CAPITOLO 3: GLI INIBITORI DEGLI HDAC.
3.1. MECCANISMO D'AZIONE DEGLI INIBITORI DEGLI HDAC.
Ad oggi la maggiore applicazione degli inibitori degli HDAC è il trattamento del cancro.
La ricerca ha posto l'attenzione sulla sintesi di specifici farmaci attivi su alterazioni molecolari presenti solo nelle cellule maligne ed assenti in quelle sane. Lo scopo è quello di individuare per ogni specifica tipologia di cancro il profilo genetico dell'alterazione. I processi molecolari che conducono all'attivazione o repressione della trascrizione sono considerati come possibili bersagli per la terapia antitumorale.
Nel tentativo di spiegare l'attività degli inibitori degli HDAC (HDACi) gli studiosi sono riusciti a spiegare molti meccanismi d'azione propri delle cellule tumorali.
Teoricamente bloccando l'attività degli HDAC, attraverso un HDACi, si favorisce la decondensazione della cromatina con un incremento generale della trascrizione genica. Gli HDACi inducono, inoltre, un aumento dell'acetilazione degli istoni. Nella realtà, tuttavia, molti geni della trascrizione subiscono una up-regulation mentre altri una down-up-regulation influenzando così molti altri meccanismi d'azione e coinvolgendo diverse proteine accessorie. Inoltre è stato dimostrato che l'attività antitumorale degli HDACi è collegata con l'acetilazione
delle proteine non istoniche, questo complica ulteriormente il meccanismo d'azione. La razionalizzazione di queste scoperte è fondamentale per poter effettuare studi clinici efficaci e sviluppare nuove terapie [6].
Il meccanismo d'azione fondamentale risulta tuttavia l'attivazione di una piccola serie di geni che regolano la proliferazione cellulare e la progressione del ciclo cellulare. In saggi in vitro gli HDACi inducono un arresto della crescita cellulare, una differenziazione o l'apoptosi delle cellule cancerose. In particolare, l'inibizione di differenti cicline o la ipofosforilazione delle proteine Rb porta all'inibizione della fase S ed il conseguente arresto del ciclo cellulare in fase G1. Questo blocca l'apoptosi e permette la differenziazione cellulare. In
alternativa le cellule tumorali possono non arrestarsi in fase G1, ma proseguire
fino alla fase G2/S ed andare incontro all'apoptosi. Nelle cellule sane invece gli
HDACi inducono un arresto del ciclo cellulare nella fase G2 riducendo la tossicità
(Figura 7) [7].
Oltre alle neoplasie, la modulazione dell'attività degli HDAC risulta interessante anche per altri campi di applicazione terapeutica. Uno di questi è l'immunomodulazione. Gli HDACi hanno azione sull'acetilazione di alcuni fattori chiave che regolano le funzioni immunologiche della cellula con la soppressione di alcune citochine proinfiammatorie come la TNFα e la IL-1, riducendo in questo modo l'insorgere dell'infiammazione.
Un'altra possibile applicazione è nell'apoplessia: gli HDACi incrementano l'espressione di proteine neuroprotettive, come Hsp70 e Bcl2, presenti nel cervello. Questa azione sulle proteine neuroprotettive può essere una possibile modalità di trattamento di pazienti colpiti da ischemia.
Dai vari studi sugli HDAC, anche utilizzando tecniche di DNA ricombinante, è emerso inoltre che, HDACi strutturalmente diversi, possono avere differenti attività su diverse classi di HDAC o persino su specifiche isoforme.
Per ultimo la modulazione delle sirtuine può essere invece utile per alleviare i cambiamenti associati all'avanzare dell'età. Le sirtuine sono collegate all'aumento della longevità, argomento di grande interesse. Ciononostante gli studi sulle loro funzioni biologiche sono ancora lontani da fornire informazioni utili per una loro applicazione terapeutica [6].
