32
MATERIALI E METODI
2.1 Descrizione clinica delle pazienti studiate
E’ stato preso in esame un totale di 127 donne caucasiche delle quali 37 affette da POF, 60 si presentavano come sospette POF e 30 presentavano familiarità per POF. I campioni di sangue, pervenuti dal 2009 al settembre 2011, provenivano dalle U:O. di Ginecologia e di Endocrinologia dell’Azienda Ospedaliera Pisana e dall’Ambulatorio di Genetica Medica della U.O. di Citogenetica. La condizione di POF veniva definita come la cessazione della funzione ovarica e l’aumento dei valori di FSH rispetto all’intervallo di normalità (>40 mlU/ml) per un periodo di tempo superiore a sei mesi. I casi POF sono stati definiti tali perché avevano presentato amenorrea prima dei 40 anni di età. Nel gruppo delle pazienti con sospetta POF sono state incluse donne con valori di FSH elevati ed alterazioni del ciclo tali da sospettare una condizione di iniziale menopausa.
Le pazienti prese in esame avevano tutte un cariotipo femminile normale (46,XX). Tutte le probande sono state considerate idiopatiche perchè nessuna di esse presentava le condizioni associate alla induzione di POF (interventi chirurgici al livello delle ovaie, precedente chemioterapia o radioterapia, patologie autoimmuni o disordini metabolici come galattosemia). L’età media delle donne in esame è 37 anni. Sono stati analizzati inoltre 100 campioni di donne omogenei per età ed etnia al gruppo POF, utilizzate come donne di controllo con funzione ovarica regolare. Tali controlli rappresentano una sezione trasversale della popolazione generale dal momento che si tratta di donne fertili e
33 con accertate gravidanze recenti. L’età media dei controlli è 41 anni. Il grafico 2.1 presenta la distribuzione dell’età dei casi e dei controlli.
Grafico 2.1: Distribuzione delle classi di età di casi e controlli
Gli intervalli di età rappresentati sono riferiti all’età in cui le pazienti sono state esaminate, l’anamnesi ha confermato la manifestazione di POF prima dei 40 anni di età. Pertanto, sebbene ci sia un certo numero casi POF anche al di sopra dei 46 anni, bisogna tener presente che la manifestazione della disfunzione è avvenuta precedentemente. La maggior parte dei controlli è rappresentato da donne con un’età superiore ai 30 anni con successo riproduttivo e gravidanze recenti.
0 10 20 30 40 50 60 70 80 18-25 26-35 36-45 46-55 >50 POF sospetti POF familiarità per POF controlli
34
2.2. Estrazione del D A genomico da sangue periferico
L'estrazione del DNA genomico da sangue periferico è stata effettuata da 350µl di sangue periferico in EDTA utilizzando l’estrattore BioRobot EZ1 (Quiagen) e il kit commerciale EZ1 DNA Blood composto da tampone di lisi cellulare (LB), da biglie magnetiche capaci di legare il DNA e da tamponi di lavaggio del DNA (WB). In tal modo il DNA liberato dalle cellule lisate con il tampone LB viene legato dalle biglie magnetiche e successivamente con l'ausilio di un magnete presente nel BioRobot, che immobilizza le biglie, il DNA viene lavato con i tamponi WB eliminando tutti i residui di lisi e i rimanenti componenti del sangue. Il prodotto finale viene eluito in 100µl di una soluzione salina presente nel kit di estrazione. La concentrazione e la purezza dei campioni è stata misurata con lo spettrofotometro NanoDrop-1000 (Thermo Scientific).
2.3 Estrazione manuale con Fenolo/Cloroformio/Isoamilico
Per i campioni che risultano omozigoti dall’analisi dell’elettroferogramma è stato necessario procedere con l’estrazione manuale del DNA per averne una quantità maggiore e più concentrata col fine di effettuare la digestione e successivo Southern Blot.
Il primo passaggio consiste in una centrifugazione a 2800rpm per 8-10min. In questo modo si può osservare la stratificazione dei linfociti all’interfaccia tra plasma ed eritrociti, che si raccolgono sul fondo. Successivamente si elimina il plasma, si aggiunge il lysis buffer (la cui composizione è indicata nella Tabella 2.2) e si centrifuga nuovamente a 2800 rpm per 8-10min.
