UNIVERSITY OF PISA
PhD Course in Basic and Developmental Neuroscience
Identification of neuronal substrates of Rett Syndrome:
electrophysiological and
neuroimaging studies
Tutors Candidate
Dr. Roberta Battini Manuela Rita Casarano Prof. Giovanni Cioni
CYCLE XXVI (2011-2013)
CONTENTS
Foreword………4
Acknowledgements………..6
1. Clinical background………..7
Core manifestations of Rett syndrome……….11
Specific variant forms of atypical RTT………..14
2. Rett syndrome and MECP2 gene………17
Genotype-phenotype correlation………..19
3. Identification of neuronal substrates of Rett syndrome: electrophysiological and neuroimaging studies………..23
3.1 Multimodal neurophysiological evaluation Introduction………...28
Subjects ………...29
Methods……….29
Results………...31
Discussion………. ………..36
3.2 Assessment of brainstem function and delineation of cardiorespiratory phenotypes Introduction………...39
Subjects………....42
Methods………....42
3.3 Voxel Based Morphometry (VBM) and Diffusion Tensor Imaging (DTI) studies Introduction………..45 Subjects………46 Methods………48 Results………...50 Discussion………....58
3.4 Cerebral blood flow in Rett patients with MECP2 mutation Introduction………..59
Subjects and Methods………...60
Results……….64
Discussion………...74
3.5 General conclusions and future prospects……….78
Appendix I: International Scoring System………..82
Appendix II: Arterial Spin Labeling………..83
Appendix III: Scientific reports around the topic of this PhD thesis………...85
Appendix IV: Scientific reports published during PhD course……….87
Foreword
The topic of this PhD thesis is the Rett syndrome (RTT, OMIM 312750), an X-linked neurodevelopmental condition characterized by loss of spoken language and hand use with the development of distinctive hand stereotypies, originally described in the 1960s by Andreas Rett [Rett1966].
The interest for RTT stems from the consideration that, although a rare disease, it is responsible for a high percentage of cases of mental retardation in children.
The clinical diagnosis has been based on consensus clinical criteria [Hagberg et al 2002], which have been slightly modified over time to reflect increased
understanding of the disease features. More than 95% of individuals with RTT
have mutations in methyl-CpG-binding protein 2 gene (MECP2), whose protein product modulates gene transcription.
Currently, no effective treatment is available for this disorder although many trials were conducted on RTT patients and animal studies have suggested that RTT symptoms are potentially reversible.
The aim of this research project, supported by TELETHON (Application GGP09196), is to identify specific biomarkers of RTT using neurophysiological and neuroimaging techniques on a group of 16 RTT girls with proven MECP2 mutation. The identification of potential biomarkers of RTT syndrome could be relevant to monitoring both the progression of disease and the response to experimental treatment.
The first part of this PhD thesis including the first two chapters describing the introduction to the RTT syndrome focused mainly on clinical and genetic aspects of the disease.
From the chapter 3 starts the second part of the work representing the main core of the research project: firstly, an introduction and overall aims and methodological approaches to the study; secondly, a detailed description of the neurophysiological and imaging studies, each organized in subsections.
The multimodal neurophysiological approach allowed us to evaluate the amount of impairment of central motor and sensory pathways in this disorder and, to our knowledge, this type of study is the first reported on RTT after the
The neuroimaging including Voxel Based Morphometry (VBM), Diffusion Tensor Imaging (DTI) and Arterial Spin Labeling (ASL) studies. VBM findings seemed to further confirm the global hypoplasia of RTT brain, already reported in literature, with preferential involvement of GM, rather than a progressive reduction of brain tissue. In DTI study our sample showed a reduction of Fractional Anisotropy in the same areas (corpus callosum, centrum semiovale..) reported in literature but only in younger RTT girls.
Finally, the application of ASL technique to RTT was the first report in literature toward an analysis performed on case-control study, also in relation to the different RTT clinical characteristics (epilepsy, cardiorespiratory phenotype, disease severity). Almost all patients showed elevated cerebral perfusion values in comparison with controls of same age and we discussed the hypothesis of a brain immaturity in RTT more than a neurodegenerative process.
The thesis concludes with a general discussion of the topic and the future perspectives on the importance to corroborate the potential role of the multimodal neurophysiological approach and of the ASL as markers of RTT disease.
Acknowledgements
Thanks to all the families and children who participated in the study.
I would like to thank professor Giovanni Cioni for the opportunity he gave me to continue my studies with a PhD course.
Many other people have contributed to my personal and professional growth, first of all Roberta Battini who guided me with patience and dedication in the complex word of rare diseases.
A special thanks goes to Laura Biagi, Giulia Valvo, Francesco Mari and dr. Giorgio Pini for their expertise and moral support, essential for the conduct of the study.
Finally, many thanks to my family, in a special way to the “evergreen” grandparents, for the help and love given to my little Alessandro.
1. CLINICAL BACKGROUND TO RETT SYNDROME
Rett syndrome (RTT-OMIM #312750) is a monogenic postnatal developmental disorder that affects normal brain development during early childhood primarily in females, with an incidence of 1 in 10,000 live births [Chahrour and Zoghbi 2007].
Although a rare disease, it is responsible for a high percentage of cases of mental retardation in children, second only to the incidence of Down syndrome.
Most of the RTT cases are sporadic (99%) and in up to 97% of classic RTT the disease is associated with mutations in the X-linked methyl-CpG-binding protein 2 (MECP2) gene [Amir et al 1999] suggesting that RTT is caused by mutations in this gene [Neul et al 2008].
RTT patients appear to reach developmental milestones seemingly normally until about 6-18 months after birth when signs and symptoms of disease begin to appear. This typically includes stereotypic hand movements, motor coordination deficits, seizures, language and learning disabilities and mild to severe cognitive impairments [Hagberg et al 1983; Nomura 2005; Chahrour and Zoghbi 2007].
The first description of RTT syndrome was reported by Andreas Rett in the 1960s [Rett 1966].
In a seminal article Hagberg et al [1983] characterized the specific clinical features and initiated the eponym by which we recognize this clinical condition. The clinical diagnosis, based on consensus clinical criteria [Hagberg et al 2002], included as essential criterion for classical RTT a documented loss, usually at the age of 1–2 years, of early acquired developmental skills, i.e. finefinger function/practical hand use, learned single words/babble, communicative behaviour, and expected personality profile. The diagnostic criteria have been modified slightly over time to reflect increased understanding of the disease features, but have retained certain key clinical elements to make the diagnosis of classic, or typical, RTT. In addition to typical RTT, it has been recognized that some individuals present with many of the clinical features of RTT, such as
regression, but do not necessarily have all of the features of the disorder. These have been termed ‘‘variant’’ or ‘‘atypical’’ RTT and have been found to cluster in some distinct clinical groups, such as preserved speech variant, early seizure variant, and congenital variant [Hagberg et al 1994].
To address some of the confusion that currently exists regarding the diagnosis of RTT, the RettSearch Consortium participated in an iterative process to come to a consensus on revised and simplified diagnostic criteria for RTT. RettSearch is an international network of clinically oriented Rett syndrome researchers, composed of experts in RTT from 13 different countries, which was initially established in 2006 through a meeting grant from the National Institutes of Health and additional support from the International Rett Syndrome Association (IRSA). Neul et al [2010] developed revised diagnostic criteria to clarify and simplify the diagnosis of typical RTT limiting the necessary criteria to the presence of regression plus four main criteria that are absolutely required for the diagnosis of typical RTT (Table 1). The clinical picture associated with typical RTT is now defined by a regression of purposeful hand use and spoken language, with the development of gait abnormalities and hand stereotypies. After the period of regression, a stage of stabilization and potentially even improvement ensues, with some individuals partially regaining skills. The authors eliminated postnatal deceleration in head growth from the necessary criteria because this feature in not found in all individuals with typical RTT [Hagberg et al 2000], but was included as a preamble to the criteria as a feature that should raise suspicion for the diagnosis.
