell’ossima 100a, previa ossidazione a chetone, seguita da riduzione stereoselettiva
a dare, dopo N-acetilazione, il derivato acetammidico 100c.
Schema 31 O OR O O OH OR' O OR O O NHAc OR' O O N OR OR' O OX O OR O O NHAc OR' 100 100a 100b 100c +
Questo approccio, ampiamente studiato nel nostro Gruppo di ricerca sia su derivati monosaccaridici che disaccaridici, non è completamente stereoselettivo e conduce ad una forte prevalenza del derivato a configurazione β-TalNAc 100b anche se variabile in funzione del tipo di sostituente all’O-6 e all’O-ossimico. Infatti, nella riduzione con LiAlH4 in Et2O dell’ossima l’attacco dell’idruro avviene sulla faccia α del doppio
Un secondo approccio ai derivati β-GalNAc 100c può essere realizzabile attraverso due consecutive reazioni con inversione di configurazione al C-2’. La prima è una semplice epimerizzazione (ossidazione-riduzione) e permette la trasformazione dell’unità β-D-galattopiranosidica in un’unità β -D-talopiranosidica, mentre la seconda è
un’amminazione con inversione dell’intermedio talopiranosidico per ottenere un derivato a configurazione 2’-ammino-D-galatto. Questa efficiente sequenza,
recentemente applicata all’alcol 98,21
L’ossidazione del derivato 98, a causa della labilità della funzione metossiisopropilica in 6’, richiede l’uso di sistemi di reazione che operano in condizioni rigorosamente neutre come il sistema ossidante tetrapropilammonioperrutenato (nPrN4+RuO4-, TPAP) in presenza di N-metilmorfolina-N-ossido (NMO), largamente
utilizzato nel nostro laboratorio. Il TPAP è un reagente dotato di una buona chemoselettività: ossida rapidamente funzioni ossidriliche primarie in presenza di secondarie e la loro ossidazione porta alla formazione di aldeidi. Il TPAP è compatibile con numerosi gruppi funzionali reattivi come: epossidi, acetali e doppi legami e, inoltre, non causa racemizzazione dei centri stereogenici in posizione α al carbonile. Le quantità di TPAP che si usano in reazioni condotte in presenza di NMO sono generalmente catalitiche, dato che l’NMO stesso, in eccesso nell’ambiente di reazione, ne rigenera le quantità consumate durante la reazione attraverso un meccanismo che non è stato ancora perfettamente chiarito.
ha fornito il derivato disaccaridico 104 con completa stereoselettività (Schema 32).
Schema 32 OGlc O O O O OC(OMe)Me 2 OGlc O O O OH OC(OMe)Me2 O OGlc O O OH OC(OMe)Me2 O OGlc O O OSO2Im OC(OMe)Me2 O O O O (MeO) 2CH O OGlc O O N3 OBn 101 NMO, TPAP 4A MS, DCM anidro 98 NaBH4, MeOH NaH 60%, Im2SO2 DMF, -30°C 1) NaN3, DMF, 100°C 2) HCl (aq) 5% 3) NaH 60%, BnBr DMF Glc = 102 (96% ) 103 (92%) 104 (77%)
L’ossidazione dell’alcol 98 è effettuata a temperatura ambiente in soluzione di CH2Cl2 anidro contenente 0.05 equivalenti di TPAP commerciale ed 1.5 equivalenti di
al fine di rimuovere l’acqua che si forma durante la reazione. In queste condizioni, dopo 4 ore a temperatura ambiente, si forma un unico prodotto a Rf maggiore che è isolato
per semplice filtrazione della miscela di reazione attraverso un triplice strato alternato di celite-gel di silice-celite ed evaporazione del solvente.
L’analisi NMR (1H, 13C) del grezzo ottenuto come sciroppo evidenzia la presenza esclusiva del 2-uloside 101 (resa 98%), ed esso può essere direttamente utilizzato nella successiva reazione senza ulteriori processi di purificazione.
Il chetone 101 presenta dati analitici e spettroscopici in accordo con quelli riportati in letteratura.22
Il 2’-uloside 101 grezzo, sottoposto ad una riduzione con idruri, fornisce in alta resa chimica e completa stereoselettività il derivato β -D-talopiranosidico 102.22 Trattando,
quindi, il composto 101 con NaBH4 in MeOH si evidenzia (TLC, 2 ore) la scomparsa
del prodotto di partenza e la formazione di un unico prodotto. Sottoponendo la miscela di reazione a consueto trattamento si ottiene un grezzo di consistenza sciropposa che per cromatografia su gel di silice fornisce l’alcol 102 puro (resa 96% calcolata da 98). L’analisi spettroscopica (1H, 13C) conferma, in accordo con quanto riportato in letteratura,22 la configurazione D-talo del composto 102, e l’avvenuta inversione di configurazione della posizione 2’ è confermata, nello spettro protonico, da un doppietto (δ 4.90) assegnato al protone anomerico che, presentando una costante di accoppiamento J1´,2´ di 2.2 Hz, conferma l’assetto assiale-equatoriale dei protoni H-1 e
H-2. La completa stereoselettività della reazione di riduzione del carbonile è attribuita, in analogia a quanto descritto nel caso delle ossime, sia alla presenza del ponte isopropilidenico sulla faccia β dell’anello piranosidico, che esercita un effetto sterico di schermo impedendo l’attacco dell’agente riducente, sia dal controllo stereoanomerico che dirige l’attacco dell’idruro da parte opposta a quella del sostituente anomerico.
In particolare, negli spettri 1H e 13C NMR, risultano diagnostici rispettivamente il doppietto a δ 5.20, attribuito al protone H-1’ (J1’,3’ 0.7 Hz), ed un
segnale a δ 197.3, attribuibile alla risonanza del C-2’ di natura chetonica.
Il derivato 102 rappresenta un ottimo precursore per la realizzazione dell’amminazione con inversione di configurazione in posizione 2’ attraverso la più classica trasformazione di una funzione alcolica in amminica mediante reazioni SN2 con
un nucleofilo azotato su un solfonato come l’imidazolo-1-solfonato (imidazilato, Imz), secondo quanto illustrato nello Schema 32. L’imidazilato è un gruppo uscente con
sostituzione nucleofila in cui altri solfonati non danno risultati soddisfacenti e risulta migliore, ad esempio dei reattivi triflati, per la maggiore maneggevolezza dei reagenti di partenza, per la semplicità delle procedure operative e per l’ottenimento di composti stabili e purificabili su silice.
La reazione è condotta a basse temperature e avviene tra l’alcossido del derivato deprotetto, generato per trattamento dell’alcol con NaH in DMF, e un eccesso di N,N-sulfurildiimidazolo (Im2SO2) in modo da portare alla formazione del desiderato
imidazilato e di un equivalente del sale sodico dell’imidazolo (Schema 33).
Schema 33 N N S N N O O N N R O N N S R O O O Reagenti: i: 1) DMF, NaH, 0°C, 2) Im2SO3, -30°C i Na + -+ sodio imidazolato imidazolo-1-solfonato
La preparazione dell’intermedio imidazilsolfonato 103 (Schema 32) è realizzata per trattamento di 102 con NaH e Im2SO2 a -30°C. Quando l’analisi TLC (2 ore) mostra la
formazione di un unico prodotto si distrugge l’eccesso di NaH per aggiunta di MeOH a -40°C. Dopo estrazione del prodotto, evaporazione dei solventi e purificazione flash cromatografica del grezzo, si ottiene l’imidazilsolfonato 103 puro (resa 92%) che presenta dati analitici e spettroscopici in accordo con quelli riportati in letteratura.21 In particolare, l’analisi NMR (1H, 13C) evidenzia i segnali della struttura imidazolica (vedi parte sperimentale), mentre il forte effetto di deschermo (13 ppm) che si evidenzia sul C-2’ (da δ 66.9 a δ 79.9) conferma l’avvenuta solfonazione del gruppo alcolico in 2’.