35 Tabella 2.2: Composizione del Lysis Buffer
Una volta eliminato il sopranatante si ripete l’ultimo passaggio 2 o 3 volte per ottenere un pellet sufficientemente chiaro. Il pellet, dopo essere stato sospeso in 1ml di TNE (Tris HCl 10 mM a pH8, NaCl 100 mM, EDTA 1 mM a pH8), viene centrifugato a 9000rpm per 7min e quindi sospeso in 600 Il di TNE, 70 Il di SDS (Sodio Dodecil Solfato) al 10% e 10Il di Proteinasi K (20 mg/ml, Invitrogen). Il campione viene così digerito a 37°C overnight oppure a 56°C per 3h.
Per l’estrazione del DNA con Fenolo/Cloroformio/Isoamilico (PCI) si procede nel seguente modo:
Si aggiunge nella provetta un ugual volume di PCI, si agita per inversione per almeno 5min, si centrifuga a 10000 rpm per 5min recuperando poi la fase superiore contenente il DNA in un’altra provetta. Il passaggio viene ripetuto una seconda volta con PCI ed una terza con Cloroformio/Isoamilico (CI). Si recupera la fase superiore contenente DNA che viene precipitato aggiungendo 2 volumi e mezzo di etanolo al 99% freddo e 1/10 di volume di sodio acetato 3M. Il DNA precipita sottoforma di una “medusa” filamentosa in sospensione. Si lascia a –80°C per 3h oppure a -20°C overnight. Si centrifuga quindi a 12000rpm per 10min a 4°C e al pellet si effettua un lavaggio in etanolo freddo al 70%. Si Componente Concentrazione
Saccarosio 0.32 M
TRIS HCl 10 mM
MgCl2 5 mM
36 conclude l’estrazione asciugando il pellet per eliminare le tracce di etanolo, quindi si risospende il pellet in TE o in acqua sterile e si conserva a 4°C. La concentrazione e la purezza del DNA è determinata con una lettura spettrofotometrica.
2.4 Polymerase Chain Reaction (PCR)
A causa dell’alto contenuto di guanina e citosina nella porzione oggetto di studio del gene FMR1, l’amplificazione mediante PCR risulta problematica. Infatti i tentativi iniziali di analisi della mutazione con la PCR non ebbero successo a causa dell'alta stabilità delle doppie eliche ad alto contenuto di CG. Accorgimenti tecnici per facilitare l’amplificazione di questi tratti di DNA sono l’uso di dimetilsolfossido (DMSO) e di 7-deaza-deossiguanosina trifosfato, che destabilizza la doppia elica del DNA e facilita la separazione delle catene antisenso.
Difatti quando è in atto l’amplificazione di uno strand di DNA che presenta un elevato contenuto di G e C si determina spesso la formazione di complessi ripiegamenti intra ed inter-catena dovuti all’elevato numero di legami a idrogeno tra guanine consecutive a livello dell’azoto presente in posizione 7 dell’anello aromatico. Tali complessi si evidenziano in elettroforesi come bande più corte rispetto ai frammenti target, queste bande rappresentano dei prodotti tronchi di PCR che hanno luogo a causa della formazione di siti di arresto di sintesi, vale a dire le forcine originatesi nel filamento che determinano un blocco dell’attività polimerasica. In particolare, è stato sperimentalmente dimostrato (Yotvat Nadel
37 et al, 1995 ) che la replicazione del sito ricco in GC del gene FMR1 subisce un arresto in vitro a causa delle strutture a forcina che si formano tra le guanine presenti sul filamento singolo ed anche di altre strutture secondarie non canoniche quali le “tetraplex” (Figura 2.3).
Figura 2.3: modelli di strutture a forcina di oligomeri CGG.
Inoltre i fenomeni di “mis-priming” e “mis-annealing” (quest’ultimo tra il templato ed il filamento complementare) causate dall’elevata Temperatura di melting (Tm) possono contribuire alla non corretta amplificazione della sequenza.