3.2. INIBITORI DEGLI HDAC DI CLASSE I E II
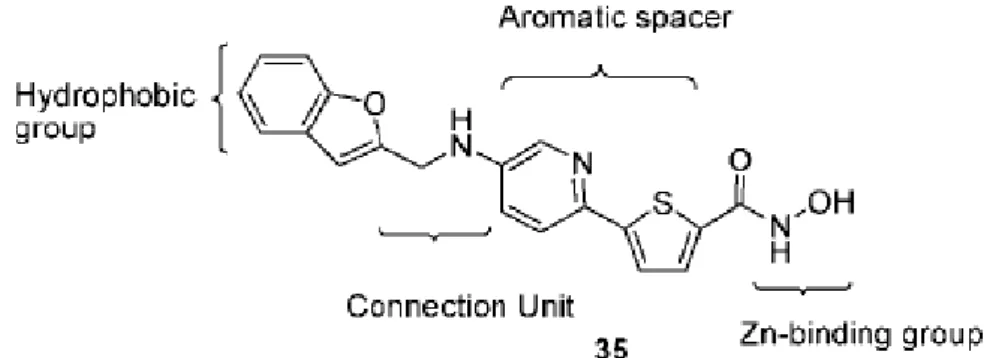
Gli HDACi degli enzimi appartenenti alle classi I e II sono caratterizzati da elementi strutturali comuni che permettono l'interazione con le differenti zone del tunnel catalitico dell'enzima. Queste caratteristiche sono riassunte in un modello farmacoforico comune per tutti i composti di questa classe. Questo comprende un cap group (CAP) capace di interagire con il margine superiore del tunnel catalitico dell'enzima, spesso congiunto ad un'unità di connessione (CU) che unisce il CAP ad uno spaziatore idrofobico (HS), che permette a sua volta alla molecola di entrare nel tunnel. All'estremità dell'HS vi è il gruppo inibitore dell'enzima (EIG o ZBG) capace di complessare lo Zn2+ presente all'estremità del sito catalitico. (Figura 8) [7].
Gli HDACi possono essere divisi in due gruppi principali a seconda della porzione deputata al legame con lo Zn: quelli che contengono come gruppo funzionale legante un acido idrossamico e quelli che non lo contengono.
Il primo gruppo è di gran lunga il più ricco e può essere ulteriormente suddiviso in sottoclassi in base al tipo del gruppo linker. Si distinguono così, acidi idrossamici con linker a catena lineare, cinnamoilica, aromatica o eteroaromatica (Figura 9).
Al secondo gruppo appartengono invece altre classi di molecole che sono state ottenute sostituendo il gruppo dell'acido idrossamico, cioè la parte che si lega allo Zn2+, con altri gruppi funzionali; queste sono: la classe dei tioli e
dei loro derivati, la classe delle benzammidi e la classe dei chetoni.
Inoltre sono riportati alcuni composti che non posseggono un gruppo Zn2+ chelante classico, ma sono comunque capaci di inibire alcune isoforme di
HDAC [10].
3.2.1. HDACi CON “ZBG” COSTITUITO DA UN ACIDO IDROSSAMICO.
In genere gli acidi idrossamici si comportano come pan-inibitori delle classi I e II.
3.2.1.a ACIDI IDROSSAMICI CON LINKER A CATENA LINEARE [10].
Una grande parte degli HDACi fino ad ora noti sono caratterizzati da un residuo di acido idrossamico collegato ad uno spaziatore lineare di cinque o sei carboni idrofobici attaccati a loro volta ad un CAP aromatico tramite una unità di connessione CU.
Il SAHA 1a (tabella 1) è il capostipite della classe e pertanto è la molecola meglio conosciuta. La sua capacità di legame agli HDAC deriva da interazioni polari con lo Zn2+ e da interazioni idrofobiche della catena carboniosa lineare
con l'interno del sito attivo.
Molti studiosi hanno lavorato sull'ipotesi che la modificazione del sistema idrofobico e dell'unità di connessione che interagiscono con l'entrata al sito catalitico dell'enzima HDAC, possa fornire inibitori più potenti e possibilmente più selettivi per alcune isoforme. Tuttavia si ritiene che la presenza di un gruppo polare nella CU non sia necessaria all'azione inibitrice, infatti gli inibitori più potenti ne sono privi.
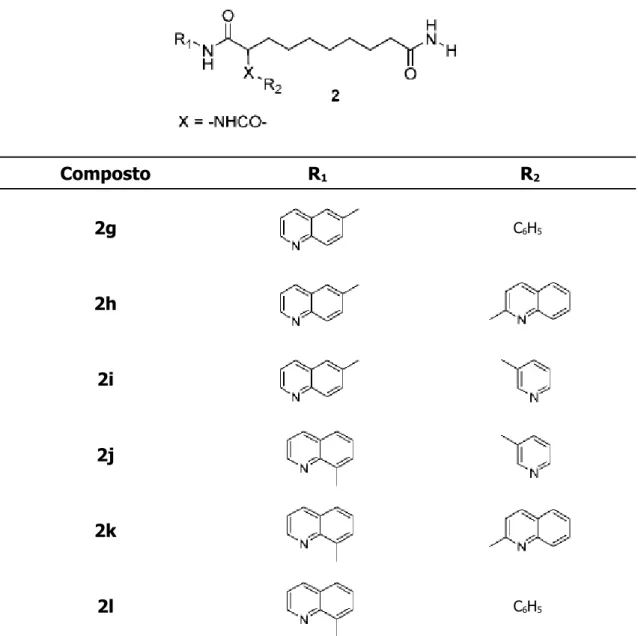
derivati strutturali di questo, in cui l'unità di connessione è α-sostituita con un gruppo ammidico. Questi composti di struttura generale 2 (tabella 2) contengono un'anilide o un'amminochinolide e generalmente mostrano un miglioramento sia nell'attività inibitoria dell'enzima che in quella antiproliferativa, rispetto ai corrispondenti derivati benzilamminici.
