Capitolo 2
2.1 Introduzione al sequenziamento del DNA
La tecnologia del DNA ricombinante, nata nei primi anni ’70, ha fornito mezzi potenti per analizzare e alterare geni e proteine e mezzi in grado di modificare, in modo preciso e predeterminato, il patrimonio genetico di un organismo. Questa tecnologia, frutto della ricerca su DNA, RNA e virus, si basa principalmente sulla disponibilità di enzimi che possono tagliare, unire e replicare il DNA e trascrivere l’RNA in DNA (trascrizione inversa). Un esempio di questi enzimi è la DNA polimerasi già introdotta precedentemente [1].
Di importanza fondamentale per l’analisi del DNA sono gli enzimi di restrizione che riescono a riconoscere sequenze di basi specifiche nel DNA a doppia elica e a tagliare entrambi i filamenti in punti precisi. Il loro utilizzo è indispensabile per analizzare la struttura dei cromosomi, in quanto riescono a isolare “frammenti di restrizione”, cioè geni o parti di DNA di interesse che possono quindi essere analizzati. È infatti più semplice lavorare su una frazione di cromosoma, più o meno grande, piuttosto che sul cromosoma intero.
Il passo successivo, in un ipotetico schema di operazioni da effettuare per analizzare il DNA, consiste nell’avere a disposizione un numero di copie elevato del frammento di interesse, per cui il frammento viene clonato, fornendo materiale illimitato per studi sperimentali [2].
La clonazione in vivo, applicazione della tecnologia del DNA ricombinante, è stata la prima a essere sviluppata e consiste nel costruire nuovi genomi, introdurli in cellule ospiti e farli replicare. Il frammento di DNA di interesse viene infatti legato covalentemente a un DNA vettore, che ha la capacità di replicarsi in modo autonomo in un ospite adatto. I vettori sono molecole di DNA che hanno origine da virus (come il fago λ), batteri e cellule di lievito (YAC). Essi accolgono varie misure di frammenti di DNA estraneo: per esempio il fago λ accoglie frammenti che hanno misura in un range 10 kb-15 kb, mentre gli YAC sono adatti per frammenti in un range di 100 kb-1 Mb [3]. La replicazione del vettore è veloce e, una volta terminata, è possibile, con opportuna procedura enzimatica, riavere il frammento iniziale replicato anche miliardi di volte in poco tempo4.
Esiste anche un altro tipo di clonazione, in vitro, detta “Polymerase Chain Reaction” o PCR. Essa può amplificare ogni sequenza di DNA voluta di qualsiasi origine (virus, batterio, pianta o uomo) centinaia di milioni di volte in alcune ore; tale compito avrebbe richiesto alcuni giorni con la tecnologia ricombinante. La PCR è estremamente preziosa in quanto facilmente automatizzabile e capace di amplificare piccole quantità di campione. La PCR sarà analizzata in dettaglio nel paragrafo 2.5.
In questo capitolo verranno analizzate alcune metodologie classiche che portano alla determinazione della sequenza delle basi in un genoma a partire dalla fase di lisi della membrana delle cellule, cui seguirà una fase dove le molecole di DNA saranno isolate e purificate; dopo che il DNA è
stato opportunamente frammentato con enzimi di restrizione, sarà quindi amplificato e sequenziato (fig. 2.1).
Inizialmente sono state usate le tecniche “blotting” che permettono di separare e caratterizzare una sequenza di basi specifica di DNA o RNA mediante ibridazione con un filamento marcato complementare; è possibile identificare anche una proteina particolare colorandola con un anticorpo specifico.
Nel 1977 sono stati messi a punto i due approcci base di sequenziamento, che permettono la determinazione di una sequenza nucleotidica non nota base per base: il metodo dei didedossi di Sanger e il metodo di Maxam-Gilbert (o metodo di taglio chimico5). Pur essendo entrambi validi, il metodo Sanger è il solo utilizzato sia per la sua semplicità sia perché è possibile automatizzarlo.
Determinante per i metodi di sequenziamento è il contributo dell’elettroforesi che permette la separazione di frammenti di restrizione che verranno opportunamente visualizzati successivamente.
5 Per approfondimenti sul metodo di sequenziamento Maxam-Gilbert consultare:
Lubert Sryer, “Biochimica”, Ed. Zanichelli, dicembre 1996, pp 140-141. Figura 2.1: passi essenziali
che portano alla determinazione della sequenza di basi in un genoma. Lisi delle cellule Purificazione DNA Amplificazione DNA Sequenziamento
2.2 Metodologie di estrazione di acidi nucleici
L’isolamento e la purificazione degli acidi nucleici sono i primi delicati passi nella maggior parte delle applicazioni di biologia molecolare.
L’ottimizzazione dell’estrazione dipende:
• dal tipo di acido nucleico che si vuole isolare (per esempio singola o doppia catena di DNA, RNA totale, mRNA…);
• dalla fonte utilizzata per l’estrazione (tessuti animali o vegetali, cellule eucariotiche o procariotiche…);
• dal materiale biologico contenente la fonte degli acidi nucleici usato per l’estrazione (organo intero, sangue, siero, plasma…); • dall’applicazione prevista post-estrazione (PCR, clonaggio,
restrizione enzimatica, Southern blotting...)
Indipendentemente dalla tecnica di estrazione usata, essa deve rispondere a due requisiti principali: la resa e la purezza, intesa sia come presenza in soluzione dell’acido nucleico in esame, sia come assenza di sostanze contaminanti che, legandosi ai reagenti in soluzione, potrebbero modificare i risultati del sequenziamento [4].
Dopo aver scelto il campione biologico da cui estrarre il materiale genetico, il percorso di estrazione e purificazione prevede quattro fasi:
1. lisi delle cellule. La dissoluzione della cellula (distruzione della membrana) è una fase delicata in quanto risultato di due eventi contrastanti: l’esigenza di frammentare il materiale di partenza e quella di non alterare gli acidi nucleici da analizzare.
I metodi tradizionali di lisi si basano su trattamenti complessi che includono la digestione enzimatica, la solubilizzazione tramite
detergente o tecniche meccaniche di spaccatura. Esistono interessanti metodi di lisi basati su shock osmotico [5] e ultrasuoni.
2. Inattivazione delle nucleasi cellulari.
3. Separazione e recupero dell’acido nucleico dalla soluzione contenente il lisato cellulare. Un primo metodo per effettuare tale operazione prevede l’uso di solventi organici (come fenolo o cloroformio), che consentono la separazione degli acidi nucleici dai contaminanti (proteine); successivamente, dopo centrifugazione, gli acidi nucleici vengono recuperati in soluzione acquosa. Sono stati ultimamente sviluppati protocolli chimici per la separazione e l’isolamento di frammenti di DNA realizzati in un singolo passaggio [6].
Una seconda metodologia di separazione prevede l’adsorbimento del DNA sulla superficie di una matrice di gel di silice in presenza di sali caotropici (in commercio si trovano diversi kit di separazione basati su questo principio).
4. Precipitazione: avviene di solito in alcool etilico o isopropanolo e permette il recupero degli acidi nucleici in forma solida. Dopo lavaggio con etanolo, si ha una valutazione quali-quantitativa degli acidi nucleici estratti e quindi la loro conservazione.
Negli ultimi anni le metodologie classiche di separazione e purificazione effettuate in un laboratorio biochimico, sono state affiancate da nuove tecniche nel tentativo di rendere le procedure automatiche o realizzabili su dispositivi microfluidici integrati.
Sono stati quindi sviluppati processi di lisi termica [7] e lisi elettrica [8] su strumenti microfluidici.
In figura 2.2 è mostrato, come esempio, l’effetto della lisi elettrica effettuata in uno strumento microfluidico realizzato su vetro. La cellula viene portata idrodinamicamente all’incrocio di due rami presenti nel dispositivo dove viene sottoposta a un campo elettrico e quindi ha luogo la
lisi, in un tempo inferiore a 66 ms. La velocità di distruzione della membrana cellulare è correlata all’intensità del campo elettrico applicato.
L’effettivo range delle forze del campo è tra 150-500 V/cm picco-picco, per campi di onda quadra in alternata a 50 Hz, con un offset in continua di 300-400 V/cm.
La combinazione di lisi chimica ed elettrica fornisce risultati migliori.
Anche nei dispositivi microfluidici, prima del sequenziamento, è essenziale che il DNA sia purificato. Le purificazioni classiche nella maggior parte dei casi prevedono, dopo il trattamento chimico, una centrifugazione. Tale procedura non è eseguibile in automatico, per cui è necessario trovare metodi alternativi di purificazione [9].
Un metodo automatico di purificazione prevede l’utilizzo di particelle magnetiche ed è per questo detto “metodo di separazione magnetica”. Esso, contrariamente alla separazione eseguita con centrifugazione, non presenta forze di taglio e quindi previene possibili danni sul materiale biologico [10].