38 Pertanto le PCR create specificamente per l’amplificazione delle sequenze ricche in GC seguono protocolli modificati che prevengono la formazione delle suddette strutture mediante l’aggiunta di molecole organiche quali il 7-deaza dGTP, un analogo del dGTP che non presenta l’azoto in posizione 7 dell’anello purinico. L’aggiunta di questo nucleotide modificato previene la formazione dei legami a idrogeno tra guanine senza però interferire nei legami di Watson e Crick riducendo così la probabilità di formazione di strutture secondarie.
Inoltre le comuni metodiche di PCR non consentono l’amplificazione di ripetizioni superiori a 110 e rilevano con difficoltà mosaici in cui sono presenti sia alleli premutati che alleli normali a causa dell’amplificazione preferenziale dell’allele normale, più corto. Questo si rivela particolarmente problematico quando il caso in esame è una femmina che presenta un mosaicismo in cui coesistono diversi numeri di ripetizioni, condizione che rende la diagnosi più problematica: infatti, se l’allele normale è presente anche in una piccola percentuale di cellule, si amplifica conducendo ad una diagnosi errata di normalità (falso negativo). Tali mosaici sono identificabili correttamente mediante analisi in Southern Blot.
Tuttavia sono altrettanto rilevanti i vantaggi che l’uso della PCR conferisce nell’analisi delle ripetizioni al locus FMR1, vale a dire la maggiore rapidità della diagnosi, il costo ridotto dei reagenti, la ridotta quantità di DNA richiesta (meno di 100 ng) e la maggiore accuratezza con cui viene determinato l’esatto numero di ripetizioni (quando inferiori a 110).
Femmine eterozigoti per una premutazione piuttosto estesa non amplificabile in PCR e femmine omozigoti per un allele normale possono generare lo stesso risultato, cioè l’identificazione di un solo allele normale. Nel primo caso la
39 preferenziale amplificazione dell’allele normale più corto può impedire l’identificazione dell’allele premutato, mentre nel secondo caso i due alleli co-migrano.
Per questo motivo, nella nostra analisi le femmine che presentavano soltanto un picco nell’elettroferogramma venivano ulteriormente analizzate mediante Southern Blot per verificare se si trattasse di un caso di omozigosi o se, oltre all’allele visualizzato, presentavano un allele espanso che per i limiti propri della PCR non era stato amplificato.
L’amplificazione delle ripetizioni CGG nel locus FRAXA è stata eseguita con il kit GC-Rich PCR System (Roche), utilizzando 50 ng di DNA ed i seguenti primer C ed F:
C: 5’-GCT CAG CTC CGT TTC GGT TTC ACT TCC GGT-3’ F: 5’-AGC CCC GCA CTT CCA CCA CCA GCT CCT CCA-3’
Il volume totale di reazione è di 10 µl ed i componenti sono riportati nella Tabella 2.4.
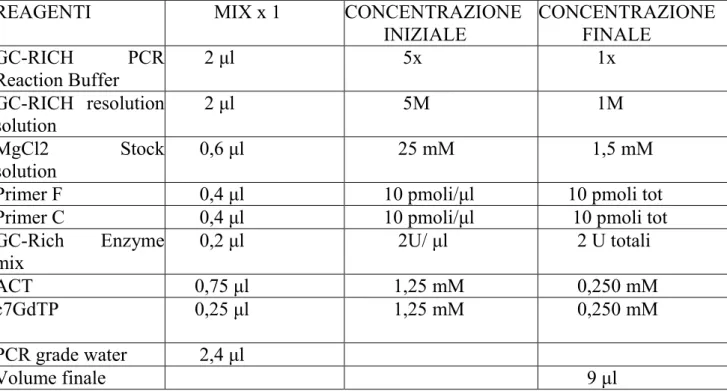
40 REAGENTI MIX x 1 CONCENTRAZIONE
INIZIALE CONCENTRAZIONE FINALE GC-RICH PCR Reaction Buffer 2 µl 5x 1x GC-RICH resolution solution 2 µl 5M 1M MgCl2 Stock solution 0,6 µl 25 mM 1,5 mM Primer F 0,4 µl 10 pmoli/µl 10 pmoli tot Primer C 0,4 µl 10 pmoli/µl 10 pmoli tot GC-Rich Enzyme
mix 0,2 µl 2U/ µl 2 U totali ACT 0,75 µl 1,25 mM 0,250 mM c7GdTP 0,25 µl 1,25 mM 0,250 mM PCR grade water 2,4 µl
Volume finale 9 µl
Tabella 2.4: Reagenti della GC Rich- PCR System
