2.1. Trasformazione di cellule di E.coli ed amplificazione del
DNA plasmidico
2.1.1. Vettori plasmidici
I vettori utilizzati per amplificare il cDNA da trascrivere per le sonde antisenso, sono i seguenti:
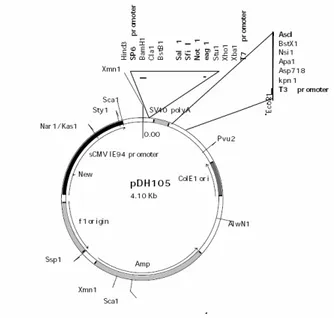
pCS105: questo plasmide contiene un sito di multiclonaggio fiancheggiato dai promotori delle RNA polimerasi SP6 e T7; inoltre contiene un gene di resistenza all’ampicillina (Fig. 13).
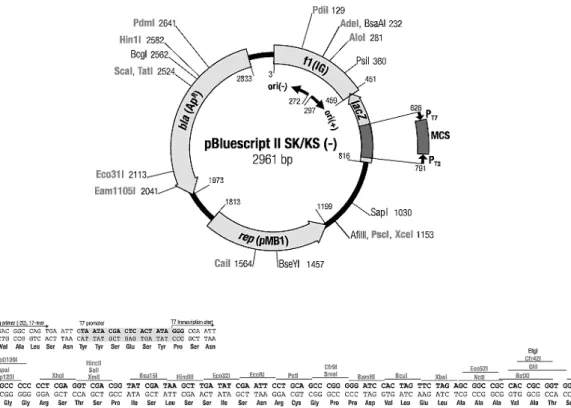
pBluescript II SK (-): questo plasmide contiene un sito di multiclonaggio fiancheggiato dai promotori delle RNA polimerasi T3 e T7; inoltre contiene un gene di resistenza all’ampicillina (Fig. 14).
pCMV-sport6: questo plasmide contiene un sito di multiclonaggio fiancheggiato dai promotori delle RNA polimerasi T3 e SP6; inoltre contiene un gene di resistenza all’ampicillina (Fig. 15).
Figura 14. Mappa di restrizione del vettore plasmidico pBluescript II SK/KS (-)
2.1.2. Cloni di ESTs
Nome ID GeneBank ID Clone
XL007 BJ046466 NIBB XL007h13 XL014 BJ046645 NIBB XL014a11 XL086 BJ083786 NIBB XL086d12 XL173 BJ100032 NIBB XL173b06 XL406 BP692972 NIBB XL406c10ex XL418A BP674881 NIBB XL418a19ex XL418D BP674937 NIBB XL4118ex XL488 BP699735 NIBB XL488j05ex XL520 BP707388 NIBB XL520j24ex E09 BC060755 I.M.A.G.E. 4969000 L11 BE575673 I.M.A.G.E. 3401626
Le ESTs sono arrivate sotto forma di DNA plasmidico adsorbito su carta da filtro, sono state amplificate, linearizzate e trascritte in base alle indicazioni disponibili.
Le ESTs da XL007 a XL173 sono clonate in pBluescript II SK(-) nei siti EcoR1 e Xho1 rispettivamente alle estremità 5’ e 3’ del cDNA.
Le ESTs da XL406 a XL520 sono clonate in pCS105 nei siti Sal1 e Not1 rispettivamente alle estremità 5’ e 3’ del cDNA.
Le ESTs E09 e L11 sono clonate in pCMV-sport6 nei siti Sal1 e Not1 rispettivamente alle estremità 5’ e 3’ del cDNA.
XL007: linearizzato con BamH1, trascritto con T7 RNA polimerasi. XL014: linearizzato con EcoR1, trascritto con T7 RNA polimerasi. XL086: linearizzato con EcoR1, trascritto con T7 RNA polimerasi. XL173: linearizzato con Not1, trascritto con T7 RNA polimerasi. XL406: linearizzato con BamH1, trascritto con T3 RNA polimerasi. XL418A: linearizzato con BamH1, trascritto con T3 RNA polimerasi. XL418D: linearizzato con BamH1, trascritto con T3 RNA polimerasi. XL488: linearizzato con BamH1, trascritto con T3 RNA polimerasi. XL520: linearizzato con BamH1, trascritto con T3 RNA polimerasi. E09: linearizzato con Sal1, trascritto con T7 RNA polimerasi. L11: linearizzato con Sal1, trascritto con T7 RNA polimerasi.