Il trattamento di 103, avente un buon gruppo uscente sul C-2’, con NaN3 a 100°C in
DMF porta, attraverso una sostituzione di tipo SN2, alla corrispondente azide,
precursore diretto dell’unità β -D-GalNAc. Allo scopo di sostituire il labile gruppo
metossiisopropilico (MIP) in posizione 6’ con un sostituente più stabile alle successive condizioni del processo sintetico, come il gruppo benzilico, il grezzo contenente l’azide è diluito con diclorometano e trattato con HCl acquoso al 5% (v/v) allo scopo di idrolizzare selettivamente il gruppo MIP in posizione 6’. Per estrazione ed evaporazione dei solventi si ottiene un grezzo di reazione che sottoposto a benzilazione classica (BnBr, NaH in DMF), seguita da purificazione flash cromatografica su gel di silice, fornisce il 6-O-benzil derivato 104 con una resa del 77% calcolata da 103.
L’azide 104, non riportata in letteratura, è stata completamente caratterizzata dal punto di vista chimico-fisico e i dati NMR, ricavati utilizzando tecniche monodimensionali (1H e 13C) e bidimensionali (DEPT-135, COSY, HETCOR), sono in accordo con la struttura proposta. In particolare, nello spettro 13C, il segnale relativo al C-2’ (δ 67.5) risulta più schermato (circa 13 ppm) rispetto al corrispondente carbonio in
103 per la sostituzione del gruppo imidazilsolfato con un gruppo azidico in questa
posizione. Inoltre, l’avvenuta inversione al C-2’ è evidenziata dai valori delle costanti di accoppiamento J1’,2’, J2’,3’ eJ3’,4’ che, passandoda 1.0, 5.7 e 5.8 Hz in 103 a 8.5, 8.2 e 5.3
Hz in 104, indicano una disposizione assiale-assiale-assiale dei protoni H-1’, H-2’ e H-3’, propria della configurazione D-galatto. La presenza del gruppo benzilico sul C-6’
è confermata, nello spettro protonico, dalla presenza di un multipletto (δ 7.38-7.27) attribuibile ai 5 protoni aromatici. Inoltre, nello spettro 13C, la perdita dei segnali relativi al gruppo metossiisopropilico (MIP) ed il valore del chemical shift del segnale relativo al C-6’ (δ 69.8), più deschermato (circa 10 ppm) rispetto al corrispondente segnale di
103 (δ 60.3), dimostra l’avvenuta rimozione del gruppo MIP e la successiva formazione
dell’etere benzilico in posizione 6’.
Il gruppo azidico è, in generale, facilmente trasformabile in gruppo acetammidico utilizzando diverse metodiche basate, in ogni caso, sulla riduzione ad ammina seguita da N-acetilazione con anidride acetica in MeOH. In precedenti ricerche a questa Tesi era stato osservato che in derivati disaccaridici contenenti unità β-GalNAc la rimozione selettiva delle funzioni isopropilideniche e la successiva ossidazione regioselettiva al C-5 dell’unità glucosidica, passaggi chiave per il proseguo della nostra sequenza sintetica, risultano poco efficienti. Sulla base di queste considerazioni è stato deciso di rimandare la trasformazione del gruppo azidico di 104 in NHAc alla fase finale della sintesi di 74, e quindi considerare 104 come il più idoneo precursore disaccaridico su cui realizzare la trasformazione dell’unità riducente di D-Glc in DNJ.
3.3. Preparazione del derivato 2-azido-
β-
D-galattopiranosil-(1
→4)-
D-xilo-esos-5-ulosico
Raggiunto il primo obbiettivo inerente la preparazione, completamente stereocontrollata, dell’azide β-D-galattopiranosidica protetta 104, ci siamo occupati dell’elaborazione dell’unità glucosidica aciclica, in modo da preparare l’intermedio
chiave 1,5-dicarbonilico tipo 97 (Schema 34) su cui effettuare, con l’ultimo processo della sequenza, la reazione di doppia amminazione riducente intramolecolare (amminociclizzazione). Schema 34 OH O O O H NHAc OH O H N H OH OH OP2 O O P1O NP OP3 O H OH OH O CHO 97 74
A tal scopo si rendeva necessaria la rimozione del gruppo protettivo al C-5 dell’unità aciclica di 104, su cui realizzare la successiva ossidazione regioselettiva. E’ da sottolineare che la presenza di diverse funzioni acido-labili sull’intermedio 104 rende indispensabile l’uso di condizioni d’idrolisi atte a mantenere il legame interglicosidico, il gruppo acetalico 2,3-O-isopropilidenico e quello dimetilacetalico che protegge il carbonile anomerico, la cui deprotezione porterebbe alla ripiranilizzazione dell’unità riducente in forma piranosica, impegnando l’OH-5 nel legame emiacetalico ed impedendone, di conseguenza, la possibilità di ossidazione selettiva.
Nell’ambito di precedenti ricerche svolte nel Laboratorio (Schema 35) era stato osservato che nella deprotezione di derivati poliacetonati del lattosio sia ossigenati (105)10 che contenenti unita β-D-TalNAc e β-D-MaNAc (105a,b),23 con soluzioni
acquose di AcOH in varie concentrazioni, compatibili con gruppi eterei ed esterei, e a differente temperatura, si ottenevano miscele di prodotti parzialmente deprotetti dove i tetraoli 106 e 106a e il triolo 106b rappresentavano sempre i prodotti maggioritari (rese 65-75%). Schema 35 O O O (MeO)2CH O O O R1 R X OBn R2 Y OH O O (MeO)2CH OH O O R1 R X OBn R2 Y 105: R,R1=O2CMe2; Y=OBn; R2=X=H
105a: R1=R=OH; R2=Y=H; X=NHAc
105b: R2=OH; R=OBn; R1=Y=H; X=NHAc
106: R=R1=OH; Y=OBn; R2=X=H
106a: R1=R=OH; R2=Y=H; X=NHAc
106b: R2=OH; R=OBn; R1=Y=H; X=NHAc
Questi studi hanno evidenziato, inoltre, una forte differenza di reattività fra i diversi gruppi acido-labili, evidenziando che quelli più reattivi sono i due acetonuri in 3’,4’, coinvolgente una fusione trans-diossolanica tensionata, e in 5,6, che impegna un gruppo
alcolico primario. Sensibilmente meno reattivi sono il gruppo 2,3-O-isopropilidenico, che impegna una funzione diolica secondaria aciclica, ed il gruppo dimetilacetalico, mentre il legame interglicosidico è privo di reattività in AcOH acquoso. Durante questi studi era stato evidenziato che la deprotezione selettiva dell’acetale ciclico in posizione 5,6 di 105 a dare il corrispondente 5,6 diolo in rese accettabili (45%) era possibile utilizzando il metodo detto del “glicol sacrificale”, che prevede la rimozione selettiva di gruppi isopropilidenici in soluzione diclorometanica utilizzando una reazione di transacetilazione catalizzata da acidi tra lo zucchero protetto ed un 1,2-diolo come il glicole propilenico (Figura 31).23
Figura 31 OH OH O O O O OH OH + +
Come in tutte le reazioni di transacetalazione, s’instaura un equilibrio che risente dei parametri sperimentali quali la concentrazione dei reagenti, l’uso del glicole in forte eccesso e la possibilità di sottrarre uno dei prodotti all’equilibrio. Generalmente, la scarsa solubilità del carboidrato deprotetto nei solventi clorurati, che sono generalmente quelli utilizzati, ne determina la precipitazione dall’ambiente di reazione e quindi sposta l’equilibrio verso la deprotezione. Osservazioni qualitative mostrano che, a parità di condizioni, gli acetali non anomerici vengono deprotetti più facilmente di quelli anomerici. Oltre al glicole propilenico possono essere impiegati altri tipi di glicoli come il 2,3-butandiolo, il pinacolo, l’(±)1-fenil-1,2-etandiolo, ottenendo risultati del tutto simili. Il glicole propilenico è adatto per preparazioni su larga scala a causa del suo basso costo, mentre i glicoli alto bollenti sono utili per le indagini gas-cromatografiche.