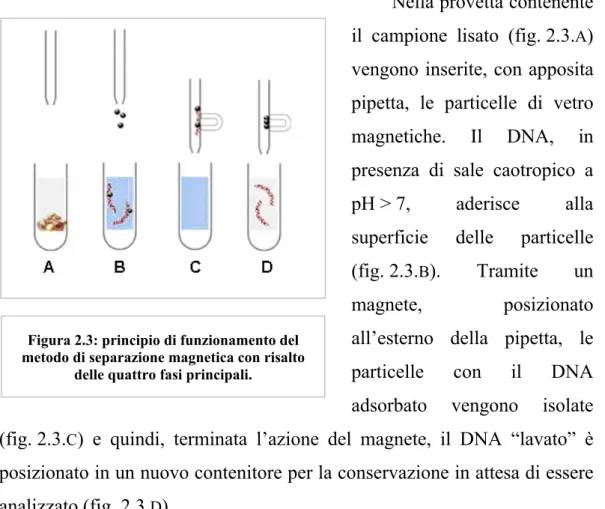
Il principio di funzionamento di questo metodo di separazione è descritto in figura 2.3 nei suoi passi principali. Una caratteristica essenziale del suo funzionamento risiede nella morfologia delle particelle: esse hanno
Figura 2.2: lisi di una cellula in un campo elettrico. [8]
Nella provetta contenente il campione lisato (fig. 2.3.A) vengono inserite, con apposita pipetta, le particelle di vetro magnetiche. Il DNA, in presenza di sale caotropico a pH > 7, aderisce alla superficie delle particelle (fig. 2.3.B). Tramite un
magnete, posizionato all’esterno della pipetta, le
particelle con il DNA adsorbato vengono isolate (fig. 2.3.C) e quindi, terminata l’azione del magnete, il DNA “lavato” è
posizionato in un nuovo contenitore per la conservazione in attesa di essere analizzato (fig. 2.3.D).
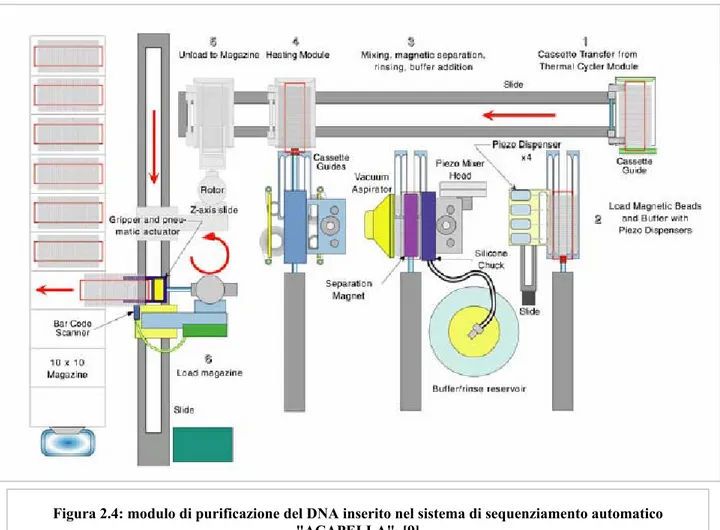
Sono stati sviluppati anche moduli di purificazione automatici che sfruttano il principio delle particelle magnetiche (fig. 2.4). Tale struttura è inserita in una apparecchiatura più complessa chiamata “ACAPELLA” che riesce a sequenziare 5000 campioni in 8 h. Per purificare, usa particelle magnetiche “Dynal streptavidin” di diametro 2.8 µm che vengono inserite con 2 µl di campione da sequenziare e un buffer opportuno in un capillare. Dopo che la soluzione è stata mescolata, viene tenuta ferma per 5 min per far completare i legami tra il DNA e le particelle magnetiche e successivamente sottoposta a un campo magnetico per 1 min. Con i capillari ancora nel campo magnetico, viene effettuata la pulizia delle particelle con una soluzione di etanolo e quindi, rimosso il campo magnetico, le particelle si separano dal DNA che è pronto per essere sequenziato.
Figura 2.3: principio di funzionamento del metodo di separazione magnetica con risalto
Il principio descritto può essere usato in dispositivi integrati dove i campi magnetici sono generati da microelettrodi planari integrati in una microcamera, oppure viene usato un campo magnetico esterno prodotto da un magnete permanente. La separazione viene effettuata velocemente e quando il campo magnetico è rimosso la demagnetizzazione delle particelle è totale e ovviamente non lascia traccia sul DNA purificato.
Figura 2.4: modulo di purificazione del DNA inserito nel sistema di sequenziamento automatico "ACAPELLA". [9]
2.3 Tecniche
elettroforetiche
L’elettroforesi è un processo elettrocinetico nel quale molecole e particelle cariche, in soluzione acquosa, sotto l’influenza di un campo elettrico, migrano in direzione del polo che ha carica opposta [11].
In campo biologico sono molte le molecole che possiedono gruppi ionizzabili (come aminoacidi, proteine e acidi nucleici) e quindi, a ogni valore di pH, sono presenti in soluzione come specie elettricamente cariche.
Per esempio, grazie alla presenza dei gruppi fosfato (PO43-), le molecole di DNA sono cariche negativamente e quindi migreranno verso il polo positivo (anodo) se sottoposte a un campo elettrico.
Nel corso degli anni sono state sviluppate varie apparecchiature per separare molecole cariche basate su principi elettroforetici.
Fondamentalmente qualsiasi apparecchiatura per elettroforesi è composta da tre componenti principali: un alimentatore, un termostato, che permette il controllo e la regolazione della temperatura, e una camera di separazione, che è la parte principale dello strumento e può essere realizzata in molti modi differenti.
Le separazioni elettroforetiche possono essere fatte in soluzioni libere come nell’elettroforesi capillare o sistemi senza fase di supporto, ma anche su mezzi stabilizzanti come gel.
Inizialmente furono utilizzate camere in cui la separazione avveniva in soluzione libera [12]. In tal caso l’efficienza di separazione era limitata dalla diffusione termica e dalla convezione. Per tali motivi, l’elettroforesi tradizionale è stata successivamente eseguita su mezzi di supporto anti-convettivi come poliacrilamide o gel di agarosio. Essa è particolarmente indicata per la separazione di molecole biologiche come
acidi nucleici e proteine. Pur essendo ancor oggi molto usata, tale tipo di elettroforesi non è vantaggiosa a causa di lunghi tempi di analisi, bassa efficienza e difficoltà nell’automazione. L’alternativa è l’elettroforesi capillare che, essendo fatta in tubi stretti anti-convettivi, non necessita del mezzo gel e quindi è realizzata nuovamente in soluzione libera.
Nella figura 2.5 è mostrata una camera di separazione (o cella elettroforetica) per elettroforesi su gel nel
modo verticale.
In tal caso il principio di funzionamento è il seguente: la miscela campione da separare va sciolta in un tampone6, con il quale va anche saturato il mezzo gelatinoso di supporto per consentire la conduzione della corrente, che si genera quando fra i due elettrodi viene applicata una differenza di potenziale. Il campo elettrico generato fa si che le componenti del campione migrino in una direzione (verso l’anodo o verso il catodo) in base alla loro carica, con velocità che dipende anche dalla loro forma e dimensione, oltre che dall’intensità di corrente. Se il campo elettrico viene tolto prima che le molecole abbiano raggiunto gli elettrodi, si ha una separazione dei singoli componenti in base alla loro mobilità elettroforetica.
6 Una soluzione si dice tampone (o buffer) se riesce a mantenere un certo valore di pH nella
soluzione, nonostante l'aggiunta di reagenti che tenderebbero a farlo variare. Nel nostro caso il Figura 2.5: cella elettroforetica per elettroforesi su
Una volta terminata la separazione dei campioni, questi vengono visualizzati mediante opportuni metodi di colorazione o rivelazione. Il metodo di separazione descritto è detto elettroforesi a zona (ZE).
Oltre a quello appena citato, esistono altri due metodi base di separazione: l’Isotacoforesi (ITP) e l’Isoelettrofocalizzazione (IEF)7. Quest’ultima tecnica è indicata per la separazione di composti anfoteri (cioè composti che hanno sia proprietà acide che basiche), come aminoacidi e peptidi, in cui sia la direzione che la velocità di migrazione dipendono dal pH e ha luogo in un gradiente di pH.
2.3.1
Principi generali di elettroforesi
La separazione per elettroforesi è basata su differenze nella velocità del soluto in un campo elettrico.
La velocità di uno ione è data da:
v = µe E (1)
dove v è la velocità dello ione, µe la mobilità elettroforetica ed Eè il campo elettrico applicato. Il campo elettrico è semplicemente una funzione della differenza di potenziale (V) applicata e della distanza (d) fra gli elettrodi:
E = d V
(2)
La mobilità assoluta8, per un dato ione e mezzo, è una costante che è caratteristica di quello ione. La mobilità è determinata dalla forza elettrica cui la molecola è sottoposta, bilanciata dalla forza frizionale dovuta al mezzo di supporto.
