CHAPTER I
Introduction
Even if the logic of the life has assumed an autonomic form since extremely remote times, its roots should be found into physics and chemistry. Nucleic acids are in the limit between chemistry and biology and their chemical properties dictate the requisites that allow life to blow out of the inanimate.
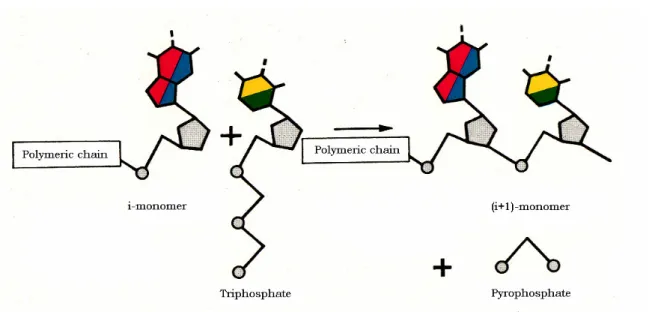
The monomer subunit of nucleic acids, the nucleotide, is a well defined chemical compound formed by three molecular residues: a phosphate group, a sugar (ribose or deoxy-ribose) and a base (purine or pyrimidine). If the monomers are present in form of rich energy (for instance triphosphates as ATP), then, they can react spontaneously to give a polynucleotide strand even in the absence of the catalytic action of enzymes (Fig. 1.1).
Fig. 1.1. Growth of a polynucleotide chain.
The above process is definitely a chemical process; however, the bases present in a strand formed as depicted above, act as the symbols of a language whose sequence enables a message to be codified. A new property is born: information which is totally absent from the physicochemical context that, instead, characterizes the interactions between atoms, molecules and the play of the different forms of energy and their transformations.
The genetic information is then enclosed and stored in the sequence of nucleic acid bases, which constitute the letters of the genetic alphabet.
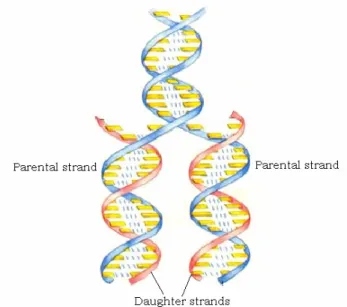
This alphabet is constituted by four bases: A (adenine), G (guanine), C (cytosine) and T (thymine) that are present in DNA. In RNA T is replaced by U (uracyl). A specific interaction is provided by hydrogen bonds which lead to formation of inter- or intra-strands base pairs of the type A···T and G···C. A second important property is arisen: complementarity. This property makes the genetic message able to be read, conserved through replication and transcription. The replication process of DNA is schematically depicted in Fig. 1.2.
Fig. 1.2. The replication process.
It should be noted that replication of a nucleic acid does not occur in an exact way since the complementary interaction is disturbed by external agents of physical nature (thermal agitation, photophysical effects) or chemical nature (chemical fluctuation, molecules able to interact with a given site of the nucleic acid). These external actions give rise to defective copies or mutants.
The auto-reproduction and the mutagen activity are two inseparable features of nucleic acids which have been able to produce messages rich of significance. Actually, both of them are the cause of the genetic evolution and, in the end, of the variety and wonderfulness of the forms of life present on the heart.
On the other hand, mutations can have externally deleterious effects on the leaving organisms. Many forms of cancer and viral infections are though to be caused by mutations induced by external attacks on DNA. Actually, the binding of small organic and inorganic molecules to DNA can interfere with the numerous processes in which DNA participates, as transcription (gene expression and protein synthesis) and replication.
There are several kinds of interactions associated with ligands that bind DNA (Mountzouris and Hurely, 1996; Wilson, 1996). Molecules able to covalently react with nucleic acids, as alkylating agents, heavy metal ions and metal complexes, can attack nucleic acids causing various kinds of damage (cleavage of the phosphodiesther bond and/or alkylation of the nucleobases). They can, in the end, lead to the failure of the correct conservation and transmission of the genetic inheritance (DNA) and the correct synthesis of proteins (RNA).
Effects of similar importance could be caused also by small molecules able to bind non-covalently to the nucleobases. Three important modes of binding have been observed where non-covalent (weak) interactions are operative: external binding, intercalation and groove binding. Agents undergoing such kinds of interaction with nucleic acids are called with different names derived from their properties or functions, these being denoted as ligands, dyes, drugs, probes, intercalators, groove binders etc.