Sulla base di questi risultati, e con lo scopo di preparare il diolo 107, una prima prova d’idrolisi del derivato 104 (Schema 36) è stata condotta con 7 equivalenti di glicole propilenico in presenza di quantità catalitiche di TsOH (20%). Dopo 2 ore a temperatura ambiente l’analisi TLC evidenzia la presenza del prodotto di partenza (Rf
0.85) e la formazione di tre macchie di uguale intensità, rispettivamente a Rf 0.62 (107),
Rf 0.51 (108) e Rf 0.38 (109), e tracce di un prodotto a Rf 0.21 che è compatibile con un
Schema 36 O O O O O O O O (MeO)2CH N3 OBn O O OH OH O O O O (MeO)2CH N3 OBn O O O O OH O O O H (MeO)2CH N3 OBn O O OH OH OH O O O H (MeO)2CH N3 OBn propandiolo, TsOH CH2Cl2 anidro + + 104 107 108 109
Dopo neutralizzazione con una soluzione acquosa satura di NaHCO3, onde evitare
l’ulteriore formazione dell’esaolo a Rf 0.21, e lavaggio della soluzione diclorometanica
con H2O allo scopo di eliminare l’eccesso di glicole propilenico, si ottiene un grezzo
che, purificato mediante flash cromatografia su gel di silice, fornisce i prodotti 106 (resa 13%) e 109 (resa 4%) ed una miscela (resa 20%), non separabile su silice, che all’analisi NMR risulta costituita dal 5,6-diolo 107 impuro per il 30% di 108, valutato sulla base delle altezze relative ai segnali dei C-1’ rispettivamente a δ 102.4 e 101.2 rispettivamente.
Vista l’impossibilità di ottenere il diolo 107 puro ed in buona resa, ci siamo indirizzati verso la preparazione del tetraolo 109 che poteva condurre al desiderato 5-chetoaldoso, in analogia con quanto osservato in precedenti ricerche per gli analoghi tetraoli lattosidici ossigenati,13 mediante ossidazione regioselettiva al C-5 come verrà discusso successivamente. A tal scopo l’idrolisi di 104 (Schema 37) è stata condotta con AcOH acquoso all’80% a 40°C, nelle condizioni applicate ai derivati 105 e 105a. Dopo 3.5 ore l’analisi TLC evidenzia la completa scomparsa del prodotto di partenza, la formazione del tetraolo 109 come prodotto largamente maggioritario e tracce dei dioli
107 e 108. La rimozione azeotropica dell’acido acetico con toluene e la successiva
purificazione flash cromatografica su silice permette di isolare, oltre al tetraolo 109 puro (resa 68%), una frazione (resa 23%) che all’analisi NMR risulta costituita da una miscela dei dioli 107 e 108 in rapporto 6:4.
Schema 37 O O O O O O O O (MeO)2CH N3 OBn O O OH OH OH O O O H (MeO)2CH N3 OBn AcOH 80% 40°C 109 (68%) 104
Mentre per il diolo 107, ottenuto sempre in miscela con 108, l’analisi NMR (1H,
13
109, isolati puri, è stato possibile effettuare una completa caratterizzazione dal punto di
vista chimico-fisico, e i dati NMR (1H, 13C) sono in accordo con le strutture proposte (vedi parte sperimentale). In particolare, gli spettri protonici consentono un facile controllo della presenza dei gruppi dimetilacetalici e degli acetali isopropilidenici, dato che i segnali relativi ai C-metili ed ai metossili sono facilmente individuabili per la loro intensità e localizzazione in zone degli spettri poco affollate: quelli relativi ai metossili a δ 3.0-3.5 e quelli relativi a C-metilici degli anelli diossolanici a δ 1.3-1.5. Esperimenti di eterocorrelazione (HETCOR) fra i chemical shifts dei carboni e quelli dei protoni permettono l’assegnazione completa dei carboni; il paragone dei valori di chemical shift dei carboni C-3’, C-4’, C-5 e C-6 negli spettri 13C dei composti deprotetti 107, 108 e
109 con quelli dei corrispondenti carboni del derivato protetto 104 (Tabella 1) permette
di verificare l’avvenuta rimozione dei gruppi isopropilidenici nelle posizione 3’,4’ e/o 5,6. La presenza dei gruppi acetalici in queste posizioni esercita, infatti, un effetto di deschermo di circa 3-5 ppm rispetto ai corrispondenti segnali dei carboni non funzionalizzati.
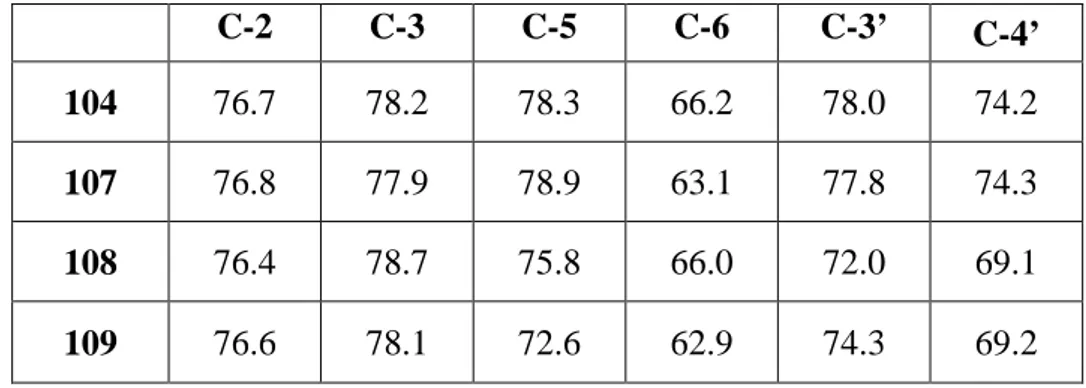
Tabella 1. Valori dei chemical shifts (ppm) dei carboni diagnostici di 104, 107, 108 e 109. C-2 C-3 C-5 C-6 C-3’ C-4’
104 76.7 78.2 78.3 66.2 78.0 74.2
107 76.8 77.9 78.9 63.1 77.8 74.3
108 76.4 78.7 75.8 66.0 72.0 69.1
109 76.6 78.1 72.6 62.9 74.3 69.2
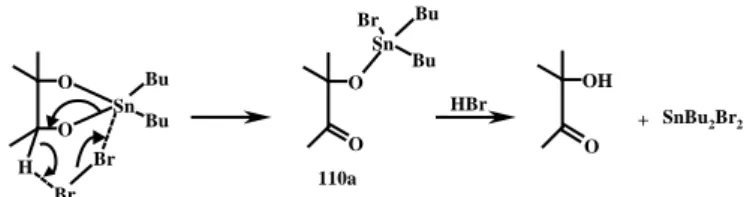
Il passaggio successivo della nostra sequenza sintetica prevede l’ossidazione regioselettiva dell’ossidrile al C-5 di 109 e, anche in questo caso, è necessario applicare metodologie che operino in condizioni blande compatibili con i gruppi acido-labili presenti sul tetraolo 109, come la funzione isopropilidenica sul C-2 e C-3, dimetilacetalica sul C-1 e glicosidica. Un efficace metodo di ossidazione regioselettiva di carboidrati ed altri polioli prevede la formazione di intermedi ciclici stannilidenici di formula generale 110 (Figura 32), seguita da apertura ossidativa dell’acetale stannilidenico con Br2 o NBS, reagenti in grado di generare un bromo elettrofilo (Br+).24
Figura 32 OH OH (C) O O (C) SnBu2 n Bu2SnO n H2O 110 + +
Gli acetali stannilidenici di tipo 110 sono generalmente ottenuti per trattamento di un 1,2- o 1,3-diolo con un equivalente di Bu2SnO in toluene ed in condizioni di
rimozione azeotropica dell’acqua che si forma durante la reazione. In genere questi derivati sono stabili, la loro manipolazione non richiede particolari precauzioni (atmosfera inerte, ambienti rigorosamente anidri) e sono ottenuti in maniera quantitativa e quindi non richiedono processi di purificazione. Essi reagiscono con vari tipi di elettrofili in solventi a bassa polarità (toluene, cloroformio) e la reazione risulta essere, nella maggior parte dei casi, completamente regioselettiva. Si pensa che una spiegazione plausibile dell’andamento della reazione sia da ricercarsi nella differenza di coordinazione che si realizza, per ragioni d’ingombro sterico, per i differenti atomi di ossigeno delle strutture oligomeriche e nelle sottili differenze di acidità e di elettronegatività delle diverse funzioni alcoliche. Il meccanismo di apertura ossidativa dell’acetale stannilidenico è illustrato nella Figura 33.