2.1.3. Preparazione di cellule competenti
Mediante il seguente protocollo le cellule di E. coli (ceppo DH5α) sono state rese competenti per la successiva trasformazione con vettori plasmidici.
• Prelevare una colonia cresciuta ON su terreno solido e inocularla in 50 ml di terreno liquido.
• Far procedere la crescita a 37°C in agitazione fino a che la densità ottica (OD) misurata a 600 nm raggiunge il valore di 0,2.
• Interrompere la crescita mantenendo 3’ in ghiaccio.
• Centrifugare in tubo sterile per 10’ a 5000 rpm a 4°C.
• Eliminare il sopranatante e risospendere in ½ del volume iniziale (circa 25 ml) di RbCl 50 mM freddo.
• Mantenere 30’ in ghiaccio.
• Centrifugare in tubo sterile per 10’ a 5000 rpm a 4°C.
• Eliminare il sopranatante e risospendere in 1/50 del volume iniziale (circa 0,8 ml) di RbCl 50 mM freddo.
Terreni di coltura
Luria-Bertani Broth (LB): NaCl 1%
bacto tryptone 1% bacto yeast extract 0,5%
Bottom Agar: agar sciolto in LB 1,5%
Antibiotici usati per terreni selettivi: Ampicillina 100 µg/µl
2.1.4. Trasformazione batterica
Con questo procedimento è possibile trasformare cellule batteriche con DNA plasmidico.
• Aggiungere 5-20 ng di plasmide ad un’aliquota di 200 µl di cellule competenti.
• Incubare in ghiaccio per 30 min.
• Sottoporre a “heat shock” a 42°C per 45 secondi.
• Incubare in ghiaccio per 5 minuti.
• Aggiungere 800 µl di LB preriscaldato a 37°C e incubare in stufa per 60 minuti.
• Piastrare le cellule su terreno solido selettivo.
• Dopo incubazione a 37°C ON, sulla piastra Petri compaiono le colonie: l’antibiotico fa sì che crescano solo le cellule che hanno assunto il plasmide poiché questo contiene il gene che conferisce la resistenza all’antibiotico stesso.
• Ogni volta che è stata effettuata una trasformazione, è stata controllata l’efficacia dell’antibiotico piastrando, in parallelo, 100 µl di cellule batteriche sottoposte a uguale trattamento in assenza di DNA plasmidico.
2.1.5. Estrazione di DNA plasmidico su piccola scala mediante lisi alcalina ("mini-prep”)
Tale metodo permette di estrarre DNA plasmidico ( 2-20 g) da cellule trasformate, per effettuare successivamente delle digestioni diagnostiche. L’estrazione è stata fatta con il kit “GenEluteTM HP Plasmid Miniprep” (Sigma®), seguendo le indicazioni fornite dal produttore.
2.1.6. Estrazione di DNA plasmidico su media scala (“midi-prep”) mediante colonne NUCLEOBOND© AX-100 (Macherey-Nagel) Per poter disporre di quantità di plasmidi adeguate alle necessità sperimentali è stato necessario amplificarli: ciò è stato possibile tramite la trasformazione di cellule batteriche competenti all’acquisizione del plasmide e la successiva estrazione del DNA plasmidico per mezzo di colonnine cromatografiche “NUCLEOBOND© AX-100” a scambio anionico seguendo le indicazioni del produttore.
2.2. Elettroforesi su gel di agarosio
Si è fatto ricorso alla corsa elettroforetica su gel di agarosio all’1% (peso/volume) in tutti i casi in cui è stato necessario verificare la completezza di una digestione, valutare la purezza di un DNA estratto, di stimare la concentrazione o la lunghezza in paia di basi del DNA nelle varie preparazioni.
• I gel sono preparati sciogliendo l’agarosio in TBE, portato alla temperatura di ebollizione.
• Prima che il gel polimerizzi si aggiunge EtBr (bromuro d’etidio) ad una concentrazione finale di 10 (g/ml).
• Il gel, lasciato brevemente a raffreddare viene colato in un lettino da elettroforesi di un apparato orizzontale.
• Una volta polimerizzato il gel è posto nell’apparato ed immerso in un tampone di corsa, TBE a pH 8.
