Capitolo III
__________________________________________________________________
3.1 Sintesi dei precursori
Per preparare i composti 20a-h, 21a-h e 22a-c, obiettivo di questo lavoro di Tesi (cfr. par. 1.3), sulla base delle retrosintesi riportate negli Schemi 5, 6, 7 del paragrafo 2.3 era in ogni caso necessario disporre degli arilalchini 49a-h, degli acidi boronici 56a-g e dello (Z)-3- bromopropenoato di metile (50).
N N N S Me3Si H H H Me H H R1 R2 R3 49b: R1=R2=R3=H 49c: R1=R2=R3=OMe 49d: R1=F, R2=OMe, R3=H 49h: R1=OH, R2=OMe, R3=H 49a 49f 49g 49e B(OH)2 MeO (HO)2B R1 R2 R3 56a: R1=R2=R3=H 56b: R1=F, R2=OMe, R3=H 56c: R1=R3=H, R2=OCF3 56d:R1=NH2, R2=R3=H 56e: R1=R2=R3=OMe 56f: R1=OH, R2=OMe, R3=H 56g Br Z COOMe 50
Per quanto riguarda gli 1-alchini, 49a, 49b, 49f e 49g sono commerciali, mentre 49c e 49d sono stati preparati a partire dalle corrispondenti aldeidi commerciali 62a e 62b, rispettivamente, seguendo una tipica procedura generale99 (Schema 8). In particolare, tali aldeidi sono state convertite negli 1,1-dibromoeteni 63a e 63b per reazione con l’ilide di fosforo ottenuta trattando il tetrabromuro di carbonio, CH2Cl2 e a 0°C, con trifenilfosfina. Dopo aver eliminato mediante cromatografia su gel di silice l’ossido di trifenilfosfina formatosi durante questa reazione, tali dibromuri sono stati fatti reagire, senza ulteriori purificazioni, con 2 equivalenti di n-BuLi esanico, in THF e a –78°C, ottenendo con ottime rese globali i desiderati alchini 49c-d, inquinati solo da tracce (< 1%) dei corrispondenti alcheni (Schema 8).
Schema 8 H O R1 MeO R2 MeO R1 R2 Br Br PPh3 (2 equiv.), CBr4 (1 equiv.), CH2Cl2, 0°C, 10 min. 62a: R1=R2=OMe 62b: R1=F, R2=H 63a: R1=R2=OMe 63b: R1=F, R2=H R1 MeO R2 H
n-BuLi (2 equiv.), THF/esano,
-78°C, 1.5 h, quindi r.t.
49c: R1=R2=OMe (81% da 62a) 49d: R1=F, R2=H (60% da 62b)
Il 2-etiniltiofene (49e) è stato invece preparato seguendo una procedura già utilizzata allo stesso scopo nel nostro laboratorio (Schema 9).100 Così il 2-iodotiofene (64a), commerciale, è stato fatto reagire a temperatura ambiente con 1 equivalente di trimetilsililetinilmagnesio bromuro (65), ottenuto per scambio idrogeno-metallo fra 49a ed etinilmagnesio bromuro, in presenza di 5 moli% di Pd(PPh3)4 ed in THF. Il trimetilsililalchino grezzo 66a così ottenuto è stato quindi trattato a 0°C con un eccesso di KOH in metanolo, per dare il desiderato 49e, con resa globale del 60% (Schema 9).
Schema 9 S S S I Me3Si H Me3Si MgBr (65) (1 equiv.) Pd(PPh3)4 (5 mol%) THF, benzene, r.t., 16 h. KOH(acq) 1M (1 equiv.),
MeOH, 0°C, 2h
64a 66a
49e
(60% da 64a)
Infine l’ultimo degli 1-alchini necessari, 49h, è stato preparato a partire dal guaiacolo (67), anch’esso commerciale. Infatti, seguendo una procedura descritta in letteratura,101 67 è stato convertito prima nello ioduro 64c, mediante una sequenza di reazioni che non prevede alcun isolamento intermedio, e quindi, in ulteriori due stadi, in 49h (Schema 10).
Schema 10 HO MeO H HO MeO SiMe3 I HO MeO I AcO MeO AcO MeO MeO HO 1)AcCl (1.1 equiv.), Py (2.3 equiv.), CH2Cl2, 0°C, 5 min 2) H3PO4(acq) 1M,0°C 1) ICl (1.2 equiv.), CH2Cl2, r.t., 24h 2) Na2S2O3, 0°C, 2h LiOH•H2O (3.5 equiv.), MeOH/H2O/THF (3:1:3), r.t., 4 h 49a (1.3 equiv.), PdCl2(PPh3)2 (4 mol%), CuI (8 mol%), r.t., 4.5 h TBAF (1.3 equiv.) THF, r.t., 20 min. 67 68 64b 64c 66b 49h (95% da 67) (76% da 64c)
Più in particolare, l’ossidrile di 67 è stato protetto mediante O-acetilazione con cloruro di acetile e piridina ed il prodotto grezzo ottenuto dopo opportuno work-up (68) è stato iodurato selettivamente con ICl in CH2Cl2, a 0°C. Si è così formato 64b che, grezzo, è stato saponificato a 0°C con LiOH in MeOH/H2O/THF, fornendo quindi 64c con resa globale del 95% dopo ricristallizzazione (Schema 10). Successivamente 64c è stato sottoposto a una reazione di cross-coupling tipo Sonogashira90 con il trimetilsililacetilene (49a), seguita dalla protodesililazione di 66b grezzo così ottenuto, effettuata con un eccesso di tetrabutilammonio fluoruro in THF (Schema 10). Tale procedura ha fornito 49h con resa dell’80% da 64c e purezza del 98%.