Figura 33 Br Br Sn O H O Bu Bu O Sn Br O Bu Bu OH O HBr 110a SnBu2Br2 +
L’attacco del reagente (Br2 o NBS) sui derivati stannilidenici avviene sull’ossigeno
anulare più impedito, promuovendo il trasferimento di idruro secondo un meccanismo ciclico e concertato dal derivato stannilidenico al bromo, con formazione di HBr e dell’intermedio chetonico 110a (Figura 33) in cui il gruppo stannossanico, a forte ingombro sterico, rimane legato sempre nella posizione meno ingombrata (C-6) determinando l’ossidazione regioselettiva dell’ossidrile secondario OH-5.
Questa metodologia ossidativa è stata applicata con successo nel nostro Laboratorio nell’ossidazione regioselettiva al C-5 dei tetraoli 10613 e 106a10 (Schema 38) a dare i
5-cheto derivato 111 (resa 88%) e 111a (resa 67%) precursori degli azadisaccaridi 65 e 30 (capitolo 1) rispettivamente.
Schema 38 OH O O (MeO)2CH OH O O OH O H X OBn Y OH O O (MeO)2CH O O O OH O H X OBn Y 1) Bu2SnO, CH3Ph, riflusso 2) CHCl3, NBS 106: Y=OBn; X=H 106a: Y=NHAc; X=H 111: Y=OBn; X=H 111a: Y=NHAc; X=H
E’ interessante notare che per 106 e 106a, nonostante la presenza di due diverse funzioni dioliche, la prima in posizione aciclica 5,6 e l’altra in 3’,4’ ciclica, la reazione risulta completamente regioselettiva se vengono utilizzate quantità equimolari o un leggero eccesso di Bu2SnO (1.1 eq) e la formazione dell’acetale stannilidenico
coinvolgente l’ossidrile primario è più favorita rispetto a quello che interessa i due ossidrili secondari.
Sulla base di queste considerazioni il tetraolo 109 (Schema 39) è stato sottoposto a reazione di stannilidenazione con quantità equimolari di Bu2SnO in toluene ed in
condizioni di rimozione azeotropica dell’acqua mediante apparecchiatura Dean-Stark. Dopo 20 ore a riflusso del solvente (140°C), si concentra la soluzione ed il derivato stannilidenico, ripreso con CHCl3, è trattato a 0°C e in assenza di luce con quantità
equimolari di NBS. Dopo 1.5 ore (TLC) si evidenzia la scomparsa di 109 (Rf 0.31) e la
formazione di due macchie prevalenti a Rf 0.44 e Rf 0.50. La successiva purificazione
flash cromatografica su gel di silice del grezzo di reazione ha fornito il 5-cheto derivato
112 (Rf 0.44, resa 28%) ed una frazione (Rf 0.50) che all’analisi 13C NMR risulta
costituita da una miscela, non separabile su silice, di due derivati disaccaridici impuri per succinimmide (circa il 15%) di difficile interpretazione.
Probabilmente la bassa resa di questo processo può essere attribuita alla presenza del gruppo azidico equatoriale che, avendo deboli caratteristiche nucleofile, potrebbe interferire in qualche modo con il bromo elettrofilo, indirizzando la reazione verso percorsi differenti. Schema 39 O O OH OH OH O O O H (MeO)2CH N3 OBn O O O OH OH O O O H (MeO)2CH N3 OBn 1) Bu2SnO, toluene 2) NBS, CHCl3
L’uloside 112, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista chimico-fisico e i dati NMR (1H, 13C) sono in accordo con la struttura proposta (vedi parte sperimentale). L’avvenuta ossidazione è confermata nello spettro
13C NMR dal segnale a δ 210.7, diagnostico per la funzione chetonica, e dalla
scomparsa del segnale relativo al carbonio alcolico in posizione 5 (δ 72.6). D’altra parte, fra i valori di chemical shift dei segnali relativi ai carboni del precursore 109 e quelli del corrispondente uloside 112 vi è un ottimo accordo, ad eccezione per quelli assegnati ai carboni C-4 e C-6 sull’unità D-xilo, che nei prodotti ottenuti risultano più
deschermati (4-5 ppm) a causa dell’effetto anisotropico del carbonile in posizione alfa. Questo effetto è evidenziabile anche negli spettri protonici, nei quali i segnali relativi ai protoni 4, 6a e 6b risuonano a campi più alti. Inoltre, la risonanza del protone H-4 (δ H-4.26, J3,4 1.8 Hz) si presenta come un doppietto deschermato evidenziando la
perdita dell’accoppiamento con il protone H-5 e, quindi, la presenza di un carbonile in tale posizione.
Questo risultato deludente ci ha spinto a modificare la nostra sequenza sintetica verso la sintesi di derivati disaccaridici, analoghi di 109, aventi l’unità β-D
-galattopiranosidica completamente funzionalizzata e quella glucosidica aciclica deprotetta selettivamente al C-5 o in entrambe le posizioni 5 e 6, su cui effettuare la reazione di ossidazione senza la complicazione dovuta alla presenza di due funzioni dioliche su unità differenti. Come primo possibile approccio alla sintesi di questi analoghi abbiamo valutato la possibilità di proteggere selettivamente il più reattivo gruppo alcolico primario del tetraolo 109, come etere o estere, in modo da ottenere i derivati 113a,b (Schema 40) che, dopo isopropilidenzione delle posizioni 3’,4’, forniscono i derivati disaccaridici 114a,b selettivamente deprotetti al C-5, entrambi precursori di 5-chetoaldosi. Schema 40 O O OH OR OH O O O H (MeO)2CH N3 OBn O O OH OR O O O O (MeO)2CH N3 OBn O O OH OH OH O O O H (MeO)2CH N3 OBn 113a: R=Tr 113b: R=COCMe3 114a: R=Tr 114b: R=COCMe3 109
La protezione selettiva di un gruppo alcolico primario, in presenza di altri gruppi alcolici secondari liberi, è un problema affrontato da tempo nella chimica dei carboidrati e risolto con numerosi metodi tutti basati sull’uso di reagenti molto ingombranti stericamente che risentono, quindi, più delle differenze di intorno chimico che della differente nucleofilia dovuta a problemi elettronici delle funzioni alcoliche. In generale, i reagenti più utilizzati sono il cloruro di tritile o il cloruro di pivaloile, che permettono di ottenere due funzionalità diverse, eterea o esterea rispettivamente.
Sfortunatamente, il tetraolo 109 si è dimostrato poco reattivo sia nei confronti dell’eterificazione con cloruro di tritile in piridina, sia nell’esterificazione con cloruro di pivaloile in piridina-diclorometano anidri. In entrambi i casi, oltre a recuperare il tetraolo 109 non reagito, si isolano i disaccaridi selettivamente protetti al C-6 113a e
113b (Schema 40) in resa dell’11 e 25% rispettivamente.
Per realizzare la completa protezione dell’unità galattopiranosidica di 109 abbiamo deciso di cambiare strategia ed effettuare l’isopropilidenazione selettiva delle posizioni 5 e 6 dell’unità aciclica, confidando nella maggiore reattività dell’ossidrile primario in posizione 6, in modo da ottenere il diolo 108 che, benzilato nelle posizioni 3’,4’ e deprotetto in 5,6, avrebbe permesso di ottenere il diolo 116 (Schema 41) su cui effettuare l’ossidazione al C-5 mediante apertura regioselettiva con NBS di un intermedio stannilidenacetalico, come descritto precedentemente per la preparazione di
112. Schema 41 O O OH OH OH O O O H (MeO)2CH N3 OBn O O O O OH O O O H (MeO)2CH N3 OBn O O O O O O (MeO)2CH N3 OBn OBn BnO O O OH OH O O (MeO)2CH N3 OBn OBn BnO O O O O O O O O (MeO)2CH N3 OBn 109 CH2Cl2 anidro PyHOTs, 2-MP 108 (66%) NaH 60%, BnBr DMF 115 (98%) AcOH 80% 40°C 116 (73%) 104 (27%) +
come catalizzatore acido,25
Il meccanismo di reazione prevede un attacco iniziale del viniletere protonato 117 (Schema 42) sul gruppo ossidrilico primario stericamente più accessibile del diolo, per dare un acetale intermedio aciclico 117a (nello schema è riportata la forma protonata), il quale subisce un attacco nucleofilo intramolecolare da parte del gruppo ossidrilico più vicino a dare l’acetale ciclico 117b.
una metodologia ampiamente utilizzata nella chimica dei carboidrati per proteggere dioli vicinali e particolarmente efficiente quando, nella reazione di isopropilidenazione, è coinvolto un ossidrile primario.