Intercalation is the most common binding mode and corresponds to the insertion of a molecule in the cavities between the base pairs at the core of the helix. This mode of interaction is typically observed for molecules having planar aromatic rings and, owing to its ability to alter the informational content, is considered as the first step in mutagenesis or carcinogenesis. The cytotoxic properties observed for DNA intercalators have led to the extensive use of these compounds as chemotherapeutic agents in the treatment of numerous diseases including cancer (Waring, 1981). Groove binding requires molecules with higher flexibility compared to intercalators, since the molecule should be able to follow the groove of the polymer and twist around the central axis of the helix (Geierstanger and Wemmer, 1995). Binding through the major groove is preferred by large binders as proteins. If the dye is positively charged, electrostatic forces do contribute to stabilize the polynucleotide-dye complexes independently of the binding mode. Recent studies have underlined that several dyes form well-defined helical aggregates located in stacks outside of the nucleic acid backbone, using DNA as a template (Seifert et al., 1999; Ogul’chansky et al., 2001; Garoff et al., 2002; Losytskyy et al., 2002; Schaberle et al., 2003) or single stranded RNA (Biver, Ciatto et al., 2006). In this external binding mode, contrary to intercalation and groove binding where base-dye interactions are of the first importance, the most important role is played by dye-dye stacking interactions.
In most cases, the binding of a ligand induces a change in the structure of the nucleic acid resulting in an alteration of the thermodynamic stability, which is
revealed, by changes of the functional properties of the polynucleotide. The features of the polynucleotide-dye interactions largely depend on the composition, sequence and nature of nucleobases. For instance, greater polarity of G-C base pairs respect to A-T base pairs also was found to play an important role in preferential binding (Müller and Crothers, 1975). However, the primary structure of the macromolecule as the nucleotide sequence is not the only parameter affecting the binding; actually, the secondary structure of the polynucleotide plays a principal role too. This is proved by the finding that several molecules, for instance daunomycin (Müller and Crothers, 1968), are able to discriminate between DNA and RNA, binding only to the former polynucleotide. At the same time, chemical modifications of the intercalating drug (polarity, charge, side chain and shape) can induce sequence selectivity (Krugh et al., 1980; Erkilla et al., 1999; Graves and Velea, 2000).
Over the past decades, there has been progress on the design of small molecules recognising binding sequences specifically in the minor groove of duplex DNA (Dervan, 2001). Understanding the features that contribute to recognition of DNA is crucial for the development of drugs, such as antivirus or antitumor agents (Hurely, 2002). A number of laboratories have also worked towards the development of small molecules that specifically bind and stabilize triplex (Rogers and Glazer, 2004) and G-quadruplex structures (Fransceschin et al., 2006) as possible tools for antigene and antisense strategies (Winters et al., 2003; Crooke, 2004) and as potential anticancer agents.
1.1 Binding of drugs to Polynucleotides
Early studies of the interaction between dyes and DNA or RNA, revealed the existence of two distinct kinds of binding processes (Waring, 1965; LePecq and Paoletti, 1967) designated by Blake and Peacocke as binding processes I and II (Blake and Peacocke, 1968). Binding I corresponds to intercalation, whereas binding II corresponds to external binding.
1.1.1 Binding process I: Intercalation
Addition of proflavine or ethidium to a nucleic acid results in a red shift of the adsorption band of the dye. This effect reveals dye-base non-covalent interactions and was denoted as “binding process I”. The process is characterized by a binding stoichiometry of more than one base pair per 1 dye molecule for simple drugs (Waring, 1965), but it can increase to 6 for more hindering ligands (Müller and
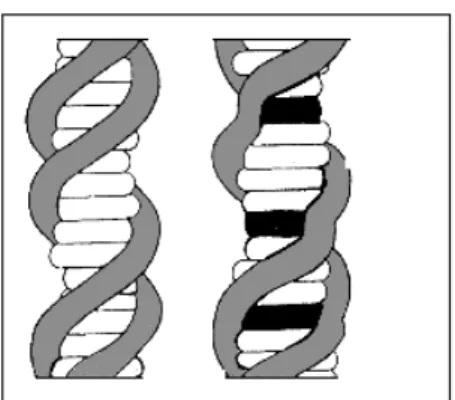
Crothers, 1968). These results agree with the intercalative mode of binding described by Lerman (1961) and have given rise to the concept of nearest neighbouring site exclusion. According to this concept the intercalated molecule makes one or more sites adjacent to the occupied one non reactive so that, under conditions of complete saturation, only a fraction of the cavities of the nucleic acid will be occupied. Such a behaviour predicts not only the reduction of the total number of the available sites but also an upward deviation from Scatchard plot that occurs corresponding to high drug saturation connected with site rearrangements (see Appendix III). The intercalation reaction involves, as the key process, the penetration of the aromatic portion of a drug molecule between base pairs (Fig. 1.3), vertical stacking interactions between the bases and the intercalating molecule being a major stabilizing feature of the formed complexes. Intercalation causes an increase of the distance between adjacent base pairs. The resulting helix distortion is compensated by adjustments in the sugar-phosphate backbone and by partial unwinding of the duplex.