In studi successivi, nei composti di formula 2 è stato modificato il gruppo X con residui di solfonammide, urea, tiourea o carbammato che hanno dimostrato di conferire alle molecole una buona attività antiproliferativa.
I derivati con struttura generale 3 e 4 (tabella 3) risultano fino a 200 volte più potenti del SAHA. Inoltre la presenza di due porzioni aromatiche sembra aumentare la probabilità di legame con il sito attivo dell'enzima tramite interazioni idrofobiche addizionali con la superficie esterna del sito stesso dell'HDAC, ne sono un esempio i composti di struttura generale 5 e 6 (tabella 3).
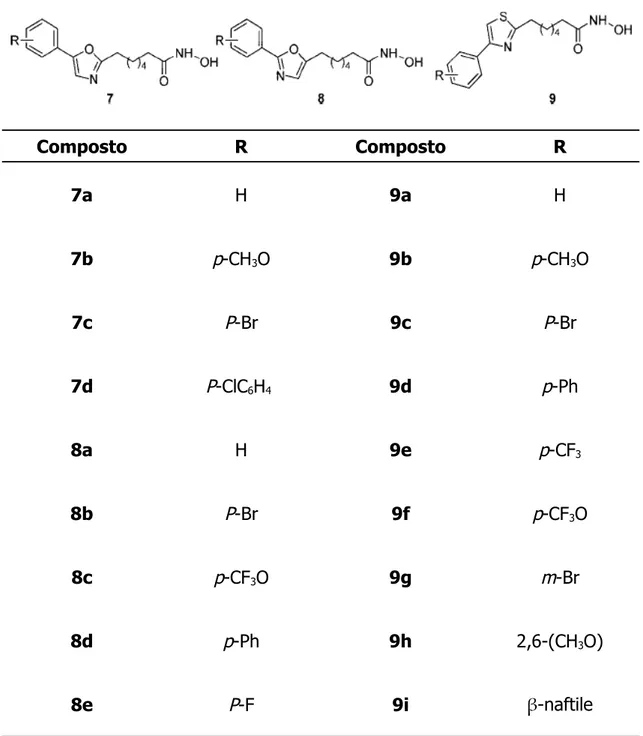
Abbot e SK Corporation hanno entrambi pubblicato studi su composti caratterizzati da un linker lineare la cui unità di connessione è un anello eteroaromatico a cinque termini, in particolare un ossazolo o un tiazolo. Ne sono esempi le molecole di formula generale 7, 8 e 9 (tabella 4). Questi composti sono molto efficaci sugli enzimi isolati, ma mostrano un notevole calo di attività nei sistemi cellulari con un'attività che diventa paragonabile a quella del SAHA.
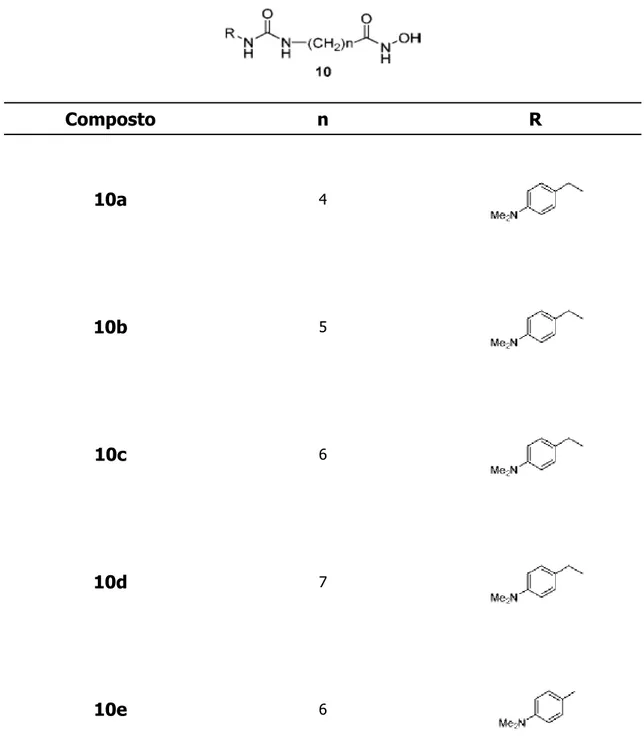
della Oxford GlycoSciences hanno portato all'introduzione nella CU di un gruppo ureidico semplice o fenil sostituito. Tali strutture hanno la formula generale 10 (tabella 5) e 11 (tabella 6) e sono efficaci in vitro, ma non in vivo. Sfortunatamente in vivo la loro efficacia diminuisce notevolmente, nel migliore dei casi la loro potenza è comparabile con quella del SAHA.