Il programma di amplificazione (Thermal Cycler GeneAmp 9700 PCR System PE Applied Biosystems) è costituito da una iniziale denaturazione a 98 °C per 3’, seguita da 14 cicli con denaturazione a 98,5 °C per 20”, “annealing” a 56 °C per 4’, che consente l’appaiamento dei primers alle sequenze complementari del campione di DNA, ed estensione a 69 °C per 6’, in cui la Taq si attacca ai primers e copia i filamenti di DNA target originando così due molecole di DNA a doppia catena. Seguono 15 cicli con denaturazione per 20” a 98,5 °C, annealing per 4’ a 56°C ed estensione a 69°C per 6’. In quest’ultima fase la temperatura di denaturazione aumenta ad ogni ciclo di 0,1 °C. Le dimensioni dell’amplificato dipendono dal numero delle triplette; l’allele più frequente con 30 ripetizioni dà un amplificato di 311 paia di basi (bp).
41
2.5 Analisi dei prodotti di PCR
Una miscela costituita da 1,5-2 µl di prodotto di PCR, 12 µl di formamide e 0.5µl di 500 Rox standard viene denaturata a 95°C per 2 minuti e successivamente caricata in un sequenziatore automatico ABI PRISM 3100 (Applied Biosystems, Foster City, CA)
Lo strumento è un analizzatore genetico automatizzato che consiste in un apparato per elettroforesi capillare collegato ad un computer che include il software con il quale vengono acquisiti e analizzati i dati.
Esso è provvisto di:
• un sistema di 16 capillari, entro i quali un polimero di acrilamide funziona da matrice per la separazione elettroforetica di singoli frammenti;
• un laser che eccita i vari cromofori legati ai prodotti di PCR ed ai frammenti dello standard quando questi passano dalla finestra di rilevamento;
• una camera CCD (charge-coupled device) che registra i segnali di emissione;
• un fotomoltiplicatore che converte in segnali elettrici;
• un pacchetto di softwares che opera la raccolta, il processamento, l’estrazione e la conversione del segnale in elettroferogramma.
Il dato in uscita è quindi rappresentato da una serie di picchi ciascuno dei quali corrisponde ad un frammento di lunghezza diversa.
Lo strumento è capace dunque di determinare la sequenza nucleotidica o misurare e quantizzare i frammenti di DNA mediante la separazione elettroforetica che avviene in base alla loro lunghezza. I campioni amplificati e
42 caricati sullo strumento vengono introdotti cineticamente nei capillari ripieni di un polimero specifico, che nel nostro caso era il POP4. I frammenti di DNA fluorescenti migrano attraverso il polimero e si separano in base al differente peso molecolare. Durante questa fase i fluorocromi legati al DNA vengono eccitati dall’Argon laser ed emettono luce a lunghezze d’onda specifiche per ciascun fluorocromo. Tale luce viene registrata dalla CCD camera in modo che tutti i tipi di fluorescenza (relativi ai differenti fluorocromi utilizzati) possono essere evidenziati contemporaneamente. Il software analizza l’intensità della luce e la memorizza come segnali elettrici per una successiva elaborazione. I frammenti di DNA vengono rappresentati come picchi su un elettroferogramma. La posizione del picco è indice del peso molecolare del frammento, l’altezza dei picchi è un indice grossolano della quantità del prodotto amplificato, il colore consente di distinguere frammenti diversi con uguale peso molecolare.
I risultati ottenuti vengono analizzati dal software GeneScan3.1, che utilizza gli strumenti di analisi genetica Perkin-Elmer ABI PRISM per determinare le dimensioni e le quantità dei frammenti di DNA attraverso un sistema automatico di rilevamento fluorescente. L’informazione contenuta nei picchi dell’elettroferogramma viene dunque convertita in profilo genetico attraverso dei numeri che indicano il numero di ripetizioni presenti in ogni allele. Per ottenere il numero delle ripetizioni a partire dalla posizione dei picchi si è proceduto con il seguente calcolo:
43 L’inserto laterale dei primers è dato dalla somma delle lunghezze dei due primers più le zone fiancheggianti la sequenza ripetuta.