• I campioni da analizzare vengono diluiti in acqua e “loading buffer”. Il “loading buffer” appesantisce il campione, facendolo andare sul fondo del pozzetto e consente al contempo di seguire la corsa elettroforetica.
• Si caricano i campioni su gel e si applica una differenza di potenziale di 50/120 V per un tempo variabile dai 5 ai 60 minuti.
• Si visualizzano infine le bande degli acidi nucleici ponendo il gel sotto raggi UV: il bromuro di etidio che si è intercalato alle basi appare, in queste condizioni, fluorescente.
• Le dimensioni dei frammenti sono stimate in presenza di marcatori con lunghezza in paia di basi (pb) e peso molecolare noti (Invitrogen 1kb DNA ladder).
Soluzioni per l’elettroforesi su gel di agarosio TBE pH 8.0 Tris base 0.089 M Acido borico 0.089 M EDTA 0.002 M “Loading buffer” 6x Glicerolo 5% Blu di bromofenolo 0.05% Xilene cianolo 0.05%
“Loading buffer” 2x denaturante per RNA EDTA 18 mM
Formammide 95 % SDS 0.025 %
Xilene cianolo 0.025% Gel di agarosio
Agarosio 1% (peso/vol) Et-Br c.f. 10 (g/ml) TBE 1X
“Marker” di lunghezza: Invitrogen 1 kb DNA ladder
2.3. Stima della concentrazione di DNA e RNA in soluzione
La stima della concentrazione dei campioni di DNA e RNA è stata effettuata mediante due metodi:
Elettroforesi su gel d’agarosio all’1%. Comparando la fluorescenza agli UV della banda di interesse con quella di marker di quantità si può stimare approssimativamente la concentrazione del campione iniziale.
Spettrofotometria UV. Questa tecnica permette di stimare precisamente la concentrazione del campione misurandone l’assorbanza alla lunghezza d’onda di 260 nm e la purezza in base al rapporto OD260/OD280 (se è maggiore o uguale a 1,8 il campione è ritenuto privo di contaminazione proteica). Lo spettrometro utilizzato è un GeneQuant pro (biochrom).
2.4. Digestione di DNA con enzimi di restrizione e
purificazione di DNA
2.4.1. Digestione di DNA con enzimi di restrizione
L’uso di enzimi di restrizione è stato necessario per effettuare digestioni diagnostiche e linearizzare plasmidi per usarli come DNA stampo in esperimenti di trascrizione in vitro.
Le reazioni di digestione sono state assemblate di norma con 3 unità/µg di DNA da digerire di enzima di restrizione e con il suo tampone specifico, facendo attenzione che il volume di enzima non eccedesse il 10% del volume finale di reazione perchè il glicerolo presente negli stock di enzimi non interferisse con l’attività enzimatica. L’incubazione è stata protratta da 2h a 5h a seconda dell’enzima e della quantità di DNA da digerire, a 37°C.
La completezza della reazione è stata valutata sottoponendo un’aliquota della miscela di reazione a elettroforesi su gel d’agarosio 1% colorato con bromuro d’etidio e confrontando la banda risultante con quella generata da un’aliquota di corrispondente DNA non digerito.
Quando è stato necessario effettuare doppie digestioni, si è assemblata una reazione unica contenente entrambi gli enzimi di restrizione.
2.4.2. Purificazione del DNA
Per purificare campioni di DNA in seguito a digestioni con enzimi di restrizione, è stato utilizzato il kit Gen Elute™PCR Clean-Up (Sigma®), seguendo le indicazioni fornite dal produttore. La risospensione finale è stata effettuata in 30 µl anziché nei 50 µl indicati dal produttore.
2.5. Trascrizione in vitro di una sonda antisenso e
purificazione di RNA
Questo protocollo è stato usato nella trascrizione di RNA antisenso marcato con DIG-UTP (digossigenina-UTP) da usare come sonda negli esperimenti di ibridazione in situ: l’RNA polimerasi necessaria (T3 o T7) è stata scelta in base alla direzione di clonaggio della sequenza di interesse nel vettore, che è stato linearizzato di conseguenza.
• Assemblare la reazione con 1-1.5 µg di DNA linearizzato, 2 µl DTT 100 mM, 4 µl transcription buffer 5x, 2 µl DIG-labeling mix (Roche), 1 µl RNase OUT (Invitrogen), 2 µl RNA polimerasi. Portare a un volume finale di 20 µl con H2O mQ.