7 Per approfondimenti su tali metodi consultare:
Reiner Westermeier, “Electrophoresis in Practice”, Ed. VCH, 1997, capitoli 2 e 3 (pp 41-56).
8 In realtà, sperimentalmente, si determina una mobilità effettiva, di valore differente da quello
della mobilità assoluta. Tale differenza è dovuta a variazioni di temperatura, al valore del pH e alla composizione (tipo e concentrazione) del buffer su cui avviene la “corsa”elettroforetica.
La forza (FE) che spinge una molecola di carica q verso uno degli elettrodi è:
FE = q E (3)
Tuttavia esiste anche una forza frizionale (FF) che ne rallenta il movimento. Tale forza è dovuta all’attrito che la molecola incontra durante il suo cammino e dipende quindi dalle sue dimensioni idrodinamiche, cioè dalla sua forma, e dalla viscosità della soluzione (η) :
FF = - 6 π η r v
(4)
(dove r è il raggio dello ione considerato, supposto sferico per semplicità) . Durante l’elettroforesi, si raggiunge uno stato di equilibrio, ottenuto quando le forze FE e FF si compensano.
A questo punto tali forze sono uguali ma opposte: q E = 6 π η r v (5)
Risolvendo quest’ultima equazione in funzione della velocità e sostituendo quindi nell’equazione (1), si ottiene la mobilità elettroforetica:
µe =
r
q
η
π
6
Da quest’ultima espressione, è evidente che specie piccole ma con alta carica, hanno grande mobilità, mentre specie grandi poco cariche hanno bassa mobilità. In conclusione avremo: v = µe E = µe d V =
d
V
r
q
η
π
6
(6)
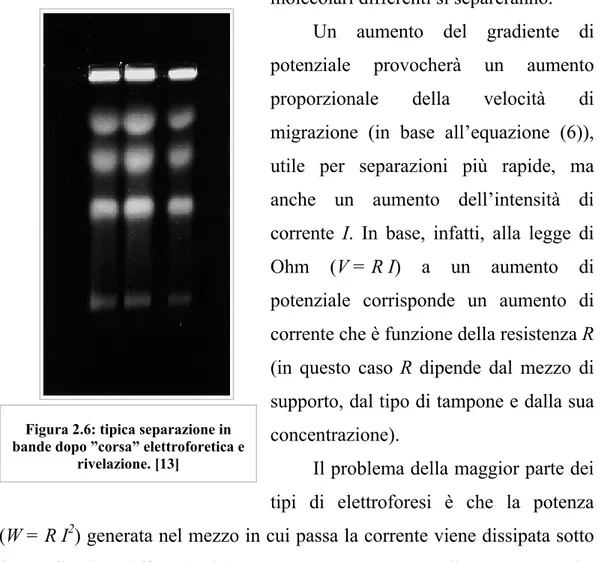
Pertanto, quando si applica una differenza di potenziale, molecole con carica elettrica totale differente inizieranno a separarsi in funzione della loro mobilità elettroforetica, formando caratteristiche “bande” (fig. 2.6): ciascuna banda corrisponde a molecole con caratteristiche diverse.
Anche molecole con carica elettrica uguale ma con dimensioni molecolari differenti si separeranno.
Un aumento del gradiente di potenziale provocherà un aumento proporzionale della velocità di migrazione (in base all’equazione (6)), utile per separazioni più rapide, ma anche un aumento dell’intensità di corrente I. In base, infatti, alla legge di Ohm (V = R I) a un aumento di potenziale corrisponde un aumento di corrente che è funzione della resistenza R (in questo caso R dipende dal mezzo di supporto, dal tipo di tampone e dalla sua concentrazione).
Il problema della maggior parte dei tipi di elettroforesi è che la potenza (W = R I2) generata nel mezzo in cui passa la corrente viene dissipata sotto forma di calore (effetto Joule) con conseguente aumento di temperatura, che deve quindi essere controllato e limitato.
Il calore, infatti, può avere i seguenti effetti negativi:
1. aumento della velocità di diffusione dei campioni e degli ioni del tampone, che determina la formazione di bande meno definite;
2. comparsa di correnti convettive, che portano al mescolamento dei campioni separati;
3. denaturazione di quei campioni che sono poco stabili alle alte temperature;
Figura 2.6: tipica separazione in bande dopo ”corsa” elettroforetica e
4. diminuzione della viscosità del tampone e quindi diminuzione della resistenza del mezzo.
Se durante l’elettroforesi venisse applicato un potenziale costante agli elettrodi, variazioni della resistenza del mezzo potrebbero portare a variazioni di intensità di corrente; in particolare, diminuzioni di resistenza porterebbero ad aumenti di corrente e quindi del calore sviluppato. Per questo motivo vengono utilizzati alimentatori che impongono una corrente costante. In tal modo vengono eliminate le fluttuazioni di calore e limitati gli effetti negativi sopra elencati.
Lo sviluppo di calore resta in ogni caso un problema. Infatti sono necessarie alte intensità di corrente per avere separazioni elettroforetiche veloci, ma ciò causa una notevole produzione di calore: è necessario quindi adottare condizioni di compromesso fra le due esigenze contrastanti. In ogni caso, sistemi di refrigerazione sono indispensabili per le apparecchiature elettroforetiche per dissipare il calore prodotto.
Un altro fattore fondamentale da considerare in una separazione elettroforetica è il fenomeno dell’elettroosmosi, o flusso elettroosmotico (EOF).
Il fenomeno è dovuto alla presenza di gruppi carichi sulla superficie del mezzo di supporto. Per esempio la carta contiene gruppi carbossilici e l’agarosio gruppi solfonati. Questi gruppi diventano ionizzati (adsorbono ioni) in tamponi basici o neutri, per cui, in un campo elettrico, saranno attratti dall’anodo. Poiché però essi sono fissati nella matrice, non potranno migrare. Tale situazione da luogo a un flusso opposto di ioni H3O+ verso il catodo che trascina con se molecole solubilizzate prive di carica. Questo è il fenomeno dell’elettroosmosi ed è additivo con l’elettroforesi, per cui accelera il movimento dei cationi e rallenta quello degli anioni (per esempio la corsa di molecole di DNA verso l’anodo viene rallentata).
Anche nell’elettroforesi capillare è presente il fenomeno dell’elettroosmosi. In questo caso la parete interna del capillare in vetro ha gruppi ionizzabili di silanolo (SiOH), che sono in contatto con il buffer durante l’elettroforesi. I gruppi silanolo si dissociano rapidamente, dando una carica negativa alla parete del capillare (fig. 2.7).
Quando il capillare è riempito con il buffer, la sua parete caricata negativamente attrae ioni carichi positivamente dalla soluzione buffer, creando un doppio strato elettrico e una differenza di potenziale (potenziale Zeta) vicino alla parete del capillare. Il doppio strato elettrico include uno strato rigido di ioni adsorbiti e uno strato diffuso. Quando è applicato un potenziale nel capillare, i cationi nello strato diffuso sono liberi di migrare verso il catodo, trascinando con se le molecole in soluzione, anche quelle di segno opposto che normalmente migrerebbero verso l’anodo (come il DNA).
Figura 2.7: fenomeno dell'elettroosmosi nell'elettroforesi capillare. [12]
2.3.2
Materiali di supporto
Il compito del materiale di supporto su cui vengono fatte “correre” le molecole da separare è quello di prevenire disturbi meccanici e correnti convettive, limitando quindi la diffusione, per fare in modo che i campioni con caratteristiche simili siano contenuti in zone ristrette e distinte fra loro e non si confondano con zone adiacenti. Tali problemi sono causati principalmente da variazioni di temperatura [13].
In generale i mezzi di supporto sono materiali porosi, che vengono opportunamente bagnati nel tampone di corsa all’interno del quale si ha l'elettroforesi. Essi possono avere sia la proprietà di adsorbire varie specie molecolari (proprietà utile per la cromatografia9) sia quella di agire come un separatore (o “setaccio”) molecolare: i pori di una determinata dimensione permettono, infatti, il passaggio di molecole di uguali dimensioni, o più piccole, ma contrastano quello di molecole di dimensioni maggiori rallentandone il cammino (fig. 2.8). Nei gel di amido per esempio sono presenti entrambi gli effetti, mentre nei gel di poliacrilamide e agarosio (non-adsorbenti), l’effetto setaccio è predominante.
9 La cromatografia è un metodo di identificazione e separazione dei componenti di miscele
Figura 2.8: effetto "setaccio" molecolare nei materiali
I primi supporti utilizzati erano costituiti da carta da filtro o da strisce di acetato di cellulosa. La carta da filtro è particolarmente indicata per la separazione di miscele di proteine se l’elettroforesi viene eseguita a bassa tensione (20 V/cm)10, mentre per la separazione di piccole molecole (aminoacidi e nucleotidi) è necessario applicare alte differenze di potenziale (200 V/cm) con notevoli problemi di sviluppo di calore. Inoltre l’adsorbimento impedisce un’alta risoluzione. Quest’ultimo problema è parzialmente superato usando strisce di acetato di cellulosa.
