1. INTRODUCTION
1.1 RNA interference
In 1998, Andrew Fire and Craig Mello discovered a new way to inhibit gene expression in C. elegans with double stranded RNAs (dsRNAs). In that work, they showed how only a few molecules of an exogenous dsRNA were capable to completely eliminate the expression of a gene with sequence homology to the introduced dsRNA. The phenomenon was called RNA interference (RNAi), and was further shown to regulate an astonishing number of gene silencing processes in many different organisms.
The technique illustrated in the 1998 paper (Fire et al., 1998) has been extensively used in reverse genetics to knock-out genes in all kinds of animal models. Moreover, RNAi has been recently used to develop antiviral therapies. It is noteworthy that for their results Andrew Fire and Craig Mello were awarded the 2007 Nobel Prize.
1.2 Brief hystory of small, non coding RNAs
The theory of a class of RNA regulating gene expression in eukaryotic cells was first developed in the late 60s (Britten and Davidson, 1969), but became old-fashioned with the discovery of the existence of a large number of transcription factors, proteins acting into the nucleus to activate or repress different genetic programs. Nevertheless, small RNAs, distinguished in small interfering RNA (siRNA) and microRNA (miRNA), do actually repress gene expression by pairing mRNAs in a Watson-Crick manner.
Long double stranded RNAs can be present in cells as the product of enzymes like RNA dependent RNA polymerase, which take RNAs as templates for transcription. Moreover, transposones and retroviral genomes can have a double stranded RNA form. Finally, naturally occurring long dsRNA could rise from bidirectional transcription and/or hybridization of overlapping transcripts. Cellular processes transform these molecular species in siRNAs capable to suppress the expression of the source nucleic
acid. This mechanism has probably evolved as a strategy to protect the genome from exogenous threats like viral infections (Baulcombe, 2002).
The second important class of small dsRNAs is constituted by microRNAs. In 1993, a group led by Victor Ambros discovered the first miRNA in C. elegans. It was shown that two tiny RNAs derived from the lin-4 (lineage abnormal-4) locus, were capable of regulating the developmental timing of worms, negatively regulating the expression of the gene lin-14 (Lee et al., 1993; Wightman et al., 1993). The mechanism proposed is a milestone in molecular biology: the 22 nucleotides (nt) long lin-4 RNA, though imperfectly, acts forming base pairs with multiple regulatory sequences in the 3’ untraslated region (3’ UTR) of the lin-14 mRNA, thereby repressing the translation of the mRNA itself. Finally, the 61 nt long lin-4 RNA, containing an hairpin structure, was described as the precursor of the smaller one (Lee et al., 1993; Wightman et al., 1993).
For seven years, the case of lin-4 was considered an exception more than a paradigm. The discovery of a second RNA regulating a later developmental transition in C. elegans, termed let-7 (lethal-7; Reinhart et al., 2000; Slack et al. 2000), and the appearance of its homologs in fly, humans and other animals led researchers to name lin-4 and let-7 as small temporal RNA (stRNA) (Pasquinelli et al., 2000). Only one year later, about 20 new genes in Drosophila, 30 in humans and 60 in worms were shown to produce small RNA similar to stRNAs: the only difference was that these molecules were specifically expressed in different cell types more than at different developmental stages.
Few years later, lin-4 became renowned as the eponym of the large class of endogenously expressed genetic regulators named microRNAs (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). Thus, microRNAs are small non-coding RNA that negatively regulate gene expression.
1.3 MicroRNA biogenesis
MicroRNAs derive from a genomic region distant from previously discovered genes, implying the existence of dedicated and independent transcription units (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). Nevertheless, a non-negligible part of miRNA genes are located into introns of pre-mRNAs.
Even transcriptional orientation seems to be maintained, suggesting that these genes, named for this reason miRTRONs, exploit the whole expression machinery of the genes in which they are located (Aravin et al., 2003; Lagos-Quintana et al., 2003; Lai et al., 2003; Lim et al., 2003). With this strategy cells can co-express proteins and miRNAs using a single transcriptional unit. In addition, and probably with the same purpose, a sizable part of miRNAs is clustered in the genome. These transcriptional units probably act as multi-cistronic primary transcripts (Lagos-Quintana et al., 2001; Lau et al., 2001). However, the majority of miRNA genes, in particular in worms and humans, are isolated and not clustered (Lim et al., 2003a, 2003b).
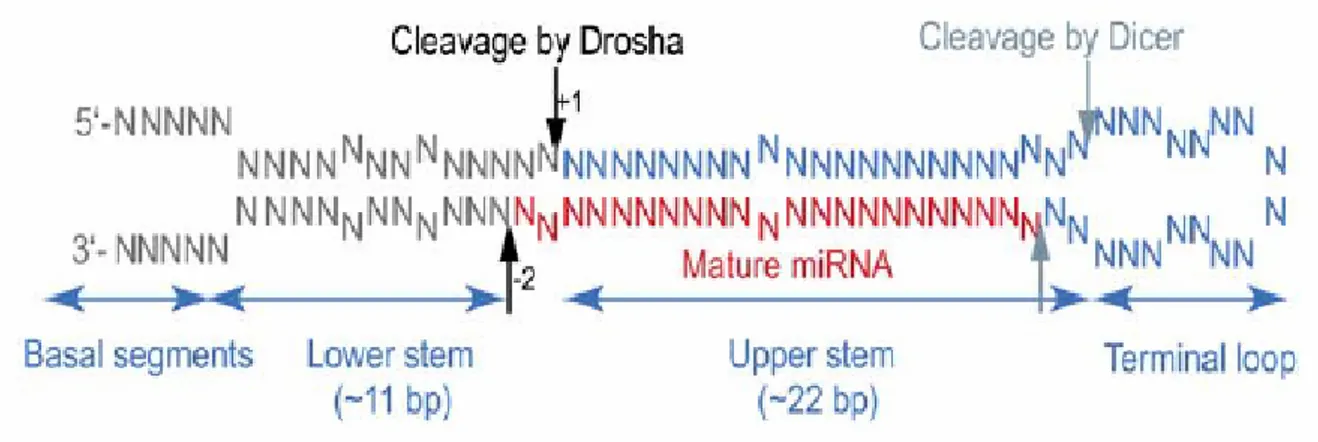
MicroRNAs are transcribed by RNA polymerase II (fig. 3) as long primary transcripts named primary-microRNAs (Cai et al., 2004; Kim, 2004; Lee et al., 2002, 2004). These precursors (pri-miRNAs) contain a 5’ cap and a 3’ poly-A tail; in addition, they undergo an intramolecular pairing interaction that originates a secondary structure containing a double stranded (ds) RNA stem of at least 33 base pairs, a terminal single stranded (ss) RNA loop that links the two helices at one side of the stem and two non-interacting segments of ssRNA to the other side of the molecule (fig.1).
Figure 1. The average face of pri-miRNA.
A pri-miRNA can be divided into four parts: a terminal loop, the upper stem, the lower stem, and basal segments. Predicted sites for Drosha cleavage and Dicer cleavage are indicated with black arrows and gray arrows, respectively.
1.3.1 Drosha
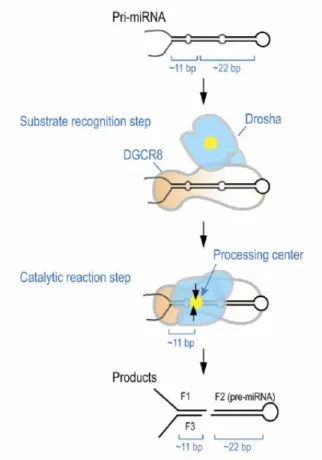
Pri-miRNAs are processed into the nucleus by the so called “microprocessor complex” (fig. 3), formed by the RNAse III endonuclease Drosha (Lee et al., 2003) and by its cofactor DGCR-8 (DiGeorge syndrome critical region-8). Each of these two proteins is individually inactive in cropping the pri-miRNA, so the formation of the complex is necessary their activity, as they both play different but crucial roles. DGCR-8 (Gregory et al., 2004; Han et al., 2004; Landthaler et al., 2004), known as Pasha in Drosophila and C. elegans (Denli et al., 2004), acts by harbouring the precursor at the two ssRNA-dsRNA junctions (SD junctions). At this stage of processing, Drosha does not interact with RNA (fig. 2).
Figure 2. An ‘‘ssRNA-dsRNA junction anchoring’’ model for the processing of pri-miRNA.
DGCR8 may play a major role in substrate recognition by directly anchoring at the ssRNA-dsRNA junction. DGCR8 also interacts with the stem of about 33 bp and the terminal loop for a full activity although the terminal loop structure is not critical for DGCR8 binding and cleavage reaction. After the initial recognition step, Drosha may transiently interact with the substrate for catalysis. The processing center (yellow circle) of Drosha is placed at about 11 bp from the basal segments. (Han et al., 2006).