Table 1. Revised Diagnostic Criteria for RTT [Neul et al 2010]
Consider diagnosis when postnatal deceleration of head growth observed.
Required for typical or classic RTT
1. A period of regression followed by recovery or stabilization 2. All main criteria and all exclusion criteria
3. Supportive criteria are not required, although often present in typical RTT
Required for atypical or variant RTT
1. A period of regression followed by recovery or stabilization 2. At least 2 of the 4 main criteria
3. 5 out of 11 supportive criteria
Main criteria
1. Partial or complete loss of acquired purposeful hand skills 2. Partial or complete loss of acquired spoken language
3. Gait abnormalities: impaired (dyspraxic) or absence of ability 4. Stereotypic hand movements
Exclusion criteria for typical RTT
1. Brain injury secondary to trauma (peri- or postnatally), neurometabolic disease, or severe infection that causes neurological problems
2. Grossly abnormal psychomotor development in first 6 months of life
Supportive criteria for atypical RTT
1. Breathing disturbances when awake 2. Bruxism when awake
3. Impaired sleep pattern 4. Abnormal muscle tone
5. Peripheral vasomotor disturbances 6. Scoliosis/kyphosis
7. Growth retardation
The evolution of RTT usually follows four stages [Hagberg et al 1992].
Stage I (early onset or stagnation period) starts between 6 and 18 months and lasts from a few months to more than a year. It is sometimes overlooked because symptoms are usually subtle. Decreased eye contact and reduced interest in toys are accompanied by delays in gross motor skills (sitting or crawling). Slowing of head growth appears as does occasional subtle hand-wringing.
Stage II (rapid destructive [regression] stage) usually begins between ages 1 and 4 years and lasts for weeks to months. Hallmarks are loss of language and motor skills and appearance of stereotypic hand movements, social withdrawal with autistic-like behaviour accompanied by extreme irritability with screaming and crying episodes, disturbed sleep, and occasional feeding difficulties. Breathing abnormalities and vasomotor changes start to appear, accompanied by gait apraxia and evolving limb spasticity. Slowing of head growth continues. Stage III (plateau or pseudostationary stage) starts between ages 2 and 10 years and lasts for years. Apraxia, motor problems, and seizures are prominent; however, there is usually improvement in behaviour, less irritability and autistic-like features, better social and communication skills, improved alertness, and longer attention span. Many girls remain in this stage for most of their lives. Stage IV (late motor deterioration stage) is characterized by reduced mobility related to increased rigidity, spasticity, and dystonic posturing accompanied by aggravation of scoliosis. Girls can lose their independent or partially dependent walking abilities. Part of these negative changes may be related to decreased motor interventions in educational and home setting environments; however, there is no decline in cognition, communication, or hand skills.
Although initially recognized only in girls, boys who meet the criteria for typical RTT have been identified [Christen et al 1995] and thus should be considered to have typical RTT.
CORE MANIFESTATIONS OF RETT SYNDROME
Stereotypic hand movements
Almost continuous repetitive wringing/squeezing, clapping/tapping, mouthing
and washing/rubbing hand automatisms during wakefulness that constitute the
hallmark of the condition. Breathing irregularities
Peculiar and disorganized breathing disturbances are the rule in these patients. They usually consist of irregularly occurring episodes of intensive hyperventilation, often disrupted by apneic breath-holding episodes usually lasting up to 30–40 seconds. The apneic pauses can be threateningly long and are often accompanied by Valsalva maneuvers. The breathing abnormalities only occur during wakefulness. Insights into hyperventilation in RTT have recently been reviewed [Kerr and Julu 1999; Julu et al 2001, 2005, 2008].
Abnormal muscular tone
Many RTT girls develop successively, through school age, a complex distal lower limb deformative pattern. The feet become more and more fixed in a rigid supinated flexed position, hampering standing and walking. This obviously results from a mixture of slowly appearing, and progressive, distal dystonia which gradually conceals the original obvious yet milder lower limb spasticity. These dystonic features are very often asymmetric, usually with right-sided domination (unpublished observations). With increasing age, the asymmetric dystonia successively results in various patterns of inverted plantar-curved malpositions of the toes, sometimes grotesque in type.
Scoliosis/kyphosis
A scoliosis of neurogenic type with a double curve deformation is a very important and threatening deformity. A double curve of this kind appears, with large variations in severity, in many classical RTT females. Characteristically, it develops successively from early school age and is more rapid and pronounced than in many other types of neurogenic scoliosis in childhood. The type of double curve most frequently seen has a longer upper curve (most often right convex) and a shorter lower one (then left convex). Floppiness, neuromuscular insufficiency, and extrapyramidal asymmetries (dystonic features) indicate the
threat of rapid development of the scoliosis. In such situations, active surgical corrections with spinal fusions should not be delayed. In contrast, in those without neurological asymmetries, deformities of the spine are usually much more benign. Among them, isolated high kyphosis deformities are not uncommon and surgical correction is then often not necessary.
Small cold hands and feet
Many RTT females display a disproportionate early deceleration in growth in their feet, more than would correlate to their frequently reduced total body size. Those with remarkably small, cold, bluish-red feet in adolescence tend to develop trophic skin and nail changes. In some, episodically occurring profuse sweating of the feet is also noted. It has been suggested that deranged autonomic regulation could explain the abnormal biology behind this [Kerr and Julu, 1999].
Intensive eye communication
Intense staring to obtain eye communication, or express wishes, is a prominent feature in most RTT girls. Usually, it appears after the end of the regression period. In schoolgirls and adolescents, this behavioural pattern has often become further strengthened. Well-functioning RTT females even tend to develop a primitive yet sophisticated “eye pointing” language as a substitute for their loss of speech and fine motor communication skills. This has led creative teachers to develop useful methods of eye communication for use in habilitation programs [Djukic et al 2012].
Feeding abnormalities— gastrointestinal symptomatology
This symptomatology is quite common and can sometimes be very alarming and threatening. Disturbances to the autonomic nervous system are considered to be of major importance as causative. Swallowing dysfunction and deranged mobility of the upper gastrointestinal tract have been well documented [Motil et al 1999]. Gastroesophageal reflux occurs frequently and constipation occurs in almost all RTT girls, but the clinical pattern is unspecific in type and has been difficult to delineate.
Disorder of posture and locomotion
The abnormal muscular tone in RTT in one aspect of the severely disorganized motor control affecting posture and movement. Perception seems to be involved as well as execution. Some girls who can walk seem to prefer to do so on
tiptoe. Truncal ataxia and jerky tremor are often observed in RTT girls. Permanent contractures of the joints develop most frequently in those with the most abnormal tone, hypotonic, hypertonic or dystonic and may occur rapidly if posture is poor [Witt Engerstrom 1990].
Disorder of voluntary movements
Voluntary movements peculiarly disabled in RTT syndrome, however some spontaneous fluent and useful movement does remain after regression and this may facilitate swimming, adjustment of the sitting position even on horseback, grasping a swing appropriately and body movement to music. The majority of people with RTT have minimal hand use and although objects of interest may be approached, studied and tapped they are not explored.
Seizures
Epileptic seizures occur in about 60% of people [Glaze et al 2010]. Other studies from Europe and Australia, reported higher rates of seizures, varying between 79% and 88% [Steffenburg et al 2001; Jian et al 2007; Pintaudi et al 2010]. Overall, these studies suggest that seizure onset before the age of 3 years is unlikely and onset after age 20 is rare. There is a tendency of remission with ageing, but in a minority epilepsy is severe and difficult to control. Seizures may be generalized or partial and several types may occur in the same patient. Episodes suggestive of infantile spasms have been described in young children; however true hypsarrhytmia has only rarely been reported from EEG studies. Drugs in general use for the control of epilepsy are frequently effective and well tolerated.