Per avere una massima risoluzione (soprattutto per proteine e acidi nucleici) è necessario effettuare l’elettroforesi su gel come amido (oggi poco usato), poliacrilamide e agarosio.
La matrice di gel ideale dovrebbe avere la misura dei pori regolabile e regolare, essere inerte chimicamente e non presentare elettroosmosi.
I gel di agarosio sono usati soprattutto quando sono necessarie analisi di molecole di grandi dimensioni (grandi frammenti di DNA a singolo e doppio filamento e proteine). L’agarosio è un polisaccaride lineare ed è uno dei costituenti dell’agar, una miscela di polisaccaridi isolati da alcune specie di alghe rosse, e viene normalmente utilizzato a concentrazioni dell’ 1% e 3%. Il gel viene formato sospendendo dell’agarosio in polvere in un tampone acquoso portato quindi all’ebollizione e lasciato raffreddare a temperatura ambiente per formare un gel rigido. Si possono ottenere gel di vario potere elettroosmotico e diverso grado di purezza. La misura dei pori è funzione della concentrazione iniziale di agarosio: si ottengono pori larghi utilizzando basse concentrazioni, mentre pori più piccoli sono ottenuti con concentrazioni più elevate. A seconda della concentrazione di agarosio (e quindi delle dimensioni dei pori) possono essere separate molecole di DNA di misure diverse (tabella 4), solitamente in un range tra 100 b e 20 kb.
10 La dimensione in centimetri (cm) indica lunghezza del gel su cui viene effettuata la corsa
Per la separazione di molecole di dimensioni inferiori (come molecole di RNA, piccoli frammenti di DNA e soprattutto proteine) è usato un gel
artificiale di poliacrilamide, prodotto dalla polimerizzazione di monomeri di acrilamide con metilene-bis-acrilamide.
I gel di poliacrilamide sono particolarmente stabili meccanicamente, chimicamente inerti e presentano piccola elettroosmosi. Inoltre è possibile determinare con precisione la dimensione dei pori (variando la percentuale di acrilamide), che sono di dimensioni inferiori rispetto a quelli dei gel di agarosio.
La preparazione delle matrici di gel deve essere particolarmente curata per evitare problemi di non omogeneità e adsorbimento molecolare [14].
2.3.3
Elettroforesi di DNA su gel di agarosio
Solitamente, per la separazione di campioni di DNA, si realizza l’elettroforesi su gel di agarosio in un apparato a configurazione orizzontale con il gel immerso nella soluzione buffer (configurazione detta “gel sottomarino”, fig. 2.9) [15]. Tale soluzione è preferita, per esempio alla configurazione verticale di figura 2.5 usata con gel di poliacrilamide per la separazione di proteine, in quanto in certi esperimenti l’elettroforesi ha tempi di esecuzione lunghi (in alcuni casi limite anche 24 h) e il gel, se non fosse immerso, tenderebbe ad asciugarsi e a spaccarsi.
TABELLA 4 : concentrazioni approssimative di gel per separare frammenti lineari di DNA di varie misure. [15] Concentrazione del gel Range di separazione (b) Agarosio – 0.3 % 60000 – 5000 Agarosio – 0.7 % 20000 – 800 Agarosio – 0.9 % 7000 – 500 Agarosio – 1.2 % 6000 – 400 Agarosio – 1.5 % 4000 – 200 Agarosio – 2.0 % 3000 –100 Agarosio – 4.0 % 500 – 10 Acrilamide – 4 % 1000 – 800 Acrilamide – 10 % 500 – 25 Acrilamide – 20 % 50 – 1
Il gruppo fosfato di ciascun nucleotide porta una singola forte carica negativa che è molto più grande di ciascuna delle cariche presenti sulle basi. In tal modo il rapporto carica su massa di tutti i polinucleotidi è indipendente dalla composizione della base. Si dimostra inoltre che il rapporto carica su massa è circa lo stesso per tutti i polinucleotidi e quindi, in un campo elettrico, migrerebbero con la stessa mobilità verso l’elettrodo.
Ne consegue che il principale fattore di separazione per gli acidi nucleici è l’effetto “setaccio” molecolare: le molecole piccole si muoveranno più velocemente di quelle più grandi incontrando minor difficoltà nel passare attraverso i pori del gel.
La separazione avviene quindi grazie al differente peso molecolare che hanno i vari frammenti di DNA all’interno del campione e alla loro forma. Si ottiene già una buona separazione per molecole che hanno peso molecolare che differisce dell’1%.
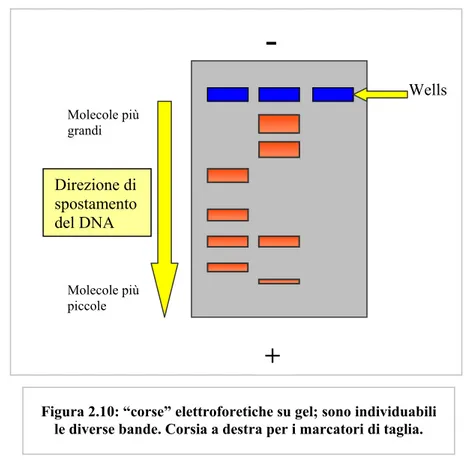
Quando la separazione è completata, il DNA è visualizzato bagnando il gel in una soluzione diluita di tintura di bromuro di etidio (EtBr) fluorescente. Il bromuro di etidio tende a legarsi al DNA e quindi, dopo che il gel viene lavato per eliminare la tintura non legata, se stimolato da raggi ultravioletti (300-350 nm) emette una luce a 500-650 nm, evidenziando la presenza di DNA. Il gel con le bande fluorescenti viene infine fotografato (fig. 2.6 e fig. 2.10).
Per poter stabilire la lunghezza delle varie specie molecolari presenti nel campione è conveniente posizionare sul gel, accanto al campione, un marcatore di taglia. Nel caso del DNA si tratta di una serie di frammenti di
Figura 2.9: schema semplificato di un apparato orizzontale per elettroforesi su gel. [11]
lunghezza nota e definita. Al termine dell’elettroforesi, il confronto tra la mobilità del campione rispetto al marcatore di taglia, permetterà di determinare con buona approssimazione le caratteristiche del campione.
I pesi molecolari del DNA di ciascuna banda sono determinati dalla distanza della banda dal suo punto di origine. L’equazione:
D = a – b log M
descrive la relazione tra peso molecolare M e distanza D (a e b sono costanti in funzione del tempo di elettroforesi, del buffer e della concentrazione del gel).
In commercio si trovano molti sistemi “submarine” per elettroforesi su gel per la separazione di acidi nucleici11. Essi variano fra loro per dimensioni del gel e numero di “well” e quindi per numero di “corse” che è possibile fare contemporaneamente (fig. 2.10).
-
Wells Molecole più grandi Molecole più piccole Direzione di spostamento del DNA-+
Figura 2.10: “corse” elettroforetiche su gel; sono individuabili le diverse bande. Corsia a destra per i marcatori di taglia.
Per esempio esiste un formato piccolo di gel 7 x 10 cm con 28 wells e un formato più grande 15 x 15 cm con 124 wells [16]. Anche le tensioni applicate sono variabili a seconda dello strumento utilizzato: per un gel di dimensioni 15 x 15 cm è consigliato un voltaggio in un range di 20-200 Volt per cm (a seconda delle applicazioni).
In figura 2.11 è mostrato un sistema per elettroforesi su gel che consente fino a 96 corse elettroforetiche contemporanee in 90 min. Le sue dimensioni sono di 23.5 x 7.5 cm, mentre il volume di buffer necessario per il suo funzionamento è di 850 ml [17].
2.3.4
Pulsed field gel electrophoresis (PFGE)
L’elettroforesi su gel di agarosio è il mezzo più comune per separare campioni di dimensioni fino a 60 kb.
Con l’introduzione della PFGE (Schwartz e Cantor, 1984), il limite di risoluzione aumenta considerevolmente, arrivando a separare campioni fino a 10 Mb, cioè si riescono a separare cromosomi interi [18].
Durante la normale elettroforesi a campo elettrico continuo, il DNA che ha dimensioni comprese nel range 30-50 kb migra con la stessa mobilità, indipendentemente dalla grandezza; ciò è visibile sul gel come una singola larga banda diffusa.
Si osserva tuttavia che se il DNA è forzato a cambiare direzione durante l’elettroforesi, per mezzo di alterazioni periodiche dell’angolo del campo elettrico applicato, i frammenti di differente misura cominciano a
Figura 2.11: modello JBS-96 – mini-sistema Submarine su gel ultra-largo.
separarsi l’uno dall’altro. Infatti dopo ogni riorientamento del campo elettrico, il DNA di misura più piccola comincerà a muoversi nella nuova direzione più velocemente di quello di misura maggiore. In tal modo il DNA più grande resta indietro, separandosi dal DNA più piccolo.
