Risultati e discussione
3. RISULTATI E DISCUSSIONE
3.1 Sintesi tradizionale dell’intermedio Z-Asp(OtBu)-Trp-Phe-Dap(Boc)-Leu-OMe
Uno dei progetti in corso di studio all’interno dell’azienda Menarini Ricerche è la sintesi del Nepadutant 1 (Schema 3.1).
L’intermedio sintetico più importante per la costruzione del Nepadutant è il pentapeptide 3.1, che risulta essere costituito da una catena lineare di cinque amminoacidi opportunamente protetti, di cui quattro naturali, l’acido L-aspartico (Asp), L-triptofano (Trp), L-fenilalanina (Phe), L-leucina (Leu) ed uno non naturale, l' acido (S)-2,3-diamminopropionico (Dap) (Schema 3.1).
OMe N H O NH Boc HN O N H O H N O N H Z O OtBu N H O 3.1 1Nepadutant HN HN H N O O O N H O N H HN O NH O O NH O O HN OH OH OH O
Schema 3.1 Nepadutant e intermedio pentapeptide 3.1.
La sintesi del pentapeptide 3.1 inizialmente sviluppata dalla Menarini Ricerche è lineare e prevede il coupling sequenziale degli amminoacidi che ne
Risultati e discussione
costituiscono la catena peptidica (Schema 3.2) a partire dalla L-leucina metilestere. Gli amminoacidi sono protetti sul terminale amminico con un gruppo benzilossicarbonilico (Z), che viene successivamente rimosso con il metodo classico di idrogenazione catalitica.
L’amminoacido non naturale Z-Dap-OH (3.2) viene protetto in catena laterale come N-Boc, ottenendo l’amminoacido Z-Dap(Boc)-OH (3.3) e viene trasformato in estere attivo Z-Dap(Boc)-OSu (3.4) mediante l’utilizzo della DCC e HO-Su. L’estere 3.4 così ottenuto viene fatto reagire con l’estere metilico della L-leucina come cloridrato H-Leu-OMe · HCl (3.5), in DMF in presenza di N-metilmorfolina come base, con formazione del dipeptide Z-Dap(Boc)-Leu-OMe (3.6). Lo step successivo consiste nella rimozione del gruppo Z per idrogenazione con Pd/C in THF a dare il dipeptide H-Dap(Boc)-Leu-OMe (3.7).
Il composto deprotetto 3.7 viene fatto reagire con la fenilalanina attivata, Z-Phe-OSu (3.8), composto commercialmente disponibile già attivato come estere succinimmidico. Il coupling viene effettuato in DMF in presenza della base N-metilmorfolina come nel coupling precedente e il peptide che si forma, Z-Phe-Dap(Boc)-Leu-OMe (3.9), viene sottoposto a idrogenazione con Pd/C in DMF per rimuovere il gruppo Z dal residuo amminoacidico della fenilalanina della catena peptidica. Il tripeptide deprotetto H-Phe-Dap(Boc)-Leu-OMe (3.10) viene fatto reagire, in DMF, con l’amminoacido Z-Trp-OSu (3.11) ottenendo il tetrapeptide Z-Trp-Phe-Dap(Boc)-Leu-OMe (3.12). La successiva idrogenazione (H2, Pd/C)
per la rimozione del gruppo Z dal residuo di triptofano è condotta in N-metilpirrolidone come solvente, per la scarsa solubilità del peptide deprotetto H-Trp-Phe-Dap(Boc)-Leu-OMe (3.13), che viene infine condensato con l’amminoacido OSu (3.14) per dare il pentapeptide Z-Asp(OtBu)-Trp-Phe-Dap(Boc)-Leu-OMe (3.1).
Risultati e discussione NH2 O OH HN Z Z-Dap-OH (3.2) NH O OH HN Z Boc (Boc)2O Z-Dap(Boc)-OH (3.3) CH2Cl2 DCC, HOSu NH Boc O OSu HN Z Z-Dap(Boc)-OSu (3.4) H2N O OMe HCl H-Leu-OMe HCl (3.5) Z HN HN O OMe NH Boc O DMF, NMM Z-Dap(Boc)-Leu-OMe (3.6) H2(Pd/C) H2N HN O OMe NH Boc O H-Dap(Boc)-Leu-OMe (3.7) Z-Phe-OSu (3.8) N OMe H O NH Boc HN O N H Z O O N H Z OSu Z-Phe-Dap(Boc)-Leu-OMe (3.9) H2(Pd/C) DMF, NMM OMe N H O NH Boc HN O H2N O H-Phe-Dap(Boc)-Leu-OMe (3.10) Z-Trp-OSu (3.11) N H O OSu HN Z H2(Pd/C) OMe N H O NH Boc HN O N H O HN N H O Z-Trp-Phe-Dap(Boc)-Leu-OMe (3.12) Z DMF, NMM NMP Z-Asp(OtBu)-OSu (3.14) OMe N H O NH Boc HN O N H O H N O N H Z O OtBu N H O H-Trp-Phe-Dap(Boc)-Leu-OMe (3.13) Z-Asp(OtBu)-Trp-Phe-Dap(Boc)-Leu-OMe (3.1) O N H Z O OtBu OSu OMe N H O NH Boc HN O N H O H2N N H O NMP
Schema 3.2 Schema di sintesi del pentapeptide 3.1 già sviluppato dalla Menarini
Risultati e discussione
Il processo sintetico sopradescritto che conduce al pentapeptide 3.1, è risultato sufficientemente efficace tanto da essere scalato e trasferito su scala pilota. Ma proprio in questa fase sono stati evidenziati problemi tecnici che hanno reso la sintesi del pentapeptide 3.1 alquanto laboriosa e poco adatta ad un ulteriore scale-up. I problemi incontrati sono principalmente conseguenza del fatto che, nella procedura sintetica che conduce al pentapeptide 3.1, viene fatto un largo uso del gruppo protettivo Z per proteggere l’amminoacido che di volta in volta viene aggiunto alla catena peptidica in costruzione. L’uso del gruppo Z è giustificato dal fatto che è interesse sintetico primario impedire o ridurre al minimo possibile ogni eventuale processo di racemizzazione, che potrebbe provocare la formazione di un pentapetide 3.1 finale in forma non diastereoisomericamente pura. I problemi legati al gruppo Z emergono al momento del suo allontanamento, che classicamente viene condotto mediante idrogenolisi catalitica (H2, Pd/C), e non
tanto per l’esecuzione delle reazioni stesse, quanto per l’isolamento del prodotto di reazione deprotetto. A questo scopo viene inizialmente condotta una filtrazione per allontanare il catalizzatore dalla soluzione contenente il prodotto deprotetto. Tutte le filtrazioni sono risultate lente per l’elevata viscosità della miscela di reazione, con intasamento del filtro, ed anche poco efficaci, in quanto notevoli quantità residue di catalizzatore rimangono nella soluzione filtrata. Considerato che, come si può vedere dallo Schema 3.2 e dalla discussione collegata, nella sintesi del pentapeptide 3.1, per ben tre volte il gruppo Z deve essere necessariamente allontanato, questi aspetti costituiscono un problema non indifferente, che rende difficile e laboriosa una tecnica così semplice in laboratorio.