• Incubare 2h a 37°C.
• Prelevare 1µl del volume di reazione e conservare in ghiaccio per la successiva corsa elettroforetica di controllo.
• Aggiungere 2µ di DNase/RNase free (Invitrogen) e incubare per 15’ a 37°C.
• Aggiungere 1 µl EDTA 0.5 M, 1/10 Vol di NH4Ac, 1 Vol isopropanolo. Rimescolare e lasciare 20-40’ a -20°C.
• Centrifugare per 15’ a 13000 rpm a 4°C e rimuovere il sovranatante.
• Lavare con EtOH 75% (non meno di 100 µl) e centrifugare 3’ a 13000 rpm RT.
• Eliminare il sovranantante e risospendere in 22 µl H2O mQ.
• Prelevare 1 µl del campione e caricarlo in un gel di agarosio 1% con “loading buffer” tipo II assieme al campione prelevato in precedenza e a soluzioni di tRNA a concentrazione nota usate come marcatori di quantità.
• L’assenza della banda del DNA stampo successiva al trattamento con DNasi indica il buon fine della reazione; il confronto con i marcatori da’
2.6. Ibridazione in situ su embrioni interi (whole mount)
2.6.1. Embrioni di Xenopus laevis
Gli embrioni di Xenopus laevis per gli esperimenti sono stati ottenuti mediante fecondazione in vitro.
Prima di essere operato per la rimozione dei testicoli, il maschio è stato anestetizzato immergendolo in una soluzione 0.1% di MSS (metansulfonato dell’estere etilico dell’acido 3-amminobenzoico) disposta in ghiaccio per abbassare rapidamente il metabolismo dell’animale. I testicoli si possono conservare per 3-5 giorni a 4°C immersi nella soluzione per testicoli. Dopo l’operazione il maschio viene sacrificato. Lo stoccaggio avviene secondo la vigente normativa veterinaria.
Le femmine di Xenopus laevis sono state pre-stimolate con 100 UI di Folligon Intervet per uso veterinario da 4 a 11 giorni prima della deposizione e con 1000 UI di Profasi HP 5000 Serono (gonadotropina corionica) 10-12 h prima della deposizione: entrambi gli ormoni sono stati somministrati mediante iniezione nel sacco perilinfatico.
Le uova da fecondare sono state ottenute esercitando una leggera pressione sull’addome degli animali e raccolte in piastre Petri: questa operazione può essere ripetuta a intervalli di 1 ora. La fecondazione è stata effettuata bagnando le uova con una sospensione ottenuta sminuzzando un frammento di testicolo in MMR 1X e lasciandole in poco MMR 0.1X..
Dopo almeno 30 minuti o quando comunque fosse ben rilevabile la rotazione corticale degli embrioni, effetto dell’avvenuta fecondazione, gli embrioni sono stati privati del loro rivestimento gelatinoso ricoprendoli di soluzione degelificante e lasciandoveli 5-10 minuti e comunque fino a che non fosse evidente la perdita dl suddetto rivestimento. La soluzione degelificante è stata eliminata sciacquando gli embrioni in MMR 0,1x.
Gli embrioni vengono fatti sviluppare in MMR 0.1x fino allo stadio desiderato, valutato secondo i criteri di Nieuwkoop e Faber (Nieuwkoop et al., 1967a; Nieuwkoop et al., 1967b).
Gli embrioni sono stati di norma fissati in MEMFA 1h a RT o ON a 4°C e conservati in metanolo 100% a -20°C.
Soluzioni per embrioni di Xenopus laevis MMR NaCl 0.1 M KCl 2 mM MgSO4 1 mM CaCl2 2 mM HEPES 5 mM pH 7.8 EDTA 0.1 mM Soluzione degelificante DTT 3.2 mM Tris-HCl 0.2 M pH 8,8 Soluzione per il testicolo MMR 1x
Siero di agnello inattivato al calore 10% Gentamicina 20µg/ml MEMFA MOPS 0.1 M pH 7.4 EGTA 2 mM MgSO4 1 mM Formaldeide 3.7%
Generalmente si preparano “stocks” sterili di una soluzione 10x dei Sali (MEM) che viene diluita e addizionata di formaldeide al momento dell’uso.