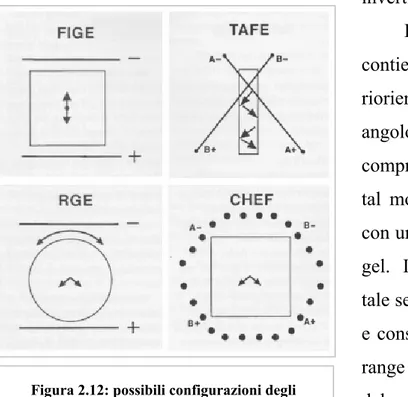
Solitamente gli strumenti disponibili per effettuare la PFGE si dividono in due categorie. L’equipaggiamento più semplice è progettato per la “Field Invertion Gel Elecrophoresis” (FIGE) che lavora invertendo periodicamente la polarità degli elettrodi durante l’elettroforesi (fig. 2.12). Poiché la tecnica FIGE fa subire un riorientamento a 180º, il DNA perde del tempo perché deve tornare indietro ogni volta che il campo elettrico viene
invertito.
L’altra categoria contiene strumenti che riorientano il DNA di un
angolo generalmente compreso fra 96º e 120º. In
tal modo il DNA si muove con un percorso a zigzag nel gel. In condizioni ottimali tale separazione è più veloce e consente risoluzione in un range di misura più ampio del caso precedente. Le tecniche TAFE (“Transverse Alternating Field Electrophoresis”), RGE (“Rotating Gel Electrophoresis”) e CHEF (“Contour–Clamped Homogeneous Electric Field”) sono usate per riorientare il DNA e di esse è disponibile la strumentazione. La difficoltà principale è quella di riuscire a effettuare la separazione mantenendo il
Figura 2.12: possibili configurazioni degli elettrodi di unità PFGE usate comunemente. [18]
separazione fatta. Per risolvere tale problema, per esempio, sia la tecnica RGE che la CHEF usano un campo elettrico omogeneo e il gel posizionato orizzontalmente. Mentre però la tecnica CHEF cambia la direzione del campo elettrico per riorientare il DNA elettronicamente, cambiando la polarità di una matrice di elettrodi, l’RGE ha un campo elettrico fisso e, per muovere il DNA in una nuova direzione, ruota il gel (fig. 2.12).
Parametri più importanti per una separazione fatta con la PFGE:
• Intensità del campo elettrico: il suo valore è il compromesso fra il tempo di separazione e la risoluzione di una classe di campioni di misura particolare. Per esempio per DNA fino a 2000 kb sono necessari 4-6 V/cm per un tempo di 1-2 d.
• Durata del campo elettrico in una direzione (“pulse time”); è una funzione del DNA da separare: tempi lunghi permettono la separazione di DNA più grandi. Per esempio a 5.4 V/cm, cromosomi di 1.6 e 2.2 Mb sono separati con un “pulse time” di 90 s.
• Angolo di riorientamento. • Temperatura.
2.3.5
Elettroforesi Capillare (CE)
Con il termine CE, è indicata l’elettroforesi capillare ad alta risoluzione (“High Performance Capillary Electrophoresis” o HPCE).
La versatilità dell’elettroforesi capillare permette, tuttavia, di poter usare tale metodologia per la separazione di un’ampia gamma di composti biologici (come proteine, peptidi, aminoacidi, acidi nucleici…) e quindi si possono distinguere diversi tipi di elettroforesi capillare ad alta risoluzione a seconda del principio base usato per la separazione.
È possibile distinguere: la “Capillary Zone Electrophoresis” (CZE), basata sulla diversa mobilità delle particelle cariche in soluzione libera; la “Capillary Gel Electrophoresis” (CGE), che effettua la separazione in base alle dimensioni e alla carica; la “Micellar Electrokinetic Chromatography” (MEKC), la “Capillary Isoelectric Focusing” (CIEF) e la “Capillary Isotachophoresis” (CITP). Un generico sistema per elettroforesi capillare è schematizzato in figura 2.13.
L’elettroforesi avviene all’interno di un tubo (capillare) stretto, di solito realizzato in silice fusa o teflon, avente diametro interno nel range di 25-75 µm e diametro esterno di 350-400 µm, rivestito da uno strato
Questo tipo di realizzazione permette di minimizzare i problemi derivanti dallo sviluppo di calore che, tradizionalmente, limitano le tecniche elettroforetiche, in quanto causa di gradienti di temperatura non uniformi, cambiamenti locali di viscosità e conseguenti zone allargate.
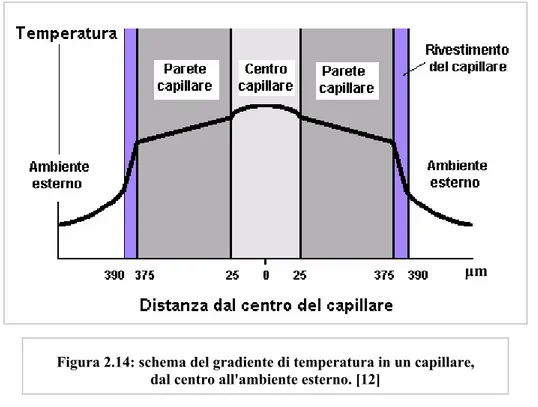
Il calore generato dal passaggio della corrente elettrica causa un aumento di temperatura che è funzione delle dimensioni dei capillari, della conduttività del buffer e della tensione applicata. Si hanno temperature sensibilmente elevate quando la potenza generata è maggiore di quella dissipata. La dissipazione termica del calore attraverso le pareti del capillare può dar luogo a temperature più alte nel centro del capillare piuttosto che alle pareti (fig. 2.14). Tali gradienti di temperatura causano differenze locali di viscosità nel buffer e quindi una migrazione non uniforme.
Si dimostra che per limitare i gradienti di temperatura, è conveniente usare capillari con raggio interno piccolo e grande raggio esterno. Infatti il volume interno piccolo limita la quantità di calore generato, mentre l’alto rapporto tra superficie interna e volume aiuta a dissipare il calore generato
Figura 2.14: schema del gradiente di temperatura in un capillare, dal centro all'ambiente esterno. [12]
attraverso la parete del capillare. Il grande diametro esterno invece è vantaggioso in quanto consente di ridurre le proprietà isolanti del poliamide e migliora il trasferimento di calore verso la periferia del capillare. È anche importante la rimozione del calore all’esterno del capillare, che può essere realizzata con un sistema di ventilazione opportuno.
È possibile ottenere alte efficienze di separazione utilizzando campi elettrici elevati (nel range 100-500 V/cm). La lunghezza del capillare non influisce sull’efficienza del processo, ma gioca un ruolo importante sul tempo di migrazione e quindi sulla durata dell’analisi. La situazione ideale consisterebbe nell’applicare un potenziale il più alto possibile, utilizzando un capillare il più corto possibile. Tuttavia ci sono delle limitazioni pratiche: quando la lunghezza del capillare diminuisce, infatti, la quantità di calore che deve essere dissipata aumenta, a causa della diminuzione della resistenza elettrica del capillare. Nello stesso tempo la superficie disponibile per la dissipazione del calore diminuisce. Gli effetti dovuti al calore pongono quindi un limite pratico all’utilizzo di capillari molto corti. Inoltre, più è alto il potenziale applicato, più alta diviene la corrente che attraversa il capillare e quindi è maggiore la quantità di calore generata. Diventa necessaria la scelta di un compromesso fra il potenziale applicato e la lunghezza del capillare. Comunemente si utilizzano potenziali di circa 10-30 kV e capillari lunghi 25-75 cm (fig. 2.15).
25-75 µm 350-400 µm
Figura 2.15: dimensioni possibili di un capillare per
Il funzionamento di una apparecchiatura per elettroforesi capillare, come quella di figura 2.12, è il seguente: una piccola quantità di soluzione contenente il campione (tra 1 e 50 nl) viene iniettata nel capillare, che contiene un buffer appropriato, dall’estremità anodica. Per effettuare la separazione viene applicata una differenza di potenziale tra le due estremità del capillare. Come precedentemente analizzato (paragrafo 2.3.1) all’interno del capillare, oltre all’attrazione dei poli sulle molecole di segno opposto, si ha un flusso elettroosmotico (EOF) verso il catodo. Caratteristica fondamentale del flusso elettroosmotico nel capillare è che il suo profilo è praticamente piatto (fig. 2.16).
Il profilo piatto del flusso è vantaggioso dal momento che non contribuisce direttamente alla dispersione del soluto (cosa che avverrebbe nel caso di flusso laminare dove le componenti che si trovano al centro del tubo migrano ad una velocità maggiore di quelle che si trovano ai bordi). In figura 2.16 si osserva che la quantità di flusso elettroosmotico scende rapidamente nei pressi della parete a causa dell’attrito. Dal momento che tale strato si estende poco nella soluzione, il suo effetto è poco importante ai fini della separazione (si avrebbero problemi con diametri interni del capillare maggiori di 200 µm). Ulteriore beneficio derivante dalla presenza dell’EOF è che esso causa la migrazione di tutte le specie, indipendentemente dalla carica, nella stessa direzione, cioè verso il catodo.