Inoltre nella sintesi si fa largo uso di DMF, un solvente classificato come teratogeno, che dovrebbe essere ridotto al minimo indispensabile per una sintesi su scala industriale.
L’isolamento dei peptidi è generalmente condotto per precipitazione con soluzioni acquose di bicarbonato, ed un altro problema evidenziato su scala pilota è la difficoltà di filtrazione dei prodotti, ottenuti spesso sotto forma di gel. Come conseguenza anche l’essiccamento richiede tempi lunghi, in quanto i solidi inglobano quantità elevate di solvente.
Risultati e discussione
All’aumentare del peso molecolare i peptidi diventano meno solubili e nello stadio di sintesi del tetrapeptide H-Trp-Phe-Dap(Boc)-Leu-OMe (3.13) ci sono rischi di perdite di prodotto per precipitazione sul catalizzatore.
Un ultimo inconveniente osservato nella sintesi tradizionale è dovuto al fatto che il pentapeptide finale Z-Asp(OtBu)-Trp-Phe-Dap(Boc)-Leu-OMe (3.1) presenta residui elevati di palladio derivanti dagli stadi precedenti e deve essere effettuata una purificazione per trattamento con carbone a caldo, seguito da filtrazione e cristallizzazione. Tale procedura permette di ottenere il prodotto con le caratteristiche di purezza desiderate, ma con una perdita di resa del 15-20%. Considerando tutti questi aspetti, si può ben capire come la sintesi inizialmente individuata non abbia le caratteristiche adeguate per un trasferimento su larga scala.
Per questo motivo era iniziata, all’interno del Dipartimento di Sviluppo Chimico della Menarini Ricerche, un’indagine tesa ad individuare una via sintetica alternativa che conducesse al pentapeptide 3.1 o ad un suo analogo senza dover rimuovere alcun gruppo protettivo benzilossicarbonilico (Z). Un eventuale analogo si poteva differenziare da 3.1 solo per il diverso tipo di gruppi protettori presenti ed al tempo stesso doveva essere in grado di proseguire la sintesi verso il Nepadutant. Questa indagine si era concentrata nell’individuazione del nuovo pentapeptide TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15) (Schema 3.3), che possiede tutte le caratteristiche di cui sopra (corretta sequenza degli amminoacidi e presenza di appropriati gruppi protettori per il proseguimento della sintesi) e per il quale era già stata proposta la sequenza sintetica. La nuova procedura (Schema 3.3) non prevedeva mai la rimozione del gruppo Z dal sistema peptidico in costruzione e questo potenzialmente poteva permettere di eliminare o almeno ridurre i punti critici fino ad allora evidenziati.
Lo scopo della mia tesi è stato quello di verificare la fattibilità della sintesi prevista per il pentapeptide 3.15, sia in termini di realizzazione dei singoli stadi, sia in termini di resa dei singoli passaggi e di integrità configurazionale degli intermedi sintetici e del pentapeptide finale 3.15.
Per questo scopo la mia attività sperimentale è stata svolta in una prima parte presso il Dipartimento di Scienze Farmaceutiche e in un secondo momento presso i laboratori della Menarini Ricerche a San Piero a Grado (PI). Nella prima parte
Risultati e discussione OH NH N H Boc Z O OSu NH N H Boc Z O HCl H-Leu-OMe HCl (3.5) H2N O O Me OMe N H O O NH Z HN Boc O Me N H O O NH Z H2N OSu O N H Boc OMe N H O NH Z O HN O N H Boc OMe O NH Z O HN O H2N OH OBn O H2N O OH OB n O N H O TFA TFA-Asp(OBn)-OH (3.25) OSu OBn O N H O TFA TFA-Asp(OBn)-OSu (3.26) N H H2N O OH H-Trp-OH (3.27) N H O OH H N O N H TFA O O Bn TFA-Asp(OBn)-Trp-OH (3.17) Boc-Dap(Z)-OH (3.18) Boc-Dap(Z)-OSu (3.19) Boc-Dap(Z)-Leu-OMe (3.20) H-Dap(Z)-Leu-OMe (3.21) Boc-Phe-OSu (3.22) Boc-Phe-Dap(Z)-Leu-OMe (3.23) H-Phe-Dap(Z)-Leu-OMe (3.16) OMe N H O NH Z HN O N H O H N O N H TFA O O Bn N H O TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15) H-Asp(OBn)-OH (3.24)
Risultati e discussione
ho curato essenzialmente l’aspetto sintetico, mirando all’ottenimento del prodotto finale di nostro interesse, il pentapeptide 3.15. Nella seconda parte ho invece curato l’importante aspetto del controllo strutturale dei singoli intermedi e del prodotto finale e riesaminato ogni singolo stadio anche dal punto di vista delle rese, ove necessario.