Il risultato ottenuto viene normalizzato rispetto al genotipo di una femmina con un numero di ripetizioni studiato tramite sequenza.
2.6 Amplificazione mediante il kit AmplideX FMR1 PCR (Asuragen)
Alcuni campioni sono stati analizzati mediante AmplideX FMR1 PCR; Il kit, fornito dalla ditta Asuragen, è stato disegnato appositamente per superare le difficoltà sopra descritte nell’amplificazione del sito ricco in GC del gene FMR1. Permette infatti di risolvere il problema della zigosità mediante un’elevata risoluzione e la possibilità di rilevare anche le mutazioni complete con un numero di ripetizioni superiori a 200. Il kit presenta dei reagenti che permettono di scegliere tra due differenti tipologie di PCR, cioè la CGG RP PCR, che prevede l’utilizzo di un terzo primer complementare alla sequenza dei repeats, e la gene specific PCR.
La quantità di DNA genomico da utilizzare è pari a 20-80 ng, pertanto occorrono 2 µl di un campione con concentrazione dai 10 ai 40 ng/ µl.
La tabella 2.5 riporta i vari reagenti utilizzati per la preparazione del mix di reazione e le rispettive quantità.
44 Tabella 2.5: Reagenti della PCR AmplideX FMR1 PCR (Asuragen)
Il protocollo di amplificazione è il seguente:
CGG repeat primed PCR
• Denaturazione a 95°C per 5 minuti
• 10 cicli a 97 °C per 35 secondi, 62 °C per 35 secondi e 68 °C per 4 minuti
• 20 cicli a 97 °C per 35 secondi, 62°C per 35 secondi, 68 °C per 4 minuti ( più ulteriori 20 secondi di estensione per ciclo)
• estensione a 72 °C per 10 minuti
Gene-Specific PCR
• denaturazione a 98 °C per 5 minuti
• 25 cicli a 97 °C per 35 secondi, 62 °C per 35 secondi, 72 °C per 4 minuti
• estensione 72 °C per 10 minuti
Si procede con l’analisi dei prodotti di PCR mediante elettroforesi capillare in sequenziatore ABI prism 3130, utilizzando il polimero POP 7; la configurazione del
Volume (µl)
Componenti Gene specific PCR CGG RP PCR
GC-Rich Amp Buffer 11,45 11,45
FMR1 F, R FAM Primers 0,50 0,50
FMR1 CGG Primer 0,50 0,50
Diluente 1,00 0,50 CG-Rich Polymerase mix 0,05 0,05
DNA 2,00 2,00
45 sistema di analisi prevede inoltre l’utilizzo di capillari di 36 cm. Per la preparazione del mix 11 µl di Hi-Di Formamide vengono miscelati a 2 µl di ROX 1000 Size ladder (quantità necessari per la corsa di un campione) ed aggiunti a 2 µl di PCR (nella piastra di sequenziamento da 96 pozzetti), si procede poi con la denaturazione per 2 minuti a 95 °C ed il caricamento della piastra nello strumento.
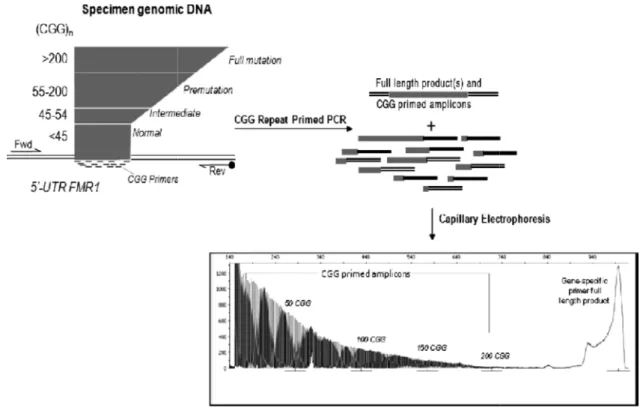
Entrambe le tecniche permettono di individuare il numero totale di ripetizioni dalla lunghezza del picco risultante dall’elettroferogramma, tuttavia nel caso della CGG RP PCR saranno visualizzabili anche i picchi relativi agli ampliconi specifici, derivati dalle diverse combinazioni tra il repeat primer ed il primer reverse. I picchi RP sono pertanto separati tra loro da 3 paia di basi ed il profilo di amplificazione permette anche di capire il numero e la posizione delle interruzioni AGG. I frammenti generati dal primer interno sono più piccoli rispetto a quelli dell’intero repeat ed hanno ovviamente lunghezza variabile, sono identificati come picchi di intensità inferiore. La presenza di un’interruzione AGG può essere visualizzata dalla perdita di intensità di segnale poiché il primer interno non è in grado di appaiarsi ad un tratto CGG che sia interrotto da una tripletta AGG.