Figura 2.16: profilo di flusso elettroosmotico (EOF) in un capillare paragonato a un flusso laminare. [12]
Anche gli anioni migreranno tutti verso il catodo dal momento che il flusso elettroosmotico è più grande, di almeno un ordine di grandezza, della loro mobilità elettroforetica. In conclusione, anioni, cationi ed elementi neutri in un capillare migrano tutti nella stessa direzione se sottoposti a elettroforesi.
I cationi migrano più velocemente, dal momento che l’attrazione elettroforetica verso il catodo e l’EOF sono diretti nella stessa direzione e quindi si sommano.
In vicinanza del catodo le molecole attraversano una finestra dove ne viene rivelato il passaggio attraverso varie tecniche.
I risultati della CE sono presentati sotto forma di elettroferogramma, che mette in relazione la risposta del rivelatore in funzione del tempo di migrazione. Nella figura 2.17 è mostrato un semplice elettroferogramma risultato della separazione di una miscela di tre componenti di soluti (uno cationico, uno neutro e uno anionico).
Figura 2.17: semplice elettroferogramma. I picchi indicano il passaggio dei prodotti di separazione, l'intensità della risposta serve
2.4 Sequenziamento di Acidi Nucleici
2.4.1
Blotting di DNA, RNA e proteine
Un frammento di restrizione contenente una specifica sequenza di basi può essere identificato ibridandolo con un filamento marcato di DNA complementare.
Una miscela di frammenti di restrizione viene separata tramite elettroforesi su gel di agarosio, quindi si denatura il DNA portandolo nella forma a singola catena e lo si trasferisce su un foglio di nitrocellulosa per capillarità (“blotting”) (fig. 2.18).
I vari frammenti di DNA mantengono sulla nitrocellulosa le stesse posizioni che avevano nel gel. Le posizioni possono essere determinate esponendo il foglio a un filamento di DNA a singola elica di sequenza nota (detta “probe” o sonda) marcato radioattivamente (per esempio con 32P).
La sonda si lega con un filamento complementare e l’autoradiografia12 rivela quindi la posizione dell’insieme ibridato (cioè del complesso frammento di restrizione-sonda). In questo modo si può identificare un frammento particolare in mezzo a milioni di altri frammenti. Questa tecnica molto efficace prende il nome di Southern blotting in quanto ideata da Edwin Southern.
La stessa procedura di analisi può essere seguita per individuare specifiche sequenze di RNA. La tecnica di analisi dell’RNA è stata chiamata Northern blotting. Infine la Western blotting è una tecnica usata per rivelare una particolare proteina colorandola con uno specifico anticorpo.
Southern, Northern e Western blotting sono noti anche come blotting di DNA, RNA e proteine [19, 20].
2.4.2
Metodo dei didesossi di Sanger
Il DNA può essere sequenziato generando frammenti di DNA la cui lunghezza dipende dall’ultima base della sequenza. L’insieme di tali frammenti può essere generato attraverso una interruzione controllata della replicazione enzimatica. Questo metodo è stato sviluppato da Fredrick Sanger e dai suoi collaboratori.
Si realizza la stessa procedura, contemporaneamente, in quattro miscele di reazione distinte (A, G, T e C). In ciascuna di esse, si usa l’enzima DNA polimerasi per realizzare il complemento di una particolare sequenza di una molecola di DNA a singolo filamento. La sintesi è cominciata da un frammento (detto primer), solitamente ottenuto tramite
metodi chimici di sintesi, che è complementare alla parte di sequenza nota da altri studi.
Ciascuna miscela contiene, oltre ai quattro desossiribonucleotidi trifosfato (dATP, dTTP, dCTP, dGTP) marcati radioattivamente, anche un piccolo quantitativo di un analogo 2´-3´-didesossi (ddNTP). In ognuna delle quattro miscele si ha un differente ddNTP: per esempio nella miscela A, ddATP (fig. 2.19), in quella G, ddGTP… L’incorporazione di questo analogo blocca la crescita ulteriore della catena.
La concentrazione del ddNTP in ciascuna miscela è abbastanza bassa in modo che la terminazione della catena possa avvenire solo occasionalmente. La DNA polimerasi inserirà talvolta il nucleotide giusto e altre volte il ddNTP analogo, bloccando la reazione e quindi la crescita della catena. Nella miscela A di figura 2.19, la crescita della catena è interrotta ogni volta che viene inserito un ddATP (l’unico “analogo” presente in questa miscela); saranno quindi prodotti frammenti di varie lunghezze, tutti terminanti con ddATP. Si osserva che il didesossi analogo di dATP è inserito solo dove nella sequenza di DNA da analizzare è presente una T; nella miscela A, i frammenti di differenti lunghezze corrispondono alla posizione di T.
Le quattro miscele di frammenti di terminazione della catena (una per ogni analogo didesossi), dopo denaturazione, vengono sottoposte a
Figura 2.19: strategia del metodo di Sanger nella miscela contenente ddATP (miscela A).
elettroforesi su gel di poliacrilamide in differenti “corsie” e la sequenza delle basi del DNA complementare a quella cercata viene letta dall’autoradiogramma delle quattro linee (fig. 2.20).
Questo tipo di sequenziamento è “manuale” in quanto la lettura dell’autoradiogramma è affidata a un tecnico.
Il metodo di Sanger è adatto per essere automatizzato, passando a una rivelazione per fluorescenza.
È infatti possibile marcare il primer con un composto fluorescente diversamente colorato per ciascuna delle quattro miscele di reazione di terminazione (per esempio si può associare un composto che emette una luce blu per A, uno che emette una luce rossa per C…); ciascun primer quindi emette fluorescenza a lunghezza d’onda diversa ed è facilmente individuabile. Eseguendo l’analisi esattamente come nel caso di sequenziamento manuale con quattro “corsie” per l’elettroforesi (e quindi quattro miscele distinte), l’indagine mediante fluorescenza produce un risultato analogo al caso precedente in cui però le bande separate di DNA sono rivelate dalla loro fluorescenza come essa emerge dal gel; la sequenza dei loro colori porta direttamente alla sequenza delle basi (fig. 2.21). In questo modo possono essere determinate sequenze lunghe fino a 500 basi.
In alternativa, è possibile marcare i ddNTP, ciascuno con una differente etichetta fluorescente. Quando viene usato questo metodo, tutte e quattro le miscele del caso precedente possono coesistere in un’unica
Figura 2.20: autoradiogramma del sequenziamento manuale di Sanger.
C T A G G C A T T A G C G
l’elettroforesi, evitando il rischio di variabilità elettroforetica fra una corsia e l’altra (fig. 2.22).
La rivelazione fluorescente è migliore perché elimina l’uso di reagenti radioattivi. La marcatura più usata attualmente è quella basata sui terminatori di catena chiamati “big-dyes terminator”, che consistono di didesossinucleotidi marcati con molecole con sistema di trasferimento di energia da un donatore a un accettore [21].
L’introduzione della marcatura fluorescente permette di passare dal sequenziamento manuale a quello automatico che prevede una corsa elettroforetica su gel di poliacrilamide, su supporto a lastra o a capillare.
In entrambi i casi la migrazione dei vari frammenti è seguita rilevando le emissioni in fluorescenza a diverse lunghezze d’onda dei diversi fluorocromi dopo l’eccitazione provocata dal laser. Le emissioni vengono raccolte e analizzate da una camera CCD (charge coupled device) che elabora i diversi segnali di fluorescenza con elevata sensibilità.
Figura 2.21: indagine mediante fluorescenza di frammenti oligonucleotidici prodotti con il metodo dei dideossi. I primer di
La sequenza delle bande di DNA marcato viene visualizzata in un unico grafico detto elettroferogramma, caratterizzato da una successione di picchi di quattro colori diversi, che corrispondono alle emissioni fluorescenti dei diversi fluorocromi, ogni volta che i vari frammenti di diversa lunghezza nucleotidica raggiungono, lungo la corsa elettroforetica, la posizione del rilevatore (fig. 2.23).
Figura 2.23: esempio di elettroferogramma. Rispetto al caso precedente sono associati colori differenti alle basi. In questo caso ad
A è associato il colore verde, a C il blu, a G il nero (trasformazione Figura 2.22: indagine mediante fluorescenza di frammenti
oligonucleotidici prodotti con il metodo dei dideossi. È possibile passare da una elettroforesi a quattro corsie a una a singola corsia.
G C A T T A G C G C T A G
Come precedentemente visto, l’introduzione dell’elettroforesi capillare per la separazione dei frammenti marcati ha consentito un notevole aumento della processività. Sono stati inoltre sviluppati modelli di sequenziatori automatici che sono in grado di eseguire corse elettroforetiche multiple su apparecchi multicapillari.