25 Schema 42 OH R OH C+ OMe O R OH O+ Me H O R O + - MeOH - H+ 117 117a 117b
Il tetraolo 109 è stato, quindi, trattato con 2-MP (Schema 41) in CH2Cl2 anidro in
presenza di tosilato di piridinio (10%). Dopo 45 minuti a temperatura ambiente l’analisi TLC evidenzia la scomparsa del prodotto di partenza e la formazione di un prodotto principale a Rf 0.62 e di un prodotto minoritario a Rf 0.85. La neutralizzazione della
miscela con una soluzione acquosa satura di NaHCO3 seguita da estrazione con CH2Cl2
e purificazione flash cromatografica su gel di silice fornisce il desiderato diolo 108 (Rf
0.62) ed il triacetonuro 104 (Rf 0.85) in resa rispettivamente del 66 e 27%, puri
all’analisi NMR (1H, 13C) ed aventi parametri spettrali identici a quelli dei campioni ottenuti precedentemente.
La successiva benzilazione (BnBr, NaH in DMF) delle funzioni ossidriliche libere del diolo 108 permette di ottenere, dopo consueto work-up della miscela di reazione e purificazione flash cromatografica su gel di silice, il tri-O-benzil derivato 115 con una resa del 98% (Schema 41).
Visto che la reazione di acetonazione selettiva con 2-MP del tetraolo 109 fornisce prevalentemente il derivato deprotetto 108 abbiamo deciso di effettuare la reazione di benzilazione direttamente sul grezzo, in modo da evitare una purificazione cromatografica. In queste condizioni il derivato 115 è isolato in resa complessiva del 73% (calcolata da 109).
Il tri-O-benzil derivato 115, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista chimico-fisico e i dati NMR (1H, 13C) sono in accordo con la struttura proposta (vedi parte sperimentale). L’avvenuta benzilazione in posizione 3’ e 4’ è confermata, nello spettro protonico, dalla presenza di un multipletto (δ 7.47-7.27) attribuibile ai 15 protoni aromatici dei tre sostituenti benzilici; inoltre, nello spettro 13C, i segnali relativi al C-3’ (δ 81.3) e al C-4’ (δ 73.8) di 115 risultano deschermati di circa 5-9 ppm rispetto ai corrispondenti segnali (δ 72.6, 69.1) di 108 a conferma dell’avvenuta eterificazione in queste posizioni.
Il composto 115 è stato, quindi, sottoposto ad idrolisi acida controllata in modo da realizzare la rimozione selettiva del più reattivo gruppo isopropilidenico in posizione 5,6 effettuando un’accurata valutazione dei tempi di reazione mediante analisi TLC e fermando la reazione quando il rapporto fra i prodotti formatosi risulta a favore del desiderato diolo 116 (Schema 41). Il trattamento di 115 con AcOH acquoso all’80% a 40°C permette, dopo 2 ore e rimozione azeotropica dell’acido acetico con toluene seguita da purificazione flash cromatografica su gel di silice, di recuperare il prodotto di partenza non reagito (15%) e di isolare il diolo 116 ed il tetraolo 116a (Figura 34) in resa del 73 e 11% rispettivamente
Figura 34 OH OH OH OH O O (MeO)2CH N3 OBn OBn BnO 116a
Il diolo 116, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista chimico-fisico e i dati NMR (1H, 13C) sono in accordo con la struttura proposta (vedi parte sperimentale). In particolare, nello spettro 13C, i segnali relativi al C-5 (δ 72.9) e al C-6 (δ 63.9) risultano schermati di circa 3-5 ppm rispetto ai corrispondenti segnali (δ 77.8, 66.1) di 115, a conferma dell’avvenuta rimozione del gruppo isopropilidenico in posizione 5,6.
Anche i dati NMR del tetraolo 116a sono in accordo con la struttura proposta (vedi parte sperimentale); in particolare la rimozione dei due acetali isopropilidenici in posizione 5,6 e 2,3 è confermata, nello spettro protonico, dall’assenza di segnali relativi
In accordo con la procedura descritta precedentemente per l’ossidazione del tetraolo
109, il diolo 116 è stato sottoposto a reazione di stannilidenazione con quantità
equimolari di Bu2SnO in toluene ed in condizioni di rimozione azeotropica dell’acqua
mediante apparecchiatura Dean-Stark. Dopo 20 ore a riflusso del solvente (140°C), si concentra la soluzione ed il derivato stannilidenico, ripreso con CHCl3, è trattato a 0°C
e in assenza di luce con quantità equimolari di NBS. Dopo l’aggiunta si lascia evolvere la reazione a temperatura ambiente, fino a che l’analisi TLC (1 ora) evidenzia la scomparsa di 116 e la formazione di un unico prodotto a Rf più alto. La successiva
purificazione flash cromatografica su gel di silice del grezzo di reazione ha fornito il 5-cheto derivato 118 in resa dell’89% (Schema 43).
Schema 43 O O OH OH O O (MeO)2CH N3 OBn OBn BnO O O O OH O O (MeO)2HC N3 OBn OBn BnO OH OH O OH O O N3 OBn OBn BnO OHC 1) Bu2SnO, toluene 2) NBS, CHCl3 116 118 (89%) TFA 90% 119 (quantitativa)
L’uloside 118, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista chimico-fisico e i dati NMR (1H, 13C) sono in accordo con la struttura proposta (vedi parte sperimentale). L’avvenuta ossidazione è confermata, nello spettro
13C, dal segnale a δ 210.8, diagnostico per la funzione chetonica, e dalla scomparsa del
segnale relativo al carbonio alcolico in posizione 5 (δ 72.9). Anche nel caso del 5-uloside 118 i carboni C-4 e C-6 sull’unità D-xilo risultano più deschermati (circa 5 ppm)
dei corrispondenti carboni del diolo 116 a causa dell’effetto anisotropico del carbonile in posizione alfa. Questo effetto è evidenziabile anche negli spettri protonici, nei quali i segnali relativi ai protoni H-4, H-6a e H-6b risuonano a campi più alti.
Una volta preparato il derivato chetonico 118 la tappa successiva della nostra sequenza sintetica ha previsto la rimozione della funzione dimetilacetalica e isopropilidenica con conseguente esposizione della funzione aldeidica e formazione del derivato 1,5 dicarbonilico 119 (Schema 43), selettivamente glicosilato in posizione 4, sul quale applicare la doppia amminazione riducente intramolecolare.
Per realizzare l’idrolisi acida delle funzioni acetaliche è indispensabile utilizzare condizioni sperimentali che consentano di rimuovere questi gruppi protettivi senza scindere il legame glicosidico. Anche se il prolungato riscaldamento con soluzioni
acquose di AcOH può essere sufficiente a realizzare questa operazione, si è scelto di utilizzare l’acido trifluoroacetico (CF3COOH) acquoso al 90% a temperatura ambiente,
che riduce fortemente i tempi di reazione, può essere allontanato con facilità per semplice co-evaporazione con toluene a causa della sua elevata volatilità e non è in grado di scindere i legami glicosidici.