2.6.2. Microiniezione di embrioni di Xenopus laevis
Per gli esperimenti di microiniezione sono stati usati embrioni pigmentati in cui è possibile distinguere il polo animale da quello vegetativo e i blastomeri dorsali da quelli ventrali.
Al momento della microiniezione gli embrioni degelificati vengono trasferiti in una piastra Petri del diametro di 5 cm sul fondo della quale è stata fissata una reticella di plastica, con maglie di 1 mm, che ne limita gli spostamenti. Inoltre gli embrioni sono immersi in una soluzione di Ficoll al 4% (peso/volume) sciolto in MMR 0.1x: il Ficoll è uno zucchero viscoso, permette agli embrioni di mantenere la forma sferica durante la microiniezione e ne limita la perdita di citoplasma successivamente.
Gli embrioni microiniettati sono stati lasciati sviluppare ON in MMR 0.1x-ficoll 4%, trasferiti in MMR 0.1x finchè gli embrioni di controllo non inietttati non avessero raggiunto lo stadio desiderato e quindi fissati.
Le microiniezioni sono state eseguite con un microiniettore Drummond Nanoject, che consente l’iniezione di volumi compresi tra 4.6 nl e 73.6 nl ad incrementi discreti. Gli aghi sono stati preparati per tiratura a caldo a partire da capillari forniti da Drummond: la loro bontà è stata controllata allo stereoscopio. Prima di essere montati sul microiniettore gli aghi sono stati riempiti di olio minerale con una siringa. Il caricamento degli aghi con la soluzione da iniettare è eseguito dal microiniettore stesso.
Durante gli esperimenti sono stati microiniettati alternativamente 200 pg
di β-galattosidasi, 200 pg di β-galattosidasi più 50 pg di Xrx1 e 200 pg di β-galattosidasi più 70 pg di Xrx1.
2.6.3. Microdissezione di embrioni
Per studiare l’espressione di geni endodermici è stato necessario sezionare sagittalmente gli embrioni per rendere facilmente accessibile l’endoderma alle successive manipolazioni.
Sono state preparate piastre Petri da 6 cm colando agar all’1% in H2O sul fondo fino ad ottenerne uno strato di 2-3 mm su cui è stato praticato un forellino del diametro degli embrioni da microdissezionare.
Gli embrioni fissati 30-40’ in MEMFA 1x poi lavati 5’in PBS 1x sono stati adagiati nel forellino della piastra, contenente PBS 1x, in modo da bloccarne eventuali spostamenti e sono stati sezionati sagittalmente con l’ausilio di una lama per microdissezione.
2.6.4. Reazione cromogenica della β-galattosidasi
Negli esperimenti di microiniezione è stato co-iniettato mRNA di β-galattosidasi nucleare come marcatore: ciò ha permesso di valutare la bontà
della microiniezione attraverso la reazione catalizzata dall’enzima, che in presenza di un substrato cromogeno produce un precipitato colorato. Come substrato cromogeno è stato utilizzato il Salmon-gal, che produce un precipitato di colore rosso, seguendo il seguente protocollo:
• Fissare gli embrioni microiniettati in MEMFA per 30-40’.
• 2 x 5’ lavaggi in PBS 1x (si può lasciare a 4°C fino a 48 h).
• 2 x 15’ lavaggi in “Wash solution” (o ON a 4°C).
• Lasciare 5’ in soluzione di ferri.
• Rivelare in 0,5 ml totali di soluzione di ferri contente 5 µl di stock Salmon-gal, incubando a 37°C. Controllare periodicamente la rivelazione.
Soluzioni per la reazione cromogenica della β-galattosidasi Soluzione di ferri K3Fe(CN)6 30 mM K4Fe(CN)6.3H2O 30 mM PBS 1x Stock Salmon-gal
Salmon-gal (Sigma) 5% in metanolo Wash solution (conservare a 4°C) PBS pH 7.4 1X
Sodio deossicolato 0,01% Nonidet P40 0,02%
MgCl2 2mM
2.6.5. Ibridazione in situ su embrioni interi (whole mount)
L’ibridazione in situ su embrioni interi permette di studiare il “pattern” di espressione di uno o più geni nei diversi stadi embrionali di Xenopus laevis mediante l’ibridazione di sonde marcate con DIG-UTP ai trascritti presenti nei tessuti; il protocollo seguito è sostanzialmente quello descritto da Niehrs (Gawantka et al., 1995):
gli embrioni vengono posti in vials da 4 ml RF (RNase free) dove non indicato diversamente riempire la vial fino ai 4/5.