3.2 Sintesi alternativa dell’intermedio pentapeptidico TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe
Il nuovo pentapeptide 3.15, rispetto al precedente pentapeptide 3.1 preparato dalla Menarini Ricerche, presenta l’ammino gruppo del residuo aspartico protetto come trifluoroacetammide (TFA) invece che sotto forma di uretano (Z) ed il gruppo carbossilico della catena laterale dello stesso residuo aspartico protetto come estere benzilico invece di estere t-butilico. Il gruppo amminico della catena laterale dell’amminoacido non naturale 2,3-diamminopropionico è ora protetto con il gruppo Z invece che con il gruppo Boc (Schema 3.4).
OMe N H O NH Z HN O N H O H N O N H TFA O OBn N H O TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15) OMe N H O NH Boc HN O N H O H N O N H Z O OtBu N H O Z-Asp(OBn)-Trp-Phe-Dap(Boc)-Leu-OMe (3.1)
Schema 3.4 Pentapeptide 3.1 e nuovo pentapeptide 3.15.
Risultati e discussione
La via alternativa sintetica dell’intermedio pentapeptidico 3.15 (Schema 3.3), sperimentata durante il mio percorso di tesi, consiste nella sintesi non lineare della sequenza amminoacidica che ne costituisce la struttura.
Vengono prima preparati separatamente il tripeptide H-Phe-Dap(Z)-Leu-OMe (3.16), costruito in modo lineare, ed il dipeptide TFA-Asp(OBn)-Trp-OH (3.17). Successivamente i due segmenti vengono condensati mediante opportuna attivazione del gruppo carbossilico a dare il pentapeptide TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15), desiderato.
La sintesi segmentale, oltre a non presentare passaggi di rimozione del gruppo protettore Z, potenzialmente potrebbe avere il vantaggio di una migliore solubilità degli intermedi dipeptide 3.17 o tripeptide 3.16 in solventi organici, permettendo la riduzione dell’utilizzo della DMF, generalmente impiegata per la sintesi di peptidi a più alto peso molecolare che risultano più insolubili.
3.2.1 Sintesi del tripeptide H-Phe-Dap(Z)-Leu-OMe (3.16)
La sintesi del tripeptide 3.16, prende origine dall’attivazione dall’amminoacido non naturale diammino propionico Boc-Dap(Z)-OH (Schema 3.5).
3.2.1 a) Sintesi dell’estere attivo Boc-Dap(Z)-OSu (3.19)
L’amminoacido Boc-Dap(Z)-OH (3.18), prodotto commerciale, viene attivato come estere succinimmidico in CH2Cl2 mediante l’utilizzo della DCC e
dell’HOSu ottenendo l’amminoacido attivato 3.19 con elevata resa (97%) (Schema 3.5). OH O N H Boc NH Z CH2Cl2 DCC, HOSu OSu O N H Boc NH Z Boc-Dap(Z)-OH (3.18) Boc-Dap(Z)-OSu (3.19)
Risultati e discussione
L’estere attivo 3.19 Boc-Dap(Z)-OSu viene preparato sciogliendo l’amminoacido 3.18 nel solvente di reazione fino a completa dissoluzione e successivamente aggiungendo l’HOSu. La sospensione viene raffreddata a 0° C e a questa temperatura viene aggiunta la DCC. Dalla miscela di reazione precipita in maniera molto copiosa la DCU che viene eliminata per filtrazione. Il composto viene isolato evaporando il solvente sotto vuoto, ottenendo l’estere attivo 3.19 come un solido bianco amorfo.
3.2.1 b) Sintesi del dipeptide Boc-Dap(Z)-Leu-OMe (3.20)
Il coupling tra l’amminoacido attivato 3.19 e l’estere metilico della L-leucina come cloridrato H-Leu-OMe · HCl (3.5) è stato effettuato inizialmente in DMF, utilizzando la N-metilmorfolina come base (Schema 3.6).
NH Z O OSu HN Boc + HCl HN Boc HN O OMe NH Z O NMM DMF o CH2Cl2 H-Leu-OMe HCl (3.5) Boc-Dap(Z)-OSu (3.19) Boc-Dap(Z)-Leu-OMe (3.20) H2N O OMe
Schema 3.6 Sintesi del dipeptide Boc-Dap(Z)-Leu-OMe (3.20).
La reazione viene effettuata a temperatura ambiente e sciogliendo, fino a completa solubilizzazione, l’estere attivo 3.19 in DMF e aggiungendo di seguito l’amminoacido 3.5 e la N-metilmorfolina. Il dipeptide Boc-Dap(Z)-Leu-OMe (3.20), così ottenuto, è stato isolato mediante precipitazione per aggiunta di una soluzione di NaHCO3 0.5 N alla miscela di reazione. Il dipeptide 3.20 viene poi
filtrato ed essiccato sotto vuoto (resa 80%).
Considerato che l’estere attivo 3.19 era risultato solubile in CH2Cl2 ci è
sembrato interessante effettuare la reazione di coupling in questo solvente in modo da eliminare la DMF. In queste nuove condizioni di reazione si osserva la precipitazione dell’HOSu insolubile in CH2Cl2. In questo caso, per isolare il
Risultati e discussione
prodotto, si procede effettuando dei lavaggi della soluzione organica con NaHSO4
5% e con NaCl soluzione satura ed infine evaporando la soluzione organica. Anche in queste condizioni il dipeptide 3.20 viene ottenuto con buone rese (75%). Questo permette, quindi, di effettuare il coupling in CH2Cl2 e di eliminare la DMF
come solvente di reazione.