Drosha homologs belong to the RNAse III endonuclease family. Either class II and III of these enzymes have two RNAse III domains (RIIID) and a double stranded RNA binding domain (dsRBD). Class I proteins possess a RIIID and a dsRBD and are typically found in bacteria and yeast. Drosha endonucleases are grouped in class II, while Dicer enzymes form the third class (discussed later).
RNAse III proteins share the characteristic trait of generating two-nucleotides-long overhangs at the 3’ end of the processed dsRNA (Kim, 2005; Tomari and Zamore, 2005). For this task, two RNAse III domains of the protein act together as a single processing center, forming an intramolecular dimer in class II and III proteins: the C-terminal RIIID (RIIIDb), near the dsRBD, cleaves the 5’-strand of the miRNA, while the other (RIIIDa) acts upon the 3’-end of the resulting molecule (Han et al., 2004; Blaszczyk et al., 2001; Zhang et al., 2004).
After the interaction of DGCR-8 with the pri-miRNA, Drosha is able to bind the molecule with its dsRBD to situate the processing center at about 11 bp from the SD junction. The terminal loop is not necessary for the whole reaction, but the two ssRNA strands on the other side appear to be crucial for it.
The resulting product, named “precursor microRNA“ (pre-miRNA) is made up by a 22 nucleotides long dsRNA stem with a ssRNA terminal loop, for a total of about 65-70 nucleotides (Han et al., 2006). The pre-miRNA is translocated into the cytoplasm by an exportin 5/RanGTP complex (fig. 3), which recognises the precursor by its 3’ overhangs (Bohnsack et al., 2004; Lund et al., 2004; Yi et al., 2003). It is in this cellular compartment that small non coding RNAs exert their action.
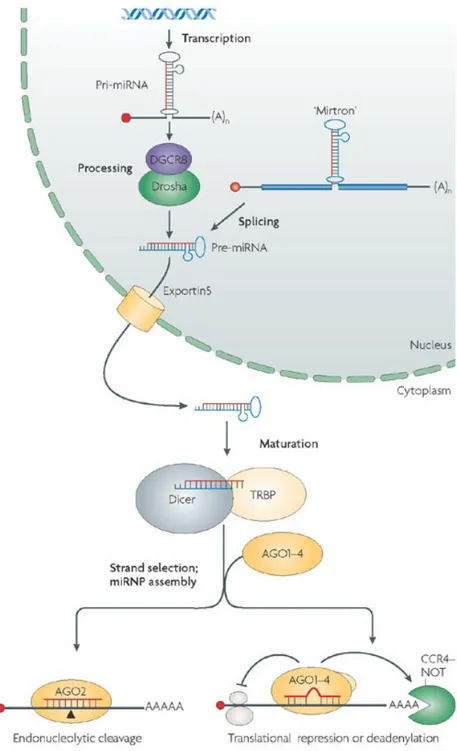
Figure 3. The Biogenesis of miRNAs.
MicroRNAs are transcribed as capped and poly-adenilated transcripts (pri-miRNAs) that are successively processed by Drosha-DGCR-8 complex. Intronic miRNAs (miRTRONs) bypass this cleavage step.
Resulting pre-miRNAs are exported into cytoplasm and cleaved by Dicer. So called mature miRNAs, in turns, are incorporated into RISC particles, where acts in post-transcriptional gene silencing (PTGS).
1.3.2 Dicer
Pre-miRNAs are processed by a second endonuclease called Dicer (fig. 3), a class III RNAse III enzyme. Like Drosha, Dicer acts in concert with a dsRNA binding protein, the so-called tar-binding protein (TRBP) in humans, known in Drosophila with the name of Loquacious (Loqs).
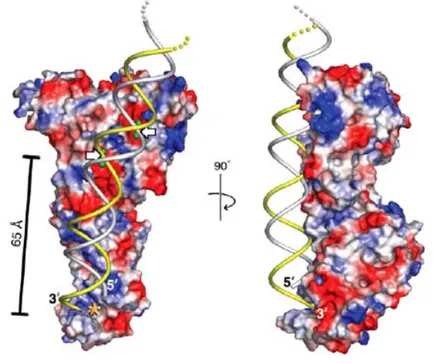
Dicer protein is formed by a PAZ domain, that binds the 2 nucleotides 3’ overhangs, a domain of unknown function (for this reason called DUF 283), two RNAse III domains (RIIIDa and RIIIDb), a dsRBD and an helicase domain, not necessary for RNAi but useful for the stabilization of RNA molecules. The distance between the PAZ domain and the cleavage site of the enzyme (formed by the two RNAse III domains, as previously told for class III RNAse III endonucleases) is 65 Å. This distance is fundamental, as it determines the length of the cleavage product, matching the previously predicted 25 nucleotides long mature miRNA. For such a peculiarity, Dicer has been termed as a molecular ruler that measures and cuts 25 nucleotides from the end of a dsRNA, bound by the PAZ domain (fig. 4; MacRae et al., 2006).
Figure 4: A model for dsRNA processing by Dicer.
Front and side views of a surface representation of a protozoan Dicer with modelled dsRNA. Red and blue symbols represent acidic and basic protein surface charges, respectively. Putative catalytic metal ions are shown as green spheres. White arrows point to scissile phosphates. Asterisk denotes a pocket formed by a PAZ domain, binding the 3’ overhang. (MacRae et al., 2006).
Ultimately, inhibition of gene expression by microRNAs and RNAi by long dsRNA converge in a common pathway: the generation of 22 nt long dsRNAs by the RNAse III Dicer. Thus, the two biological pathways can be considered as a single one. Indeed, there are no molecular differences between processed siRNAs and mature miRNAs.
The function of the whole RNAi pathway is the inhibition of gene expression by operating upon mRNAs molecules. To perform this task, siRNAs or mature miRNAs are incorporated into a multimeric protein complex called RNA induced silencing complex (RISC). Small RNAs in RISC particles acts by directing the complexes to pair to the target mRNAs. In the case of perfect hybridization of RNA with the target, the latter is destroyed; if some mismatch occurs, as in numerous animal miRNAs, a translational silencing takes place.
1.3.3 Slicer
The endonuclease degrading mRNAs within the RNAi pathway was called “Slicer” and remained elusive for some time. Then, after cristallographic studies on bacterial proteins, Slicer was identified with Argonaute-2 (Ago2), a member of Argonaute protein family. Ago2 was shown to be the only member of the family capable of reconstituting an effective RISC activity in the presence of a dsRNA. In facts, the two partners Ago2 and dsRNA, are sufficient to act as a “minimal” RISC complex. The entire Slicer activity resides in the Piwi domain, which contains an RNAse H fold, competent for catalysis. RNAse H proteins usually cleave RNA-DNA hybrids, acting on the central nucleotide of RNA if the DNA segment is short. Ago2 behave like this, but using an RNA instead of a DNA oligonucleotide. The Piwi domain, also, is crucial in binding single stranded siRNA or miRNA by sequestering the first 5’ nucleotide. The PAZ domain is active in Ago2 protein by binding the 3’ overhangs of the small RNA incorporated in the complex.
1.3.4 RISC loading
A critical step in the assembly of an active RISC complex is the conversion of a double stranded small RNA into a single stranded one, competent to incorporation. The two strands of mature miRNA are not equivalent, as only one of them is specifically loaded into RISC. This filament is termed as the “guiding” strand (antisense to the mRNA), while the other is called the “passenger” (sense) strand.
The mechanism specifying the identity of the incorporated strand relies on the thermodynamic properties of the RNA duplex: the 5’ end of the guiding strand is less stable than 5’ end of the passenger strand, either in mature miRNAs or in siRNAs. In such a manner, RISC can control the selection of the proper RNA strand (Khvorova et al., 2003).
Loading of dsRNAs onto RISC could rely on the unwinding properties of an unknown ATP-dependent RNA helicase, acting in separating guiding to passenger strands and thus facilitating the incorporation of the correct one. Many possible proteins in RNAi pathway may have this activity, but none of this have been confirmed to act for the primary role. In the last years, it has been shown how the passenger strand of a siRNA is the first target of Ago2 itself. In facts, Slicer activity of RISC complexes cleaves the sense strand of the 22-25 nt long RNA duplex, releasing predicted 9 and 12 nt fragments carrying 5’-phosphate and 3’-hydroxyl termini (Matranga et al. 2005; Rand et al., 2005), as shown for all RISC cleavage products (Martinez and Tuschl, 2004).
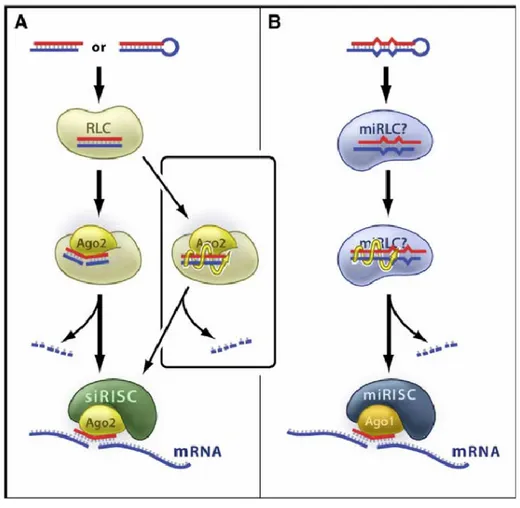
Figure 5. Mechanisms of Guide RNA Loading and Activation within RISC.