Per quanto riguarda i necessari acidi boronici, 56a-g, solo 56f, che contiene un ossidrile fenolico libero, non è un prodotto commerciale. Usualmente acidi idrossiarilboronici come 56f vengono preparati da corrispondenti idrossiarilalogenuri mediante sequenze costituite da: i) protezione dell’ossidrile; ii) scambio alogeno metallo; iii) trattamento dell’arilmetallo così ottenuto con un trialchilboronato e, infine, iv) deprotezione dell’ossidrile.a,102 Per evitare una tediosa sequenza di questo tipo, abbiamo deciso di preparare, in luogo di 56f, il
a A nostra conoscenza esiste un solo esempio di sintesi di un acido idrossiarilboronico effettuata in modo convenzionale senza protezione dell’ossidrile, ma il lavoro che la riporta contiene pochi e confusi dettagli sperimentali.102f
corrispondente estere boronico 56h, e di utilizzare a questo scopo la procedura in un solo stadio descritta molto recentemente da Wang et al. per la sintesi di un simile estere idrossiarilboronico.103 Pertanto lo ioduro 64c, preparato come descritto nello Schema 10, è stato fatto reagire a 80°C con un leggero eccesso del diboronato 69, in presenza di 3 mol% di PdCl2(dppf) e di un eccesso di KOAc in DMSO, ottenendo 56h con resa del 62% dopo isolamento mediante MPLC su gel di silice. Questo estere non è stato poi convertito nel corrispondente acido boronico, ma è stato impiegato con buoni risultati nelle reazioni di Suzuki descritte nel paragrafo 3.
Schema 11 O B O OB O B O O I MeO HO HO MeO (1.2 equiv.) PdCl2(dppf) (3 mol%), KOAc (3 equiv.), DMSO, 80°C, 6h 64c 56h 69
Per quanto riguarda infine lo (Z)-3-bromopropenoato di metile (50), tale composto non è stato isolato bensì preparato e utilizzato in situ così come riportato nel paragrafo 3.2.
3.2 Sintesi dei aril-2-en-4-inoati 33b,c,e del
5-trimetilsilil-2-en-4-inoato 33d e dei
7-aril-2-en-4,6-diinoati 48a,b,c.
Tenendo conto delle retrosintesi delineate negli Schemi 5, 6 e 7 (cfr. par. 2.3) a questo punto era necessario preparare gli eninoati 33b-e e gli endiinoati 48a-c. R1 MeO R2 COOMe Me 3Si COOMe 33b: R1=R2=OMe 33c: R1=F, R2=H 33e: R1=OH, R2=H Z Z R1 R2 R3 COOMe Z 48a: R1=R2=R3=H 48b: R1=R2=R3=OMe 48c: R1=OH, R2=OMe, R3=H 33d
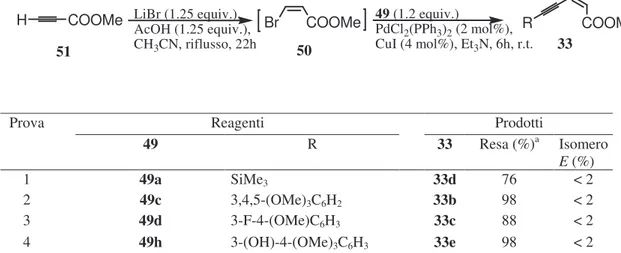
I composti 33b-e sono stati ottenuti con rese elevate seguendo una pratica procedura generale one pot messa a punto da Lu et al.91d e già adottata con successo nel nostro laboratorio104. Come riportato nella Tabella 1, questa procedura prevede di trattare a caldo il propiolato di metile (51) con eccessi di LiBr e AcOH in acetonitrile, ottenendo così una miscela di reazione contenente lo (Z)-3-bromopropenoato di metile (50) stereoisomericamente puro. Tale miscela viene quindi trattata a temperatura ambiente, in sequenza, con 2 mol% di PdCl2(PPh3)2, 1,2 equivalenti dell’opportuno 1-alchino 49 ed un eccesso di trietilammina. Così facendo i desiderati esteri eninoici 33b-e sono stati ottenuti con rese dal 76 al 98% e purezze stereoisomeriche maggiori del 98% (Tabella 1).
Tabella 1 Sintesi degli eninoati di metile 33b-e
H COOMe LiBr (1.25 equiv.), AcOH (1.25 equiv.), Br COOMe R COOMe
CH3CN, riflusso, 22h
49 (1.2 equiv.)