Trattando, quindi, il 5-cheto derivato 118 (Schema 43) con CF3COOH al 90% a
temperatura ambiente, l'analisi TLC evidenzia, dopo 3 ore, la scomparsa del prodotto di partenza. L’evaporazione dei solventi, seguita da ripetute co-evaporazioni con toluene per eliminare completamente l'acido trifluoroacetico, conduce ad un grezzo di consistenza sciropposa costituito da più macchie che si presentano, nella TLC, come zone più intense di un’unica strisciata. Anche l’analisi NMR (1H, 13C) del grezzo di reazione contenente 119 (resa quantitativa), sebbene evidenzi la scomparsa dei segnali caratteristici del gruppo isopropilidenico e della funzione dimetilacetalica, è resa difficile per la complessità degli spettri. Ciò è spiegabile considerando la natura del disaccaride 119, che esiste come miscela di tautomeri in equilibrio tra loro derivanti da emiacetalizzazione dei gruppi alcolici sulle singole funzioni carboniliche (chetonica e aldeidica), la cui quantità in equilibrio è dipendente, in generale, da più fattori, quali l’acidità della silice, i solventi utilizzati nell’eluizione delle TLC e quelli usati nella registrazione degli spettri. Il grezzo di idrolisi non è sottoposto a processi cromatografici poiché, trattandosi di una miscela di tautomeri dello stesso prodotto, la successiva reazione di amminociclizzazione avrebbe consentito lo spostamento dell’equilibrio verso la forma più reattiva ottenendo la trasformazione di 119 nell’azadisaccaride più facilmente isolabile, evitando così perdite di prodotto per decomposizione o assorbimento irreversibile su silice. Resta, inoltre, da sottolineare che il grezzo d’idrolisi può essere conservato per alcune settimane a bassa temperatura (-18°C).
3.4. Amminociclizzazione del derivato 1,5-dicarbonilico 119
Un efficace metodo generale per trasformare i derivati esos-5-ulosici in piperidine poliossidrilate (1-desossiazazuccheri) in buone rese e, in alcuni casi, con completa stereoselettività prevede, come discusso nel capitolo 1 (paragrafo 1.5.3), una reazione di
ampiamente applicata a composti 1,5-dicarbonilici sia mono- che disaccaridici.10,13 La reazione prevede l’uso di ammoniaca o di un ammina primaria in presenza di riducenti in ambiente debolmente acido. Come sistema riducente si può utilizzare H2 e
catalizzatore, secondo le condizioni delle normali idrogenazioni catalitiche, oppure si può procedere per via chimica con l’uso di idruri. L’idruro di scelta per queste reazioni è il cianoboroidruro (NaCNBH3) che, in condizioni debolmente acide, ha una
chemoselettività molto elevata per la riduzione dei doppi legami imminici, mentre è capace di ridurre i doppi legami carbonilici solamente a pH molto più bassi. Basandoci su quanto descritto per la preparazione del noto azadisaccaride 65a13 (Schema 13, pag. 28), l’amminociclizzazione di 119 è stata condotta con NaCNBH3 in MeOH a 60°C
impiegando un’ammina primaria lipofila avente un sostituente facilmente rimuovibile quale la benzilammina.
Il meccanismo comunemente accettato per questo tipo di reazioni (Schema 44) prevede un primo attacco dell’azoto amminico sul più reattivo gruppo aldeidico del chetoaldoso 120, con successiva eliminazione di una molecola d’acqua a formare l’intermedio imminico 120b, il quale è ridotto ad opera del NaCNBH3 all’ammina 120c.
L’intermedio 120c ciclizza poi attraverso l’attacco dell’azoto sul gruppo chetonico e, per successiva eliminazione di una molecola d’acqua, si forma il catione imminio ciclico 120f che, nello stadio stereo-determinante della reazione, subisce l’attacco nucleofilo di uno ione idruro a dare gli azapiranosi epimeri al C-5 121a,b.
Schema 44 O O H OH O H OR R'O O N OH O H OR R" R'O H H+ O N H OH O H OR R'O R" N OH O H O CH2OR' OR R" H H+ N OH O H O H CH2OR' OR R" H N OH O H OR R" R'O H N O H OH R" OR' OR R'O O C H N H OH OH O H OR R" N O H OH OR R" OR' R"-NH2 . . . . : . . 120 120a 120b 120c 120d 120e 120f 121a 121b + + +
I risultati ottenuti nel Laboratorio per questo tipo di reazioni hanno evidenziato che, mentre le reazioni di amminociclizzazione di derivati dell’L-arabino-esos-5-ulosio 120 (OH-4 assiale) con differenti ammine primarie conducono sempre a miscele dei due 1-desossi-azapiranosi epimeri 121a (stereochimica D-galatto) e 121b (stereochimica L
-altro), le reazioni nelle medesime condizioni di derivati D-xilo-esos-5-ulosici (OH-4
equatoriale), quale è anche il composto 119 di nostro interesse, portano con una selettività molto elevata e spesso completa agli imminopiranosi a configurazione D
-gluco (121a, OH-4 equatoriale).
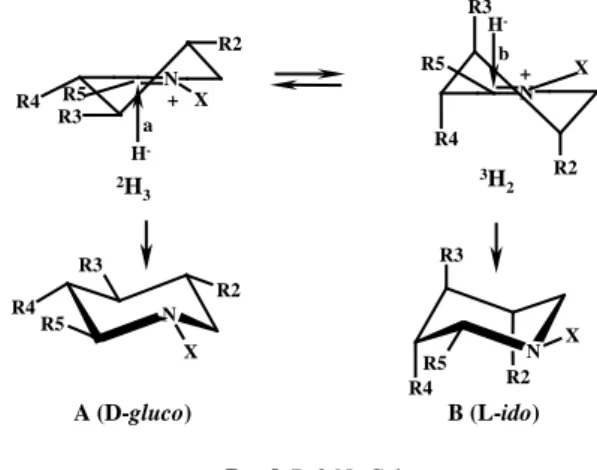
Una razionalizzazione di questa particolare stereoselettività nella reazione di amminociclizzazione porta a stabilire una sua dipendenza da fattori che possono influenzare la reattività relativa degli ioni imminio ciclici, che sicuramente, a causa della loro elevata flessibilità dovuta alla presenza del doppio legame C=N, si trovano in un equilibrio molto veloce fra i due conformeri a semisedia 2H3 e 3H2 (Figura 35), che,
verosimilmente, devono avere energie non molto diverse.
Figura 35 N R2 X R3 R4 R5 N R2 R3 R5 R4 N R2 R3 R5 X R4 N R3 R2 X R5 R4 H -2H 3 X H -3H 2 A (D-gluco) B (L-ido) a b R4= β-D-2-N3-Gal + +
Supponendo che il trasferimento di idruro abbia uno stato di transizione più simile ai prodotti che ai reagenti, la reattività relativa dei due ioni imminio, che determinerà la distribuzione finale dei prodotti, dipenderà dalle interazioni che si svilupperanno negli stati di transizione a conformazione “quasi”-sedia fra i diversi gruppi sostituenti. E’, inoltre, noto che la riduzione degli ioni imminio con idruri è una reazione sotto controllo stereoelettronico, e prevede un attacco da una posizione pseudo-assiale per raggiungere, nello stato di transizione, un assetto antiperiplanare fra il doppietto che si forma sull’N e l’incipiente legame σ fra l’idruro ed il C imminico. Su queste basi, la
spiegata considerando che l’attacco assiale sulla conformazione 3H2 (strada b, pro-L
-ido), meno stabile del conformero alternativo 2H3, presenta, oltre ad una interazione
sfavorevole fra il sostituente ossigenato R3 e l’idruro che attacca, una serie di interazioni 1,3-syn-diassiali, come ad esempio quella fra i sostituenti R4 ed R2. L’attacco sul conformero 2H3 (strada a, pro-D-gluco), in cui tutti i sostituenti sono pseudo-equatoriali,
coinvolge uno stato di transizione di più bassa energia. Evidentemente, nella serie arabino, ove il C-4 ha orientamento opposto, viene meno l’interazione negativa precedentemente descritta, ottenendo un livellamento dell’energia dei due stati di transizione con conseguente diminuzione della stereoselettività della reazione di amminociclizzazione.
Sulla base di queste premesse il 5-chetoaldoso disaccaridico 119 (Schema 45) a configurazione D-xilo è stato sottoposto a reazione di doppia amminazione riducente
intramolecolare in MeOH anidro utilizzando il cloridrato della benzilammina (1.0 eq) in presenza di NaCNBH3 (2.2 eq). Dopo 65 ore a 60°C, l’analisi TLC evidenzia la
scomparsa del prodotto di partenza e la formazione di un prodotto nettamente prevalente. La miscela di reazione, concentrata a pressione ridotta e purificata mediante flash cromatografia su gel di silice, fornisce l’azadisaccaride 122 in resa del 58% calcolata a partire dall’uloside 119.