(A) In Drosophila, perfectly base-paired siRNAs proceed through the RISC-loading complex (RLC) and engage Ago2. The precise details of the assembly of this complex are unknown. Passenger-strand cleavage by Ago2 precedes and presumably facilitates siRNA strand dissociation (left), leading to the formation of siRNA-programmed RISC (siRISC) that goes on to target the cleavage of specific mRNA. A slower, cleavage-independent “bypass” pathway (right) also generates active siRISC, perhaps by unwinding of the siRNA (yellow spiral arrow). The passenger strands are degraded by cellular nucleases.
(B) In Drosophila, imperfectly base-paired miRNA precursors generally assemble into Ago1-containing miRNA-programmed RISC (miRISC), perhaps through an miRISC-loading complex (miRLC). In this case, the bulges and mismatches in the duplex prevent passenger-strand cleavage, and the active miRISC forms via the cleavage-independent route.
(Preall and Sontheimer, 2005).
The other known human Argonaute proteins Ago1, Ago3 and Ago4, complex with single stranded small RNAs in spite of lacking the endonuclease activity (Liu et al. 2004; Meister et al., 2004). Thus, researchers have proposed another and slower “bypass” mechanism for RISC assembly independent from cleavage.
MiRNAs seems to be loaded in RISC primarily by this system, probably involving proteins competent to unwind the precursor.
Finally, there are a number of biochemical proofs demonstrating an involvement of the RNAse III Dicer in RISC assembly. For instance, an heterotrimeric complex formed by the human proteins hDcr (Dicer), TRBP and hAgo2 can reconstitute entire RNAi activity in the presence of small RNAs (Gregory et al., 2005). Moreover, dsRBD of Dicer directly interacts with Piwi domain of hAgo2, probably performing important functions in passing the small RNA from a protein complex to another. Once the small RNA is loaded into RISC, the multimeric complex can target mRNAs to inhibit their expression.
1.4 Sequence specificity
The hallmark of RNAi is a sequence specific inhibition of target mRNAs due to pairing to a small RNA incorporated into a protein complex. To regulate this phenomenon, cells have developed an essential machinery: only seven of the 22-25 nucleotides of the integrated miRNA or siRNA are really critical for binding to the target mRNA.
In particular, the nucleotides 2 to 9 at the 5’ end are capable to nucleate the interaction with the target. On this basis, this crucial region has been named the “seed sequence”. The adjacent 3’ region subsequently pairs to the transcript, making the binding more or less stable. Once such interaction occurs, the complex translocates into cellular regions called “P-bodies”, sites with high concentration of proteins with destabilizing activity upon mRNAs. As mentioned above, only if most of the small RNA sequence interacts with the target, the latter is cleaved by RISC complex. After this step, the 3’ fragment is destroyed into the cytoplasm by the exonuclease Xnr1, while the 5’ one follows another destiny; a short polyU tail is added to its 3’ end, decapping occurs and the RNA fragment is degraded by several 3’-to-5’ exonucleases (for reviews, see Bartel, 2004; Zamore and Haley, 2005 and references therein).
Animal miRNAs rarely lead to mRNAs cleavage. In facts, frequent mismatches taking place outside the seed region make the interaction with the target less stable; for this reason, the silencing mechanism requires multiple binding sites in 3’ UTR of
mRNAs, and miRNAs acts in a cooperative manner (Doench and Sharp, 2004). The lack of accuracy in the interaction from miRNAs and mRNAs is a fundamental characteristic of gene silencing with small RNAs. Therefore, only one miRNA can control the translation of hundreds of transcripts, and a small fraction of molecules can easily and efficiently regulate the expression of about a third of the whole genome.Instead of mRNA destruction, gene silencing by miRNA activated RISCs consists in the block of cap-dependent initiation of translation of targeted mRNAs. That way, ribosomes are stalled, with an obvious decrease in protein synthesis. The particles are further translocated in P-bodies, where they oligomerize to be stored or slowly interact with the mRNA decay machinery (Chu and Rana, 2006).
However, as mentioned above, there is usually no cleavage of transcripts, albeit some researchers reported a certain level of mRNA instability. This is not an universal rule, as lin-4 and let-7 in C. elegans actually lead to target destruction: all is dictated by the degree of complementar match between small RNA and its target.Different organisms including Drosophila possess specialized proteins for the interaction with siRNAs or miRNAs. For instance, Drosophila has dedicated Dicer and Argonaute proteins for different small RNAs. It is remarkable that mice and humans have only one copy of Dicer in the genome (Dcr-1), probably because the presence of long dsRNA in mammalian cells would trigger defensive pathways based upon interferon.
In addition, these RNAs are uncommon also for the reason that RNA dependent RNA polymerases are not present in vertebrates, unlike in Drosophila. It is probably for this reason that mammals have preferentially retained the microRNA pathway of gene silencing. Nevertheless, Slicer activity is also preserved in mammalian cells, as confirmed by the presence of Ago2 in the human genome.
1.5 Role of RNAi in heterochromatin formation
The term heterochromatin defines silent non coding DNA formed by long arrays of short repeats organized in tandem and having a head-to-tail orientation. These arrays, also known as satellites, are concentrated in centromeric and pericentromeric regions and are often interrupted by retrotransposable elements.
Recently, RNAi machinery has been demonstrated to be crucial in eukaryotic cells in keeping these genomic regions silent: briefly, a nuclear complex would be the equivalent of cytoplasmic RISC. The RNA-induced transcriptional silencing (RITS) complex would use siRNA generated by Dicer from long dsRNA derived in turn from the pairing of products from transcription on either strands of centromeric satellite DNA. Then, RITS complex is thought to recruit chromatin remodelling complexes that have the final effect of silencing sequences homologous to the correlated siRNA and transforming them into inactive heterochromatin (Lippman and Martiensen, 2004 for a review). Defects in heterochromatic DNA result in chromosomal instability and then aneuploidy. For this reason, RNAi plays a fundamental role in maintaining the integrity of the genome. However, this effect seems to be cell type specific, as Dicer depleted embryonic stem cells (ES cells) are viable (Kanellopoulou et al., 2005; Murchison et al., 2005), while the same mutation in chicken DT40 fibroblasts causes a severe growth arrest. (Fukagawa et al., 2004)
1.6 Loss of Function of Dicer in different animal models
The peculiar activity of Dicer in cutting dsRNAs makes this protein as the “central relay station” of the different RNAi pathways. For this reason, researchers have extensively exploited this kind of bottleneck to understand the function of small RNAs in complex organisms. Moreover, the availability of a precise and detailed structure of this RNAse III enzyme (McRae et al., 2006) allowed ablation of regions of the protein crucial for catalytic activity. Initially ablation of Dicer function was achieved in flies and in vertebrates as mice and zebrafishes.
The loss of function of Dicer have been successfully performed in Zebrafish by means of N-ethyl-nitrosurea (ENU) mutagenesis (Wienholds et al., 2005). Mutant fishes exhibited a strong regressive phenotype with general developmental arrest at 15 days post-fertilization, not attributable to specific organ failure. At this age, a strong decrease in miRNAs levels occurs physiologically. The presence of maternal transcripts of dicer1 explain the overall correct formation of the body plan and organs, as the production of miRNAs is retained in mutant fishes at the time of gastrulation, neurulation and organogenesis. In order to eliminate zygotic and maternal processing of pre-miRNAs,
dicer1 was mutated also in the germ cells of parent fishes by replacing them with a germ line deriving by mutant donors. Intercrossing of these fishes generated the so called maternal-zygotic mutant for dicer (MZdicer). This mutant displayed a general delayed development, though axis formation and regionalization were not affected. Nevertheless, severe defects in various morphogenetic processes were observede. In particular, MZdicer mutants showed a shortening of the embryo and an accumulation of cells in the rostral region, owed to gastrulation defects leading to a reduced extension of the axis. Development of central nervous system, somitogenesis and cardiogenesis were affected too and blood circulation was severely impaired. It is noteworthy that the derailed brain morphogenesis in MZdicer mutant fishes could be rescued with the injection of a neural specific miRNA, miR-430 (Giraldez et al., 2005).