3.2.1 c) Sintesi del dipeptide deprotetto H-Dap(Z)-Leu-OMe (3.21)
Lo step seguente consiste nella rimozione del gruppo Boc dal residuo dell’ amminoacidico (S)-2,3-diamminopropionico del dipeptide 3.20 (Schema 3.7). La reazione viene effettuata sciogliendo il dipeptide in una miscela di CH2Cl2/CF3COOH, preparata a parte. Si ottiene il dipeptide deprotetto
H-Dap(Z)-Leu-OMe (3.21) che isolato risulta essere un solido.
OMe N H O O NH Z H2N H-Dap(Z)-Leu-(OMe) (3.21) OMe N H O O NH Z HN Boc CF3COOH CH2Cl2 Boc-Dap(Z)-Leu-OMe (3.20)
Schema 3.7 Sintesi del dipeptide deprotetto H-Dap(Z)-Leu-OMe (3.21).
3.2.1 d) Sintesi del tripeptide deprotetto Boc-Phe-Dap(Z)-Leu-OMe (3.23)
Lo step seguente consistente nel coupling fra il dipeptide 3.21 e l’amminoacido attivato Boc–Phe-OSu (3.22), commercialmente disponibile, con formazione del tripeptide Boc-Phe-Dap(Z)-Leu-OMe (3.23), viene effettuato in
DMF, utilizzando la base N-metilmorfolina, come nella sintesi tradizionale (Schema 3.8).
Risultati e discussione OMe N H O O NH Z H2N OMe N H O NH Z O HN O N H Boc O N H Boc OSu DMF + Boc-Phe-OSu (3.22) Boc-Phe-Dap(Z)-Leu-OMe (3.23) H-Dap(Z)-Leu-(OMe) (3.21)
Schema 3.8 Sintesi del tripeptide Boc-Phe-Dap(Z)-Leu-OMe (3.23).
La reazione di condensazione viene effettuata aggiungendo ad una soluzione del dipeptide deprotetto 3.21 in DMF l’amminoacido Boc-Phe-OSu (3.22) e l’N-metilmorfolina. Il tripeptide 3.23 viene isolato mediante precipitazione per aggiunta di una soluzione di NaHCO3 0.5 N.
La reazione è stata ripetuta anche senza l’impiego della base N-metilmorfolina, ottenendo il tripeptide 3.23 con un ottima resa (>91%) come nella sintesi tradizionale. Quindi, in questo caso, è possibile effettuare la reazione di coupling senza l’impiego della base.
3.2.1 e) Sintesi del tripeptide de protetto H-Phe-Dap(Z)-Leu-OMe (3.16)
Lo step finale che porta alla formazione del tripeptide H-Phe-Dap(Z)-Leu-OMe (3.16), consiste nella rimozione del gruppo Boc dal residuo amminoacidico della
L-fenilalanina del tripeptide 3.23 (Schema 3.9).
OMe N H O NH Z O HN O N H Boc Boc-Phe-Dap(Z)-Leu-OMe (3.23) OMe N H O NH Z O HN O H2N H-Phe-Dap(Z)-Leu-OMe (3.16) CH2Cl2 CF3COOH
Risultati e discussione
La reazione viene effettuata sciogliendo il composto 3.23 in una miscela CH2Cl2/CF3COOH. Procedendo come descritto per la deprotezione del dipeptide 3.20, si ottiene, dopo evaporazione del solvente organico, il tripeptide 3.16 in
forma di resina con resa di circa 80%.
3.2.2 Sintesi del dipeptide TFA-Asp(OBn)-Trp-OH (3.17)
La sintesi del dipeptide TFA-Asp-Trp-OH (3.17) inizia dalla reazione di protezione del gruppo amminico dell’acido aspartico H-Asp(OBn)-OH come derivato trifluoroacetoammidico.
3.2.2 a) Sintesi dell’amminoacido protetto TFA-Asp(OBn)-OH (3.25)
Seguendo una procedura già riportata in letteratura,13 la protezione viene effettuata per trattamento dell’acido aspartico monobenzilato H-Asp(OBn)-OH (3.24) con fenil trifluoroacetato (3.28), ottenendo il composto TFA-Asp(OBn)-OH (3.25) (Schema 3.10). OH O H2N O OBn O CF3 O OH O N H TFA O OBn +
H-Asp(OBn)-OH (3.24) Fenil trif luoroacetato (3.28) TFA-Asp(OBn)-OH (3.25)
Schema 3.10 Sintesi dell’intermedio 3.25 TFA-Asp(OBn)-OH via fenil
trifluoroacetato.
Il fenil trifluoroacetato (3.28) viene preparato di fresco distillando una soluzione di fenolo e anidride trifluoroacetica e raccogliendo la frazione con p.eb. 145°C, 14 (Schema 3.11).
Risultati e discussione OH CF3 O F3C O O CF3 O O +
Fenil trif luoroacetato (3.28)
Schema 3.11 Sintesi del fenil trifluoroacetato (3.28).
Successivamente l’acido aspartico benzil estere 3.24 [H-Asp(OBn)-OH] viene aggiunto al feniltrifluoroacetato e la miscela di reazione è riscaldata alla temperatura di 135°C, temperatura alla quale l’acido aspartico si solubilizza. Dopo raffreddamento il solido precipitato viene filtrato a dare l’ammide TFA-Asp(OBn)-OH (3.25), che viene purificata dal fenolo, sottoprodotto di reazione, effettuando numerose triturazioni con etere di petrolio. In alternativa l’ammide
3.25 è stata purificata per estrazione acido/base, ottenendo l’amminoacido
protetto 3.25 in buona resa (87%).
Poichè il metodo utilizzato e appena descritto prevede la fase di riscaldamento iniziale che avrebbe potuto determinare anche una parziale racemizzazione dell’amminoacido, abbiamo pensato di effettuare la protezione dell’amminoacido H-Asp(OBn)-OH utilizzando l’etil trifluoroacetato in MeOH, secondo un metodo alternativo che non prevede alcun riscaldamento15. Il protocolloprevede il trattamento dell’acido aspartico monobenzilato in metanolo con l’etil trifluoroacetato in presenza di trietilammina (2 eq). (Schema 3.12).