Schema 45 OH OH O OH O O N3 OBn OBn BnO OHC OHO N OH O N3 OBn OBn BnO OH Ph BnNH2.HCl NaCNBH3 MeOH anidro 119 122 (58%)
Con l’ottenimento dell’azadisaccaride 122 è stata confermata sia la struttura del disaccaride 1,5-dicarbonilico 119, sia l’andamento stereochimico della reazione di amminociclizzazione, analogo a quanto riportato per i derivati 71a,b (Schema 15, pag. 29) a configurazione D-xilo10 e non influenzato dalla tipologia dell’unità non riducente β-D-2’-N3-Gal.
L’azadisaccaride 122, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista chimico-fisico e i dati NMR (1H, 13C) sono in accordo con la struttura proposta (vedi parte sperimentale e Figure 38-46, pag. 75). La presenza
dell’azoto piranosico facilita la distinzione dei protoni legati ai carboni in alfa all’azoto (H-1ax, H-1eq e H-5), che risuonano a campi più alti (δ 2.75-1.92) rispetto a quelli in alfa ad un ossigeno. Nel caso degli spettri 13C, invece, l’azoto anulare provoca lo spostamento del chemical shift dei carboni ad esso legati (C-1 e C-5) verso campi più bassi. Nello spettro protonico dell’azadisaccaride 122, quindi, risulta diagnostica la presenza di tre gruppi di segnali, integranti ciascuno per un protone, a campi più alti rispetto a quelli legati ad un carbonio ossigenato: due doppi doppietti accoppiati fra loro a δ 1.92 (H-1ax) e 2.75 (H-1eq), di cui quello maggiormente schermato con costanti di accoppiamento molto alte (J1ax,1eq 11.2 Hz e J1ax,2 10.1 Hz) è il protone H-1 assiale, e un
doppio tripletto a δ 2.27 attribuibile al protone H-5, avente il valore di J4,5 molto alta
(pari a 9.7 Hz). Lo spettro 13C NMR del derivato 122 è caratterizzato, oltre che dal segnale relativo al C metilenico in 6 a δ 57.9, da due segnali a campi più alti di quelli comunemente presenti negli aldopiranosidi: un metilene a δ 56.3 ed un metino a δ 67.0 dovuti ai due carboni legati all’azoto anulare, rispettivamente il C-1 ed il C-5. Particolarmente indicativi, ai fini della determinazione della configurazione dell’unità azapiranosica, sono gli alti valori delle costanti di accoppiamento J2,3 8.8 Hz, J3,4 8.8 Hz
e J4,5 9.7 Hz, che indicano una relazione assiale-assiale-assiale-assiale dei protoni H-2,
H-3, H-4 e H-5 in accordo con una configurazione D-gluco e non L-ido del nucleo azapiranosico.
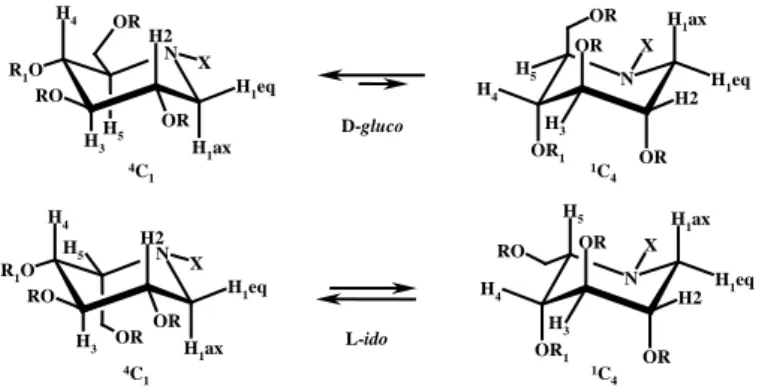
Inoltre, i valori delle costanti di accoppiamento indicano una netta preferenza per la conformazione 4C1 avente tutti i sostituenti disposti in posizione equatoriale (Figura 36).
Figura 36 OR1 H1ax OR H4 H1eq H5 H2 H3 X N OR OR H1ax H4 H2 H3 H1eq R1O H5 X N RO OR OR OR1 H1ax H5 OR H4 H1eq H2 H3 X N OR RO H1ax H4 H2 H3 H1eq R1O X N RO H5 OR OR 4C 1 1C4 D-gluco 4C 1 1C4 L-ido
Per poter disporre dell’azadisaccaride completamente deprotetto 74 per gli appropriati saggi biologici, la parte conclusiva della Tesi prevede sia la completa
acetammidico. La deprotezione completa di 122, in cui tutti i gruppi protettivi sono benzilici, non può essere realizzata attraverso debenzilazione catalitica, che provocherebbe anche la riduzione del gruppo azidico ad ammina impedendone poi la sua selettiva N-acetilazione in presenza del gruppo amminico secondario libero dell’unità DNJ. Si è reso necessario, quindi, effettuare la riduzione del gruppo azidico ad ammina con un reagente selettivo che non rimuova i gruppi benzilici, come ad esempio il sistema NiCl2·6H2O-NaBH4. La riduzione di 122 (Schema 46), condotta in
MeOH con NiCl2·6H2O e NaBH4 a temperatura ambiente, ha evidenziato, dopo 20
minuti (TLC), la scomparsa del prodotto di partenza e la formazione della corrispondente ammina a Rf inferiore, che non viene purificata ma immediatamente
sottoposta a reazione di N-acetilazione in MeOH e Ac2O a temperatura ambiente. Dopo
15 ore, evidenziata (TLC) la scomparsa dell’ammina e la formazione di due prodotti a Rf 0.42 e 0.76, si elimina il reagente per co-evaporazione con toluene e la purificazione
flash cromatografica su gel di silice del grezzo fornisce 123 (Rf 0.42) e 124 (Rf 0.76)
puri in resa rispettivamente del 37 e 55% calcolata da 122.
Schema 46 OHO N OH O N3 OBn OBn BnO OH Ph OHO N OH O NHAc OBn OBn BnO OH Ph OHO N OH O NHAc OBn OBn BnO OAc Ph 1) NiCl2.6H2O; NaBH4; MeOH anidro 2) Ac2O, MeOH + 122 123 (37%) 124 (55%)
Gli azadisaccaridi 123 e 124 sono stati completamente caratterizzati dal punto di vista chimico-fisico e i dati NMR (1H, 13C) sono in accordo con la struttura proposta (vedi parte sperimentale). In particolare, la presenza della funzione acetammidica nella struttura di 123 è confermata, nello spettro protonico, da un doppietto (δ 6.65, J2’,NH
10.0 Hz,) e da un singoletto a δ 1.89 (3H), attribuiti rispettivamente al protone ammidico (NH) e al metile acetammidico, e, nello spettro 13C, da due segnali, uno a δ 171.3, diagnostico per un carbonile di tipo ammidico, e l’altro a δ 23.4 relativo al metile acetilico. Inoltre, nello spettro 13C, il gruppo NHAc provoca lo spostamento del chemical shift del carbonio ad esso legato (C-2’) verso campi più alti (da δ 64.5 in 122 a δ 52.9 in 123).
Nel caso del derivato 124 nello spettro 13C si evidenzia, oltre a tutti i segnali relativi alla funzione acetammidica in posizione 2’, la presenza di due segnali a δ 171.7 e δ 21.1, attribuiti rispettivamente al carbonile ed al metile del frammento acilico. Inoltre,
nello spettro protonico, fra i valori di chemical shift dei segnali relativi ai protoni dell’azadisaccaride 124 e quelli del derivato 123 vi è un ottimo accordo, ad eccezione per quelli assegnati ai protoni H-6a e H-6b sull’unità azapiranosica, che risultano più deschermati (0.3-0.8 ppm) a causa dell’effetto anisotropico del carbonile, confermando la presenza di un gruppo acetato in posizione 6 di 124.