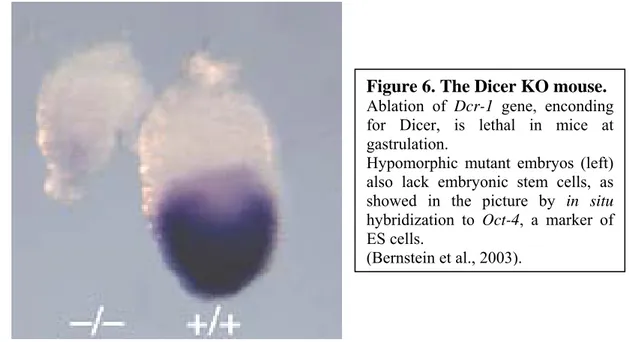
In the mouse, the knock-out of Dicer is lethal. In facts, viable littermates produced by heterozygous mating are not genetically Dicer null. Live nulls were found during the early development, at embryonic day 7,5 (fig. 6), about at the onset of gastrulation. These embryos were abnormal, small and necrotic. In addition, Dicer1 KO did not express the primitive streak marker T (Brachyury), implying the arrest of morphogenesis before the normal estabilishment of the body plan at gastrulation. At E7,5 the number of mutant embryos was also 50% lower than the expected Mendelian ratio, likely because of the occurrence of earlier degenerative phenomena. Finally, Dicer ablated embryos displayed a loss in ES cellst, as shown by lack of signal in in situ hybridization experiments tracking Oct4, a gene for a transcription factor required for ES cell maintenance and cell fate decision (Bernstein et al., 2003). Further studies have been performed upon Dicer ablated ES cells, demonstrating that these cells do not contribute to chimeras if injected into blastocysts, do not form teratomas if transplanted in nude mice and, if differentiated in culture, form embryoid bodies lacking endodermal or mesodermal lineages (Kanellopoulou et al., 2005; Murchison et al., 2005). Additionally, an increase in pericentromeric satellite repeats expression in Dicer KO cells has been demonstrated.
Figure 6. The Dicer KO mouse.
Ablation of Dcr-1 gene, enconding for Dicer, is lethal in mice at gastrulation.
Hypomorphic mutant embryos (left) also lack embryonic stem cells, as showed in the picture by in situ hybridization to Oct-4, a marker of ES cells.
(Bernstein et al., 2003).
Actually, bidirectional transcription in these regions (Rudert et al., 1995) can lead to the formation of a long dsRNA competent for Dicer cleavage, with the generation of siRNAs capable of silencing the transcription of the region itself, as occurs in S.pombe (Reinhart and Bartel, 2002). The deficiency in centromeric heterochromatin expression silencing is shown also by different pattern of methylation in these regions. Surprisingly, this feature does not affect genomic stability of ES cells, probably because they use mechanisms different from those of differentiated cells to maintain chromosome stability.
1.7 Conditional knock-outs of Dicer
The early lethality observed Dcr-1 KO mice made impossible for researchers to study later effects of the ablation in the development and adult homeostasis of complex tissues. To overcome this obstacle, scientists developed mouse lines in which one or more exons of the Dcr-1 gene, coding for crucial domains of the protein, were “floxed”, and so flanked by inserted loxP sites. This genetic modification allows the excision of the genomic region between the floxed sites by means of the activity of a Cre recombinase. For this, loss of function of Dicer will occur only in the presence of this
enzyme, whose expression can be in turn regulated in space (tissue specificity) and time.
In the Dicer conditional KO (CKO) line, the exon 23 of Dicer gene has been floxed. This region codes for most of the second RNAse III domain of the protein, and its excision leads to a complete impairment of processing activity, due to loss of the above mentioned intramolecular interaction. Crossing of CKO mice with a Cre line, specific for germ cells (beta-actinCre) resulted in mice that, for further cross breeding, produced a progeny identical to Dicer KO mouse (Harfe et al., 2005). The same authors also obtained selective deletion of Dicer from limb mesoderm by crossing the conditional line with a transgenetic expressing Cre recombinase in the mesodermal lineage of either forelimb and hindlimb (prx1cre). The progeny displayed a reduction in limb size caused by apoptosis of Shh-expressing cells in the limb mesoderm. Interestingly, all cell types and limb structures were correctly specified in CKO animals, without apparent defects in structure patterning. (Harfe et al., 2005).
Using similar approaches, different groups have selectively inactivated the gene dcr-1 in various mouse organs and tissues including lung, heart, kidney, cartilage, skeletal muscle and skin. Embryos lacking Dicer in the developing heart exhibited pericardial edema and a poorly developed ventricular myocardium, dying at E12,5 for heart failure (Zhao et al., 2007).
In lungs, Dicer was shown to be required for normal developmental branching, as its inactivation transforms the morphology of this organ at E15,5 from the characteristic tree-like arborization to a series of large fluid-filled sacs within each lobe. Natural occurring cell death is also increased and occurs ectopically. Obviously, mutant mice die at birth (Harris et al., 2006).
Finally, Dicer was shown to play a fundamental role in the morphogenesis of the locomotion system: skeletal muscles lacking an active Dicer protein are reduced because of hypoplasia (O’Rourke et al., 2007), while skeletal development is severely affected due to a decrease in chondrocyte proliferation and a premature differentiation of these cells in postmitotic hypertrophic chondrocytes (Kobayashi et al., 2008).
All these studies demonstrate the fundamental role of Dicer and, therefore, of miRNAs in the morphogenesis of most body organs. However, it is noteworthy that the the Dicer KO organisms generated so far exhibit and impairment in the proliferation
and/or in the apoptotic processes acting during development, rather than defects in the specification of the different cell types characteristic of the organ studied.
1.8 Role of Dicer and miRNAs in the Central Nervous System
The high social impact of neurodegenerative diseases in the world drives scientist attention onto yet uncovered mechanisms of disorders of the central nervous system (CNS). In this view, researchers have tested the hypothesis that the novel class of discovered small RNA (microRNAs) could play specific functions in the physiology and balance of this complex system in health and disease Previous results on MZdicer mutant zebrafishes (described above) shed light onto involvement of RNAi pathways in neural development. These animals exhibit an impairment formation of the neural tube, and absence of the constrictions that generate the ventricles subdividing brain into distinct regions Retinal and spinal chord development are affected too, and axonal pathfinding is quite perturbed. Nevertheless, overall gene patterning appears to be preserved (Giraldez et al., 2005).
Mosaic flies harboring the homozygous mutation of dcr-1 in eye and antennal immaginal discs show half sized eyes with disturbed organization of ommatidial facets and the loss of sensory bristles over the eye surface (Lee et al., 2004).
In the last two years, the use of transgenic lines harbouring the expression of Cre recombinase in different neuronal cell types allowed to delete the function of Dicer in several mouse CNS areas, including s the cortex, the striatum, the hippocampus, the cerebellum and the substantia nigra of the midbrain.
Midbrain dopaminergic neurons play a critical role in locomotion and complex behaviours and their function is lost in Parkinson’s disease. Dicer has been shown to be required for the terminal differentiation and/or maintenance of these neurons, as the ventral tegmental area and the substantia nigra are practically devoid of them in 8-weeks old Dicer CKO mice. Accordingly, these animals show markedly reduced locomotion, a typical parkinsonian trait. In addition, a specific miRNA (miR-133b) has been shown to regulate the expression of Pitx-3, a transcription factor required for dopaminergic differentiation. Pitx-3, in turn, is capable of activating the transcription of this miRNA, thus generating a negative feedback circuit (Kim et al., 2007).
Dopaminergic neurons send inputs to dopaminoceptive striatal neurons, which in turn make synapses with basal ganglia neurons. The latter are impaired in a multiplicity of neurodevelopmental, neurodegenerative and additive disorders. Removal of Dicer in basal ganglia neurons leads to reduced life span for wasting starting at 8 weeks of age. Moreover, CKO mutants exhibit motor deficits reminiscent of neurodegenerative disorders such as Huntinton’s disease. Finally, reduction of brain and neuronal cell size, astrogliosis and behavioural defects similar to those observed in Rett syndrome have been in these KO mice. However, neurons innervated by dopaminergic cells of the basal ganglia survive the mutation (Cuellar et al., 2008).
The function of microRNAs has been investigated in the cerebellar tissue as well. Conditional ablation of Dicer in Purkinje cells leads to a slow but progressive degeneration of these neurons, which provokes in turn a severe ataxic phenotype at the age of 3 months and later (Schaefer et al., 2007).
Finally, the function of Dicer has been investigated in excitatory forebrain neurons of the cortex and hippocampus. Targeted ablation of the enzyme causes microcephaly and enlargement of lateral ventricles due to a 5,5 fold increase in early postnatal apoptosis. The phenotype is not linked to cortical delamination or neuronal migration defects. Furthermore, significant alterations in dendritic branching, dendritic spine length and axonal pathfinding occur in CKO animals (Davis et al., 2008).
In summary, a wealth of data show that miRNAs are essential for the maintenance of genetic programs required for proper CNS morphogenesis and function.
1.9 miRNAs and retina
Little is known about the role of miRNAs in the eye and particularly in the retina. A great effort was done to identify small RNAs specific to neural tissues in general. Expression profiling of the CNS revealed high specific hybridization signals in arrays for a sizeable number of miRNAs (Babak et al, 2004; Barad et al., 2004; Miska et al., 2004; Sempere et al., 2004; Shingara et al., 2005; Thomson et al., 2004). Among these, 45 are expressed ubiquitously in the mouse CNS, while at least 63 exhibit differential expression in specific regions (Bak et al., 2008). A more focused study describes 78 miRNAs expressed in the adult mouse retina, 21 of which appear to be retinal specific (Xu et al., 2007). The authors identify also a retinal specific cluster, formed by miR-96, miR-182 and miR-183, which displays a diurnal variation in the expression pattern. This cluster could be important in the regulation of retinal circadian rhythms Using different approaches, other groups have shown the cell type specificity in the expression patterns of several eye and retinal miRNAs (Ryan et al., 2006; Karali et al., 2007). Nevertheless, very little is known about the overall function of miRNAs and their putative targets in retinal development and adult homeostasis.