• Reidratare gli embrioni conservati in etanolo a -20°C, con passaggi di 5’ ciascuno in:
Etanolo 75 % PBTw 25 % Etanolo 50 % PBTw 50 Etanolo 25 % PBTw 75 PBTw 100%
• Aggiungere 0,5 ml/vial di soluzione di proteinasi K in PBTw 1:2000; incubare 5’ esatti senza agitazione (omettere questo passaggio e successivi lavaggi in PBTw con embrioni emisezionati o con espianti).
• Effettuare due brevi lavaggi con PBTw con agitazione manuale.
• Sostituire il PBTw con paraformaldeide al 4% in PBS, 1 ml per vial; incubare 20’ a RT con agitazione manuale occasionale (omettere questo passaggio e successivi lavaggi in PBTw con embrioni emisezionati o con espianti).
• Effettuare due brevi lavaggi con PBTw con agitazione manuale, quindi 4x5’ lavaggi in PBTw con agitazione orizzontale.
• Sostituire il PBTw con 0,5 ml/vial tampone di ibridazione al 50% in PBS, incubare 3’ quindi sostituire con tampone di ibridazione 100%, incubare 3’.
• Sostituire con 0,5 ml/vial di tampone di ibridazione 100% e preibridare 2-3 h a 60°C.
• Denaturare 40 ng, 50 ng o 60 ng di sonda di RNA marcato (a seconda dell’intensità del segnale di ciascuna sonda) in 10 µl di H2O distillata per 2’ a 95°C, passare rapidamente in ghiaccio e aggiungere 600 µl di tampone di ibridazione (concentrazione finale della sonda: 83.3 ng/µl). Aggiungere la miscela alle vials e ibridare ON a 60°C.
• Preparare una soluzione SSC 2x/0.1% CHAPS e lasciarla ON a 37°C.
• Preparare una soluzione SSC 0.2x/0.1% CHAPS e lasciarla ON a 60°C.
• Recuperare la miscela con la sonda e sostituirla con 1 ml/vial 50% tampone di ibridazione/50% (SSC 2x/0.1% CHAPS); incubare per 10’.
• Incubare 2x30’ con 1ml/vial di SSC 2x/0.1% CHAPS preequilibrato a 37°C.
• Incubare 2x30’ con 1 ml/vial SSC 0.2x/0.1% CHAPS preequilibrato a 60°C.
• Sostituire con 50% (SSC 0.2x/0.1% CHAPS)/50% TBS 1x a RT per 5’, quindi TBS 1x per 5’.
• Sostituire con 0,5 ml/vial di blocking buffer, incubare 2h a 4°C con agitazione orizzontale.
• Diluire l’anticorpo AP Fab Anti-DiG (Roche) 1:2500 in blocking buffer e incubare 2h a 4°C con agitazione orizzontale.
• Sostituire il “blocking buffer” con 0,5 ml/vial di soluzione di anticorpo diluito e incubare 4 h a RT con agitazione verticale.
• Eliminare la soluzione di anticorpo e effettuare 5x1h lavaggi in TBSx a RT (uno può essere ON a 4°C) con agitazione orizzontale.
• Effettuare 2x5’ lavaggi con 1 ml/vial di APB.
• Sostituire l’APB con 0,5 ml/vial di “BM Purple AP substrate precipitaing” (Roche) e incubare a RT al buio (incubando a 14°C il segnale risulta più pulito, ma è necessario un tempo più lungo; a 4°C la reazione si blocca): sostituire il substrato quando necessario e quando la marcatura è adeguata sostituire il “BM Purple” con TBSx per 10’ per bloccare la reazione.
• Fissare gli embrioni in MEMFA 1 h a RT oppure ON a 4°C e conservarli in etanolo a -20°C.
La fosfatasi alcalina coniugata all’anticorpo scinde il substrato cromogenico (“BM Purple”) generando un precipitato colorato che evidenzia le zone in cui la sonda si è ibridata, dove il gene in esame è stato trascritto.