CF3 O C H2 O H3C + OH O N H TFA O OBn OH O H2N O OBn MeOH NEt3 H-Asp(OBn)-OH (3.24) TFA-Asp(OBn)-OH (3.25)
Schema 3.12 Sintesi dell’amminoacido protetto TFA-Asp(OBn)-OH (3.25) via etil
trifluoroacetato.
Dopo evaporazione del solvente di reazione, il residuo viene ripreso in diclorometano, lavato acido/base, per eliminare il cloridrato di trietilammina, ed il
Risultati e discussione
composto protetto 3.25 viene isolato per concentrazione della soluzione seguita da ripetute triturazioni con esano.
3.2.2 b) Sintesi dell’estere attivo TFA-Asp(OBn)-OSu (3.26)
L’attivazione dell’acido aspartico protetto 3.25 (TFA-Asp(OBn)-OH) come estere succinimmidico, per dare il derivato attivato 3.26 (TFA-Asp(OBn)-OSu) è stata effettuata via DCC e HO-Su in CH2Cl2. Dalla miscela di reazione precipita
la DCU, che viene filtrata e il composto 3.26 viene isolato dopo evaporazione del solvente di reazione (Schema 3.13).
OH O N H TFA O OBn OSu O N H TFA O OBn DCC, HOSu TFA-Asp(OBn)-OH (3.25) TFA-Asp(OBn)-OSu (3.26) CH2Cl2
Schema 3.13 Sintesi dell’estere attivo TFA-Asp(OBn)-OSu (3.26).
3.2.2 c) Sintesi del dipeptide TFA-Asp(OBn)-Trp-OH (3.17)
Lo step successivo prevede la reazione tra l’estere attivo TFA-Asp(OBn)-OSu (3.26) e l’amminoacido L-triptofano (3.27) per ottenere il dipeptide TFA-Asp(OBn)-Trp-OH (3.17) (Schema 3.14). OSu O N H TFA O OBn + N H O OH H2N N H O OH H N N H TFA O OBn DMF NMM TFA-Asp(OBn)-OSu (3.26) H-Trp-OH (3.27) O TFA-Asp(OBn)-Trp-OH (3.17)
Risultati e discussione
Il coupling viene effettuato in DMF, a temperatura ambiente, in presenza della base N-metilmorfolina (1 eq). Una volta terminata la reazione, la soluzione viene evaporata al rotavapor ed alla pompa da vuoto per eliminare la DMF. Il composto che si ottiene viene sciolto in AcOEt. L’isolamento del dipeptide TFA-Asp(OBn)-Trp-OH (3.17) viene condotto per estrazione acido/base del prodotto grezzo seguito da evaporazione del solvente. Si ottiene un prodotto solido con buona resa (72%) e purezza cromatografica (78.5%).
All’analisi 1H-NMR il dipeptide 3.17 risulta essere costituito da una miscela di due diastereoisomeri in rapporto 80/20. Il dato è stato confermato mediante analisi HPLC, utilizzando una colonna con fase stazionaria specifica per la separazione di isomeri strutturali. Nel corso della reazione si era pertanto verificato un indesiderato, anche se possibile, processo di epimerizzazione che andava a compromettere l’efficacia dell’intero processo sintetico alternativo a cui stavamo lavorando, in quanto avrebbe condotto ad un pentapeptide 3.15 anch’esso non diastereoisomericamente puro.
Tentativi di purificazione del dipeptide 3.17 mediante cristallizzazione frazionata avevano condotto al dipeptide 3.17 puro solo in una occasione per precipitazione da una soluzione in AcOEt per aggiunta di esano. Sfortunatamente la procedura si era dimostrata non riproducibile. E’ stato pertanto necessario approfondire lo studio della sintesi del dipeptide 3.17 al fine di comprendere lo stadio critico per l’integrità configurazionale e di introdurre opportune modifiche per eliminare o almeno ridurre il processo di epimerizzazione.
3.2.2 d) Studio del processo di epimerizzazione
Il primo step della sintesi del dipeptide 3.17 è la protezione dell’acido aspartico monobenzilato 3.24 come trifluoroacetammide che è stata effettuata con due protocolli diversi, di cui uno prevede una reazione a temperatura elevata (135°C), mentre l’altro prevede l’utilizzo di un eccesso (2eq.) di trietilammina.
In linea di principio entrambe queste procedure potrebbero comportare una parziale racemizzazione al centro chirale dell’amminoacido. In effetti i trifluoroacetil amminoacidi sono stati poco utilizzati nella sintesi peptidica, perchè i primi tentativi in cui veniva utilizzata l’anidride trifluoroacetica per la
Risultati e discussione
protezione comportavano perdita dell’integrità stereochimica. Abbiamo pertanto deciso di determinare la purezza ottica dell’amminoacido protetto TFA-Asp(OBn)-OH (3.25) e, a tale scopo, abbiamo preparato il corrispondente enantiomero a partire dall’acido D-Aspartico monobenzilato, prodotto disponibile in commercio. L’acido H-DAsp(OBn)-OH è stato protetto seguendo la procedura che prevede l’utilizzo di fenil trifluoroacetato, ottenendo il composto TFA-DAsp(OBn)-OH (ent-3.25). I due N-trifluoroacetil amminoacidi enantiomeri sono stati quindi analizzati mediante HPLC con una colonna chirale e sono state individuate le condizioni per separare i 2 isomeri ottici con ottima risoluzione. La purezza enantiomerica dell’amminoacido protetto 3.25 [TFA-Asp(OBn)-OH] è risultata molto elevata sia nel caso in cui viene preparato via fenil trifluoroacetato ( ee > 99%), sia nel caso in cui viene preparato mediante etil trifluoroacetato ( ee 100%).