The overall occurrence of seizures in RTT is probably overestimated. Indeed, video-EEG recordings suggest that paroxysmal non epileptic manifestations such as motor activity, twitching, jerking, falling forward, as well as episodes of staring, laughing, breath holding, and hyperventilation are often misdiagnosed as seizures [Glaze et al 1998]. The consequence may be overtreatment and pseudo drug-resistance.
Seizure severity and drug resistance are difficult to assess. This is due in part to difficulties in differential diagnosis between epileptic and non epileptic seizures and in part to the variation in severity observed at different ages [Guerrini et al 2012].
Although clinical epilepsy is not universal the EEG is usually very abnormal, lacking normal rhythms and poorly responsive with bursts of generalized slow high amplitude waves, sometimes with spikes, accentuated during rest and sleep. In early regression the EEG may be within normal limits or shows some immaturity and slight slowing. In late regression and early post-regression the EEG became grossly abnormal in the majority of cases [Cooper et al 1998]. Cortical reflex myoclonus, manifested as multifocal, arrhythmic, and asynchronous jerks involving distal limbs, is frequently observed in Rett syndrome and exhibits particular neurophysiologic features, including a prolonged intracortical delay of the long-loop reflex [Guerrini et al 1998].
SPECIFIC VARIANT FORMS OF ATYPICAL RTT
A variety of specifically defined ‘variant’ or ‘atypical’ forms of RTT have been recognized that have distinct clinical features. Some of these have been recognized in only a small number of cases, making it difficult to make any clear statement concerning the defining clinical features. According to the recently revised diagnostic criteria [Neul et al 2010] the variant forms are: the Preserved Speech variant [Zappella 1992], the Congenital variant [Rolando et al 1985] and the Early Seizure variant [Hanefeld et al 1985].
Preserved Speech Variant
The preserved speech variant is the milder form of RTT: affected girls show the same stages of this condition and by the second half of the first decade are making slow progress in manual and verbal abilities. They walk without help, and may be able to make simple drawings and write a few words. Most of them can speak in sentences. Autistic behaviour can often be observed. Zappella described 29 preserved speech variant patients with MECP2 mutations and compared their phenotype with that of 129 classic RTT cases [Renieri et al 2008]. The study described microcephaly in the 27.5% of patients, microsomia in the 32% and low weight in the 25%. It is important to note that 7% were macrocephalic, some girls were overweight and 11% of patients showed tall
stature. Hand stereotypies were present as in the classic form but less intense and less evident in some periods of development. Hand use was much better than in the classic form but some degrees of dyspraxia were usually present. Epilepsy was significantly less frequent in this variant: 41% versus 70-80% of the classic form. Language was always present and was limited to single words or to simple phrases or complex phrases. The Preserved Speech variant (PSV) is due to MECP2 mutations. However, the kind of mutation differed statistically from the mutation type present in classic RTT. Only two patients carried an early truncating mutation that, in turn, was present in about 60% of classic RTT cases [Philippe et al 2006; Sampieri et al 2007]. In addition, it has been noted a significant presence of late-truncating mutations followed by missense mutations, especially the p.R133C, in the PSV group in comparison with classic RTT, where the trend was completely the opposite, with the majority being early truncating mutations followed by missense and late-truncating mutations.
In the original article describing the first cases, the name of preserved speech variant was initially suggested by reviewers. Increasing experience has shown that language is not ‘‘preserved” but represents a later improvement as well as a subsequent advance in hand use: a clear example of progress in a manual and verbal abilities, in contrast to classic RTT where these symptoms do not improve.
Congenital variant
The congenital variant was initially described by Rolando in 1985. In this form, girls are floppy and retarded from the very first months of life, present severe postnatal microcephaly before 4 months, regression in the first 5 months and specific movements abnormalities such as tongue stereotypies and jerky movements of the limbs [Neul et al 2010].
Only few cases being reported with MECP2 mutations [Monros et al 2001; Smeets et al 2003]. Ariani et al [2008] demonstrated that FOXG1 is a gene responsible for this variant RTT syndrome. FOXG1 encodes forkhead box protein G1, FoxG1 (formerly brain factor 1 [BF-1]), a transcriptional factor with expression restricted to fetal and adult brain and testis. FoxG1 interacts with the transcriptional repressor JARID1B and with global transcriptional corepressors
of the Groucho family. The interaction with these proteins is of functional importance for early brain development [Tan et al 2003; Yao et al 2001].
Early seizure variant
Hanefeld described female patients with atypical RTT and infantile spasms [Hanefeld, 1985]. This variant was extended to include those with seizures of early onset before regression [Goutieres and Aicardi, 1986]. X-linked cyclin-dependent kinase-like 5 (CDKL5) associated encephalopathy is a recently described X-linked disorder with this RTT phenotype. Patients with CDKL5 mutations show a similar clinical course: they had seizures in the first months of life and subsequently developed recognizable RTT features. The onset of seizures is before regression, about 5 months of live, and can be characterized by infantile spasms or refractory myoclonic epilepsy [Neul et al 2010].
2. RETT SYNDROME AND MECP2 GENE
RTT is currently considered a monogenic X-linked dominant disorder due to mutations in the MECP2 gene. In 1999 Amir et al discovered that mutations in the MECP2 gene were associated both with rare familial cases of RTT as well as with the more common sporadic occurrences of typical RTT.
Methyl-CpG-binding protein 2 (MECP2) gene has a pivotal role in neuronal
maturation throughout the central nervous system (CNS) [Della Sala et al 2014]. MECP2 was initially considered a transcriptional repressor whose potency depends on the density and location of methyl-CpGs near a promoter [Nan et al 1997].
MECP2-mediated gene silencing could occur through chromatin modification mediated by MECP2 interaction with Sin3A/HDACI or Ski/NcoR/HDACII repression complexes [Jones et al 1998; Nan et al 1998; Kokura et al 2001]. These enzymes remodel chromatin, which then becomes inaccessible to the transcriptional machinery [Nan et al 1998]. Further studies showed that MECP2, in addition to its role as a global repressor, acts as a splicing regulator. The authors identified the RNA-binding protein Y box-binding protein 1 (YB1), a principal component of messenger ribonucleoprotein particles that controls multiple step of mRNA processing, as a MECP2 binding partner. More specifically, in MECP2-deficient neurons the splicing is altered, and aberrantly spliced transcripts can arise [Young et al 2005]. Another surprising finding resulted from genome wide analysis of gene expression in MECP2 mutants showing that MECP2, contrary to expectation, could activate the expression of many genes directly or indirectly. In support of this idea, a biochemical interaction between MECP2 and the transcriptional activator CREB was reported [Chahrour et al 2008]. Recent work demonstrates that MECP2 binds broadly throughout the genome, suggesting that MECP2 functions more as a global regulator of transcription and chromatin remodeling rather than a sequence-specific transcription factor [Skene et al 2010; Cohen et al 2011]. One gene that has consistently shown expression changes when MECP2 is absent is BDNF. Chang et al. [2006] reported that BDNF protein levels are
decreased rather than increased in brains of MECP2 null mice. A direct test of BDNF action in MECP2 mutants showed that BDNF overexpression improved the locomotor function of MECP2 mutant mice in the dark cycle running wheel assay and increased firing of layer 5 of cortical neurons [Chang et al 2006; Greenberg et al 2009]. Furthermore, exogenous BDNF application to brainstem slices was shown to rescue synaptic dysfunction in Mecp2 null mice [Kline et al 2010]. Other data supporting a link between MECP2 and BDNF include the effect of environmental enrichment (EE) on animal model of RTT [Kondo et al 2008; Nag et al 2009; Lonetti et al 2010].