Da questo profilo si puo dedurre l’eventuale presenza di una mutazione completa anche qualora il frammento fosse troppo lungo ed amplificato con difficoltà (e pertanto il picco della mutazione completa non visualizzabile), grazie alla presenza di questi picchi intermedi (figura 2.6)
Figura 2.6:Funzionamento della CGG Repeat Primed PCR ed elettroferogramma che rappresenta il numero delle ripetizioni CGG, la zigosità allelica e la quantità e la posizione delle interruzioni AGG. (Amplidex FMR1 PCR kit, user manual)
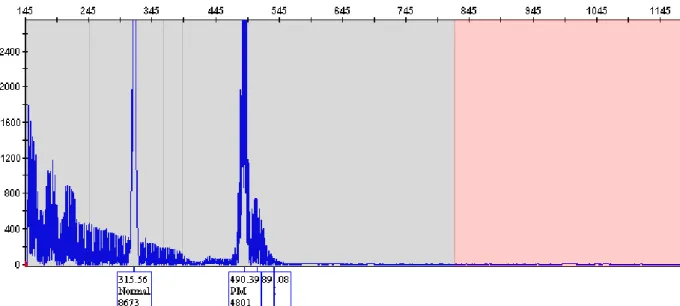
Lo standard ROX 1000 Size Ladder deve presentare all’elettroferogramma 21 picchi.
I picchi presenti tra le prime due linee verticali (tra 245 e 400 bp) sono indice di normalità, quelli che cadono nello spazio compre
rappresentano alleli nel range della zona grigia, i picchi presenti nell’ultimo quadrante grigio rappresentano gli alleli premutati mentre quelli nel quadrante rosa (più di 820 bp) indicano una
nto della CGG Repeat Primed PCR ed elettroferogramma che lle ripetizioni CGG, la zigosità allelica e la quantità e la posizione Amplidex FMR1 PCR kit, user manual)
00 Size Ladder deve presentare all’elettroferogramma 21
prime due linee verticali (tra 245 e 400 bp) sono indice di cadono nello spazio compreso tra la seconda e la terza linea nel range della zona grigia, i picchi presenti nell’ultimo
resentano gli alleli premutati mentre quelli nel quadrante dicano una mutazione completa(figura 2.7)
46 ettroferogramma che uantità e la posizione
troferogramma 21
bp) sono indice di nda e la terza linea esenti nell’ultimo elli nel quadrante 7).
47 Figura 2.7: Elettroferogramma della CGG Repeat Primed PCR (Amplidex FMR1 PCR kit, user manual)
Figura 2.8:Visualizzazione delle interruzioni AGG (Amplidex FMR1 PCR kit, User manual) Con questa tecnica viene accuratamente valutato anche il numero di interruzioni AGG (Figura 2.8) che può essere diverso anche in soggetti che hanno lo stesso numero di ripetizioni. Le interruzioni AGG conferiscono una maggiore stabilità al DNA e riducono il rischio di espansione nella generazione successiva.
48
2.7 Digestione enzimatica e Southern Blot
L’analisi in Southern Blot richiede un’alta quantità del DNA estratto (5-10 microgrammi). L’esecuzione necessita di più giorni poiché consta di varie tappe e richiede una corsa elettroforetica sufficientemente prolungata in modo da permettere il riconoscimento di piccole premutazioni. E’ la tecnica d’elezione per l’identificazione di tutte l’espansioni, delle mutazioni complete, dello stato di metilazione dei mosaicismi e delle delezioni.