PdCl2(PPh3)2 (2 mol%), CuI (4 mol%), Et3N, 6h, r.t.
51 50 33
aResa in prodotto isolato
Può essere interessante rilevare che 33d è risultato inquinato da una piccola quantità (circa il 3%) del diino derivante dall’omocoupling del trimetilsililacetilene (49a), che non siamo stati in grado di eliminare.
Per quanto riguarda gli endiini 48, in primo luogo abbiamo provato a prepararli seguendo la via B decritta nello Schema 7, che prevede l’impiego di una reazione di Cadiot-Codkiewicz. Infatti questo tipo di approccio sulla carta sembrava il più pratico e veloce. Pertanto il trimetilsilileninoato 33d è stato convertito sia nel corrispondente bromuro 53 sia nel corrispondente 1-alchino 55 (Schema 12). Nel primo caso, seguendo una classica procedura generale,9433d è stato fatto reagire con un leggero eccesso di N-bromosuccinimmide in presenza di 20 moli% di AgNO3. Nel secondo caso, invece, 33d è stato trattato con TBAF secondo un tipico protocollo di protodesililazione (Schema 12).
Schema 12 COOMe Me3Si COOMe H COOMe Br TBAF, THF, r.t., 20 min. NBS, acetone/H2O (10:1), AgNO3 (20 mol%), -50°C, 9h, quindi r.t. 3h 53 55 33d 29% 31%
Prova Reagenti Prodotti
49 R 33 Resa (%)a Isomero E (%) 1 49a SiMe3 33d 76 < 2 2 49c 3,4,5-(OMe)3C6H2 33b 98 < 2 3 49d 3-F-4-(OMe)C6H3 33c 88 < 2 4 49h 3-(OH)-4-(OMe)3C6H3 33e 98 < 2
Come è riportato nello Schema 12, pur avendo utilizzato condizioni molto più controllate di quelle previste nella procedura standard (-50°C anziché 0°C o temperatura ambiente)94,105, purtroppo la resa di 53 è stata solo del 29%, a causa della formazione di una grande quantità di prodotti secondari.b Non molto migliore è stata la resa (31%), sempre a causa della formazione di numerosi sottoprodotti.
Nonostante ciò abbiamo effettuato alcune prove di cross-coupling secondo Cadiot-Codkiewicz, condotte in condizioni classiche93 (Schema 13).
Schema 13 H OMe MeO MeO COOMe Br COOMe CuCl (6 mol%), NH2OH•HCl (0.34
equiv.), EtNH2(acq), MeOH, 0°C, 5 min.
48b: R1=R2=R3=OMe
CuCl (6 mol%), NH2OH•HCl (0.34
equiv.), EtNH2(acq), MeOH, 0°C, 2h 55 70 48a H 48a: R1=R2=R3=H (51%) 53 49b COOMe CuCl (6 mol%), NH2OH•HCl (0.34
equiv.), EtNH2(acq), MeOH, 0°C, 5 min.
(40%) OMe MeO MeO 53 49c
In particolare 49b e 49c sono stati fatti reagire a 0°C con il bromoalchino 53, in presenza di 6 moli% di CuCl, 0.34 equivalenti di NH2OH·HCl e 14 equivalenti di EtNH2 acquosa (Schema 13), ottenendo, rispettivamente, 48a con resa del 51% e 48b con resa del 40%.
Successivamente è stata anche effettuata una prova corrispondente ad una disconnessione opposta, ovvero l’1-alchino 55 è stato trattato, sempre in analoghe condizioni, con l’1-bromoalchino 70 (già disponibile nel nostro laboratorio) (Schema 13). In questo caso però, si è formato quasi esclusivamente il diino 71, derivante da un omocoupling di 70, e solo tracce del desiderato 48a.
b Prove pilota effettuate a temperature maggiori hanno fornito grezzi di reazione ancora più sporchi.
Considerando i mediocri risultati ottenuti sia nei cross-coupling riportati nello Schema 13 che nella preparazione degli esteri 53 e 55 (Schema 12), abbiamo deciso di abbandonare questa strada e di provare a preparare i composti 33 desiderati attraverso la via A delineata nello Schema 7.
71
In primo luogo sono stati quindi preparati i necessari diini 72a-c, seguendo un classico protocollo di etinilomologazione di alchini106 (Tabella 2). Così gli alchini 49b, 49c e 49h sono stati trattati con 2.5 equivalenti di (Z)-1,2-dicloroetilene, 8 moli% di CuI, 4 moli% di Pd(PPh3)4 e 5 equivalenti di n-BuNH2, in benzene e a temperatura ambiente (Tabella 2). I cloroenini 73a-c così ottenuti, già durante la concentrazione dei corrispondenti grezzi di reazione, si sono rivelati estremamente sensibili a luce e calore, come del resto già riportato in letteratura per 72a107. Pertanto i relativi grezzi di reazione parzialmente concentrati sono stati filtrati su Celite®, per eliminare la maggior parte dei metalli presenti, prima di rimuovere completamente il solvente. Quindi i residui così ottenuti sono stati sottoposti senza ulteriori purificazioni alle successive reazioni di eliminazione con tetrabutilammonio fluoruro (TBAF) in THF (Tabella 2). Tabella 2 Sintesi dei diini 72a-c
Ar
H Ar Cl
H
Ar
(Z)-1,2-dicloroetene (2.5 equiv.) CuI (8 mol%), Pd(PPh3)4 (4 mol%) n-BuNH2 (5 equiv.), C6H6, r.t. 22h TBAF (2 equiv.) THF, r.t. 49 72 73
a Rese in prodotti isolati.