In fact, when first Lerman proposed the intercalation model (Lerman, 1961), he observed that addition of dyes (proflavine or acridine orange) to DNA, resulted in a marked change in the viscosity and sedimentation coefficient of the polymer. These observations led him to postulate that dyes of the acridine family induce alterations in the nucleotide secondary structure through an intercalative type of binding which was later confirmed (Waring, 1968; Sobell et al., 1977; Lippard, 1978). Such a binding would result in a lengthening of the duplex structure. These studies were the first in citing the linkage between perturbations of the DNA and frameshift mutagenesis.
Fig. 1.3. Intercalation model as proposed by Lerman representing the secondary structure
of ordinary DNA (left) and DNA containing intercalated molecules (right) (the occupied sites are in black).
The viscometric method is generally considered as the more convenient to probe intercalation. Neverthless, many other techniques can provide interesting information on DNA structure alterations due to intercalation.
Autoradiographic studies by Cairns on the proflavine/DNA system (Cairns, 1962) had showed what confirmed only later by studies on viscosity, sedimentation and electric dicroism (Cohen and Eisenberg, 1969; Hogan et al., 1979). The x-ray diffraction and high resolution NMR methods of DNA/drugs complexes confirmed indeed a length increase in the separation of base pairs by 3.4Å. In contrast, electric dichroism measurements suggest a range of possible helix length increase (2.0 ÷ 3.7Å) per intercalated drug, depending on the drug tested. Some general information about the relative orientation dye/DNA axis is available from flow and electric discroism suggesting that the drugs are not exactly perpendicular but slightly tilted (Lerman, 1961; Hogan et al., 1979). More specific information about base-dye overlap and intercalation site is available from NMR experiments on mixture of drugs with synthetic oligo or polynucleotides.
Although the knowledge of the geometry of the complexes between intercalating agents and DNA is of great importance, the thermodynamic properties provide quantitative information on the driving force of the interaction. The affinity of this mode of binding, obtained from binding equilibria, kinetics and calorimetry, is relatively high (∆G°= -6 ÷ -9 kcal mol-1) while enthalpy contribution is negative ( -5 ÷ -10 kcal mol-1 range for mono positively charged dyes) (Li and Crothers, 1969; Quadrifoglio and Crescenzi, 1974; Bresloff and Crothers, 1975). Rarely, usually when uncharged drugs are involved, positive enthalpy variations of small entity (0 ÷ 2 kcal mol-1) can be encountered (Wakelin and Waring, 1974).
Intercalation usually leads to an increase in Tm (midpoint of the thermal transition profile), not only for polynucleotides (Waring, 1974; Patel, 1979; Patel, 1980) but also for synthetic oligonucleotides (Patel and Canuel, 1976; Patel, 1980) and dinucleotides (Patel and Shen, 1978; Young, 1980) as a consequence of duplex structure stabilization. Both the nature of the dye and the nucleic acid sequence determine together the extent of duplex stabilization. For example, ethidium bromide (EB) stabilizes poly(dA·dT) more than proflavine (Patel, 1979; Patel, 1980) whereas the system EB/poly(A)·poly(U) is more stabilized than EB/poly(I)·poly(C) (Waring, 1974). Nucleic acid stabilization is manifested at the intercalation site by a coupling of the drug and its adjacent base pairs (Robinson et al., 1980) such that the relative internal motions of these base pairs are slowed (Hogan and Jardetzky, 1980).
The principal biological effects of the intercalating drugs (Gale et al., 1972; Neidle, 1979; Schwartz, 1979; Graves and Velea, 2000; Haq, 2002) are inhibition of cell growth, cell death and cell transformation. Since these effects are noted to inhibit rapidly proliferating cells (Schwartz, 1979), many intercalating drugs find uses as antibacterical, antiparasitic and antitumor agents. Nevertheless, binding of intercalators to DNA can interfere with numerous processes and cause (Gale et al., 1972; Neidle, 1979):
1) inhibition of DNA dependent enzymes that hinders the replication, transcription and reparation of the polynucleotide. In most cases these enzymes are inhibited by competition for DNA sites with the intercalating drug;
2) frameshift mutagenesis that disturb the reading order of genetic code. It is the deletion or the addition of base pairs to DNA, resulting in a shift of the codon-reading frame and usually loss of the active product;
3) damage to DNA. Several intercalating drugs have been shown to produce single strand breaks in the DNA.
The ability of intercalating dyes to unwind closed circular DNA is based on sedimentation (Bauer and Vinograd, 1968), viscosity (Watson and Bauer, 1977), electron microscopy (Liu and Wang, 1975) and gel electrophoretic measurements (Keller, 1975). The “unwinding angle” of different potential intercalators was determined and it is defined as the degree of the unwinding of the DNA duplex as a given drug molecule is bound.