Soluzioni per ibridazione in situ su embrioni interi PBS NaCl 8 g KCl 0.2 g Na2PO4.16H2O 1.44 g KH2PO4 0.24 g pH 7.4 TBS NaCl 8 g KCl 0.2g Tris base 3 g pH 7.4
SSC 20x NaCl 3.0 M Na citrato 0.3 M PBTw 0.1% PBS 1x Tween 20 0.1% TBSx TBS 1x Triton X-100 0.1% Paraformaldeide 20%
• Sciogliere 5 g di paraformaldeide in 100 ml H2O RF a 60°C
• Chiarificare con 10 µl NaOH 10 N, portare a volume e filtrare.
• Conservare a -20°C. Una volta scongelata usare entro un mese. Tampone di ibridazione
Formammide 50% SSC 5x
RNA di torula 1 mg/ml Eparina 100 µg/ml
Roche Bocking reagent 1 % Tween 20 0.1 %
CHAPS 0.1 % EDTA 10 mM
Soluzione stock di Proteinasi K 20µg/µl APB (“Alcaline phosphatase buffer”) Tris HCl pH 9.5 100 mM MgCl2 50 mM NaCl 100 mM Tween 20 0.1 % Tetramisole 2 mM “Egg extract”
• Omogenizzare embrioni conservati a -20°C in 1 volume di PBS 1x
• Centrifugare 10’ a 4°C a 12000 rpm, recuperare la fase acquosa.
• Centrifugare 5x10’ a 4°C a 12000 rpm recuperando la fase acquosa.
• Denaturare a 56°C per 15’, centrifugare per 10’ a 12000 rpm a 4°C.
“Blocking buffer” TBSx 1.6 ml
Siero di agnello inattivato al calore 300 µl Egg extract 100 µl
Roche blocking reagent 400 µl.
2.6.6. Depigmentazione degli embrioni (“bleaching”)
Per evidenziare meglio il segnale dell’ibridazione gli embrioni sono stati sottoposti a depigmentazione:
• Sostituire l’Etanolo in cui sono conservati gli embrioni con Etanolo 70% in H2O per 5’ a RT in agitazione orizzontale.
• Sostituire con 50% Etanolo/ 50% SSC 1x per 5’ a RT in agitazione
orizzontale.
• Sostituire con “bleaching solution” e porre le vials in agitazione orizzontale sotto una lampada ad incandescenza, controllando costantemente la depigmentazione.
• Fermare la depigmentazione passando in metanolo 70% per 5’ e successivamente in Etanolo 100%. Conservare gli embrioni a -20°C.
“Bleaching solution” H2O2 1%-3%
SSC 0.5x
2.7. Ibridazione in situ su sezioni
2.7.1. Sezioni al criostato
Gli embrioni devono essere fissati e crioprotetti per mantenere la morfologia cellulare originaria e infine inclusi in una resina per permettere il taglio al criostato.
• Trasferire gli embrioni in tubi da batterio.
• Fissare in paraformaldeide 4% in PBS 1x 8ml/tubo per 2 ore a RT in agitazione orizzontale.
• Sostituire con saccarosio 20% in PBS 1x e lasciare in agitazione orizzontale finchè gli embrioni non affondano oppure ON a 4°C.
• Trasferire il numero desiderato di embrioni in appositi stampi di plastica e ricoprire di O.C.T™.
• Orientare gli embrioni nel senso del taglio.
• Effettuare un congelamento rapido appoggiando i blocchetti in una vaschetta contenente etanolo a -80°C per 15-20’.
• Asciugare bene il fondo e conservare a -80°C fino al momento del taglio.
• Lasciare i blocchetti a -20°C per circa 30’ per ammorbidire la resina e poi tagliare al criostato sezioni dello spessore di 12 µm.
• Fare aderire le sezioni ottenute al vetrino avvicinandolo parallelamente ad esse, l’adesione avviene elettrostaticamente.
• Lasciare asciugare all’aria i vetrini per circa 1 ora a RT, conservarli a – 80°C.
2.7.2. Ibridazione in situ per sezioni al criostato
Il protocollo seguito è sostanzialmente quello descritto da Casarosa (Casarosa et al., 2003):
• Scongelare i vetrini a RT almeno 30’.
• Pre-riscaldare la soluzione di ibridazione a 65°C per fluidificarla.
• Diluire la sonda a 0.1-1µg/ml (la concentrazione va valutata sperimentalmente per ogni sonda) nella soluzione di ibridazione, denaturarla 5-10’ a 70°C poi mettere subito in ghiaccio per qualche minuto.