Very interestingly, a subset of miRNAs seems to be modified in different mouse models of Retinitis pigmentosa (Loscher et al., 2007, 2008). As aforesaid, Dcr-1 null eyes of Drosophila display major defects, while MZdicer mutant zebrafishes have a retinal phenotype. Finally, involvement of post-transcriptional gene silencing (PTGS) is reported to orchestrate cell cycle progression in retinal development (Decembrini et al., 2006).
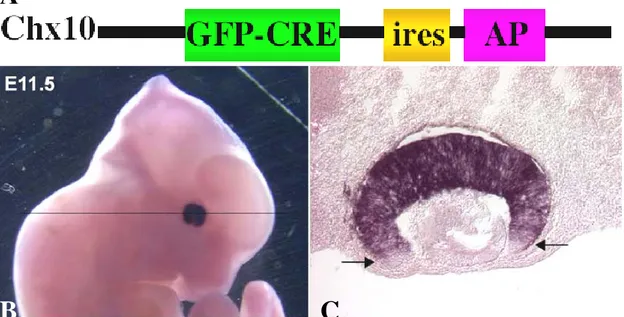
For these reasons, we decided to analyze the effects of ablating Dcr-1 gene, selectively in the retinal tissue. In the mouse, the Dcr-1 gene is the only one encoding for a Dicer protein. We developed a collaboration with the laboratory of Brian Harfe, at the University of Florida, who had recently crossed Dicer conditional knock-out mice (referred as Dicer flox/flox) with Chx10 BAC transgenic mice. The latter express the Cre recombinase in the retina only. In particular, in this strain the expression of recombinase is driven by a promoter of Chx10, a gene encoding for a transcription factor expressed in retinal progenitors during development and in bipolar cells plus a small subset of Müller glial cells in the adult life. Cre is fused with GFP for an
immediate visualization of cells expressing the transgene. Finally, an internal ribosome entry site (IRES) element downstream the GFP-Cre cassette allows the concomitant expression of placental alkaline phosphatase to label cell membrane by means of simple histochemical procedures (Rowan and Cepko, 2004).
A
B
C
Figure 7. The Chx10 BAC transgene.
(A) Structure of the Chx10 BAC. Cre recombinase is fused with GFP. In addition, to visualize the morphological features of Cre expressing cells, an IRES element permits the concomitant expression of a membrane-bound isoform of alkaline phosphatase (AP).
(B, C) Transgenic expression of Cre in retinal progenitors. (Modified from Rowan and Cepko, 2004).
In this study, we show that retinal deletion of Dicer leads to early loss of lamination and retinal degeneration, accompanied by a severe impairment of visual function. Thus, the observed phenotype resembles other models of human retinal degeneration. None of the studied rodent models of retinal disease parallels precisely the uncommon, simultaneous degeneration of both photoreceptors and inner retinal neurons detected in Dicer CKO mice. In spite of that, similarities could be noticed to other examples of inherited photoreceptor degeneration. In particular, photoreceptors appear to be the first and the most affected neurons, as occurs in Retinitis pigmentosa (RP) and cone-rod dystrophy (CRD).
Actually, modifications in miRNA transcriptome was recently observed in mouse models of autosomal recessive RP (arRP). In addition, we observed a remarkable preservation of the innermost retinal populations, particularly of the ganglion cell layer, at a time when the overlying layers were undergoing major structural remodelling. A previous study from our laboratory demonstrated morphological preservation of retinal ganglion cells several months after the loss of photoreceptors in the rd10 mouse, a model of human arRP (Mazzoni et al., 2008). The relevance of such study resides in the fact that various therapeutic approaches for retinal degeneration rely upon the viability and function of ganglion cells, and particularly those based upon epiretinal prosthetic stimulation and ectopic expression of light sensitive molecules. For all these reasons, after studying the Dicer CKO mouse, we turned our attention to retinal ganglion cell morphological preservation in a similarly aggressive example of retinal degeneration, the rd1 mouse, a long-known model of arRP. The underlying hypothesis is that, in this disease too, ganglion cells constitute an intrinsically stable population of retinal neurons.
1.10 A quick overview of retinal organization
The human eye is the body organ with the highest number of mutations, many of them occurring in the retina. As discussed below, many genes are known to cause retinal diseases, and yet an equally high number of them are thought to be unidentified. For this reason, it is particularly relevant to search for retinal specific, candidate disease genes.
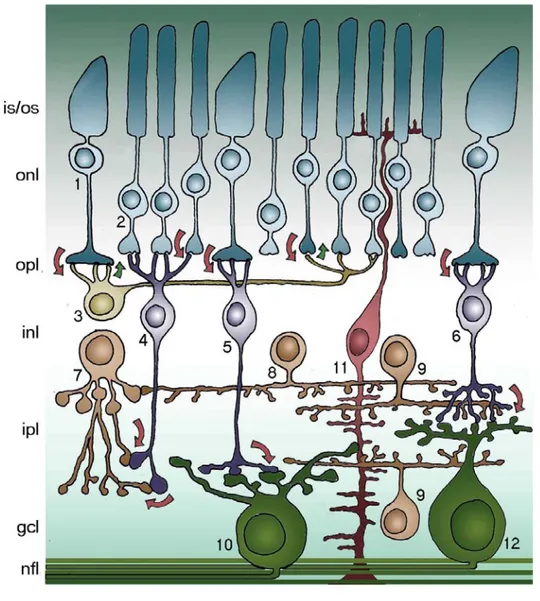
Figure 8. Schematic representation of the mouse retina.
1. cones (125,000); 2. rods (4,5*106); 3. horizontal cells (17,000); 4. rod bipolar cells (200,000); 5. cone bipolar cells ON; 6. cone bipolar cells OFF (total CB 400,000); 7. AII amacrine cells; 8. dopaminergic amacrine cells; 9. cholinergic amacrine cells; 10. ON ganglion cells; 11. Müller cells; 12. OFF ganglion cells (50,000 GCs total). is/os photoreceptor inner/outer segments; onl outer nuclear layer; opl outer plexiform layer; inl
The main function of the retina is the conversion of light into electrical stimuli and the propagation of these to higher visual centers of the brain. This is achieved through the capture from rod and cones photoreceptors of a spectrum of electromagnetic waves (called for this reason visible light) with photopigments included in highly specialized cellular compartments, termed outer segments. Photopigments are in complexes with proteins called opsins. Light causes a conformational change in these complexes that, in turns, trigger the activation of a cascade resulting in the closure of cGMP gated cation channels. This leads to a hyperpolarization of the photoreceptor membrane that in turn is transformed into a change in the release of their neurotransmitter (the aminoacid glutamate) at the synaptic complex established with is second order neurons, bipolar and horizontal cells. The first split the signal into ON and OFF responses by means of opposite electrical activities in presence of the same light input. In addition, bipolar cells transmit the signal to amacrine and ganglion cells. Horizontal cells increase contrast sensitivity by inhibiting the surrounding photoreceptors. Amacrine cells play several (and still partially unknown) modulatory actions, while ganglion cells (the only projection neurons of the retina), send action potentials with their axons to thalamus and, by that, to the visual cortex.
Besides the 5 classes of above mentioned neurons, glial cells are also represented in the retina: Müller cells have a radial arrangement and are contiguous to all the other retinal components, including blood vessels. Astrocytes are restricted to the innermost retinal layer in close association to ganglion cells and blood vessels. Microglial cells reside in the retina and participate in response to injury and inflammatory processes.
Retinal neurons and glia are spatially organized in a laminated arrangement. The cell bodies of rod and cones form the outer nuclear layer (ONL), while the somata of horizontal, bipolar and amacrine cells plus Müller glia constitute the inner nuclear layer (INL). Cell processes of neurons make synaptic contacts in the outer and inner plexiform layers (OPL and INL, respectively). Ganglion cell bodies form the homonymous layer (ganglion cell layer or GCL) together with displaced amacrine cells, in a 1:1 ratio .