1.1.2 Binding process II: External binding
The second mode of binding, found for nucleic acid-drug complexes, is weaker than the first one (∆G < 5 kcal mol-1). This is related to non intercalative modes of interaction leading to external complexes whose driving force of formation was thought to be primarily electrostatic (LePecq and Paoletti, 1967) and therefore nowadays called external binding. That the nature of external binding should not be purely electrostatic was soon recognized by the fact that this binding mode as well is associated to changes of the absorption spectra. In contrast, the formation of electrostatic ion-pairs does not alter the spectral features of the free ligands, due to the identical values of the respective extinction coefficients (Prue, 1966). Moreover, the affinities of binding mode II, although smaller than those of binding mode I, are definitely higher than those of electrostatic binding (∆G < 3 kcal mol-1) (Diebler et al., 1987).
Information about binding mode II was obtained by studies at low dye/polynucleotide ratio of proflavine binding to different polymers (Li and Crothers, 1969; Schmechel and Crothers, 1971) indicating that the enthalpy changes vary from less than -1.0 kcal mol-1 until -10 kcal mol-1, whereas the entropy term was found to display positive or negative values. These findings show that this mode of binding is very sensitive to the chemical and conformational characteristics of the polynucleotide, thus confirming that other forces, in addition to the electrostatic attraction, are at work in binding process II. Equilibrium and kinetic studies on the binding of proflavine to single stranded poly(A) revealed that the dye can bind externally to the polynucleotide backbone forming long stacks around the polymer chain stabilized by dye-dye interactions. This kind of binding was also observed to occur in DNA/intercalator systems after that the DNA cavities have been occupied by the intercalator (Waring, 1965; LePecq and Paoletti, 1967; Blake and Peacocke, 1968).
In such a case, the negative phosphate residues along the polymer backbone act as a template for external binding of the dye (Bradley and Wolf, 1959; Ogul’chansky et al., 2001). Despite being weaker than intercalation, this process has shown to play also a very important role in the biological implications of the drug-polynucleotide interaction (Li and Crothers, 1969; Bengtsson et al., 2003) and different drugs have interestingly shown different behaviours related to their stereochemical properties and shapes (Erkilla et al., 1999; Greguric et al., 2002; Nguyen et al., 2004).
It should be noted that external binding requires flat aromatic molecules able to give rise to extended dye-dye non covalent interactions. Since intercalators fulfil this requirement, it turn out that intercalation and external binding could be both observed with the same dye. Under these circumstances the prevalence of one binding mode over the other will depend on the relative concentrations of the reaction partners, in other words on the CP/CD ratio.
At high values of this ratio only few dye molecules are bound to the polymer backbone and they are located apart from each other, so dye-dye interaction is hampered but not base-dye interaction. Under these conditions binding mode I will dominate. The reverse is true for low values of CP/CD since, under these circumstances, the dye molecules initially electrostatically bound to phosphates and present in excess, tend to stack together externally to the polymer backbone. This kind of binding is revealed by a blue shift of the dye spectrum. A clear example of how binding mode I and II can be identified and separately
characterized in their thermodynamic (Dourlent and Helène, 1971) and kinetic (Biver, Ciatto et al., 2006) aspects is provided by the poly(A)/proflavine system.
1.1.3 Electrostatic binding
Most of the ligands that undergo interactions with nucleic acids are positively charged. Hence, electrostatic interaction with the phosphate groups on the polymer backbone is unavoidable. However, the strength of this interaction could not be measured separately from the other kinds of binding. Nevertheless, simple calculations based on the electrostatic theory of electrolytes and experiments on metal-ion binding to polynucleotides strongly suggest that the electrostatic binding should be weak. In effect, it has been found that Mg2+ and Ni2+, that bind outer-sphere to the phosphate residues of poly(U), exhibit a binding constant of 50 M-1 (Diebler et al., 1987) at 25°C corresponding to ∆G°=-2.4 kcal mol-1. Note that, since coulombic attraction is highly non specific, the electrostatic binding should be independent on the chemical and conformational characteristics of polynucleotides. If the electrostatic effect could not be measured separately from other polynucleotide-ligand interactions, its presence is revealed by the sensitivity of the overall binding process to salt effects.
The binding of a positively charged drug to DNA produces a release of bound counter-ions into the bulk solution (Record et al., 1978). This release results in a favourable force, which drives the complex formation reaction towards completion. The attraction between the ligand and DNA decreases with increasing the ionic strength and thus the value of the overall binding constant (K) typically decreases. It can be demonstrated (see Appendix IV) that the ratio dlogK/dlog[Na+] is a constant (m), obtained as the slope of a double logarithmic plot of binding constant versus monovalent salt concentration. The m value is equal to the product m’ψ where m’ is the number of ion pairs formed by the ligand with DNA and ψ is the fraction of counter-ions associated with each phosphate in the polynucleotide backbone. ψ depends on the interphosphate distance and normally is 0.88 for double stranded DNA (Record et al., 1978). From the K values, determined at different salt concentrations, is therefore possible to obtain direct information about the ligand charge. Binding constants at different ionic strengths have been determined for the systems investigated in this thesis.