La digestione viene effettuata overnight a 37 °C utilizzando 7-10 µg di DNA ad alto peso molecolare, estratto manualmente con la tecnica Fenolo-Cloroformio (paragrafo 2.2), in un volume finale di 50 µl overnight a 37 °C. Vengono utilizzati 50 U di EcoR1 (Biolabs) e 40 U di EAG1 (Biolabs), in presenza di Buffer EAG (Biolabs) ed MgCl2 10 mM
La digestione con EcoRI libera un frammento di 5,2 Kb che comprende il promotore ed il primo esone contenente il repeat; l’aggiunta dell’enzima EagI (sensibile alla metilazione) taglia il cromosoma X non metilato dando 2 frammenti di 2,4 Kb e di 2,8 Kb, al secondo dei quali si lega StB12.3.
EcoRI EagI EcoRI
(CGG)n
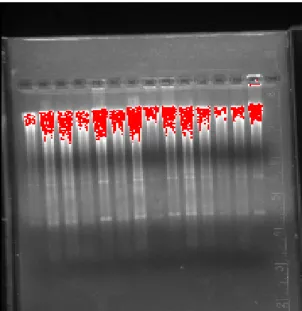
49 Per il controllo della digestione 4 µl di campione con l’aggiunta di 1 µl di blu di Bromofenolo (5x) vengono caricati su gel di agarosio all’1% e corsi per circa 30 minuti. L’immagine 2.9 rappresenta una corsa elettroforetica di frammenti digeriti.
Figura 2.9: Fotografia di una corsa elettroforetica di frammenti digeriti
Il gel viene preparato allo 0,8% in TAE (Tris basico, Acido acetico, EDTA) 1X. Vengono aggiunti al gel ancora in fase fluida15 µl di bromuro di etidio, una molecola che emette fluorescenza una volta esposta agli UV. Essa si intercala tra le basi del DNA e presenta uno spettro di eccitazione con un massimo a 302 nm con una fluorescenza circa 10 volte maggiore rispetto alla molecola libera.
Normalmente occorrono circa 30 minuti per la solidificazione del gel, in seguito esso viene posto nella camera elettroforetica e nei pozzetti viene caricata la digestione.
50 Inizialmente il voltaggio applicato è di 100 volts per 10 minuti, necessario per la fuoriuscita dei campioni dai pozzetti. Successivamente si applica un voltaggio di 67 volts per 1 ora e 37 volts overnight per una durata totale della corsa di 29 ore e 15’.
Il gel viene immerso per 8 minuti, in agitazione, in una soluzione depurinante composta da 243,75 ml di acqua e 6,25 ml di HCl al 37%, trattamento che consente di frammentare il DNA e, quindi, migliorare l’efficacia del trasferimento.
Il passaggio successivo è rappresentato da due lavaggi di 15 minuti ciascuno in 250 ml di soluzione denaturante composta da 250 ml di NaCl 3 M, 50 ml di NaOH 5M e 200 ml di acqua; seguono due lavaggi di altri 15 minuti ciascuno in un tampone di neutralizzazione dell’NaOH (250 ml di NaCl 3M; 125 ml di Tris HCl 2M, pH 7,2, 124 ml di H2O e 1 ml di EDTA 0,5M).
Si procede dunque con l’allestimento del Blot, il tampone di trasferimento utilizzato è il tampone ipersalino SSC in concentrazione 10X, adatto per le membrane di nylon ed il protocollo utilizzato è quello del trasferimento capillare.
Preibridazione e Ibridazione dei filtri
I filtri vengono prima incubati con una soluzione di preibridazione e successivamente ibridati con una soluzione di Ibridazione.