1.11 Ganglion cells diversity of structure and function
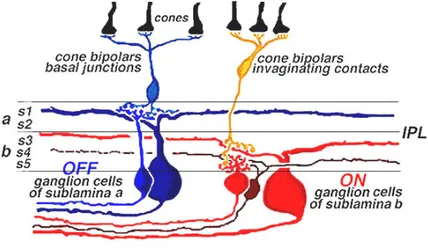
Since the time of Santiago Ramon y Cajal’s pioneeristic studies on Golgi stained retinas, it was clear that retinal ganglion cells represent a highly heterogeneous population of neurons. Classification of RGCs has been a multistep process: in the 70’s the existence of large and small field ganglion cells, termed α and β respectively (Boycott and Wassle, 1974) was recognized. Few years later, two morphological sublaminae of the IPL, termed a and b, were associated to neural circuits processing respectively OFF and ON signals, associated to light decrements or increments. Following the architectural principle according to which different functional properties are spatially segregated in the IPL, α and β RGCs, in turn, could be divided into ON- and OFF-center cells depending on their response to light; morphologically, dendrites of OFF-center RGCs stratify in sublamina a, while processes of ON-center RGCs stratify in sublamina b of the IPL (fig. 9) (Famiglietti and Kolb, 1976; Nelson et al., 1978).
Figure 9. Schematic organization of ON- and OFF- center ganglion cells into sublamina a and b.
(From http://webvision.med.utah.edu/GCPHYS1.HTM, after Nelson et al., 1978).
Progress of histological and imaging techniques made the classification of RGCs more and more precise. Presently, it is recognized that, in mammals, the number of RGC types varies from 10 to 20 cell: monkeys are reported to have 8 eight types of
RGCs, cats and rabbits 7, while rats have 13 types. Depending on the classification method, mice were reported to have from 11 to 17 types. Morphological equivalents are shown to be present in each mammal, implying an high conservation of RGCs among species. Moreover, the theory of the correlation between morphology and function has been shown to fit particularly well for RGCs.
For instance, large dendritic field cells (termed α in cats, parasol in monkeys, A and C in mice) are thought to be implicated in rapid movement processing, while cells with little and bushy dendritic arborizations (known as β in cats, midget in monkeys, and B in mice) seem to be involved in fine contour perception. Thus, in RGCs differential functional properties are strictly associated to their characteristic dendritic morphology (for a review, see Sernagor et al., 2001).
Before eye opening, the majority of RGCs are bistratified, and so responding to light onset and offset (from here the name ON-OFF RGCs). Refinement of dendritic arborizations occurs postnatally: one or more branches in a sublamina are reabsorbed, while branches in the other sublamina become more complex. This phenomenon, also known as pruning, converts immature bi-stratified ON-OFF RGCs into mature mono-stratified ON or OFF mature cells. Underlying mechanisms are not known, although a requirement of glutamatergic (Bodnarenko and Chalupa, 1993, Bodnarenko et al., 1995; Bisti et al., 1998) and cholinergic inputs (Bansal et al., 2000) is required. Recently, visual stimulation, known to increase BDNF levels released from higher centers, was also shown to play a role (Tian and Copenhagen, 2003; Landi et al., 2007). In addition, BDNF was shown to promote elongation of new branches in ON cells (Liu et al., 2007), while another neurotrophin, NT-3, not influenced by visual experience, was demonstrated to be involved too in laminar refinement. Moreover, NT-3 overexpression prompts formation of shorter, but more complex, dendritric trees in ON-OFF RGCs selectively. In the mouse retina, laminar refinement and dendritic outgrowth of RGCs seem to complete at about 1 month of age, albeit differential growth pattern was observed in ON, OFF and ON-OFF RGCs. In particular, ON RGCs grow both before and after eye opening, while ON-OFF RGCs expand mainly before eye opening ad OFF RGCs after eye opening (Liu et al., 2009).
Different morphological classification systems of RGCs have been proposed. The groups of Leo Chalupa and Richard Masland independently adopted a computer assisted
cluster analysis, in which numerous morphometric features of RGCs were measured and compared, allowing grouping of cells in different clusters (Coombs et al, 2006; Kong et al., 2005).A previous simpler classification methods was developed by the group of Shigang He. Only three morphological characteristics were analyzed: dendritic tree area, body area and depth of stratification. Conversely to the above mentioned studies, this method is based on the identification of the shape of RGCs, as it was the case in the past with Golgi stained specimens.
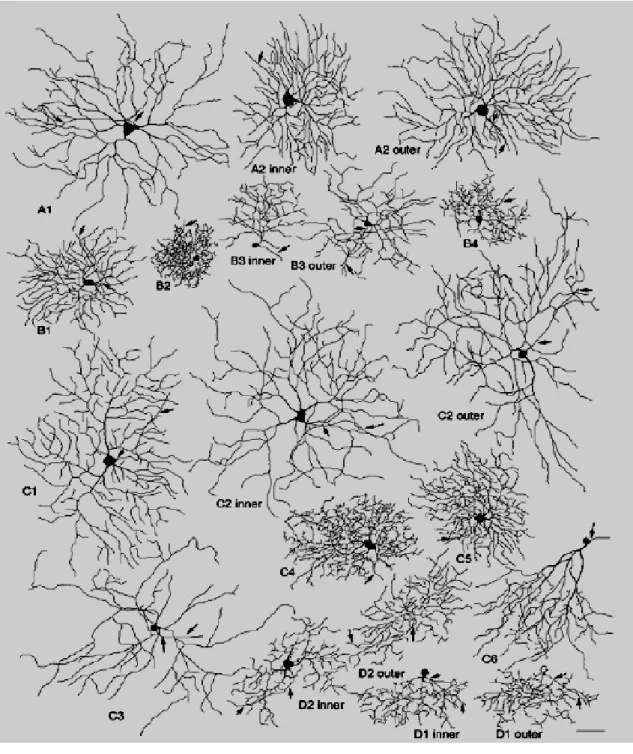
Briefly, cells with large soma and large dendritic trees are classified as RGCA ,
while cells with a small to medium sized soma and small to medium sized trees as RGCB ; the other two types comprise cells with a small to medium-sized body but
medium to large-sized dendritic arbors, termed RGCC, and, finally, bi-stratified cells,
classified as RGCD. These four classes are subdivided in turn in several subtypes for a
total number of 17 groups (fig. 10; Sun et al., 2002).The recent molecular identification of several types of RGCs conferred a more functional significance to groups in classification systems. For instance, RGCs immunoreactive for the adhesion protein JAM-B were shown to be comprised in C6 type of Sun’s classification. Interestingly, these cells have highly polarized dendritic trees all with the same orientation. This feature likely confers the cells directional selectivity to upward motion (Kim et al, 2008). With respect to directional sensitivity, it is well known since the 90’s that a large subset of mature bi-stratified cells (class RGCD of Sun’s classification) fire bursts of
axon potentials only if the visual stimulation moves along the preferred direction, while the same neurons are inhibited if motion occurs in the opposite (or null) direction (Yang and Masland, 1992, 1994).
Finally, melanopsin expressing RGCs can be assigned in group C3 of Sun’s classification. These cells are intrinsically photosensitive by means of expression of an opsin-like protein (i.e.melanopsin) that do not participate to image formation, but are required to sense the quantity of light in the process of photo-entrainment of the suprachiasmatic nucleus (SCN), the primary circadian pacemaker in mammals and many other vertebrates (Provencio et al., 1998; Hattar et al., 2002).
Figure 10. Schematic representation of different types of mouse retinal ganglion cells.
From Sun et al. 2002. This classification method is based upon visual recognition more than computer assisted algorithms. Arrows indicate axons. Scale bar is 50 µm.
1.12 Retinitis Pigmentosa
Human retinal degenerations comprise inherited diseases caused by the progressive loss of photoreceptors that eventually lead to blindness. More than 100 genes have been shown to be implicated in the development of these diseases. For the homogeneity of symptoms and course, a subset of them has been termed Retinitis pigmentosa (RP).
Typical RP displays a primary degeneration of rod photoreceptors, successively followed by the death of cones. RP is one of the major cause of blindness and visual handicap in the world; it hits a person on 4000, for a total of more than a million people affected worldwide.RP can be inherited as autosomal-dominant (adRP; 30-40% of cases), autosomal-recessive (arRP; 50-60% of cases) or X-linked (XLRP; 5-15% of cases). Digenic and maternal (mitochondrial) inheritance patterns have been reported too.
At least 45 genes involved with RP have been identified. These genes account for less than 60% of all the RP patients. Thus, for more than 40% of RP cases the underlying genetic alteration remains unknown.
In approximately 20-30% of patients, RP is part of a syndromic disease, the most frequent (10-20% of all cases) being represented by Usher’s syndrome (US), in which RP is associated with hearing impairment. This symptom can be profound and progressive up to deafness and associated with vestibular ataxia (US type I); or moderate and stationary (US type II); finally, normal hearing could also be present until adulthood (US type III). Alterations in 11 known genes cause this disease. Depending on the mutation, some of them can cause RP without hearing defects or hearing impairment without RP.
Another frequent RP associated disease (5-6%) is Bardet-Biedl syndrome (BSS). In this case, visual impairment might occur simultaneously to obesity, cognitive defects, polydactily, hypogenitalism and severe kidney disease. Ten genes linked to BBS have been identified so far.
the rhodopsin gene (RHO) are responsible for 25% of autosomal dominant cases of RP. The USH2A gene might cause about 20% of the recessive form of the disease. Finally, the RPGR gene is involved in about 70% of the cases of X-linked RP (see Hartong et al., 2006 for a review).