1.1.4 Groove binding
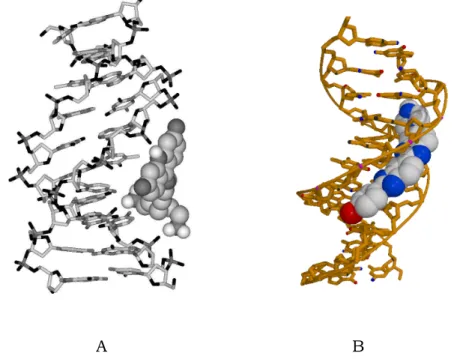
The ligands able to interact with high affinity with the grooves of DNA have been intensively investigated in recent years. The prevailing form of double helical DNA, the B form, consists of two complementary, antiparallel polydeoxyribonucleotide strands associated and stabilized by specific hydrogen bonds between complementary nucleotide bases and by base-base vertical stacking interactions. In the helical grooves, defined by sugar phosphate backbone of paired double helix, the edges of the heterocyclic bases are exposed. The structure of the B–DNA double helix is characterised by a shallow, wide major groove and a deep, narrow minor groove (Saenger, 1984).
Fig. 1.4. Schematic drawing of a (A) major and (B) minor groove DNA complexes.
The chemical features of the molecular surfaces presented by a given DNA sequence in either groove are distinct, forming the basis of molecular recognitions by small molecules and proteins. While the majority of proteins bind to the major groove (Gao et al., 1992; Kamitori and Takusagawa, 1992; Guan et al., 1993; Hannon, 2007), smaller non-covalent DNA binding drugs are thought to act preferentially via the minor groove. The different groups of minor groove binding dyes, commonly classified as groove binders, include natural products such as antibiotics and antiviral agents neotripsin and distamycin, as well as a large number of synthetic molecules, among which DAPI, berenil and Hoechst 33258 are the most known. Small molecules recognizing the minor groove of B-DNA are developed as agents for modulation of gene expression in chemical genetics
(Zimmer and Wähnert, 1986; Gottesfeld et al., 2000; Reddy et al., 2001). Most minor groove binders posses several structural features as a positive charge, curvature and flexibility of the molecule, and hydrogen bond donor and acceptor groups that make these molecules specially suitable to interact with complementary groups on the floor of the groove. Extensive biological data have demonstrated that these molecules bind in preference double stranded compared to single stranded polynucleotides (Cosa et al., 2001), have a DNA sequence selectivity for A/T–rich segments (Neidle, 1997) and, upon binding, exert smaller perturbations on DNA structure than intercalators. Since the groove binding molecules are as a rule elongated and ribbon like, one ligand positioned along DNA helix usually occupies at least 3-5 base pairs (Kumar et al., 1993). Hence, the AT specificity requires a sequence of at least 3-5 AT base pairs. Interactions between minor groove drugs and DNA generally involve hydrogen bonds linking the functional groups of the two components. These can be direct bonds or bonds mediated by water molecules or counter ions. Van der Waals contacts also play an important role. The pronounced AT selectivity is due, at least in part, to complementarity between the inherently narrow groove produced by these sequences and the narrow cross section of the drug molecules (Neidle, 1997) and seems to be related to the need to maximise Van der Waals contacts (Kennard, 1993). The extreme mutual adaptation of both the DNA and the dye on binding is such to maximise the various interactions, which stabilise the resulting complex, interactions ranging from highly directional hydrogen bonds to non specific Van der Waal forces (Kennard, 1993). Electrostatic interactions between a positively charged ligand and the DNA phosphate backbone is another important factor. The stability of the groove complexes decreases noticeably increasing the ionic strength of the solution in agreement with a greater exposure to external agents respect to intercalators. Moreover, linear dichroism measurements show that the grove binding molecules bind with an angle of around 45° compared to the B-DNA axis (Kubista et al., 1987) while intercalators bind with an angle close to 90° (Carlsson et al., 1994). Binding constant values for intercalators that bind to DNA by hydrophobic, Van der Waals and electrostatic forces usually do not exceeded 107 M-1. Complexes of groove binding molecules like netropsin and distamycin A, stabilized by hydrogen bonds may have K values as large as 108-109 M-1 (Kolesnikova et al., 1998).