In base a questo risultato è stato quindi possibile dedurre che il processo di epimerizzazione che porta alla formazione dei due diasteroisomeri del dipeptide
3.17 avviene nella successiva reazione di coupling tra l’amminoacido protetto 3.25 e il triptofano 3.27.
Nel tentativo di ridurre la formazione del diastereoisomero indesiderato, abbiamo provato ad effettuare il coupling in condizioni diverse.
Inizialmente l’attivazione è stata effettuata mediante l’impiego dell’EDC al posto della DCC, ma in questo caso la reazione risulta molto più lenta ed il dipeptide ottenuto, TFA-Asp(OBn)-Trp-OH (3.17), mostra un rapporto diastereoisomerico peggiore (45/55).
Nonostante sia noto che i peptidi protetti come acil ammidi sono maggiormente soggetti a racemizzazione rispetto ai rispettivi carbammati, in letteratura 16 sono riportate reazioni di coupling via cloruro acilico in condizioni controllate per diversi N-trifluoroacetil amminoacidi e il dipeptide finale è ottenuto con alta ritenzione di configurazione. La condensazione tra il cloruro acilico e il secondo amminoacido avviene in condizioni di Schotten-Baumann a pH neutro. L’attivazione può essere effettuata mediante il reattivo di Vilsmeier a bassa temperatura (-10°C) ottenendo peptidi con valori molto elevati di purezza ottica (percentuale di epimerizzazione inferiore allo 0.5%) o utilizzando cloruro di
Risultati e discussione
ossalile con DMF catalitica, a temperatura ambiente, che provoca solo una leggera racemizzazione (2-3%).
Abbiamo applicato la procedura descritta in letteratura via cloruro di ossalile per la sintesi del dipeptide TFA-Asp(OBn)-Trp-OH (3.17), operando a bassa temperatura (0°C), ma anche in queste condizioni abbiamo osservato una epimerizzazione praticamente completa (55/45).
Questi dati hanno evidenziato l’estrema criticità della formazione del legame peptidico del dipeptide 3.17.
Poichè i risultati migliori erano stati osservati utilizzando, come metodo di attivazione dell’acido carbossilico, la formazione dell’estere attivo dell’N-idrossisuccinimmide, abbiamo deciso di studiare l’effetto dei vari parametri di reazione sulla stereochimica del prodotto. In particolare abbiamo provato a investigare l’effetto del solvente, della temperatura e della base.
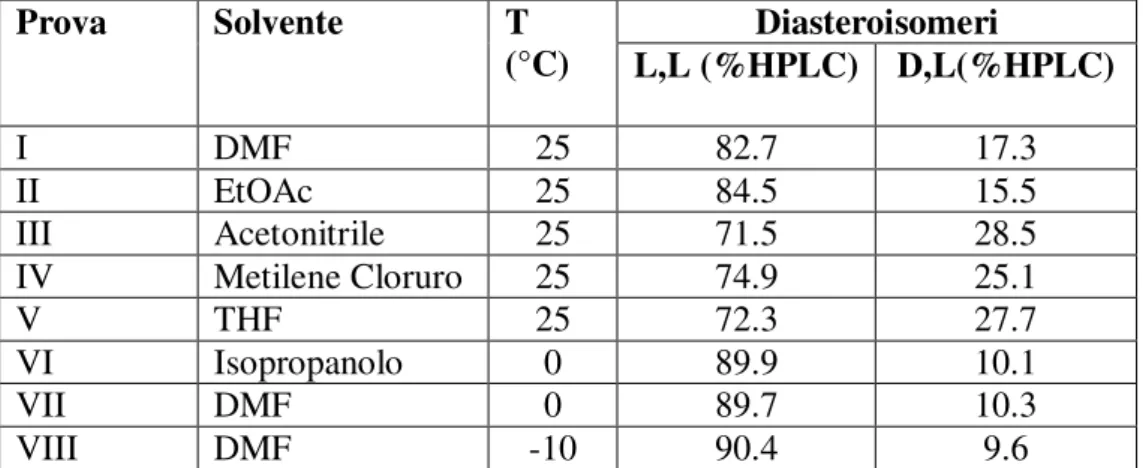
In Tabella 3 sono riportate le prove di coupling condotte in condizioni analoghe, aggiungendo l’estere attivo TFA-Asp(OBn)-OSu 3.26 ad una sospensione di L-triptofano 3.28 con 1 equivalente di N-metilmorfolina come base, ma utilizzando diversi solventi ed operando sia a temperatura ambiente che a bassa temperatura. Tutte le reazioni sono a completezza dopo 1-2 ore di agitazione ed il dipeptide è solubile ad eccezione di quando viene utilizzato l’isopropanolo come solvente.
Tabella 3.1 - Effetto del solvente e della temperatura nella reazione di coupling
di TFAAsp(OBn)OSu con HTrpOH (1.05 eq) in presenza di N -metilmorfolina (1.05eq.)
Prova Solvente T Diasteroisomeri
(°C) L,L (%HPLC) D,L(%HPLC) I DMF 25 82.7 17.3 II EtOAc 25 84.5 15.5 III Acetonitrile 25 71.5 28.5 IV Metilene Cloruro 25 74.9 25.1 V THF 25 72.3 27.7 VI Isopropanolo 0 89.9 10.1 VII DMF 0 89.7 10.3 VIII DMF -10 90.4 9.6
Risultati e discussione
Come si può osservare a temperatura ambiente risultati simili a quelli ottenuti con la DMF sono osservati in AcOEt, mentre il rapporto diastereoisomerico è inferiore, intorno a 70/30, con altri solventi quali acetonitrile, diclorometano e THF. Una variabile importante è la temperatura: i risultati infatti mostrano una minore percentuale di racemizzazione al diminuire della temperatura, con rapporto tra i due epimeri del dipeptide di 90/10 a 0°C sia utilizzando DMF che isopropanolo come solvente. La reazione in isopropanolo avviene in sospensione ed ha l’inconveniente della scarsa solubilità sia del pecursore L-triptofano che del prodotto dipeptide 3.17. Un’ulteriore diminuzione di temperatura a -10°C non è accompagnata da un aumento consistente dell’epimero desiderato.