The P23H mutation in the rhodopsin gene sequence results in a gain-of-function form of disease in which the rhodopsin protein gets the ability to create ER and cytoplasmic protein aggregates. These, in turns, activate cell death pathways. (Saliba et al., 2002; Rutkowski and Kaufman, 2004; reviewed in Mendes, 2005)
Another dominant mutation of rhodopsin is G90D, also found to be implicated in a family with congenital stationary night blindness (CSNB). Transgenic mice expressing this mutation develop gradual photoreceptor degeneration, curiously rescued by eliminating native rhodopsin. The mutated form is probably unstable and acquires the capability of activating the phototransduction cascade spontaneously, even in absence of light stimuli (Rao et al., 1994; Sieving et al., 1995, 2001).
Retinitis Pigmentosa GTPase Regulator or RPGR is a protein of unknown function normally localized to the connecting cilium of rod and cone photoreceptors. A gradual loss of photoreceptors is described in RPGR-KO mice, with the concomitant reduction in the ERG amplitude.
Other well known mutations are described to cause RP. For instance, the Tubby-like protein 1 (tulp1) gene has been implicated in arRP. Mice null for this gene exhibit a very rapid degeneration of rods and cones, with a concomitant loss of ERG. In particular, ectopic accumulation of rhodopsin occurs at an early age in tulp1 -/- mice. Tulp1 protein play a key role in transporting rhodopsin from the cell body to the rod outer segment (Hagstrom et al., 1998, 1999, 2001; Ikeda et al., 2000). to interact with cytoskeleton (Xi et al., 2007). Finally, a very recent study showed that Tulp1 is required for the correct development of photoreceptor synapse (Grossman et al., 2009).
RP1 is a photoreceptor specific gene involved in an autosomal dominant form of RP. The RP1 knock-out mouse exhibit disorganized outer segment discs causing a progressive loss of rods. Evidences of rhodopsin mislocalization indicate that this gene is required for rhodopsin transport and for the normal morphogenesis of rod outer segments (Guillonneau et al., 1999; Pierce et al., 1999; Sullivan et al., 1999; Gao et al., 2002).
1.12.1 Many causes, one disease, or, different mutations, same
symptoms
The high genetic heterogeneity in RP is the probable reason for the difference in the onset of the disease. Most patients suffer of difficulties in dark adaptation and night blindness during adolescence and loss of mid-peripheral visual field in young adulthood. With time, the disease causes loss of peripheral vision, tunnel vision and, eventually, total blindness, usually around the age of 60. A number of patients, however, remains asymptomatic until adulthood. Early symptoms are correlated with degeneration of the rod population, responsible for achromatic vision in dim light (scotopic vision). The restriction of the visual field is due to the preferential distribution of rods in the human peripheral retina (fig. 11).
On the contrary, loss of visual acuity and loss of central vision are the result of the secondary degeneration of cone photoreceptors, deputed to chromatic vision in high intensity of light (photopic vision) and highly concentrated in the central retina, where the fovea is located.
Figure 11. Loss of peripheric vision in advanced RP.
Tunnel vision (right) is a typical example of eye field degradation in RP patients (From http//www.pocklington-trust.org.uk/Templates/Internal.asp?NodeID=89018).
Another typical feature of RP is the attenuation of retinal vasculature, detectable by fundus examination. By means of simple inspection ophthalmologists can find the presence of pigments in the retina, caused by penetration of retinal pigment epithelial (RPE cells) in the inner retina. The name retinitis pigmentosa originates from this distinctive hallmark (fig.12).
Figure 12. Penetration of RPE into the inner retina in RP.
Fundus views of the retina of a normal human subject (left) and of a RP patient (right). A typical brown pigmentation might be present in the pathological condition. Pigment deposits, named bone spicules for their typical shape, are responsible for the name of the disease. A clear attenuation of retinal blood vessels is also visible.
(From http.//www.snof.org/maladies/pigmentaire.html/ pigment clumps)
Finally, RP patients are affected by lipofuscinosis, as assessed by optical coherence tomography (OCT).
The clinical diagnosis for RP might be difficult. In the modern world, artificial light allow cone-driven vision also during the nighttimes, making patients unaware of their visual deficits at initial stages. Moreover, no difficulties with normal daily activity occur in people with a visual field reduced to 50°, whether the normal is about 180° wide. Finally, reduction of visual acuity might be experienced only after the loss of more than 90% of foveal cones.
1.12.2 Mechanisms of photoreceptor degeneration in RP
Many disease genes for RP encode for protein of the rod phototransduction cascade. Recessive mutations are thought to interfere with the phototransduction pathway causing the loss of function of one of its crucial components. For instance, mutations in the rod-specific cGMP phosphodiesterase subunits A and B (PDE6A and PDE6B) cause the increase of cGMP levels in the outer segment of rods. The consequent effect is that light-sensitive cGMP gated channels remain open, determining uncontrolled influx of cations like sodium and, most importantly, calcium. Exceedingly high levels of calcium cannot be buffered by cellular machineries and calcium increase is probably the primary trigger of intrinsic apoptotic pathways in mutant rods, in this and other null mutations. Either cytochrome C and ER pathways seem to be involved, as indicated by the presence of the Apoptosis Inducing Factor (AIF) and the active form of caspase-12, respectively, within degenerating rods (Sanges et al., 2006).
As regards for autosomal dominant forms, most likely causes are mutations converting the native protein into a toxic one. Such toxicity can be attributable to interference with metabolism via formation of intracellular aggregates or from defects in structural integrity and protein trafficking in the photoreceptor outer segment.
Other then in genes for phototransduction proteins, RP mutations occur in genes involved in the visual cycle pathway, in which the regeneration of retinaldehyde (vitamin A) takes place in the pigmented epithelium (RPE). Some dominant mutations, finally, occur in housekeeping genes as PRPF31, PRPF8 and PRPF3 (precursor mRNA processing factors) coding for spliceosomal components, provoking surprisingly an RP phenotype rather then a systemic diseases.
One of the most important and still unexplained traits of RP is the degeneration of cone photoreceptors in sight of mutations in rod specific genes. For this reason, death of cones is considered indirect, and thus secondary to rod degeneration. Several mechanisms have been proposed, like the release of toxic substances by degenerating cells, the loss of retinal hoemeostatic mechanisms and/or the lack or intraretinal survival signals going from rod to cones .
Very recently the group of J. A. Sahel, from Paris, discovered a factor secreted by rods in the interphotoreceptor matrix. As suggested by its name, the so called Rod
derived Cone Viability Factor (RdCVF) exhibits the ability to promote survival of cone photoreceptors. In this view, the secondary death of cones would occur for the lack of trophic factors only when the vast majority of rods have degenerated and, consequently, could not produce adequate amounts of RdCVF and possibly other unidentified survival molecules (Léveillard et al., 2004).
According to another theory, secondary death of cones would result as a consequence of oxidative stress. Choroidal vasculature provides for a large amount of oxygen consumed by photoreceptors, that have a the high metabolic demand. In RP, upon extensive rod death, the same level of oxygen becomes available to a much smaller number of photoreceptors, mainly consisting ofcones. Hence, these cells are exposed to excess oxygen and react with the formation of reactive oxygen species (ROS), known to induce apoptotic responses (Komeima et al., 2006, 2008).
1.12.3 Therapeutic strategies for RP
The emerging knowledge upon biological mechanisms underlying retinal degeneration occurring in RP prompted researchers to develop various different therapeutic strategies in the attempt to treat or, at least, to delay the disease.
Gene therapy represents probably the only way to really ”cure” the disease with the specific correction of underlying mutations. Exogenous introduction of correct sequences could be performed in loss of function forms of RP, such as the autosomal recessive forms and the X-linked RP, where a lack of functional proteins is responsible for the pathology. This strategy has been performed different times in various animal models as rd and rds mice and RCS rats, with the resulting slow down of degeneration of photoreceptors. The most impressive results were obtained in the RPE65-/- Briard dog, a model of Leber congenital amaurosis (LCA) presenting defects in retinoid metabolism and virtually born blind. A subretinal injection of adeno-associated viruses (AAV) containing the wild type RPE65 cDNA was capable to restore visual function (Acland et al., 2001).
Conversely, mutations leading to a gain-of-function phenotype, referred as autosomal dominant, gene therapy can be used to inhibit the expression of the aberrant allele with ssRNA antisense oligonucletides or ribozymes (Farrar et al., 2002).
In particular, the use of hammerhead RNAs delayed considerably the onset of degeneration in the transgenic P23H rat, a model of rhodopsin mutation diffused in North America (Lewin et al., 1998; LaVail et al., 2000). Gene therapy has also been successfully performed to slow down apoptosis of photoreceptors, with the transfer of the bcl-2 anti-apoptotic gene. Finally, recent introduction of bacterial channel opsins in ganglion cells (Bi et al., 2006) and bipolar cells (Lagali et al., 2008) and the ectopic expression of melanopsin in ganglion cells (Lin et al., 2008) bypassed the lost function of photoreceptors in mutant mice by conferring visual sensitivity to retinal neurons acting downstream and successfully restored some visual function to the treated animals.