Prova Reagenti Tempo di desililazione
(h) Prodotti 49 Ar 72 Resa da 49 (%)a 1 49b C6H5 6 72a 59 2 49c 3,4,5-(OMe)3C6H2 6.5 72b 46 3 49h 3-(OH)-4-(OMe)C6H3 6 72c 40
Comunque anche i diini così preparati sono risultati molto labili sia alla luce che al calore, nell’ordine 72a << 72c < 72b. A questa labilità, e non a una bassa conversione, sono imputabili le rese modeste con cui sono stati ottenuti 72b e 72c (Tabella 2): tali composti hanno subito una parziale degradazione anche durante le procedure di isolamento, per quanto siano state effettuate nelle condizioni più veloci e blande possibili. Abbiamo inoltre notato che lo stesso TBAF utilizzato per l’eliminazione (Tabella 2) è in grado di decomporre 72b e 72c. Infatti, protraendo la durata delle reazioni di eliminazione di 73b e 73c oltre lo stretto necessario, si osserva una rapida scomparsa dei relativi prodotti 72b e 72c, rispettivamente, inizialmente formatisi.
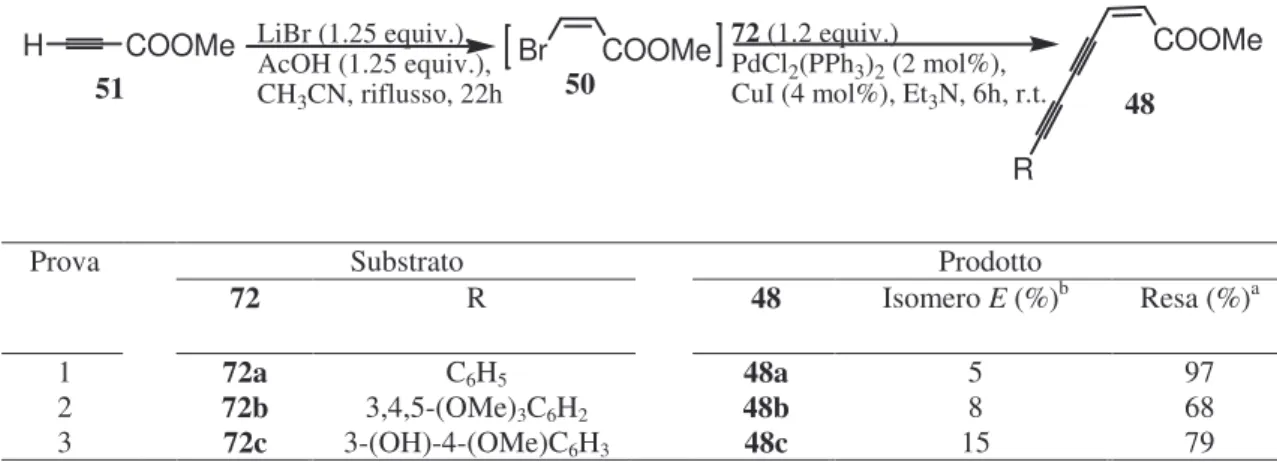
Pertanto i diini 72a-c sono stati rapidamente sottoposti alla successiva reazione di Sonogashira necessaria per ottenere i desiderati endiini 48a-c (Tabella 3), effettuata seguendo la stessa procedura one pot utilizzata per preparare gli eninoati 33 (Tabella 1). Infatti i suddetti diini 72a-c sono stati fatti reagire con lo (Z)-3-bromoacrilato di metile (50), preparato in situ come già descritto (Tabella 1), in presenza di una miscela catalitica a base di Pd e Cu e di Et3N (Tabella 3).
Tabella 3 Sintesi degli endiinoati 48
H COOMe LiBr (1.25 equiv.), AcOH (1.25 equiv.), Br COOMe
CH3CN, riflusso, 22h 72 (1.2 equiv.) PdCl2(PPh3)2 (2 mol%), CuI (4 mol%), Et3N, 6h, r.t. COOMe R 51 50 48
aRese in prodotto isolato
bPercentuali dell’isomero E nei prodotti grezzi
Come risulta dalla Tabella 3, tutti i composti 48 desiderati sono stati ottenuti con rese in prodotti isolati almeno buone, ma, in ogni caso, si sono formate quantità significative dei corrispondenti isomeri E. Purtroppo non è stato possibile eliminare completamente tali isomeri durante la purificazione mediante MPLC, a causa sia di una scarsa separazione, sia della tendenza a stereomutare
Prova Substrato Prodotto
72 R 48 Isomero E (%)b Resa (%)a
1 72a C6H5 48a 5 97
2 72b 3,4,5-(OMe)3C6H2 48b 8 68
per effetto della luce e del calore, mostrata da questi composti, e soprattutto da 48c.