The features of the groove binding process clearly show that this kind of interaction is completely different from both the external binding and the
electrostatic binding described above. Although electrostatic interactions are present in groove binding (as in all kinds of polynucleotide-ligand interactions), on the other hand, the dye-dye stacking interactions, characteristic of the external binding are absent here. The forces that stabilize the groove-bound complex are hydrogen bonds, dye-base weak interaction, in addition to electrostatic attraction. The concurrent action of those forces can make groove-bound complexes more stable than intercalates, as demonstrated by the binding affinities (∆G=-(8÷10) kcal mol-1) which are higher compared to intercalation (∆G = -(6÷8) kcal mol-1) (Chaires, 2006).
1.2 Studies on DNA/drugs interactions
1.2.1 The x-ray technique and theoretical calculations
The power of the x-ray technique lies in its ability to define the stereochemistry of crystallised molecules, providing detailed information on the structural aspects of binding that are unavailable by other methods. The x-ray studies give a wealth of information about the conformational variability and flexibility, the influence of base sequence and the role of water and counter-ions not only of DNA but also of drug/DNA complexes. These studies would lead to rules governing sequence specific binding and to the correlation between DNA structure, sequence and activity.
The analysis of the x-ray structure of DNA/drug complexes made a relatively slow start but progress has been particularly rapid and currently a lot of complexes have been characterised. Comparative studies, where the same drug is bound to different oligonucleotide sequences or the same sequence is bound to a series of different drugs containing designed modifications, let us to understand the principle of sequence recognition and the mutual influence of the two components of complexation.
The first diffraction studies were made on fibres of the DNA/proflavine complex (Lerman, 1961) but scarce information about structure details were obtained, owing to the intrinsic disorder of the fibres. Later, single crystals were obtained that provided a more detailed structure of the molecular system under study. Since the difficulty in obtaining single crystals increases as the length of the polynucleotide is increased, data on the insertion of intercalating molecules into the polynucleotide double helix are generally extrapolated from studies based on more simple systems. At present, a multitude of dye/oligonucleotide complexes has been synthesized, crystallised and their structures have been resolved via the
diffraction technique with a 1.1 Å resolution (Neidle, 1997; Adams et al., 2002). To help the extrapolation to longer sequences several molecular models have been built in an attempt to rationalize the many experimentally observed features of interaction. Calculations are done with the objective of determining the theoretical basis for the various effects associated with the different dye/polymer interactions. Sequence specificity has been studied by both quantum mechanical and potential energy techniques. Because of the obvious complexity and consequently large amount of computational time involved, theoretical studies also have to be limited to short oligonucleotides models. Even if measurements and theoretical calculations provide a better understanding of the structural questions, the combination of these techniques with solution methods is required for a complete characterization of the system under investigation.
1.2.2 Solution techniques
If several methods are commonly used to provide insight into the binding modes of small molecules to DNA (Suh and Chaires, 1995), most of our ideas about the way in which dyes can interact with different nucleic acids arise from solution studies. Solution techniques are important to determine the extent of such a binding, but they also provide a tool for comparing differences caused by the use of various dyes, DNA sequences or solution environments. Comparing the results, it is possible to determine the types of forces that are involved in the interactions and put forward some suggestions for modifying the drug molecules in order to make them more specific for certain base sequences. Though solution techniques have a limited ability to discern the conformational features of the reaction site, they are however important to confirm some of the general characteristics learned from crystal structures and to extend their range to other drugs and sequences. Optical methods as absorbance and fluorescence spectroscopy, hydrodynamic experiments, linear (LD) and circular (CD) dichroism analysis and atomic force microscopy provide indirect evidence to support or refute suspected binding modes. On the other hand, NMR experiments do provide specific information about base-drug overlaps since both intercalation and groove binding can cause changes in the chemical shift of atoms involved or located near to the binding sites.
Absorbance and fluorescence. Upon the interaction of a ligand with a nucleic acid, absorbance and fluorescence spectra of the former are altered. The most common optical changes displayed are shifts of the absorption spectra to longer
wavelength (bathochromic shift) and decrease of the molar extinction coefficient (hypochromic effect) of the UV-visible spectrum of the dye/polynucleotide complex. On the other hand, fluorescence enhancement or quenching is the basis of the wide use of such dyes as specific stain or contrasting agents in optical microscopy. Analysis of the concentration dependent spectral effects can be used to monitor quantitatively the binding process. The use of different polynucleotides, with appropriate base sequences, can underline possible site or sequence specific binding and highlight the correlation between DNA structure, sequence and activity.
Circular dichroism. The two dichroism techniques, circular and linear, can be useful to obtain some general information about drug orientation relative to the DNA helical axis.