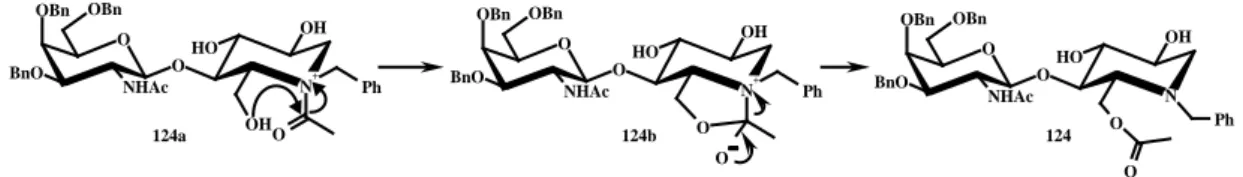
Una probabile spiegazione della formazione dell’azadisaccaride 124, in condizioni in cui l’acilazione di gruppi alcolici è poco probabile, risiede nella formazione della specie cationica 124a (Figura 37) che, attraverso un intermedio ciclico stabile, è in grado di promuovere uno shift d’acile dall’azoto anulare al vicino OH-6.
Figura 37 O O H N+ OH O NHAc OBn OBn BnO OH O Ph O O H N+ OH O NHAc OBn OBn BnO O O Ph O O H N OH O NHAc OBn OBn BnO O O Ph 124a 124b 124
Il 6-O-acil derivato 124 è stato sottoposto a reazione di peracetilazione in modo da ottenere un derivato che ci permetta di effettuare un’attribuzione completa dei segnali sia protonici che del carbonio. E’ noto, infatti, che gli spettri 1H e 13C NMR, pur permettendo un’immediata identificazione dei sostituenti, presentano delle difficoltà quando si tenta un’interpretazione completa dei segnali protonici piranosici che risuonano tutti in una zona molto ristretta dello spettro (2-4 ppm). Molto spesso l’acetilazione degli eventuali ossidrili presenti nella struttura facilita l’attribuzione completa dei segnali protonici, in quanto consente di identificare ed attribuire i protoni in alfa agli acetili che, essendo deschermati dall’effetto anisotropico del carbonile, si spostano a campi più bassi dello spettro semplificando la zona a campi più alti. Anche nel caso degli spettri 13C s’incontrano diverse difficoltà quando si cerca di attribuire i carboni piranosici, che risuonano tutti tra 55 e 85 ppm. Anche in questo caso, mentre la sostituzione degli ossidrili con gruppi eterei provoca un effetto di deschermo solo sui carboni in alfa permettendo l’identificazione del carbonio interessato, la presenza di gruppi esterei provoca un effetto di deschermo sui carboni in alfa ed un effetto di schermo su quelli in beta.
Il disaccaride 124 è, quindi, solubilizzato in una miscela di Py-Ac2O (2:1 v/v) e
maggiore. Il trattamento della reazione e la purificazione flash cromatografica su gel di silice fornisce gli acetati 125 e 126 in resa rispettivamente del 37 e 36%.
Gli azadisaccaridi 125 e 126 sono stati completamente caratterizzati dal punto di vista chimico-fisico e i dati NMR sono in accordo con la struttura proposta (vedi parte sperimentale). Schema 47 OHO N OH O NHAc OBn OBn BnO OAc Ph OAcO N OAc O NHAc OBn OBn BnO OAc Ph OHO N OAc O NHAc OBn OBn BnO OAc Ph Ac2O, Py + 124 125 (37%) 126 (36%)
In particolare, lo spettro protonico di 125 evidenzia la presenza di tre singoletti (δ: 2.01, 1.98 e 1.87), integranti ciascuno per tre protoni, relativi ai tre metili acetilici. La presenza di due gruppi esterei in posizione 2 e 3 è confermata dalle risonanze dei protoni H-2 e H-3, che risultano più deschermati rispetto agli stessi protoni presenti nel 6-O-acetato 124. Nel caso dello spettro protonico del derivato 126, oltre alla presenza di due singoletti (δ: 2.03, 1.93) integranti ciascuno per tre protoni, attribuiti ai due metili acetilici, si osserva uno spostamento di chemical shift del solo protone H-2 verso campi più bassi (da δ 3.31 a δ 4.65). Inoltre, un singoletto a δ 4.87, attribuito ad un protone ossidrilico, conferma l’assenza del gruppo acetato in posizione 3.
Considerando che il 6-O-acetato 124 è facilmente trasformabile nel derivato 123 attraverso reazione di desacetilazione, il grezzo proveniente dalla sequenza sintetica riduzione e N-acetilazione di 122 e contenente gli azadisaccaridi 123 e 124 è stato sottoposto a reazione di transesterificazione in THF-MeOH (1:3.5 v/v) e con quantità catalitiche di MeONa (Schema 48). Dopo 5 ore a 0°C si evidenzia (TLC) la completa trasformazione dell’azadisaccaride 124 nel derivato 123, e la purificazione flash cromatografica su gel di silice del grezzo, ottenuto dopo neutralizzazione con NaHCO3,
fornisce l’azadisaccaride 123 (resa 85% calcolata da 122), che presenta parametri spettrali identici a quelli del campione ottenuto precedentemente.
Schema 48 OHO N OH O NHAc OBn OBn BnO OH Ph OHO N OH O NHAc OBn OBn BnO OAc Ph OHO N OH O NHAc OBn OBn BnO OH Ph MeONa, MeOH-THF + 123 124 123 (85%)
La preparazione del 4-O-[2’-acetammido-2’-desossi-β-D
-galattopiranosil]-1,5-didesossi-1,5-imino-D-glucitolo (74) è ottenuta, in maniera quantitativa, attraverso la deprotezione di 123 per idrogenolisi in presenza di Pd/C in metanolo (Schema 49), contenente un eccesso di HCl (pH 3) in modo da evitare sia l’avvelenamento del catalizzatore per la formazione di un’ammina secondaria che la reazione di N-metilazione, frequente nei processi di idrogenolisi catalizzati da palladio su substrati come le 1-dessossi-nojirimicine. Dopo filtrazione del catalizzatore ed evaporazione dei solventi a pressione ridotta si ottiene un grezzo che, all’analisi NMR (1H, 13C), risulta costituito esclusivamente dal composto desiderato 74 sottoforma di cloridrato.
Schema 49 OHO N OH O NHAc OBn OBn BnO OH Ph OHO N H OH OH NHAc O O H OH OH 123 HCl/MeOH (1%) Pd-C (10%) MeOH anidro x HCl 74 (quantitativa)
Il composto 74, non riportato in letteratura, è stato caratterizzato dal punto di vista chimico-fisico e gli spettri 13C NMR (CD3OD) sono concordi con la struttura assegnata
(vedi parte sperimentale).
Con la preparazione di 74 è stato raggiunto lo scopo prefissato in questa seconda parte di Tesi. Il mimico disaccaridico β-D-GalNAc-(1→4)-DNJ (74), insieme al
disaccaride β-D-GlcNAc-(1→4)-DNJ (75), la cui sintesi è stata condotta in ricerche
parallele a questo lavoro, verrà testato dal Prof. Vladimir Kren dell’Accademia Ceca delle Scienze di Praga che ne valuterà l’attività agonista nei confronti dei recettori NKR-P1 (ratto) e CD69 (umano) delle cellule Natural Killer. I risultati dei test biologici condotti su 74 e 75 andranno a completare quelli ottenuti10 con i disaccaridi a struttura β-TalNAc-(1→4)-DNJ (29, Figura 22, pag. 16) e β-ManNAc-(1→4)-DNJ (30, Figura 22, pag. 16), permettendo così di ricavare delle valutazioni sulla relazione fra struttura molecolare ed attività biologica di questi mimici disaccaridici, in cui un’unità di 1-dessossi-nojirimicina (DNJ) risulta glicosilata in 4 attraverso un legame β-glicosidico con le 4 più comuni N-acetil-D-esosammine (TalNAc, ManNAc, GalNAc e GlcNAc).
20
Barili, P.L.; Berti, G.; Catelani, G.; D’Andrea, F.; De Rensis, F.; Falcini, P.; Carbohydr. Res. 1997, 298, 75-84.
21
Guazzelli, L.; Catelani, G.; D’Andrea, F.; Giannarelli, A.; Carbohydr. Res. 2009, 344, 298-303.
22
Attolino, E.; Catelani, G.; D’Andrea, F.; Eur. J. Org. Chem. 2006, 5279-5292.
23
Catelani, G.; D’Andrea, F.; Puccioni, L.; Carbohydr. Res. 2000, 324, 204-209.
24
David, S.; Hanessian, S.; Tetrahedron, 1985, 41, 643-663.
25