The neurobiological role of MeCP2 has become clearer with the generation of mouse models that reproduces the pathological signs of RTT. In fact, after a period of apparently normal development, male MeCP2 –KO mice (MeCP2 -/y) manifest severe locomotor and neurological dysfunctions by 4-5 weeks of age, tremor, seizures, weight loss and premature death by 10 weeks [Moretti et al 2006]. Embryonic MeCP2 deletion only in neurons, using conditional gene targeting, resulted in a phenotype similar to the ubiquitous knockout [Guy et al 2001; Chen et al 2001], demonstrating that MeCP2 dysfunction in the brain is of pivotal importance for RTT. Neurons are smaller, less complex and more densely packed in MeCP2 -/y mice but show no sign of neurodegeneration, suggesting that the neurological defect could stem from subcellular abnormalities, like subtle alteration of synaptic connectivity. Indeed, MeCP2 -/y mice exhibit altered dendrites and synaptic organization [Kishi et al 2004; Chao et al 2007; Moretti et al 2006], abnormalities in basal synaptic transmission and plasticity as well as an incorrect balance between neuronal excitation and inhibition [Chao et al 2007; Moretti et al 2006]. Interestingly, by restoring the proper expression of MeCP2 in null-mutant mice, behavioral symptoms were reversed even in adult animals [Guy et al 2007; Giacometti et al 2007; Tropea et al 2009; Gadalla et al 2011], implying that neurons might not be permanently damaged.
GENOTYPE-PHENOTYPE CORRELATION
Using a battery of modern mutation detection assays mutations in MECP2 can be found in 95 to 97% of individuals with typical RTT. Importantly, even using the best methodologies, 3 to 5% of individuals who strictly meet clinical criteria for RTT do not have an identified mutation in MECP2, indicating that a mutation in this gene is not required to make the diagnosis of typical RTT. The situation is more dramatic in atypical cases, with only 50 to 70% having identified mutations in MECP2 [Percy et al 2007].
A few RTT male cases, resulting from mosaicism for MECP2 mutations, have been reported [Christen et al 1995]. Male germline MECP2 mutations cause either severe encephalopathy with death at birth (usually in brothers of classical RTT females) or X-linked recessive mental retardation (XLMR). To date the wide phenotypic heterogeneity associated with MECP2 mutations in females (from classical RTT to healthy carriers) has been explained by differences in X chromosome inactivation. However, conflicting results have been obtained in different studies, with both random and highly skewed X-inactivation reported in healthy carrier females. Consequently it is possible that mechanisms other than X-inactivation play a role in the expressivity of MECP2 mutations. To explain the phenotypic heterogeneity associated with MECP2 mutations Renieri et al [2003] proposed a digenic model in which the presence of a "mutated" allele in a second gene, leading to a less functional protein, determines the clinical severity of the MECP2 mutation. The model was supported by the identification of the same mutation in XLMR and RTT cases. According to the authors the carrier mothers of XLMR families are clinically asymptomatic and present balanced X chromosome inactivation. Therefore the same mutation arising in different genetic backgrounds can cause XLMR in males, remain silent in the carrier females and cause classic RTT in females. MECP2 mutations account for approximately 70-80% of classic RTT cases. MECP2 negative cases might result from mutations in noncoding regions of MECP2 gene. Alternatively, these cases might be due to mutations in other genes (locus heterogeneity). This hypothesis is supported by the identification of several chromosomal rearrangements in MECP2 negative patients with RTT and RTT-like phenotypes. MECP2 is considered a general transcriptional repressor.
However, conditional mouse mutants with selective loss of Mecp2 in the brain develop clinical manifestations similar to RTT, indicating that MECP2 is exclusively required for central nervous system function. The involvement of MeCP2 in methylation-specific transcriptional repression suggests that MECP2 related disorders result from dysregulated gene expression. Studies on gene expression have been performed in mouse and human brains. A relatively small number of gene expression changes were identified. It is possible that MECP2 causes dysregulation of a very small subset of genes or that very subtle changes in many genes cause the neuronal phenotype [Renieri et al 2003]. Since MECP2 mutations were discovered to be the primary cause of RTT, a significant progress has been made in the MECP2 research, with respect to the expression, function and regulation of MECP2 in the brain and its contribution in RTT pathogenesis.
MECP2 mutations or altered expression are also associated with a spectrum of neurodevelopmental disorders such as autism spectrum disorders with recent links to fetal alcohol spectrum disorders. Collectively, MeCP2 relation to these neurodevelopmental disorders highlights the importance of understanding the molecular mechanisms by which MeCP2 impacts brain development, mental conditions, and compromised brain function. To date, there have been intensive efforts in designing effective therapeutic strategies for RTT benefiting from mouse models and cells collected from RTT patients. Despite significant progress in MeCP2 research over the last few decades, there is still a knowledge gap between the in vitro and in vivo research findings and translating these findings into effective therapeutic interventions in human RTT patients. It is essential to emphasize that RTT is not synonymous with mutations in MECP2 and at our present level of knowledge remains a clinical diagnosis. RTT occurs with and without mutations in MECP2, and, furthermore, mutations in MECP2 have been identified in individuals who lack the clinical features of either classic or variant RTT [Hagberg et al 2002].
MeCP2 binds to methylated cytosines in DNA to either activate or repress transcription and contains three functional domains: (1) a methyl-binding domain (MBD) on the N-terminus allowing binding to DNA, (2) a nuclear localisation sequence allowing trafficking of MeCP2 to the nucleus and (3) a transcriptional repression domain (TRD), which modulates gene transcription.
At present, 1013 distinct MECP2 mutations have been documented, resulting in 738 unique amino acid changes spread throughout these three functional domains (RettBASE: IRSF MECP2 Variation Database, http://mecp2.chw.edu.au).
Even within patients carrying mutations of the same gene, symptoms can vary substantially depending on the type of mutation and the pattern of X chromosome inactivation.
The pathogenic mutations of MECP2 include eight C>T transition mutations (T158M, R168X, R255X, R270X, R306C, R294X, R133C and R106W) accounting for 69% of mutation positive cases [Christodoulou and Grimm 2003]. There is evidence of a relationship between genotype and clinical severity, with some mutations having either a milder or a more severe clinical course [Leonard et al 2003; Smeets et al 2003; Colvin et al 2004]. Robertson et al [2006] reported some differences amongst the behaviour patterns of cases with the well-known common mutations: fear/anxiety was more commonly reported in those with R133C and R306C; those with the R294X mutation were more likely to have mood difficulties and body rocking but less likely to have hand behaviours and to display repetitive face movements. In contrast, hand behaviours were more commonly reported in those with R270X or R255X. There has been consensus in recognising that R133C, R294X, R306C and 3’ truncations are less severe and that T158M, R168X, R255X, R270X and large deletions are more severe [Leonard et al 2003; Colvin et al 2004; Schanen et al 2004; Charman et al 2005; Neul et al 2008; Bebbington et al 2008; Halbach et al 2012].
On the whole, large-scale analyses of other MECP2 mutation types and symptomatology have been challenging due to small participant sample sizes, variable diagnostic criteria and the cross-sectional nature of the phenotypic data. Additionally, atypical RTT has received relatively little attention due to small participant cohorts.
In a recent study Cuppadah et al [2014] analysed the largest cohort of individuals with RTT to date, 1052 genotyped participants, divided into typical and atypical presentations, at several time points.
The authors found novel genotype–phenotype associations for both typical and atypical RTT and demonstrated that clinical severity increases with age for most
mutation types and show that ambulation, hand use and age at onset of stereotypies are strongly associated with overall disease severity.
The data confirmed all studies already reported, and also identified R106W, R168X, R255X, R270X, splice sites, large deletions, insertions and deletions as being mutations associated with a more severe disease course in typical RTT. While previous reports have concluded that T158M is a more severe mutation, Cuppadah et al. demonstrated that T158M is characterised by a disease course of intermediate severity. The latter finding is in line with a study demonstrating that the severity of T158M and R168X is a function of the degree of X-chromosome inactivation (XCI) skewing [Archer et al 2007].