• Aggiungere 120µl di sonda per vetrino e mettere il coprioggetto. Appoggiare orizzontalmente i vetrini in una camera umida contenente 2 fogli di carta “Whatman” 3MM bagnati con 50% Sali-50% formammide. Lasciare ibridare ON a 65°C.
• Preparare la washing solution e lasciarla a 65°C ON. • Trasferire i vetrini in un portavetrini verticale.
• Fare un primo lavaggio con la washing solution di 15’ a 65°C per togliere il coprioggetto.
• Lavare 3x30’ a 65°C con la washing solution pre-riscaldata.
• Lavare 2x30’ a RT con il MABT 1x.
• Mettere 1ml per vetrino di blocking solution lasciare per 1-2 ore a RT senza coprioggetto nella camera umida con carta “Whatman” 3MM imbevuta di H2O.
• Diluire l’anticorpo AP-Fab anti-Dig (Roche)1:2500 in blocking solution.
• Aggiungere 150µl di anticorpo per vetrino, mettere il coprioggetto e incubare a RT nella camera umida ON.
• Trasferire i vetrini nel portavetrini verticale e fare 5 lavaggi in MABT per 30’ a RT.
• Lavare 2x10’ con l’AP-buffer a RT.
• Diluire 18µl l’NBT/BCIP (Roche) in 1ml AP-buffer e mettere 150µl di soluzione per vetrino. Aggiungere il coprioggetto, lasciare in rivelazione a
RT nella camera umida al buio (per rallentare la reazione mettere a 14°C o 4°C). Cambiare la soluzione ogni 24 ore. Alternativamente all’NBT/BCIP usare 200µl di “BM Purple” senza diluirlo. È stato scelto per ogni sonda il substrato che dava risultati migliori.
• Fermare la reazione quando la colorazione ha raggiunto una intensità sufficiente lavando i vetrini 5’ in PBS 1x.
• Aggiungere 1ml per vetrino di “Hoechst” staining diluito 1:1000 in PBS 1x lasciare 5’ a RT senza coprioggetto al buio per marcare i nuclei cellulari.
• Lavare 5’ in PBS 1x poi montare il coprioggetto con “Aqua Poly/Mount”.
• Lasciare asciugare per circa 30’ a RT e poi conservare a 4°C. Soluzioni per ibridazione in situ su sezioni
10x Sali (1L) NaCl 114g TrisHCl pH7.5 14.04g Tris base 1.34g NaH2HPO42H2O 7.8g Na2HPO4 7.1g 0.5M EDTA pH8 100ml 100x Denhardt’s
Albumina di siero bovino 2% peso/volume Ficoll 2% peso/volume Polivinilpirrolidone 2% peso/volume Soluzione di ibridazione Sali 1x Formammide 50% Solfato di destrano 50% rRNA 1mg/ml Denhardt’s 1x MABT Acido maleico 100 mM NaCl 150 mM Tween20 0.1% Blocking solution
Blocking reagent 2%
Siero di agnello inattivato al calore 20% pH 7.5 Washing solution SSC 1x Formammide 50% Tween-20 0,1%
2.8. Fotografie
Le fotografie degli embrioni interi o emisezionati sono state realizzate mediante una fotocamera digitale CoolSNAP-cf montata in asse focale ad uno stereoscopio Nikon SMZ1500 con l’ausilio di una coppia di fibre ottiche.
Le fotografie delle sezioni sono state realizzate mediante un microscopio Nikon Eclipse E600 integrato di fotocamera.
Il trattamento delle immagini è stato realizzato con l’ausilio del software Adobe Photoshop CS3.
2.9. Strumenti bioinformatici
Per le analisi bioinformatiche stati utilizzati gli algoritmi BLASTn e BLASTx disponibili presso l’NCBI (National Center for Biotechnology Information) all’indirizzo http//blast.ncbi.nih.gov/Blast.cgi. Questi algoritmi hanno permesso il confronto delle sequenze immesse con tutte quelle contenute nelle banche dati dell’ NCBI e disponibili in Rete. I risultati degli allineamenti di sequenza hanno permesso di cercare in base all’omologia, sequenze, anche di altri organismi, che fossero simili ai trascritti analizzati e dessero quindi indicazioni sulla possibile identita’ e funzione.