3.3 Sintesi dei 5-iodopiranoni 29b-d e 47a-b;
tentativi di sintesi di 29e e 47c.
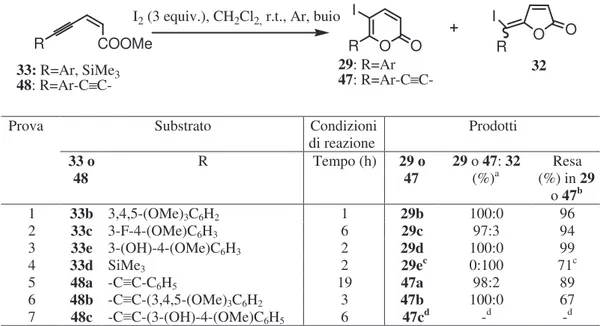
Disponendo finalmente dei necessari eninoati 33b-e ed endiinoati 48a-c, abbiamo potuto procedere con le reazioni di iodolattonizzazione programmate (cfr. par.2.3 e Schemi 5, 6 e 7). I risultati ottenuti trattando a temperatura ambiente i composti 33 e 48 sopra citati con 3 equivalenti di I2 in CH2Cl2 sono riportati nella Tabella 4.
Tabella 4 Sintesi dei 5- iododpiranoni 29 e 47
R COOMe R O O I O O I R +
I2 (3 equiv.), CH2Cl2, r.t., Ar, buio 33: R=Ar, SiMe3
48:
R=Ar-C≡C-29: R=Ar
47: R=Ar-C≡C- 32
arapporti piranone/furanone nel prodotto grezzo. brese in prodotti isolati regioisomericamente puri
cQuesta resa si riferisce alla miscela E/Z di furanoni formatasi
dIn questa prova si è formata una complessa miscela di prodotti (vedi testo)
Come mostrato nella Tabella 4, le reazioni di iodolattonizzazione effettuate sugli eninoati 33b-e e sugli endiinoati 48a-b hanno fornito i desiderati 5-iodopiranoni 29b-d e 47a-b, rispettivamente, con ottime rese in prodotti isolati regioisomericamente puri (prove 1-3 e 5-6). Può essere comunque interessante notare che nei grezzi di reazione relativi alle prove 2 (R=3-F-4-(OMe)C6H3) e 5 (R=feniletinil) erano presenti piccole quantità (3 e 2%, ripettivamente) di un sottoprodotto che, all’analisi glc-ms, mostrava la stessa massa del prodotto principale ma una diversa frammentazione. Con tutta probabilità tali sottoprodotti
Prova Substrato Condizioni
di reazione Prodotti 33 o 48 R Tempo (h) 29 o 47 29 o 47: 32 (%)a (%) in Resa 29 o 47b 1 33b 3,4,5-(OMe)3C6H2 1 29b 100:0 96 2 33c 3-F-4-(OMe)C6H3 6 29c 97:3 94 3 33e 3-(OH)-4-(OMe)C6H3 2 29d 100:0 99 4 33d SiMe3 2 29ec 0:100 71c 5 48a -C C-C6H5 19 47a 98:2 89 6 48b -C C-(3,4,5-(OMe)3C6H2 3 47b 100:0 67 7 48c -C C-(3-(OH)-4-(OMe)C6H5 6 47cd -d -d
corrispondono ai due furanoni 32c e 32d (vedi paragrafo 2.2), formatisi per una non completa regioselettività delle reazioni.
O O O O I I F MeO 32c 32d
Invece, nelle stesse condizioni, il trimetilsilil-2-en-4-inoato di metile (33d) ha fornito esclusivamente una miscela dei due furanoni stereoisomeri E e Z-32e (Tabella 4, prova 4). Infatti nel prodotto grezzo di reazione erano presenti solo due prodotti, isolati con rese del 24 e del 48%, che alla glc-ms risultavano avere la massa attesa sia per 29e che per 32e e frammentazioni identiche. Inoltre gli spettri IR di questi due composti sono risultati perfettamente sovrappaonibili e caratterizzati da una banda del carbonile situata a 1792 cm-1, una zona tipica per i carbonili furanonici (i carbonili piranonici danno luogo ad assorbimenti situati intorno ai 1710 cm-1). O O O Me3Si I O I Me3Si E-32e Z-32e
Questo risultato ha quindi mostrato l’impossibilità di accedere ai prodotti desiderati sulla base della retrosintesi riportata nello Schema 6. Quindi abbiamo abbandonato questa via e, considerandolo non interessante per i nostri scopi, non abbiamo neanche assegnato la configurazione ai due furanoni ottenuti nella prova 4 (Tabella 4).