It’s well known that RTT syndrome may manifest as a large variety of phenotypes ranging from very severe to mild disease. Since there is a weak correlation between the mutation type in the Xq28 disease-gene MECP2/X inactivation status and phenotypic variability, this disease can be used as a model to unveil the complex nature of a monogenic disorder. Grillo et al [2013] used whole exome sequencing to analyse the functional portion of the genome of two pairs of sisters with RTT syndrome. Although each pair of sisters had the same MECP2 mutation and balanced X-inactivation, one individual from each pair had a typical form and could not speak or walk, and had a profound intellectual deficit, while the other individual had a Preserved Speech variant, could speak and walk and had a moderate intellectual disability. In addition to the MECP2 mutation, each patient has a group of variants predicted to impair protein function. The classical RTT girls, but not their milder affected sister, have an enrichment of variants in genes related to oxidative stress, muscle impairment and intellectual disability and/or autism. On the other hand, a subgroup of variants related to modulation of immune system, exclusive to the PSV patients are driving toward a milder phenotype. Grillo et al. [2013] demonstrated that genome analysis has the potential to identify genetic modifiers of RTT syndrome, providing insight into disease pathophysiology.
3. IDENTIFICATION OF NEURONAL SUBSTRATES OF
RETT SYNDROME: ELECTROPHYSIOLOGICAL AND
NEUROIMAGING STUDIES
This research is part of a Telethon (Application GGP09196) multidisciplinary clinical studies on RTT going in parallel with animal experiments on MeCP2-KO mice conducted by CNR of Pisa.
The principal aim of this project is to identify specific biomarkers of RTT in patients with MECP2 mutation. This should shed light on new alteration underlying the disease and could give to clinician and to researchers markers to be used to monitor disease progression objectively and to test the efficacy of clinical trials.
Up to date neurophysiological evaluations have been widely applied in the study of RTT to provide information about the character and extent of involvement of the nervous system pathways but they were performed on “clinical” RTT and sometimes with conflicting results.
Various neuroimaging techniques were also used by investigators internationally to characterize the in vivo pathology of RTT and to understand fundamental questions about the biological basis of this disease.
In this project we have performed both neurophysiological and neuroimaging studies to the same RTT patients with MECP2 mutation in order to:
evaluate the impairment of central and peripheral nervous system in RTT patient with proven MECP2 mutation and the relationship with disease severity; determine what is distinctive to RTT patients respect to control subjects in terms of brain anatomy and cerebral perfusion with particular regard to specific phenotypes.
Study design
The patients were recruited from a Tuscan Region database including more than 110 children with RTT syndrome and from patient database of the
Department of Developmental Neuroscience of Fondazione Stella Maris Scientific Institute (IRCCS) (Pisa, Italy).
The inclusion criteria were: a) patients who met the revised diagnostic criteria for typical and atypical RTT [Neul et al. 2010]; b) patients positive for MECP2 mutation. We included patients with various level of clinical severity and with a wide range of age.
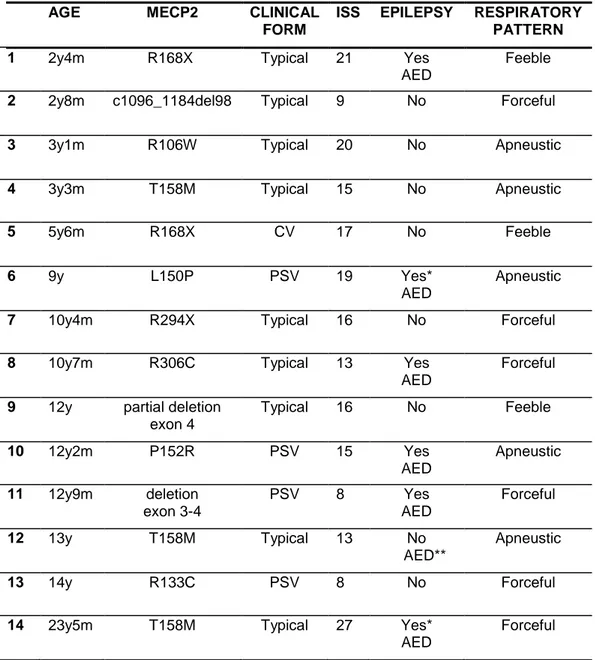
We enrolled 16 girls aged 2.4-23.5 years with a mean age of 9.3 years + 5.6; six of them were diagnosed in the Department of Developmental Neuroscience of IRCCS Fondazione Stella Maris before or during the time of project.
The sample was heterogeneous for MECP2 gene mutation and included 12 distinct types already reported in RTT population except for a novel mutation (L150P).
According to the revised diagnostic criteria [Neul et al 2010] the patients were classified as typical (n=10) and atypical RTT (n=6) consisted in Preserved Speech variant (n=4) and Congenital variant (n=2).
The degree of disease severity was quantified using the International Scoring System (ISS) [Kerr 2001] (see Appendix I). ISS consists of 21 items regarding the typical characteristic of RTT divided in five subscales: Growth and Development, Muscle-Skeletal appearance, Movement, Mental-Cortical and Brainstem-Autonomic. Each item score ranges from 2 to 0 as follows: 2 severe abnormality, 1: mild abnormality, 0: no abnormality. The higher grade of clinical severity corresponds to 40 points. On the basis of ISS scoring the patients were divided into three levels of severity: mild (scoring between 0 and 15 pts), medium (scoring between 16 and 20 pts) and severe (scoring > 20 pts). In our sample the severity of disease was defined as mild (n=7), medium (n=5) and severe (n=4).
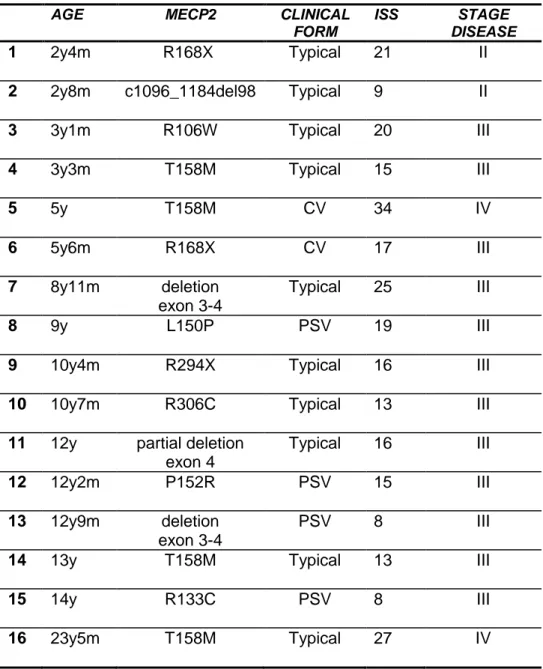
Table 1 shows the main characteristics of the sample.
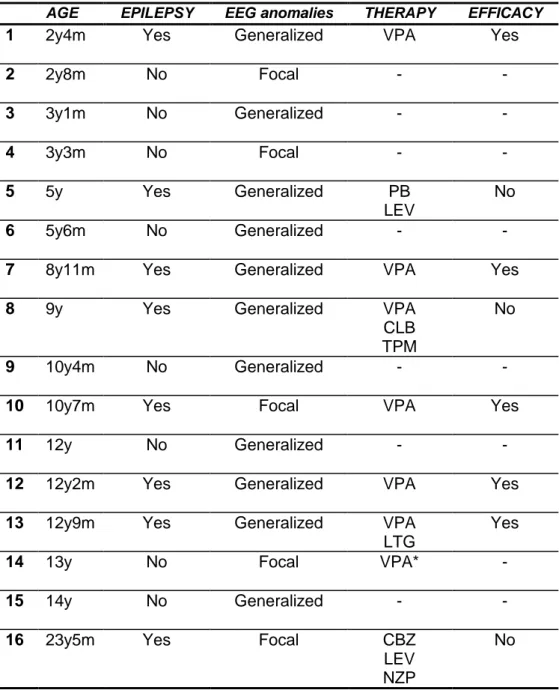
Eight out of sixteen patients had epilepsy (Epi-RTT) and were treated with antiepileptic drugs in mono- or poli-therapy. RTT 5, RTT 7 and RTT 16 presented drug resistant seizures. The epileptiform discharges were classified as generalized and focal anomalies (Table 2). The other eight patients were grouped in the non-epileptic group (NoEpi-RTT).