51 Soluzione di preibridazione:
Il volume totale per un filtro è di 20 ml ed i reagenti sono i seguenti:
• 0,4 ml di Tampone fosfato NaH2PO4 1M pH 6,8
• 2 ml di SDS all’1%
• 5 ml di SSC 5X
• 4 ml di Denhart 10X
• 0,2 ml di DNA di sperma di salmone, 0,1 mg/ml
• 8,4 ml di H2O autoclavata Soluzione di ibridazione:
• 8 ml di Formamide al 40%
• 0,4 ml di tampone fosfato NaPi 20 mM
• 2 ml di SDS all’1%
• 5 ml di SSC 5X
• 40 ml di Denhart 10X
• 0,2 ml di DNA di sperma di salmone
• 4,36 ml di H2O autoclavata
Il filtro viene inserito nell’ apposito tubo da ibridazione, vengono aggiunti 20 ml di soluzione di preibridazione (preriscaldata a 37°C) ed è incubato poi per 4 ore a 42°C.
La marcatura della sonda con fosforo radioattivo viene effettuata mediante kit Redprime Amersham seguendodo questo protocollo:
52
• 300 ng di sonda StB12.3 (concentrata 25 ng/ µl), vengono diluiti in buffer TE composto da tris HCl in concentrazione 10 mM e pH8.0 ed EDTA 1mM per un volume totale di 45 ml.
• Il DNA viene denaturato per 5 minuti a 95-100 °C e successivamente messo in ghiaccio
• Dopo centrifugazione il DNA denaturato viene aggiunto alla provetta di reazione del kit.
• 5 µl di fosforo radioattivo 32P dCTP vengono aggiunti al mix ed il tutto incubato per almeno 30 minuti minuti a 37 °C
• Mediante colonnina da filtro realizzata con pasteur e sephadex superfine il mix viene purificato
• La reazione viene stoppata aggiungendo 5 µl di EDTA 0,2 M.
• Il purificato viene denaturato per 5 minuti e poi posto 2 minuti in ghiaccio.
Si procede dunque con la reazione di ibridazione che prevede l’aggiunta della sonda marcata a 20 ml di soluzione di ibridazione nel tubo con il filtro e lasciando il tutto in incubazione O.N. a 42 °C.
Lavaggio del filtro ed esposizione
La soluzione di lavaggio ha un volume totale di 100 ml per un filtro ed è composta da:
• SSC 0,5X,
• SDS allo 0,1%
53 Vengono effettuati 4 lavaggi di 15 min, il primo a temperatura ambiente ed i successivi a 60°C.
Infine il filtro viene asciugato e posto a contatto con una pellicola autoradiografica che viene successivamente sviluppata.
Analisi dei risultati
Le bande che si presentano sul filtro di nylon in seguito allo sviluppo permettono di distinguere i soggetti che presentano gli alleli normali da quelli che portano premutazione o mutazione completa, infatti le bande di 2.8 kb e 5.2 kb rappresentano gli alleli normali presenti, rispettivamente, sul cromosoma X non metilato e su quello metilato (inattivo). L’espansione completa è rappresentata da uno “smear” mentre la premutazione da una banda al di sopra di 2.8kb e di 5.2 kb, cioè 2,8 +Delta e 5,2+Delta. Pertanto da queste bande è possibile capire se è presente una premutazione o una full mutation ed anche se è maggiormente metilato il cromosoma X che porta un’eventuale premutazione oppure quello senza premutazione in base all’intensità delle bande. E’ possibile risalire al numero di ripetizioni a partire dalla distanza in centimetri percorsa dai frammenti utilizzando un sistema di riferimento cartesiano in cui sulle ascisse sono riportati i centimetri percorsi dai frammenti e sulle ordinate, in scala logaritmica, il numero di bp (espresso in kb) corrispondente. Per interpolazione, avendo come riferimento le distanze percorse dai frammenti di 2,8 kb e 5,2 kb, viene calcolato il valore delta (differenza in bp del frammento con premutazione) che diviso per 3 e sommato al numero di ripetizioni dell’allele normale fornisce il numero di ripetizioni dell’allele premutato.
54 Nelle femmine premutate un “bias” di inattivazione del cromosoma X può dare un pattern di bande atipico (2,8Kb+Delta e 5,2 Kb oppure 2,8 Kb e 5,2 Kb+Delta), inoltre femmine in cui i due alleli differiscono di molto tra loro possono apparire come premutate, in tal caso una misura accurata degli alleli è data solo dall’analisi in PCR.