2.4.3
Sequenziamento del DNA per ibridazione con
gruppi di nucleotidi (microarray di DNA)
La capacità di sintetizzare nuove catene oligonucleotidiche aventi una sequenza di basi desiderata, ha aperto nuove possibilità di sequenziamento del DNA. Per esempio è possibile usare un oligonucleotide a singolo filamento di sequenza nota per cercare una sequenza complementare (sempre a singolo filamento) in una catena molto lunga di DNA, o al limite in un genoma, opportunamente marcata; se il filamento marcato troverà la sua sequenza complementare (nota), si avrà ibridazione, ovvero si avrà la formazione di una molecola di DNA a doppio filamento individuabile in quanto marcata [22].
A partire dal 1995 l’introduzione dei DNA microarray ad alta densità ha rivoluzionato le metodiche di sequenziamento. Il principio che sta alla base della tecnica dei microarray è il fatto che molecole di acido nucleico marcate in soluzione sono in grado di ibridarsi, con elevata sensibilità e specificità, con sequenze immobilizzate su un supporto solido, permettendo così la misurazione contemporanea di molte sequenze diverse (fig. 2.24).
Anche se esistono diverse tipologie di microarray di acidi nucleici, le tre classi principali sono:
1. microarray di cloni di DNA micropipettati: i cloni di DNA sono preparati in anticipo e poi impressi sulla superficie di un
vetrino da microscopio con speciale iniettore. Essi hanno il vantaggio di avere un costo relativamente basso.
2. microarray di oligonucleotidi prefabbricati: si sfrutta la tecnologia dei semiconduttori per posizionare su microelettrodi oligonucleotidi di sequenza nota.
3. microarray di oligonucleotidi sintetizzati in situ: si ha la combinazione di tecnologie diverse (per esempio la fotolitografia e la sintesi chimica degli oligonucleotidi) per sintetizzare direttamente sull’array gli oligonucleotidi. I vantaggi di tale metodo sono l’alta densità (fig. 2.24) e il fatto di non dover usare cloni di DNA o prodotti di PCR.
500 mila celle con sonde diverse su ciascun Gene Chip array Attuali misure di un Gene Chip
Milioni di catene di DNA posizionate su ciascuna cella
Sonda DNA su Chip lunga 25 basi
Le innovazioni che hanno reso possibile la tecnologia dei microarray sono l’uso di supporti solidi non porosi (come per esempio il vetro), molto versatili ai fini della miniaturizzazione e dell’individuazione dei marcatori fluorescenti, e la sintesi ad alta densità spaziale di oligonucleotidi su vetrini molto sottili con tecniche che usano maschere fotolitografiche usate nella tecnologia dei semiconduttori oppure tecniche di ink-jet.
Numerose sono le applicazioni della tecnologia dei microarray; le principali sono l’analisi su larga scala dell’espressione genica e la ricerca di variazioni della sequenza del DNA (ricerca di mutazioni).
2.5 Funzionamento e applicazioni della PCR
Nel 1984 Kary Mullis mise a punto un metodo ingegnoso detto reazione a catena della polimerasi (“Polymerase Chain Reaction” o PCR) che permette di amplificare specifiche sequenze di DNA, non note a priori.
Si considera una doppia elica di DNA consistente in una sequenza “bersaglio” da amplificare, contenuta in due regioni di cui conosciamo la sequenza nucleotidica (dette “regioni fiancheggianti”)
Figura 2.25:Campione di DNA che può essere amplificato con la PCR.
(fig. 2.25); sotto queste condizioni è possibile ottenere con la PCR milioni di copie della sequenza bersaglio.
La PCR viene fatta aggiungendo a una soluzione che contiene il campione di DNA da amplificare, le seguenti componenti:
• una coppia di primer che ibridano le sequenze che fiancheggiano il bersaglio. Ciascun primer solitamente è costituito da un numero di nucleotidi che va da 20 a 30;
• tutti e quattro i desossiribonucleotidi trifosfato (dNTP);
• una DNA polimerasi stabile al calore; viene usata la Taq DNA polimerasi proveniente da Thermus aquaticus, un batterio che vive in sorgenti calde;
• una opportuna concentrazione di sali minerali.
Un ciclo di PCR è costituito dalle seguenti tre fasi (fig. 2.26):
1. Separazione dei filamenti (denaturazione). I due filamenti della molecola di DNA stampo sono separate, portando la soluzione a
94-95°C per 30 s.
2. Ibridazione dei primer (detta anche fase di “annealing” o ancoraggio). La soluzione viene rapidamente raffreddata a 56°C per permettere a ciascun primer di ibridarsi con un filamento di DNA. Un primer si lega alla terminazione 3´ del DNA bersaglio su una catena e l’altro primer si lega sulla terminazione 3´ della catena bersaglio complementare.
3. Sintesi del DNA. La soluzione viene scaldata a 72ºC, temperatura ottimale per la Taq polimerasi che allunga entrambi i primer nella direzione della sequenza bersaglio perché la sintesi del DNA è nella direzione da 5´ a 3´. La polimerizzazione viene fatta procedere per circa 1 min. La sintesi ha luogo su entrambi i filamenti e si estende sopra la sequenza bersaglio [23].
Al termine della sintesi avremo due molecole identiche al campione iniziale. È possibile quindi iniziare un nuovo ciclo in cui le due molecole appena create servono da stampo e al termine del quale il numero di molecole identiche ai campioni iniziali è raddoppiato (fig. 2.27).
Figura 2.27: amplificazione dei campioni di DNA ottenuta con la PCR. [23]
Idealmente, procedendo come sopra, dopo n cicli, la sequenza iniziale è quindi amplificata 2n volte: dopo 20 cicli l’amplificazione porta ad avere più di un milione di copie e dopo 30 cicli più di un miliardo di copie. Ciascun ciclo può durare anche solo circa 2 min, per cui queste operazioni sono eseguite in poco più di 1 h (Tabella 5).
In realtà il numero di campioni di DNA (N) incrementa secondo la relazione:
N = (1 + E(n))n
dove E è un fattore di efficienza in funzione di n. Per n < 20, E è circa 1, mentre per n > 20, il fattore di efficienza si abbassa e quindi si ha un effetto plateau del processo [24].
Altre caratteristiche essenziali della PCR sono:
• il bersaglio può essere molto più grande dei primer tanto che bersagli di 10 kb sono stati amplificati con la PCR;
• la PCR è altamente specifica e l’unico DNA a essere amplificato è quello situato fra i primer;
• la PCR è estremamente sensibile ed è possibile identificare e amplificare una singola molecola di DNA.
Con l’applicazione della PCR, il sequenziamento e la clonazione sono stati molto semplificati. In medicina la PCR può fornire importanti
Tabella 5: parametri tipici per una amplificazione con PCR.
Passo Temperatura Tempo Numero di Cicli Denaturazione Iniziale 94 ºC 5 min 1 Denaturazione 94 ºC 30 s Annealing 56 ºC 30 s Estensione 72 ºC 1.5 min 35 Estensione Finale 72 ºC 10 min 1
identificare rapidamente la presenza di virus e batteri: per esempio si può rivelare la presenza del virus HIV-1 della immunodeficienza umana. Inoltre la PCR è una metodologia per la diagnosi precoce per certi tipi di cancro, in quanto è possibile identificare le mutazioni a carico di alcuni geni per il controllo della crescita cellulare.
La PCR trova anche applicazione nella genetica, nella medicina forense e legale, nella botanica…
Le applicazioni cliniche della PCR saranno ulteriormente migliorate da una nuova tecnologia: la “real time PCR” (RT-PCR). Essa permette non solo di quantizzare l’amplificato ottenuto ma anche di visualizzare la cinetica di amplificazione. La RT-PCR rappresenta uno strumento per migliorare la diagnostica e fornire risultati attendibili e in tempo reale.
Riferimenti bibliografici capitolo. 2:
[1] Jeremy M. Berg, John L. Tymoczko, Lubert Stryer, “Biochemistry-fifth edition”, Ed. W.H.Freeman and Company, 2002. Ind. internet:
http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowTO C&rid=stryer.TOC&depth=2. Collegato il 24 settembre 2003.
[2] Primer on Molecular Genetics, DOE Human Genome 1991-92 Program Report, giugno 1992, pp 20-22.
[3] Robert Kaiser, “The Biomedical Engineering-handbook”, CRC PRESS, IEEE PRESS, 1995, Cap. 99 : Tools for Genome Analysis, pp 1489-1501.
[4] A.Focà, A.G.Lamberti, “Metodiche estrattive per la preparazione dei campioni”, Roche_Diagnostics pubblicazioni, EsaDia No. 13, pp 50-53.
[5] S.Cunha, T.Odijk, E.Süleymanoglu, C.L.Woldringh, “Isolation of the Escherichia coli nucleoid”, Biochimie, No. 83, 2001, pp 149-154. [6] Kamal Datta, Ronald D. Neumann, Thomas A. Winters, “A protocol
for separation and isolation of small and/or large DNA fragments with high yield using CLA4B Sepharose”, Analytical Biochemistry, No. 317, 2003, pp 284-287.
[7] T. Smekal, D.Rhine, D.Weston, P.Grodzinski, “Design, Fabrication and testing of thermal components and their integration into a microfluidic device”, 2002 IEEE, giugno 2002, pp 1039-1045.