Table 1. Main characteristic of our RTT sample
AGE MECP2 CLINICAL
FORM
ISS STAGE DISEASE
1 2y4m R168X Typical 21 II
2 2y8m c1096_1184del98 Typical 9 II
3 3y1m R106W Typical 20 III
4 3y3m T158M Typical 15 III
5 5y T158M CV 34 IV 6 5y6m R168X CV 17 III 7 8y11m deletion exon 3-4 Typical 25 III 8 9y L150P PSV 19 III
9 10y4m R294X Typical 16 III
10 10y7m R306C Typical
13 III
11 12y partial deletion
exon 4 Typical 16 III 12 12y2m P152R PSV 15 III 13 12y9m deletion exon 3-4 PSV 8 III
14 13y T158M Typical 13 III
15 14y R133C PSV 8 III
16 23y5m T158M Typical 27 IV
Legend. CV: congenital variant; PSV: preserved speech variant; ISS: International Scoring System
Table 2. EEG findings and epilepsy in our RTT patients
Legend. VPA: valproic acid; PB: phenobarbital; LEV: levetiracetam; CLB: clobazam; TPM: topiramate; LTG: lamotrigine; CBZ: carbamazepine; NZP: nitrazepam; *: treatment for behaviour disorder
AGE EPILEPSY EEG anomalies THERAPY EFFICACY
1 2y4m Yes Generalized VPA Yes
2 2y8m No Focal - - 3 3y1m No Generalized - - 4 3y3m No Focal - - 5 5y Yes Generalized PB LEV No 6 5y6m No Generalized - -
7 8y11m Yes Generalized VPA Yes
8 9y Yes Generalized VPA
CLB TPM
No
9 10y4m No Generalized - -
10 10y7m Yes Focal VPA Yes
11 12y No Generalized - -
12 12y2m Yes Generalized VPA Yes
13 12y9m Yes Generalized VPA
LTG
Yes
14 13y No Focal VPA* -
15 14y No Generalized - -
16 23y5m Yes Focal CBZ
LEV NZP
Healthy control subjects
RTT patients were studied with an observational case-control study. For the electrophysiological studies healthy age-matched volunteers were used. The MRI database of IRCCS Stella Maris was used to select younger female controls in order to match RTT patients on age. They underwent an MRI examination because of various reasons (including headache, head trauma, cataract, dizziness). Inclusion criteria were the lack of neurological, behavioural or developmental disorders. The older controls were healthy volunteers.
The study was approved by the Ethical Committee of our Institute and informed written consent was obtained from the parents of each patient. Each patient was submitted to:
1. Electrophysiological studies - motor evoked potentials (MEP)
- visual evoked potentials flash (VEP-f) - somatosensory evoked potentials (SSEP) - neuroscope
2. MRI examination
- Voxel Based Morphometry (VBM) - Diffusion Tensor Imaging (DTI) - Arterial Spin Labeling (ASL)
3.1.
MULTIMODAL
NEUROPHYSIOLOGICAL
EVALUATION
(In preparation)
Introduction
Neurophysiological evaluations have been applied in the study of patients with Rett syndrome (RTT) to provide information about central, peripheral, and autonomic nervous system involvement [Glaze 2005].
Motor evoked potentials (MEP) using transcranial magnetic stimulation (TMS) were performed in RTT to investigate the excitability of corticospinal pathway [Eyre et al 1990; Heinen et al 1996; Nezu et al 1998; Guerrini et al 1998]. All authors found abnormal reduction of central motor conduction time (CMCT) as index of cortical hyperexcitability, possibly due to a loss of inhibitory synaptic control [Yoshikawa et al 1991; Yamanouchi et al 1993; Guerrini et al 1998]. Somatosensory Evoked Potentials (SEP) studies performed in a series of RTT individuals, showed delayed central conduction time (CCT) [Bader et al 1987; 1989a; Guerrini et al 1998] or normal CCT [Gorke W 1989]; Kimura et al [1992] studied the changes of cortical and subcortical involvement with the age and hypothesized a possible degenerative process in a later period of the disease. These findings suggested the involvement of the sensory system in RTT, as well as the motor cortex; the enlarged or “giant” amplitude SEP reported in a subset of RTT patients [Yoshikawa et al 1991; Guerrini et al 1998] together with a prolonged intracortical delay of the long-loop reflex with prolonged C-reflex latency [Guerrini et al. 1998] confirmed the suspect of cortical hyperexcitability supposed in this disorder.
Visual evoked potentials (VEP) were also studied in RTT with conflicting results: Verma, Kalmanchey and Saunders [1987; 1990;1995] reported normal results respect to control subjects, whilst Bader described increased latency of P100 in RTT patients [1989b].
Up to date the reported neurophysiological studies on RTT were performed before the identification of MECP2 as causative gene of the syndrome.
The aim of our study is to evaluate the involvement of central and peripheral nervous system in a sample of RTT patients with definite MECP2 mutations using a multimodal approach that includes VEP, SEP, Long Loop Reflexes (LLR) and TMS. We selected 16 patients with different levels of clinical impairment and a wide range of age in order to verify the role of these variables on the neurophysiological findings.
Subjects
The patients were recruited from a Tuscan Region database including more than 110 children with RTT and from patient database of the Department of Developmental Neuroscience of Fondazione Stella Maris Scientific Institute (IRCCS) (Calambrone, Pisa, Italy). Mandatory inclusion criteria were: a) patients who met the revised diagnostic criteria for typical and atypical RTT [Neul et al. 2010]; b) patients positive for MECP2 mutation.
We enrolled 16 girls aged 2.4-23.5 years (mean age 9.3 years + 5.6); MECP2 mutations consisted of 12 distinct types already reported in RTT population except for a novel mutation (L150P in patient 8).
The clinical form was identified according to the revised diagnostic criteria [Neul et al 2010]; the severity of disease was quantified using the International Scoring System (ISS) [Kerr 2001] (see Appendix I), the patients were divided into three levels of severity: mild (scoring between 0 and 15), medium (scoring between 16 and 20) and severe (scoring between 20 and 40).
Table 1 summarizes the main characteristics of the sample.
RTT patients were studied with an observational case-control study and healthy age-matched volunteers were used.
Methods TMS
Magnetic stimulation was performed with a high-power Magstim 200 magnetic stimulator (Magstim Co., UK) to elicit motor evoked potentials (MEP) through a circular coil with a mean diameter of 90 mm, held over the motor cortex at the optimum scalp position. The EMG signal was recorded using surface-recording electrodes placed contralateral over the abductor pollicis brevis (APB). The coil edge was positioned 1-2 cm lateral to the 7th cervical spine (C7) to stimulate the
cervical spinal roots. During the TMS tests the patients were seated. The conditions of facilitation were ascertained visually using the resting phase and the action-phase of the surface EMG. Trials were replicated at least four times to ensure reproducibility of responses.
We recorded total motor conduction time (TMCT), central motor conduction time (CMCT), amplitude of responses and post-Motor evoked potential obtained from RTT patients and from 13 healthy subjects (mean age 9.5 years + 5.3). Statistical comparisons were performed using Student’s t test for unpaired data.
SEP
SEP were recorded from centroparietal regions (C3’ and C4’= 2 cm behind the International 10-20 system C3 and C4) at the level of C7 and from Erb’s point (EP). Scalp electrodes were referenced to the homologous contralateral area and EP. The median nerve was stimulated at the wrist with electrical rectangular stimuli lasting 100 microseconds, repeated at a frequency of 3 Hz. Blocks of 50 to 250 consecutive artifacts-free responses were averaged. Low and high pass filters were 4Hz and 2KHz. Trials were replicated to ensure reproducibility of the responses. The amplitude was measured from the preceding peak of the opposite polarity.