Purtroppo anche la prova 7 (R=(3-idrossi-4-metossifenil)etinil, Tabella 4) ha dato luogo a risultati completamente negativi. Infatti, come hanno evidenziato analisi glc e TLC effettuate sul grezzo di reazione, si è formata una complessa miscela di prodotti difficilmente separabili per cromatografia liquida. Un’analisi glc-ms di tale miscela ha permesso di relevare la presenza del desiderato 47c, ma non di identificaregli altri componenti. D’altra parte, analisi 1H e 13C NMR, effettuate sul grezzo di reazione concentrato, hanno mostrato la presenza di alcuni
segnali attribuibili al prodotto desiderato, 47c, ma anche quella di molti altri segnali, almeno altrettanto intensi, che non è stato possibile attribuire. Ipotizzando che questo risultato fosse dovuto, almeno in parte, alla presenza di un ossidrile libero nel reagente di partenza, 48c,c abbiamo deciso di proteggere l’ossidrile di
48c come butildimetilsililetere, utilizzando una procedura convenzionale con t-butildimetilsililcloruro e trietilammina (Schema 14). E’ stata così ottenuta, con buona resa dopo purificazione mediante MPLC, una miscela stereoisomerica del sililderivato 48d (Z/E = 75:25). E’ da rilevare che 48d si è mostrato molto più instabile configurazionalmente del suo precursore, 48c. Infatti dopo sole 2h a temperatura ambiente era già vistosamente stereomutato (Z/E = 60:40). D’altra parte un campione di 48d conservato per alcuni giorni a –23°C è risultato composto principalmente dall’isomero E (Z/E = 20:80). Abbiamo pertanto deciso che non era possibile preparare lo iodolattone 47c per questa via. Quindi abbiamo rinunciato a preparare, almeno per quanto riguarda questo lavoro di Tesi, anche il composto 22c (cfr. par. 1.3), visto che, sulla base di quanto programmato (Schema 7), 47c avrebbe dovuto essere utilizzato come suo diretto precursore.
Schema 14 COOMe HO MeO COOMe TBDMSiO MeO TBDMSiCl (1 equiv.), Et3N (1.25 equiv.), CH2Cl2, r.t., 30'. 48c 48d
c Comunque anche nella prova 3 (Tabella 4) il substrato 33e ha un ossidrile libero, ma l’atteso ioduro 29d in questo caso si è formato con ottima resa e completa regioselettività.
3.4 Sintesi dei 5,6-diaril-2H-piran-2-oni 20b-i e
dei 5-aril-6-ariletinil-2H-piran-2-oni 22a-b.
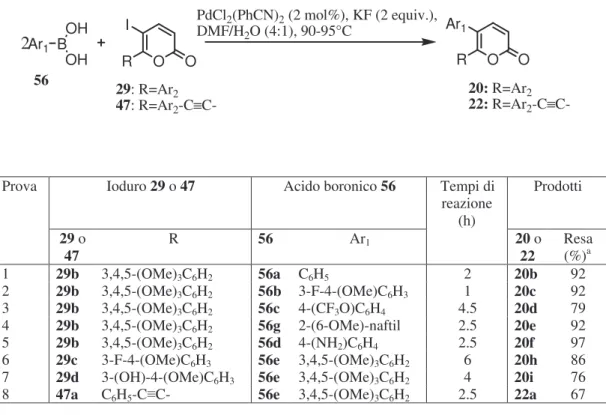
Avendo finalmente a disposizione i necessari iodolattoni 29b-d e 47a-b, abbiamo potuto procedere con la sintesi dei desiderati 5,6-diaril-2H-piran-2-oni 47a-b secondo quanto programmato (cfr. par. 1.3, Schemi 5 e 7). I composti 20b-i e 22a sono stati preparati facendo reagire gli iodopiranoni 29b-d e 47a-b con 2 equivalenti degli opportuni acidi boronici commerciali 56, utilizzando reazioni tipo Suzuki96 effettuate in presenza di 2 moli% di PdCl2(PhCN)2 come catalizzatore, 2 equivalenti di KF come base ed una miscela DMF/H2O 4:1 come solvente (Tabella 5). Abbiamo scelto queste particolari condizioni perché già impiegate con successo da Larock et al.86 per ottenere 40a a partire da acido fenilboronico e 74, un 5-iodo-2H-piran-2-one molto simile a quelli impiegati da noi. O O Ph O O I Me Ph Ph Me 40a 74
D’altra parte l’impiego del KF come base, in luogo delle tipiche basi ossigenate più usualmente utilizzate nelle reazioni di Suzuki (ad esempio carbonati, fosfati o idrossidi di metalli alcalini),96 sembrava più compatibile con la presenza di un anello lattonico nei substrati. Più avanti in questo paragrafo sarà mostrato come i nostri dubbi sulla stabilità dei substrati 29 se sottoposti a condizioni di Suzuki con basi ossigenate, anche se deboli come carbonati o fosfati, fossero fondati.
Tabella 5 Sintesi dei piranoni 5,6-diarilsostituiti e 5-aril-6-ariletinilsostituiti Ar1 BOH OH R O O I O O R Ar1 2 PdCl2(PhCN)2 (2 mol%), KF (2 equiv.), DMF/H2O (4:1), 90-95°C 29: R=Ar2 47: R=Ar2-C ≡C-20: R=Ar2 22: R=Ar2-C ≡C-56
a Resa in prodotto isolato.