Il meccanismo di racemizzazione comunemente riportato per la sintesi peptidica prevede la formazione dell’intermedio ciclico ossazolone, che in presenza di una base può perdere il protone legato all’atomo di carbonio chirale e portare alla formazione di 2 ossazoloni enantiomeri e, successivamente, ai due epimeri del peptide. Nelle reazioni di coupling del trifluoroacetil amminoacido TFA-Asp(OBn)-OSu (3.26) con H-Trp-OH (3.27)è stata utilizzata la N-metilmorfolina come base, in quantità stechiometrica, necessaria per liberare l’ammino gruppo dell’amminoacido L-triptofano presente come forma zwitterionica nel solvente organico e renderlo disponibile per la reazione con l’estere attivo 3.26. Poichè all’inizio il triptofano, anche in presenza della base, non è completamente solubile nel solvente di reazione, abbiamo ipotizzato che eventuali tracce di base libera in soluzione potessero catalizzare la racemizzazione dell’intermedio ossazolone. Abbiamo pertanto provato a sostituire l’N-metilmorfolina con il tetrabutilammonio idrossido17, che forma un sale solubile in solventi organici con l’amminoacido L-triptofano. La reazione di coupling è stata condotta a temperatura ambiente, in DMF o diclorometano come solvente, aggiungendo l’estere attivo TFA-Asp(OBn)-OSu (3.26) alla soluzione del sale dell’L-triptofano con tetrabutilammonio idrossido. I risultati ottenuti sono stati paragonabili a quelli delle reazioni in sospensione in presenza di N-metilmorfolina: il rapporto tra i due epimeri del dipeptide 3.17 risulta 81/19 in DMF e 69/31 in diclorometano.
Considerando sempre l’ipotesi che un’eventuale presenza di tracce di base in eccesso possa catalizzare la racemizzazione dell’intermedio ossazolone, abbiamo
Risultati e discussione
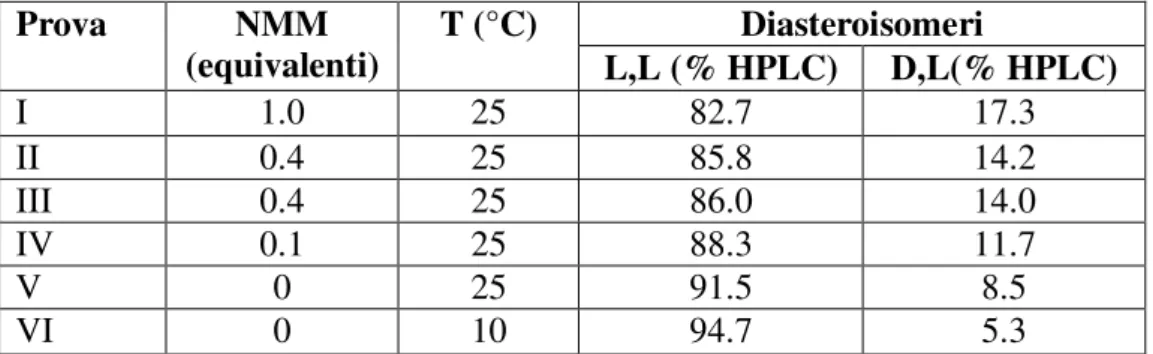
indirizzato il nostro studio verso condizioni di coupling in cui venisse impiegato un difetto di base. Un inconveniente poteva essere legato alla scarsa reattività dell’L-triptofano che, se presente come zwitterione, non ha il gruppo amminico libero e disponibile per la formazione del legame peptidico. In letteratura18 è riportato che per alcuni amminoacidi, a seconda del tipo di solvente presente nella miscela di reazione, cambia il rapporto fra la forma neutra, che può quindi reagire con l’estere attivo, e la forma zwitterionica. Poiché la DMF è riportata fra i solventi che favoriscono la forma neutra abbiamo pensato di mantenere come solvente per il coupling la DMF, di operare inizialmente a temperatura ambiente e di ridurre la quantità di N-metilmorfolina a 0.4 equivalenti. Due prove condotte nelle stesse condizioni hanno dato risultati incoraggianti e riproducibili, portando ad un dipeptide 3.17 con rapporto diastereoisomerico pari a 86/14. Riducendo ulteriormente gli equivalenti di base (0.1 eq) si conferma l’effetto positivo sulla purezza stereoisomerica, come mostrato dai risultati riportati in Tabella 3.2.
Tabella 3.2 - Effetto della base e della temperatura nella reazione di coupling di
TFA-Asp(OBn)-OSu con H-Trp-OH (1.05 eq) in DMF in presenza di N-metilmorfolina.
Infine è stata effettuata una prova in assenza di base: in queste condizioni abbiamo osservato che il trifluoroacetil amminoacido attivato 3.26 reagisce completamente con l’ L-triptofano, anche se più lentamente (4-5 ore), e si forma il dipeptide 3.17 con un rapporto diastereoisomerico di 91/9.
Il solvente fortemente polare, quindi, gioca un ruolo importante, in quanto favorisce la presenza in soluzione della forma neutra dell’amminoacido
L-Diasteroisomeri Prova NMM (equivalenti) T (°C) L,L (% HPLC) D,L(% HPLC) I 1.0 25 82.7 17.3 II 0.4 25 85.8 14.2 III 0.4 25 86.0 14.0 IV 0.1 25 88.3 11.7 V 0 25 91.5 8.5 VI 0 10 94.7 5.3
Risultati e discussione
triptofano, che reagisce con l’estere spostando continuamente l’equilibrio della specie zwitterionica verso la forma neutra.