We evaluated the responses recorded at EP (N9), C7 (N13), scalp (N20), N9-N13 interpeak time and central conduction time (CCT=latency difference between the cervical N13 and the scalp N20).
The C-reflex at rest and with muscular facilitation was sought simultaneously by recording EMG activity from the APB muscle. SEP responses of RTT patients were compared with normal values as reported by Taylor and Fagan [1988].
VEP-flash
VEP-f were performed with monocular stimulation at full field with stroboscopic light at a frequency of 1 Hz and at an intensity of 2 joules (low and high pass filters were 1 Hz and 300 Hz); the electrodes were positioned on occipital region (central and lateral). We analysed the latencies and amplitudes of N75, P100 and N145. VEP-f were performed in 13 healthy subjects (mean age 9.5 yrs + 5.3) and statistical comparisons were performed using Student’s t test for unpaired data.
Results
TMS
All patients but two (patient 5 and 9) cooperate in the examination. Neither the duration nor the amplitude of MEP differed from those of age-matched controls in any case. Regard to CMCT we did not find statistically significant difference between RTT and control group (p=0.2; RTT mean 6.0, SD 1.0; controls mean 5.5, SD 0.8) as shown in Figure 1.
Figure 1. CMCT in RTT group and control group
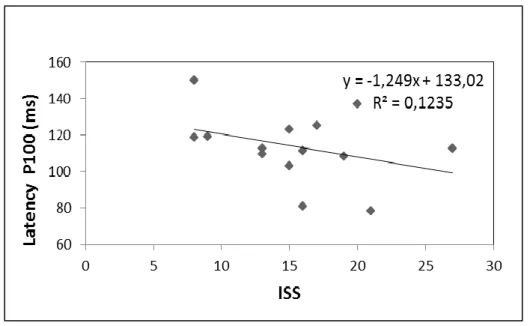
Nevertheless the progressive shortening of CMCT associated with higher levels of clinical severity (ISS score) was noticed (R2=0.3; p= 0.04) as shown in Figure 2.
Figure 2. Correlation between CMCT and ISS score in RTT group.
No statistical difference in CMCT was observed between epileptic RTT and non-epileptic RTT (p=0.3; epileptic RTT: mean 5.7, SD 0.6; non epileptic RTT: mean 6.2, SD 1.2) and no age-dependent changes of CMCT was observed (R2=0.04).
SEP
SEP were performed on all subjects, but four patients did not cooperate in the examination (patients 1,9,10,11). Three patients (patients 5,7,16) showed a multifocal, arrhythmic and asynchronous myoclonus at level of head and upper limbs.
Responses recorded at EP (N9) and C7 (N13) were normal in amplitude and latency. N9-N13 interpeak time and CCT were within normal values in all patients. Our patients did not show “giant” SEP but patient 5 presented a broad morphology of SEP. In patients 5, 7 and 16 we found the C-reflex at rest with a
In particular patient 16, evaluated during therapy with piracetam and after its suspension, showed myoclonus and C-reflex at rest only in absence of drug treatment.
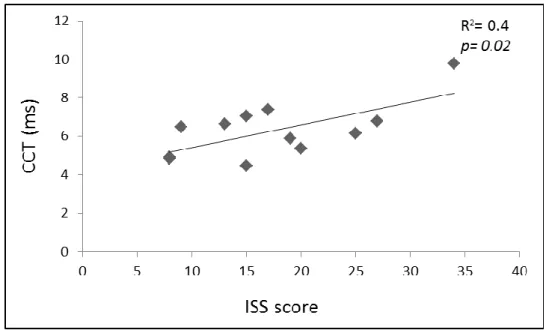
A significant correlation was found between the severity of disease (ISS score) and a longer CCT (R2=0.4; p=0.02, see figure 4); no significant correlation was found between CCT and the age (R2 =0.08).
Figure 4. Correlation between CCT and ISS score in RTT group.
We did not find significant correlation between ISS score and N9, N13 latencies (R2=0.1 and R2=0.06, respectively) or N9-N13 interpeak time (R2 =0.01).
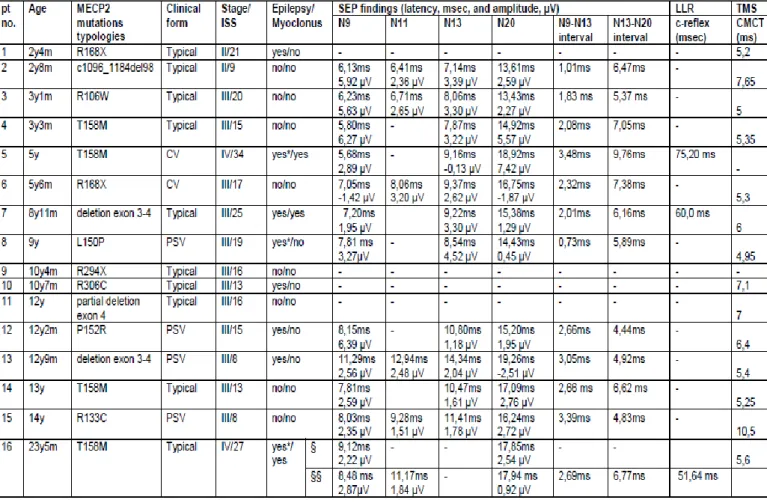
Table 1. Neurophysiological data in 16 RTT patients
Legend. LLR: Long Loop Reflexes; msec: milliseconds; µV, millivolt; §, study performed with piracetam and §§ after suspension.§, study performed with piracetam and §§ after suspension.
VEP-f
All patients but one (patient 7) cooperate in the examination. N75 and N145 amplitudes and latencies were normal in all patients. We did not find statistically significant difference of P100 latency between RTT and control group as shown in figure 5 (p=0.5; RTT mean: 113.6, SD 18.2; controls mean: 109.7, SD 9.6). No correlation was found between P100 latency and age of patients (R2=0.02) either nor P100 latency and severity of disease (R2=0.07, see figures 5 and 6).
Figure 5. Correlation between P100 latency and age in RTT group
Figure 6. Correlation between P100 latency and ISS score in RTT group.
Discussion
To our knowledge this paper reports the first application of multimodal neurophysiological study on RTT girls with definite MECP2 mutation, since previous works were published before the identification of MECP2 gene as causative of the disorder [Bader et al 1987, 1989; Eyre et al. 1990; Heinen et al. 1996; Guerrini et al. 1998; Nezu et al.1998].
We aimed to evaluate the amount of impairment of central motor and sensory pathways in this homogenous group of RTT patients positive for MECP2 mutation. Our sample represents the largest RTT group described in literature with multimodal electrophysiological approach until now.
The results obtained by TMS confirmed the previous finding that the pyramidal tract is preserved in RTT patients; we did not confirm the shortening of CMCT as reported in literature [Eyre et al. 1990; Heinen et al. 1996; Guerrini et al. 1998; Nezu et al.1998] but we found a significant correlation between a shorter CMCT and the severity of the disease (calculated with the ISS score).
Our hypothesis is that these findings could be explained by the different inclusion criteria of our study (that included only MECP2 proven mutated RTT) and by the relatively milder clinical phenotypes of our sample that includes four girls with PSV (patients 8,12,13,15) and only four severe forms with ISS score >20 (patients 1,5,7,16).
All patients but four (patients 1,2,5,16) were in the pseudo-stationary stage of disease (stage III) but presented a wide range of CMCT (4.9-7.9 ms) as well as patients in the stage II (patients 1,2). For this reason the stage of disease did not appear as an informative variable in this study.
On the contrary, our choice to classify the clinical status of our RTT patients according to ISS seems to be useful to identify a potential biomarker of disease progression.