[8] M.A.McClain, C.T.Culbertson, S.C.Jacobson, J.M.Ramsey, “Single Cell Lysis on microfluidic devices”, Oak Ridge National Laboratory:
http://www.ornl.gov/~webworks/cppr/y2001/pres/111425.pdf.
Collegato il 4 ottobre 2003.
[9] Deirdre R.Meldrum e altri, “Automated, Integrated Modules for fluid Handling, Thermal Cycling and Purification of DNA samples for
[10] Ramadan, Samper, Neuzil, Lesaicherre Marie, Meng, Kiat, Qin, Puiu, “Optomization of On-Chip Micro-Electromagnets for Biomolecular Separation”, 2002 IEEE, gennaio 2002, pp 249-254. [11] Reiner Westermeier, “Electrophoresis in Practice”, Ed. VCH
(A Wiley company), 1997.
[12] David Heiger, “High performance capillary electrophoresis”, Agilent Technologies-pubblicazione numero 5968-9963E, marzo 2000. Ind. internet: www.Chem.agilent.com/temp/rad1108F/00016811.pdf. Collegato il 22 settembre 2003.
[13] David Freifelder, “Physical Biochemistry – Applications to Biochemistry and Molecular Biology”, Ed. W.H.Freemann and Company, 1983, capitolo 9: electrophoresis (pp 276-319).
[14] Cynthia D.Furlong, David J.Beebe, “A microfabricated device for the study of the sieving effect in protein electrophoresis”, Ed. IEEE, Articolo numero: 0-7803-3811, gennaio 1997.
[15] Dr.Simpson, “Gel Electrophoresis and Photography – An Application Note”, UVP-AB-1000-02.
Ind. internet: http://www.uvp.com/pdf/ab-1000-02.pdf. Collegato il 9 settembre 2003.
[16] Dati tratti dal sito internet con indirizzo: http://www.serva.de; in particolare: http://www.serva.de/products/knowledge/051084.html. Collegato il 28 settembre 2003.
[17] Dati tratti dal sito internet con indirizzo: http://www.geneq.com; in particolare: http://www.geneq.com/catalog/en/electrhoriz.html. Collegato il 28 settembre 2003.
[18] Sean Gallagher, Barbara Joppa, Samantha Li, Scott Cole, “Pulsed Field Electrophoresis for Separation of Large DNA”, HIS Laboratories, Hoefer Scientific Instruments, 1996.
Ind. internet: http://www.nal.usda.gov/pgdic/Probe/v2n3/puls.html. Collegato il 6 settembre 2003.
[19] Jeremy M.Berg, John L.Tymoczko, Lubert Stryer, “Biochemistry-fifth edition”, Ed. W.H.Freeman and Company, 2002.
[21] I. Abbate, G. Cappiello, M.R. Capobianchi, “Sviluppo ed applicazioni del sequenziamento genico nella diagnostica macrobiologica”, Roche_Diagnostics pubblicazioni, EsaDia No. 13, pp 28-32.
[22] James Versalovic, “DNA Chip-Based Microbiology”, Clinical Biology Newsletter, Vol. 23, No. 13, pp 99-102, 2001 Elsevier Science Inc.
[23] Kary B.Mullis, “La reazione a catena della polimerasi”, Le Scienze dossier, Numero 15-Pimavera 2003, pp 22-31.
[24] G. Portella, G. Vecchio, “La PCR da ricerca avanzata a routine”, Roche_Diagnostics pubblicazioni, EsaDia No. 13, pp 4-11.
Tabelle:
Tabella 4: Dr. Simpson, “Gel Electrophoresis and Photography – An Application Note”, UVP-AB-1000-02.
Ind. internet: http://www.uvp.com/pdf/ab-1000-02.pdf. Valori della tabella confermati anche su:
http://www.serva.de/products/knowledge/051084.html.
Tabella 5: elaborata da dati raccolti da ind. internet:
http://microarray1k.aecom.yu.edu/. Collegato il 17 ottobre 2003.
Figure:
Fig. 2.1: elaborata da considerazioni fatte in Jeremy M.Berg, John L.Tymoczko, Lubert Stryer, “Biochemistry-fifth edition”,
Ed. W.H.Freeman and Company, 2002. Fig. 2.2: Oak Ridge National Laboratory:
Fig. 2.3: Elaborata dal sito della Roche Diagnostics Corporation:
http://www.roche-applied-science.com/sis/magnapure/.
Fig. 2.4: Deirdre R.Meldrum e altri, “Automated, Integrated Modules for fluid Handling, Thermal Cycling and Purification of DNA samples for High Throughput Sequencing and Analysis”, 2001 IEEE, luglio 2001, pag. 1219 (lo schema mi è stato fornito direttamente dall’autore, che ringrazio).
Fig. 2.5: Lubert Stryer, “Biochimica”, Ed. Zanichelli, elaborata da pag. 52. Fig. 2.6: David Freifelder, “Physical Biochemistry – Applications to
Biochemistry and Molecular Biology”, Ed. W.H.Freemann and Company, 1983, pag. 294.
Fig. 2.7: David Heiger, “High performance capillary electrophoresis”, Agilent Technologies-pubblicazione numero 5968-9963E, marzo 2000, elaborata da pag. 19.
Fig. 2.8: elaborata da David Freifelder, “Physical Biochemistry – Applications to Biochemistry and Molecular Biology”, Ed. W.H.Freemann and Company, 1983, capitolo 9: electrophoresis (pp 276-319).
Fig. 2.9: Reiner Westermeier, “Electrophoresis in Practice”, Ed. VCH (A Wiley company), 1997, elaborata da pag. 17.
Fig. 2.10: elaborata da considerazioni di David Freifelder, “Physical Biochemistry – Applications to Biochemistry and Molecular Biology”, Ed. W.H.Freemann and Company, 1983, capitolo 9: electrophoresis (pp 276-319).
Fig. 2.11: http://www.geneq.com/catalog/en/electrhoriz.html. Fig. 2.12: HIS Laboratories:
http://www.nal.usda.gov/pgdic/Probe/v2n3/puls.html.
Fig. 2.13, 2.14, 2.15, 2.16 e 2.17: David Heiger, “High performance capillary electrophoresis”, Agilent Technologies - pubblicazione numero 5968-9963E, Marzo 2000, elaborate rispettivamente da pag. 12, da pag. 33, da pag 16 e da pag. 22.
Fig. 2.18: elaborata da:
http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowSect ion&rid=stryer.figgrp.791. Collegato il 10 ottobre 2003.
Fig. 2.19: elaborata da:
http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowSect ion&rid=stryer.figgrp.793. Collegato il 10 ottobre 2003.
Fig. 2.20: elaborata da:
http://www.uvm.edu/~cgep/Education/Sequence.html. Collegato il
10 ottobre 2003. Fig. 2.21: elaborata da:
http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowSect ion&rid=stryer.figgrp.794. Collegato il 10 ottobre 2003.
Fig. 2.22: elaborata da:
http://www.uvm.edu/~cgep/Education/Sequence.html. Collegato il
10 ottobre 2003.
Fig. 2.23: http://www.uvm.edu/~cgep/Education/Sequence.html. Collegato il 10 ottobre 2003.
Fig. 2.24: Dal sito della Affimetrix, all’ind. internet:
http://www.affymetrix.com/corporate/media/image_library/low_res/s ingle_feature.jpg. Collegato il 20 settembre 2003.
Fig. 2.25: elaborata da Jeremy M.Berg, John L.Tymoczko, Lubert Stryer, “Biochemistry-fifth edition”, Ed. W.H.Freeman and Company, 2002.
Fig. 2.26: elaborata da Encyclopaedia Britannica, 1998.
Fig. 2.27: Kary B.Mullis, “La reazione a catena della polimerasi”, Le Scienze dossier, Numero 15-Pimavera 2003, pp 22-31, elaborata da pag. 26.
![Figura 2.2: lisi di una cellula in un campo elettrico. [8]](https://thumb-eu.123doks.com/thumbv2/123dokorg/5635266.69268/6.892.170.757.205.732/figura-lisi-di-una-cellula-in-campo-elettrico.webp)
![Figura 2.7: fenomeno dell'elettroosmosi nell'elettroforesi capillare. [12]](https://thumb-eu.123doks.com/thumbv2/123dokorg/5635266.69268/15.892.170.772.341.647/figura-fenomeno-dell-elettroosmosi-nell-elettroforesi-capillare.webp)
![Figura 2.9: schema semplificato di un apparato orizzontale per elettroforesi su gel. [11]](https://thumb-eu.123doks.com/thumbv2/123dokorg/5635266.69268/19.892.163.776.143.704/figura-schema-semplificato-apparato-orizzontale-elettroforesi-gel.webp)
![Figura 2.13: schema della strumentazione per elettroforesi capillare. [12]](https://thumb-eu.123doks.com/thumbv2/123dokorg/5635266.69268/24.892.212.730.408.926/figura-schema-strumentazione-elettroforesi-capillare.webp)