L’ultima prova riportata nella Tabella 3.2 è stata condotta a temperatura più bassa, considerando che le prove precedenti avevano mostrato un effetto positivo della diminuzione di temperatura sull’integrità stereochimica. La reazione di coupling è stata condotta in assenza di base, aggiungendo ad una sospensione di L–triptofano in DMF a 0°C l’acido attivato TFA-Asp(OBn)-OSu (3.26). La miscela di reazione è stata mantenuta per 3 ore a 0°C, ma in queste condizioni la reazione è risultata piuttosto lenta, per cui la temperatura è stata portata a 10°C per le successive 8 ore. Anche in questo caso il coupling è completo e si forma il dipeptide TFA-Asp(OBn)-Trp-OH (3.17) con un rapporto diastereisomerico 95/5.
Con quest’ultima procedura è stato preparato il dipeptide 3.17 su scala 5g da utilizzare per il successivo coupling con il tripeptide 3.16. Dopo evaporazione della DMF sotto vuoto, il residuo è stato ripreso con AcOEt e, dopo lavaggi acido base, il dipeptide 3.17 è stato ottenuto come solido con buona resa (97%) e purezza (92%). Il rapporto tra i due epimeri è risultato di 95/5, confermando che la procedura è riproducibile e permette di ottenere il dipeptide 3.17 desiderato con buona purezza chirale.
3.2.3 Sintesi del pentapeptide TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15)
La reazione di coupling tra i 2 segmenti, il tripeptide H-Phe-Dap(Z)-Leu-OMe (3.16) ed il dipeptide TFA-Asp(OBn)-Trp-OH (3.17), viene effettuata mediante attivazione del gruppo carbossilico come estere succinimmidico (Schema 3.14).
3.2.3.a Sintesi dell’estere attivo TFA-Asp(OBn)-Trp-OSu (3.29)
Come descritto per la formazione dell’estere succinimmidico 3.26 il dipeptide (3.17) è stato attivato mediante l’utilizzo della DCC e l’HO-Su in CH3CN. Viene
utilizzato come solvente di reazione l’acetonitrile al posto del diclorometano, in quanto il dipeptide (3.17) non è solubile in CH2Cl2. L’acetonitrile è comunque tra
Risultati e discussione
solubile. Dalla miscela di reazione precipita la DCU, che viene filtrata e l’estere attivo 3.29 viene isolato dopo aver evaporato la soluzione (Schema 3.14).
TFA-Asp(OBn)-Trp-OH (3.17) TFA-Asp(OBn)-Trp-OSu (3.29) DCC, HOSu N H O OH H N N H TFA O OBn O N H O OSu H N N H TFA O OBn O CH2Cl2
Schema 3.14 Sintesi dell’estere attivo TFA-Asp(OBn)-Trp-OSu (3.29).
3.2.3.b Sintesi del pentapeptide TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15)
La reazione che porta alla formazione del pentapeptide TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15) consiste nel coupling fra il tripeptide H-Phe-Dap(Z)-Leu-OMe deprotetto (3.16) e l’estere attivo TFA-Asp(OBn)-Trp-OSu (3.29) (Schema 3.15). TFA-Asp(OBn)-Trp-OH (3.29) N H O OSu H N N H TFA O OBn O OMe N H O NH Z HN O N H O H N O N H TFA O OBn N H O TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15) OMe N H O NH Z O HN O H2N H-Phe-Dap(Z)-Leu-OMe (3.16) +
Schema 3.15 Sintesi del pentapeptide TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe
Risultati e discussione
La reazione viene effettuata sciogliendo il tripeptide 3.16 in DMF e aggiungendo a 0° C l’estere attivo 3.29.
La temperatura della reazione viene in seguito portata a temperatura ambiente. La reazione di coupling è molto veloce, infatti nell’arco di 30 minuti si è già formato il pentapeptide TFA-Asp(OBn)-Trp-Phe-Dap(Z)-Leu-OMe (3.15). Una volta isolato il pentapeptide 3.15 viene ottenuto con buona purezza HPLC = 87%. La purezza diastereoisomerica del pentapeptide 3.15 è stata valutata mediante 1 H-NMR per confronto di campioni di pentapeptide sintetizzati a partire da dipeptidi
3.17 con rapporti diastereoisomerici variabili. Facendo questi confronti si è
rivelato particolarmente diagnostico il segnale della trifluoroacetammide infatti, nei campioni ottenuti partendo da dipeptidi non diastereoisomericamente puri, accanto al doppietto relativo al protone della trifluoroacetammide (9.64 ppm) è presente un altro doppietto (9.72 ppm) assegnabile alla trifluoroacetammide del pentapeptide epimero. Il rapporto tra gli integrali di questi due segnali nel pentapeptide 3.15 è analogo a quello misurato tra i corrispondenti segnali della trifluoroacetammide nel dipeptide 3.17 di partenza. La corretta attribuzione dei segnali è stata confermata dall’assenza del secondo doppietto del segnale della trifluoroacetammide nel pentapeptide ottenuto dal dipeptide diastereoisomericamente puro.
Quando è stato utilizzato un dipeptide 3.17 con il 5% di diastereoisomero indesiderato, anche il pentapeptide grezzo ottenuto 3.15 presenta effettivamente il doppietto a 9.72 ppm ed il rapporto tra gli integrali indica una quantità di epimero leggermente inferiore al 5%.
Una prova di triturazione con Etile Acetato ha permesso di migliorare la purezza diastereoisomerica del pentapeptide 3.15, riducendo la presenza di epimero a valori inferiori all’1%.
Questo risultato appare particolarmente interessante perchè individuando un opportuno solvente di cristallizzazione potrebbe essere possibile ottenere il pentapeptide 3.15 stereoisomericamente puro.