Circular dichroism is a spectroscopic method based on the fact that certain molecules interact differently with right and left circularly polarized light. To discriminate between the two forms of light, a molecule must be chiral, which includes the vast majority of biological molecules. This technique, able to discern differences between enantiomers, is highly sensitive to the three-dimensional features of the molecules, that is, to conformation (Woody, 1995). For example, DNA molecules in the A, B or Z conformation have distinguishable CD spectra that are sensitive to DNA sequence as well (Calladine and Drew, 1992). Many ligands posses achiral chromophores and thus do not exhibit any CD in solution. However, when bound to chiral nucleic acid helix structure, they acquire an induced CD (Lyng et al., 1991 and 1992) that is characteristic of their interaction with DNA (Fasman, 1996). These changes in CD can be used to determine equilibrium constants and to evidence conformational changes. Thus, CD provides information about the secondary structure of biopolymers, proteins and nucleic acids and about the binding of ligands to these type of macromolecules (Woody, 1995). The interpretation of the sign and magnitude of the induced signal is complicated since these quantities depend on the binding mode, polynucleotide sequence and orientation of the transition dipole of the ligand. Compared to intercalation between the pair bases, the minor groove binding to DNA provides a more chiral environment, affording a larger induced CD. This is most likely because a groove binder stays in contact with a larger portion of the helix and twists to follow the groove conformation. By contrast, an intercalator is in contact with only two base pairs and exhibits little or no twist (Armitage, 2005). However, more information can be extracted, for instance the electronic coupling between
multiple ligands that usually causes a splitting of the CD signals into positive and negative components (Nakanishi et al., 1994; Rodger and Nordén, 1997).
Linear dichroism. Linear dichroism (LD) can be very useful for determining DNA binding modes (Norden et al., 1992). It is defined as the difference in the adsorption of light polarized parallel and perpendicular to an orientation axis. DNA of sufficient length can become oriented in a flow cell and any specifically bound ligands will also be oriented and therefore can generate LD. Since LD signals are directly related to the orientation of the ligand’s transition dipole moment respect to the direction of the DNA helix axis, they provide information about binding geometry (Norden et al., 1992). Intercalators and groove binders give LD of opposite sing: the former, approximately perpendicular to the helix axis, have a negative LD, while the latter, closer to parallel, show positive values for the LD signal.
Viscosity and sedimentation. Hydrodinamic methods, enabling the determination of parameters that depend on the molecular dimensions as sedimentation and diffusion coefficients and viscosity of solutions, can provide information about structural variations occurring when a DNA/dye complex forms. These investigations rely on the fact that polymer lengthens to create a binding site for an intercalator, whereas no such a perturbation is required to accommodate a groove binder. This lengthening can be observed both directly, using atomic force spectroscopy (AFM) (Coury et al., 1996), and indirectly, since it produces a viscosity increase and a decrease of the sedimentation coefficient due to a decrease of the mass/length ratio of the polymer (Kernsten et al., 1966; Cohen and Eisemberg, 1969). In his pioneering studies, Lerman first used the last two observation (viscosity increase and sedimentation coefficient decrease) to support an intercalative mode of binding (Lerman, 1961; Lerman, 1964). All the above techniques are used together with cristallografy to characterize in details the binding event.
1.2.3 Binding titrations and site size determination
The titration technique has found wide application in polynucleotide/dye binding studies. Essentially, a drug solution of fixed concentration is transferred to a thermostated cuvette and the progressive absorbance or fluorescence changes are recorded after addition of serial aliquots of the poly- or oligonucleotide solution. Optical changes are normally analysed only for the dye component in terms of free dye [D] and bound dye [PD] concentrations (Peacocke and Skerrett, 1956;
Bloomfiled et al., 1974). For each of them the Lambert & Beer law has to be obeyed and the extinction coefficients (ε) or normalized fluorescence intensity must be invariant. Component polymerisation and dye self-aggregation provide non-linear concentration dependencies and they must be either avoided or included in the calculations for correct analysis of the binding. The titration technique is very flexible, being applicable to various systems and under different experimental conditions (pH, ionic strength, temperature). Fluorescence titrations are performed together with, or as an alternative to the absorption ones and usually display a much greater sensitivity.
Many methods are used for the binding analysis, the best treatment employed depending on the host/drug model used. For a simple (two-state) model, data can be fitted on the basis of the Scatchard equation (see Appendix III). Binding analysis requires the determination of the free dye concentration [D] and of the amount of bound drug per nucleotide unit (designed as r) as a function of added titrant. Scatchard model assumes a linear relationship for a r/[D] vs. r plot, that, in effect, is rarely obeyed. A better model, that takes into account occupancy of multiple binding sites, was developed by McGhee and von Hippel (McGhee and von Hippel, 1974) and Crothers (Crothers, 1968) (see Appendices II and III). An extended form of this model introduces a cooperativity factor (ω) to account for mutual interaction between bound drug molecules, with ω>1 and ω<1 corresponding to cooperative and anti-cooperative binding respectively (McGhee and von Hippel, 1974). The problem of cooperativity has been faced also by Schwarz (Schwarz, 1970) who developed a mathematical treatment which has been successfully applied to the external binding (Dourlent and Helène, 1971; Biver, Ciatto et al., 2006). The choice of the analytical model to be used depends on the system under study, but usually the best approach consists in combined iterative fitting procedures (Appendices II and III). These methods enable both the binding constant K and the binding site size n to be obtained.