The strategy as in the RPE -/- dog aforesaid has been recently used in attempt to cure LCA human patients. It is of outstanding interest that treated patients had significative improvement in their visual abilities. Particularly, after the injection patients were capable to avoid obstacle in a course and even to read several letters in distant charts (Maguire et al., 2008).
Many researchers have attempted to slow down the degeneration of rods with pharmacological neuroprotective therapies. This way, secondary degeneration of cones appears also delayed considerably with the perspective of avoiding severe visual handicap in patients.
Several trophic factors demonstrated efficacy in the protection of photoreceptors. In particular, Fibroblast Growth Factor (FGF2) and Ciliary Neurotrophic Factor (CNTF) have positive effects on cell survival, but the first leads also to retinal neovascularization, while the second has the paradoxical effect to decrease ERG responses. On the contrary, Glial Derived Neurotrophic Factor (GDNF) appeared effective in maintaining both morphology and function. The long term release of diffusible factors could be achieved with the surgical implantation in the vitreous body of encapsulated genetically engineered cells (Uteza et al., 1999; Tao et al., 2002).
Transplantation of photoreceptors and retinal progenitors seem to act more by protecting rather than by replacing the remaining cells possibly through the release of trophic factors. Nevertheless, many authors reported neurite outgrowth and even integration into the host retina, especially with subretinal injections of committed rod progenitors into the immature postnatal retina (MacLaren et al., 2006).
Grafts of bone marrow derived hematopoietic stem cells have been recently shown to integrate into the host retina, to differentiate in microglial cells and delay significantly vascular degeneration associated to neuronal death in animal models of RP (Otani et al., 2004; Sasahara et al., 2008).
The use of growth factors and morphogens such as Wnts, IGF-1, insulin and others has been also used to stimulate retinal regeneration by promoting the proliferation of quiescent progenitors included within the population of Müller glial cells (for a review, Fischer and Reh, 2003).
As a calcium overload is thought to be implicated in the degeneration of rod photoreceptors, another pharmacological approach was attempted in order to reduce RP effects. Using the calcium channel blockers D-cis-diltiazem it could be possible to inhibit the cGMP gated cationic channels located in the outer segments of photoreceptors, thus slowing down the degeneration of rods in rd1 mutant mice (Sahly et al., 1992; Frasson et al., 1999b); nonetheless, the effect of this compound turned out to be negligible in other important animal models (Pawlyk et al., 2002; Pearce-Kelling et al., 2001).
Besides experimental treatments on animals models, approaches used to slow down the disease progression in RP patients include nutritional supplementation with vitamins and integrators. In a randomized, controlled, double masked clinical trial, Berson and colleagues gave participants either vitamin A (retinyl palmitate; 15000 U.I. daily), or vitamin E (dl-α-tocopherol; 400 U.I. daily). The 4-6 years follow-up clearly demonstrated the beneficial effects of vitamin A in slowing-down the loss of visual acuity in RP patients. Vitamin E, instead, was shown to have no or even negative effects, as this compound can compete with the absorption of vitamin A itself. Supplementation with β-carotene could be ineffective as a substitute of this therapy for it is not readily and reliably converted in vitamin A.
Another assessed nutritional treatment is supplement of docohexaenoic acid (DHA), an omega-3 fatty acid found in high concentration in oily fish as tuna, salmon, herring and sardines. This treatment does not seem to have any effect in RP patients per se, but in combination with vitamin A it is thought to provide almost 20 additional years of visual preservation for patients who start this regimen in their mid-30s.
Finally, to reduce the oxidative stress caused to surviving photoreceptors by oxygen remaining unconsumed because of extensive photoreceptor death, Campochiaro and colleagues have recently shown the preservation of cone viability and function in different models by means of injections of anti-oxidants, including tocopherol, α-lipoic acid, ascorbic acid and MnTBAP (a metalloporphyrin superoxide dismutase mimetic) (Komeima et al., 2006, 2008).
Beside biological treatments, investigators are trying to restore vision by means of electronic prostheses placed into the eye, in particular either in contact with the epiretinal surface (the inner limiting membrane) or in the subretinal space. These devices are thought to replace photoreceptors by capturing light, converting it to an electrical signal and successively to propagate it to inner retinal neurons (fig. 13). In this way, loss of photoreceptors can be overcome. To date, most stimuli produced by prostheses consist in electrical impulses, but chemical stimulation with neurotransmitters has also been attempted.
Epiretinal prostheses currently tested in humans are based on external imaging devices that capture the image from the surrounding world. The last is analyzed by an external processor and obtained data are transmitted wireless to the implanted microelectronic device. This, in turn, converts the data into a pattern of electrical stimuli that propagate to ganglion cells and, more generally, to the inner retina (fig. 13C). Power sources and signal transmission can be obtained by induction coils, penetrating wires or lasers. This strategy has the advantage to dissipate the heat produced by the implant directly in the vitreous humor (fig. 14) and to make it possible the update of the software of external units. However, functionally it appears that the signal processing normally operated by second order neurons and by amacrine cells is completely lost.
Figure 13. Electronic visual prostheses.
Schematic representations of subretinal (A) and epiretinal electronic implants (B). In B: 1, camcorder mounted in a glass frame; 2, wireless transmitter;
3, extraocular junction box; 4, intraocular implant. (C). Simplified scheme of electronic epiretinal implant.
(From http://current.ucsc.edu/04-05/10-18/retina.asp).
Figure 14. Epiretinal prostheses.
Fundus photography showing an epiretinal electronic implant
On the other side, subretinal prostheses consist in an array of microphotodiodes implanted between the retina and RPE (fig. 13A). Solar cells of the array convert directly light to electricity in order to directly stimulate bipolar cells. In such a way, all the retinal processing is preserved. Yet, the low energy efficiency of current photodiodes makes it impossible to realize good prostheses without an external power source to reach threshold current levels. Moreover, proximity to retinal tissue makes the risk of thermal injury very high.
A new way to obtain good result with retinal prostheses could be suprachoroidal transretinal stimulation (STS), in which consists a stimulating electrode is placed in the space between the choroid and the sclera. This approach is non invasive and easy for surgeons, but requires a higher threshold current that is spread in a wider area, thus increasing the size of the receptive field and decreasing the resolution.
A cortical and optic nerve prostheses have been developed, but have the disadvantages to exclude all the retinal processing and so far have shown limited resolution.
Epiretinal and subretinal prostheses have been implanted chronically in human RP patients (fig. 14). As regards subretinal implants, no evidence of stimulation has been shown, while subjects have reported improvement in perception, probably for indirect effects. For epiretinal implants, the resulting vision was, at best, light perception; several subjects have shown some ability to discriminate shapes, even though requiring 20-30 seconds to do it (see Weiland et al., 2005 and references therein). Investigation in the field of artificial sight proceeds very intensely.
1.12.4 Inner retinal neurons in retinal degeneration: retinal
remodelling
The vast majority of therapeutic strategies relies on the assumption that inner retinal neurons viability and function is retained following to degeneration of the photoreceptor compartment. The last t decade of research clearly showed how this assumption is probably incorrect.
Studies in human RP patients reported, in addition of alterations of the remaining rods and cones (Li et al., 1995), the presence of a decrease of inner retinal neurons
(Santos et al., 1997), with focal loss of ganglion cells (Stone et al., 1992), that were interpreted variously.
Upon photoreceptor death, inner retinal neurons undergo a series of plastic modification generally known with the name of retinal remodeling. Regardless of the type of mutation, a common sequence of events was shown to take place in retinal degeneration, either in human and animal models. At the very initial stage of the disease, photoreceptors begin to deconstruct their synaptic terminals, sometimes sprouting axonal processes that extend even in ganglion cell layer. These processes can be often labeled by delocalized rhodopsin (Milam et al., 1996). Before undergoing apoptosis, rods can retract their spherules or, in rare cases observed in humans, migrate into the inner nuclear layer and survive. The lack of synaptic contact provokes major alterations in outer plexiform layer, as dendrites of second order neurons sprout at first, then retract to finally disappear in bipolar cells. In this stage, mislocalization of the different glutamate receptors also occurs and apoptosis eventually takes places in a not negligible subset of inner cells (Strettoi and Pignatelli, 2000; Strettoi et al., 2002; Gargini et al., 2007). Spaces left by degenerating photoreceptors are filled by penetrating RPE cells and by protrusions of Müller cells contributing to form an isolating sheet called glial scar.. The extended glial surface and the displacement of Müller cells greatly alter the normal retinal lamination. In this context, neuronal migration towards the ganglion cell layer take place, with the formation of tangles of neurons and processes called microneuromas. In such structures, some synapse are maintained and new synapses are thought to form. Fascicles of mixed processes start in this formations to run long distances in the retina (Marc and Liu, 2000; Jones et al., 2003; Marc et al., 2003; Jones et al., 2005; Jones and Marc, 2005).