In conclusion CMCT shortening is confirmed to be a neurophysiological characteristic of RTT, but its alteration depends on the level of disease severity. No significant differences between RTT patients and controls for VEP latencies and morphology were noticed; no correlation with visual evoked responses and clinical severity was found. These findings were in agreement with other authors [Verma et al 1987; Kalmanchey et al 1990; Glaze 2002] and with the preserved visual pathway typical of this disorder.
Evaluation of SEP findings in our patients did not show evidence of enlarged SEP except for patient 5 (five years old) that showed broad cortical SEP and C-reflex at rest and presented a drug resistant myoclonic epilepsy. Two RTT of our sample (patients 7 and 16, 8 years and 11 months-23 years and 5 months respectively) presented cortical myoclonus and C-reflex at rest but normal SEP. These results appeared in agreement with previous works that reported “giant” SEP in RTT only in younger girls and in patients with cortical myoclonus [Yoshikawa et al 1991; Guerrini et al 1998]. Interestingly, the patient 16 belonged to the RTT group previously studied by Guerrini et al [1998] (indicated as patient 5). This clinical and neurophysiological revaluation allowed us to confirm the effectiveness of piracetam on cortical myoclonus and to study the evolution of SEP amplitude with the age. Our study (performed at 23 years and 5 months) did not confirm the presence of “giant” SEP and borderline CCT (demonstrated at 7 years old), but showed normal cortical evoked responses at older age, whilst the C-reflex was present in both evaluations. This striking
intra-patient difference could be explained by the hypothesis that a “giant” SEP may appear in an early stage of this disorder as a marker of disease activity and cortical hyperexcitability, and then disappear in advanced stages when the activity of disease declines [Yoshikawa et al 1991].
Evaluating the CCT for all patients (calculated as the N13-N20 latency difference) we found a significant and interesting correlation between a longer interval and the clinical severity of the disease measured by ISS; no significant correlation was found between the N13-N20 findings and the age previously reported by Kimura et al [1992, 1998].
These findings confirm the role of the neurophysiological multimodal evaluation in defining the disease severity of RTT patients.
In conclusion our study on MECP2 proven mutated RTT patients confirms previous findings about the presence of several intrinsic characteristics of this disease.
In particular, it has been confirmed the role of CCT, calculated both with MEP and SEP, as a consistent biomarker of the severity of the disease.
3.2 ASSESSMENT OF BRAINSTEM FUNCTION AND
DELINEATION
OF
CARDIORESPIRATORY
PHENOTYPES
Introduction
Suggestive of brainstem and autonomic dysfunction in RTT patients are: agitation and sleep disturbance, small and narrow cold feet, abnormal awake breathing and mood disturbance. These clinical features are all directly or indirectly connected to autonomic function.
It is clear that certain features contribute more than others to the disease severity and may demand complex management systems. The brainstem features become prominent at regression and remain so throughout life as breathing abnormalities and/or dysautonomia. It is therefore reasonable to support that clinical autonomic assessment should be part of the procedure for diagnosis and follow-up in the management of the RTT disorder. Objective clinical autonomic measurements can provide reproducible results that can be used to monitor the progress of the disease.
Breathing has been noticed to be irregular in RTT girls since it was first described. Hyperventilation and prolonged breath holding are impressive respiratory anomalies and therefore are most often mentioned by parents. However, without accurate measurement, central apnoea and Valsalva’s maneuver are mistaken for breath holds, and tachypnea and rapid shallow breathing may be mistaken for hyperventilation. Subtler dysrhythmias may be missed altogether. For these reasons breathing movements could be monitored using a stretch sensitive plethysmograph placed around the chest at the level of xiphisternum. In this way respiratory movements can be recorded through an interface called the MedullaLab (MediFit Instruments Ltd, London, UK) into the microcomputer, time-locked with all other physiological markers. The shapes and rates of the breathing movements are important for characterizing the breathing rhythms.
The NeuroScope (Medifit Instruments Ltd, London, UK) is a neurophysiological technique recently developed for continuous and real-time assessment of brainstem function. Through this technique, in fact, the vital signs originating from and regulated by the brainstem are quantified and recorded synchronously for the study of both temporal and causal relationships; these signs can be classified into two categories: cardiovascular and respiratory [Julu et al 2001, 2005].
The cardiovascular vital signs recorded by the NeuroScope are: i) cardiac vagal tone (CVT) believed to be regulated by nucleus ambiguous in the caudal part of the ventrolateral medulla oblongata [Guyenet et al 1996]; ii) cardiac sensitivity to baroreflex (CSB) believed to be regulated by the commissural part of the nucleus of tractus solitarius in the lower part of the dorso-medial medulla oblongata [Kasparov et al 1999]; iii) arterial blood pressure (BP), especially the mean arterial blood pressure (MAP) known to be regulated in the rostral ventrolateral medulla oblongata [Guyenet et al 1990]; iv) heart rate (HR) which is regulated by both the nucleus ambiguous in the lower part and the cardio-accelerators in the rostral part of ventrolateral medulla oblongata [Jordan et al 1995].
The respiratory vital signs quantified and recorded by the NeuroScope are: i) breathing movements, both rate and rhythm known to be regulated by the ventrolateral brainstem from as low down as the level of first vertebrate to as rostral as the parabrachial nuclei in the pons and the dorsal respiratory group of neurones in the vicinity of the nucleus of tractus solitarius [Blessing 1997]; ii) transcutaneous partial pressures of oxygen (pO2) and carbon dioxide (pCO2) representing blood gases are also recorded. Blood gases are believed to be regulated in the medulla oblongata in conjunction with inputs from the arterial chemoreceptors [Spyer et al 2004].
A scale of vagal tone known as the Linear Vagal Scale (LVS) has been derived using atropine [Julu 1992]. This provides a non-invasive means of quantifying the only cardiovascular parasympathetic output from the brain and a very important common autonomic pathway. The CVT is also an important indicator of the integrative inhibition in the brainstem.
Since sympathetic activity is closely related to the mean arterial BP, generalized sympathetic activity can be monitored indirectly and continuously from the readings of the mean BP.
The baseline brainstem autonomic function can be defined as the autonomic activity recorded during the period when the subject is not agitated and has normal breathing rhythm with blood gases within the normal range. The brainstem autonomic functions were analysed in 72 RTT patients and11 healthy subjects [Julu et al 2005]. The CVT was about 50% lower in the RTT population compared to control, indicating significant reduction of parasympathetic activity in RTT patients. There was no significant difference in baseline sympathetic activity between RTT and control populations. The level of CVT is regulated through complex integrative processes in the nucleus tractus solitarius, nucleus ambiguous and the bulbar reticular formation, involving several supra-medullary centres. The CVT being the only inhibitory output of the cardiorespiratory integrative system serves a very important function in situation requiring fast cardiovascular responses and is a major part of the integrative inhibition in the cardiorespiratory system.
The clinical evidence showed that the baseline brainstem functions maintained by complex integrative inhibitions of neurons are most affected in the RTT disorder, while the functions maintained by pacemaker activity of neurons are preserved. Since breathing rhythm is invariably affected in RTT syndrome, there is a strong implication that rhythmogenesis of breathing is likely to be due to a complex network of neuronal inhibitions [Richter et al 1990].
Detailed analysis of 65 patients with awake breathing abnormalities revealed three major cardiorespiratory phenotypes within the Rett population: Apneustic, Forceful and Feeble breathers [Julu et al 2005].
Patients with Apneustic phenotype accumulate CO₂ because of delayed and inadequate expirations. Oral buspirone is the drug of first choice because of its effect on apneusis.
Forceful breathers usually have fixed low concentrations of partial pressure of CO₂ causing chronic respiratory alkalosis. To interrupt an episode of forceful breathing, is recommend first re-breathing into a 5-L bag attached to a facial mask that fits tightly on to a patient’s face. Long-term weaning from chronic