All polymer/ligand interactions are characterized by the site size, n, and equilibrium-binding constant, K (see Appendices II and III). The site size, n, defined as the number of ripetitive units of the polymer involved in the binding of one dye molecule under saturation conditions, is a very important parameter in polynucleotide/dye interaction studies.
A method suitable to obtain the value of this parameter consists in performing low ionic strength titrations (Zimmermann, 1986; Ciatto et al., 1999). Under these circumstances, the reaction between nucleic acid and dye becomes quantitative,
thus enabling complete saturation of the polynucleotide sites. The straight lines interpolating the initial and final part of the titration curve intersect at a point where the concentration ratio polymer/dye (CP/CD) is equal to n. The Schatchard plot, combined with McGhee and von Hippel analysis also yields the site size. Another way to obtain the dye/nucleotide binding stoichiometry is offered by the method of continuous variations. This is based on the signal changes (absorption or fluorescence) in solution when the concentration ratio CP/CD is varied while the sum (CP + CD) of concentrations is kept constant. A triangle shaped graph is obtained by plotting the signal changes as a function of the added mole fraction of ligand. The point of maximum (or minimum) yields directly the stoichiometry of the reacting system. It was first suggested by Job in 1928 for ordinary chemical reactions (Job, 1928) and later applied to biological reactions by Likussar and Boltz (Likussar and Boltz, 1971) and Cantor and Schimmel (Cantor and Schimmel, 1980). The applicability of the method to polynucleotide-dye binding has been verified (Loontiens et al., 1990) and the results were found to be in agreement with the ones derived from the McGhee and von Hippel analysis and x-ray crystallographic studies (Spink et al., 1994).
1.2.4 Kinetic analysis of drug/polynucleotide binding
Literature shows that most of the studies on interaction between dyes and nucleic acids are aimed to evaluate equilibrium properties and thermal effects. In contrast, kinetic investigations are much more limited, even if the dynamic approach is in principle more powerful than the static one since it simultaneously provides information on kinetics and equilibria. May be the reason stays on the fact that dynamic studies require non-standard methods and often home made apparatuses. It has to be pointed out that the search of correlations between the mechanism by which drug molecules interact with nucleic acids and the biological effects has been a focus of interest for a long time. It is supposed that the therapeutic efficiency of a bound drug is related to a slow rate of dissociation from the nucleic acid (Müller and Chroters, 1968). This hypothesis makes clear that in the study of interaction of DNA and other poly/oligonucleotides with small molecules, the association/dissociation kinetics are of great diagnostic importance.
The dynamic approach is concerned with the details of the reactive process and provides a more effective method to determine the mechanism of reaction. The kinetic analysis can yield forward and backward kinetic constants of every single
step of the reaction mechanism and, from combination of the rate constants, the overall equilibrium parameters can be calculated. Thermodynamics is interested only in the initial and final state of a reacting system, but not in the intermediate states and processes that can be revealed and studied by the kinetic methods. If the rates of chemical reactions vary over many orders of magnitude, classical intercalators (as ethidium bromide or proflavine) generally react in the time scale of 10-3÷10-2 seconds, whereas groove binders bind more rapidly, approaching the diffusional limit (Brensegem et al., 2001). Their rate of reactions can be investigated by means of relaxation methods and, in special cases, by flow methods as well.
By means of the kinetic method, fine details could be revealed for the interactions between polynucleotides and dyes. Results from our laboratory have shown that acridines (Wakelin and Waring, 1980; Ciatto et al., 1999; Biver et al., 2003; Biver, Secco et al., 2005) and cyanines (Biver, De Biasi et al., 2005; Biver, Pulzonetti et al., 2006) interact with DNA according to a sequential multi-step mechanism. By contrast, ethidium bromide binds to double stranded RNA according to the direct transfer model, with an interstrand dye transfer catalysed by the polymer (D’Amico et al., 2002).
It is important to note that the study of a system involving biological macromolecules and small dyes could be more complex compared to kinetic investigations of ordinary reactions, since one has to take into account additional parameters as the polynucleotide saturation degree and the site size (Jovin and Striker, 1977; Macgregor et al., 1985). Moreover, the behaviour of the system can be complicated by the presence of cooperative effects (Pecht and Rigler, 1977; Biver, Ciatto et al., 2006).