Comunque sia, le reazioni riportate nella Tabella 5 hanno fornito i prodotti desiderati con rese da buone a ottime, anche se con tempi variabili in funzione della situazione elettronica e dell’ingombro sterico dell’acido boronico utilizzato. Può essere interessante notare che analisi glc e glc-ms effettuate sui prodotti grezzi di reazione relativi alle prove 3 e 4 (Tabella 5) hanno mostrato la presenza di piccole quantità del piranone 39b, derivante da una protodeiodurazione dello ioduro 29b utilizzato come precursore. In nessun altro dei grezzi ottenuti dalle altre prove citate nella Tabella 5 abbiamo individuato simili prodotti di protodeiodurazione. O O MeO MeO OMe 39b
Peraltro la resa più bassa (67%) è stata ottenuta nella prova 8, effettuata con il 5-iodo-6-ariletinilpiranone 47b. Questa diminuzione di resa non è imputabile ad una scarsa reattività dello ioduro 47b, che dopo 2.5 h risultava scomparso dalla
Prova Ioduro 29 o 47 Acido boronico 56 Tempi di
reazione (h) Prodotti 29 o 47 R 56 Ar1 20 o 22 Resa (%)a 1 29b 3,4,5-(OMe)3C6H2 56a C6H5 2 20b 92 2 29b 3,4,5-(OMe)3C6H2 56b 3-F-4-(OMe)C6H3 1 20c 92 3 29b 3,4,5-(OMe)3C6H2 56c 4-(CF3O)C6H4 4.5 20d 79 4 29b 3,4,5-(OMe)3C6H2 56g 2-(6-OMe)-naftil 2.5 20e 92 5 29b 3,4,5-(OMe)3C6H2 56d 4-(NH2)C6H4 2.5 20f 97
6 29c 3-F-4-(OMe)C6H3 56e 3,4,5-(OMe)3C6H2 6 20h 86
7 29d 3-(OH)-4-(OMe)C6H3 56e 3,4,5-(OMe)3C6H2 4 20i 76
miscela di reazione, quanto ad una certa labilità di tale ioduro nelle condizioni di reazione. Infatti analisi glc-ms di prelievi effettuati durante la reazione hanno mostrato che, insieme al prodotto desiderato, si formavano alcuni sottoprodotti presumibilmente derivati da una degradazione di 47b.
I composti 20g e 22b sono stati invece preparati con buone rese facendo reagire 29b e 47b, rispettivamente, con il 5-(5,5-dimetil-1,2,3-diossaborinan-2-il)-2-metossifenolo (56h) (Schema 15). Schema 15 B O O O O OH MeO I R O R O OH MeO 2 56h 29b: R=3,4,5-(OMe)47b: R=3,4,5-(OMe)33CC66HH22-C ≡C-20g o 22b PdCl2(PhCN)2 (2 mol%), KF (2 equiv.), DMF/H2O (4:1), 90-95°C Prodotti R 20g 22b 3,4,5-(OMe)3C6H2 3,4,5-(OMe)3C6H2-C ≡C-Tempo (h) Resa (%) 8 4.5 67 65
Le rese ottenute delle due reazioni di Suzuki riportate nello Schema 15 sono mediamente inferiori a quelle ottenute nelle simili reazioni effettuate con acidi boronici (Tabella 5). Sono comunque ancora abbastanza buone da giustificare la nostra scelta di preparare, anziché il corrispondente acido, l’estere boronico 56h, evitando così tediose sequenze di protezione-deprotezione, e di utilizzarlo tal quale (cfr. par. 3.1). Può essere interessante rilevare che, a nostra conoscenza, le due reazioni riportate nello Schema 15 rappresentano i primi casi di impiego di un anione fluoruro come base in reazioni tipo Suzuki effettuate con esteri boronici. D’altra parte abbiamo ottenuto risultati assai peggiori di quelli riportati nello Schema 15 in alcune prove pilota condotte in condizioni analoghe a quelle riportate in letteratura per il cross-coupling fra simili 5,5-dimetil-1,2,3-diossaborinani e ioduri o triflati arilici o vinilici.108 Ad esempio, trattando 29b con 56h a 100°C in presenza di 3 moli% di Pd(PPh3)4 e 3 equivalenti di carbonato di potassio, in EtOH/toluene, si sono formate solo tracce del desiderato 20g, insieme ad una certa quantità di sottoprodotti presumibilmente derivati da una degradazione di 20g. Invece trattando 29b con 56h a 80°C in presenza di 3
moli% di Pd(PPh3)4 e 2 equivalenti di fosfato di potassio, in diossano, 20g è stato ottenuto con resa del 43% e bassa purezza chimica (ca. 85%). Infatti è risultato contaminato da vari sottoprodotti difficilmente eliminabili per via cromatografia. Tra questi abbaimo identificato, via glc-ms, il piranone 39b, derivante dalla protodeiodurazione di 29b presente insieme a vari altri composti presumibilmente originati da una degradazione di 29b, peraltro completamente scomparso durante la reazione.
3.5 Sintesi dei
5-(ariletinil)-6-aril-2H-piran-2-oni 21a-h e dei 5,6-di(ariletinil)-2H-piran-2-5-(ariletinil)-6-aril-2H-piran-2-oni
76a-b.
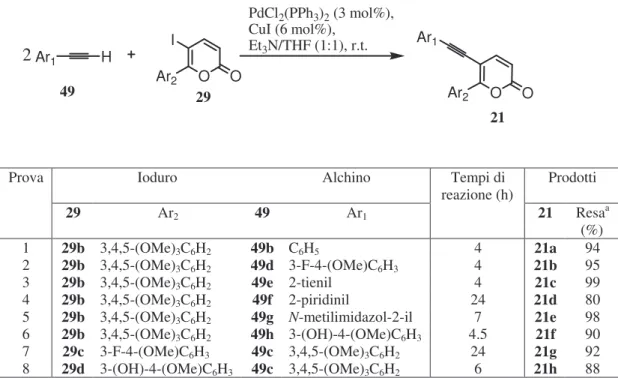
I desiderati 5-(ariletinil)-6-aril-2H-piran-2-oni 21a-h (cfr. par. 1.3) sono stati preparati, così come previsto della retrosintesi riportata nello Schema 6 (par. 2.3), mediante reazioni di cross-coupling tipo Sonogashira fra gli iodopiranoni 29b-d e gli opportuni 1-alchini 49b-h (Tabella 6). Nelle condizioni utilizzate90 (3 moli% di PdCl2(PPh3)2 e 6 moli% di CuI come catalizzatori, una miscela di Et3N e THF in rapporto 1:1 come base e solvente, temperatura ambiente e 2 equivalenti rispetto allo ioduro 29, dell’alchino 49) tutte le reazioni effettuate hanno dato luogo ai prodotti desiderati con ottime rese, anche se con tempi di reazione variabili in funzione della natura dell’alchino.
Tabella 6: Sintesi dei 5-ariletinil-6-aril-2H-piran-2oni.
O O O O Ar2 Ar2 Ar1 H Ar1 I 2 49 29 21 PdCl2(PPh3)2 (3 mol%), CuI (6 mol%), Et3N/THF (1:1), r.t.
Prova Ioduro Alchino Tempi di
reazione (h) Prodotti
29 Ar2 49 Ar1 21 Resaa
(%)
1 29b 3,4,5-(OMe)3C6H2 49b C6H5 4 21a 94
2 29b 3,4,5-(OMe)3C6H2 49d 3-F-4-(OMe)C6H3 4 21b 95
3 29b 3,4,5-(OMe)3C6H2 49e 2-tienil 4 21c 99
4 29b 3,4,5-(OMe)3C6H2 49f 2-piridinil 24 21d 80
5 29b 3,4,5-(OMe)3C6H2 49g N-metilimidazol-2-il 7 21e 98 6 29b 3,4,5-(OMe)3C6H2 49h 3-(OH)-4-(OMe)C6H3 4.5 21f 90 7 29c 3-F-4-(OMe)C6H3 49c 3,4,5-(OMe)3C6H2 24 21g 92 8 29d 3-(OH)-4-(OMe)C6H3 49c 3,4,5-(OMe)3C6H2 6 21h 88 a Resa in prodotti isolati.
D’altra parte, prove pilota effettuate seguendo una recentissima modifica della reazione di Sonogashira, che evita l’impiego di sali rameosi e fosfine,109
hanno avuto esiti assai peggiori. Infatti facendo reagire 49b con 29b in presenza di Pd(OAc)2 come catalizzatore, di Bu4NOAc come base e di THF come solvente, così come previsto da tale procedura, dopo 22h di reazione 21a è stato ottenuto con resa del 50% e accompagnato da ingenti quantità di 71, derivante dall’omocoupling dell’alchino 49b utilizzato.
71
Infine, sebbene non fosse stato programmato all’inizio di questo lavoro di Tesi e allo scopo di verificare se anche i 5-iodo-6-ariletinilpiranoni 47 hanno la stessa reattività dei più semplici 5-iodo-6-arilpiranoni 29 in reazioni di Sonogashira, abbiamo effettuato anche le due reazioni riportate nello Schema 16. In particolare gli alchini 49b e 49c sono stati fatti reagire con 47a, nelle stesse condizioni di Sonogashira utilizzate per le prove riportate nella Tabella 6. Come risulta dai dati riportati nello Schema 16, i rispettivi prodotti 76a-b si sono formati con rese sufficienti, ma in media significativamente inferiori a quelle riportate nella Tabella 6.
Schema 16 O O Ar1 I Ar1 O O H PdCl2(PPh3)2 (3 mol%), CuI (6 mol%), Et3N/THF (1:1), r.t. 49 47a 76 49 49b 49c Ar1 C6H5 3,4,5-(OMe)3C6H2 Tempo (h) 2.3 4.5 76 76a 76b Resa (%) 67 82 2