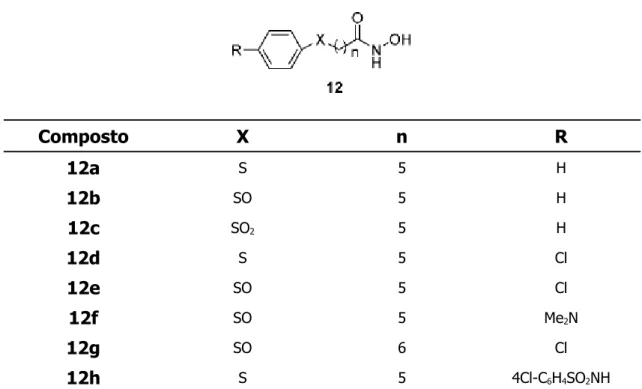
In alcuni composti l'atomo di S è stato introdotto come solfossido o solfone nell'unità di connessione. Marson [11] e collaboratori hanno descritto la
sintesi e l'efficacia di derivati arilsulfinilici e arilsulfanilici di idrossammidi esanoiche; composti di formula generale 12 (tabella 7). In questa serie il linker ottimale risulta quello sostituito da cinque metileni e i solfossidi sono generalmente più potenti dei solfoni; l'attività cellulare tuttavia è in genere deludente.
Un'altra classe che presenta nell'unità di connessione un atomo di zolfo è costituita dagli acidi idrossamici che hanno come CAP una base uracilica. Questi composti di formula 13, detti UBHA (tabella 8), sono stati testati su tre enzimi deacetilasici del mais, gli HD2, HD1B di classe I e HD1A di classe II. In questo caso la lunghezza ottimale del linker si è dimostrata di quattro o cinque unità metileniche e l'introduzione di una porzione fenilica, benzilica o 2-feniletilica al C6 dell'uracile aumenta la loro attività. Alcuni di questi composti, testati sugli HDAC1 dei topi, hanno prodotto un blocco del ciclo cellulare nella fase G1, su cellule tumorali di leucemia mieloide U937.
sulla struttura della cisteina con formula generale 14 (tabella 9). Dei trentasette composti testati, solo sei hanno mostrato buona attività nei confronti di linee cellulari di melanoma umano, con valori paragonabili a quelli del SAHA.
Il gruppo Menarini si è concentrato nello studio di composti di formula generale 15 (tabella 10) introducendo come gruppo linker una piperidina 4-alchil sostituita. In questi composti il gruppo funzionale costituito dall'acido idrossamico è conservato ad un'estremità della molecola, mentre dall'altra parte è legato un gruppo aromatico semplice. La geometria tridimensionale dell'anello aromatico è variata attraverso l'uso di differenti tipi di CU come ammidi, solfonammidi, carbammati ed uree. Gli studi di attività enzimatica hanno dimostrato che questi composti sono inibitori sia su cellule HeLa che nel carcinoma del colon.
La stessa compagnia ha lavorato su una nuova classe strutturale di molecole derivate dal SAHA: gli acidi n-alchilidrossamici ω-sostituiti con sistemi triciclici caratterizzati da un anello centrale a sette termini. Sono rappresentati dalle molecole aventi formula generale 16 (tabella 10) ed hanno dimostrato di avere una certa attività.
3.2.1.b ACIDI IDROSSAMICI CON LINKER CINNAMOILICO [10].
Studi su composti in cui è stato inserito un linker di tipo cinnamoilico, hanno messo in evidenza l'importanza di questo gruppo nell'attività inibitoria di
questi composti.
Nel 2002 Novartis ha pubblicato una serie di acidi cinnamoil idrossamici amminometil sostituiti, fra i quali i composti 1n e 1c (Figura 10 e tabella 1) sono entrati entrambi nella fase II di sperimentazione clinica, e l'1c è attualmente in fase III.
Successivamente è stata progettata una nuova classe di HDACi contenenti il linker cinnamoilico ed una CU costituita da una solfonammide. Questa classe di composti è suddivisa in due sottoclassi: le solfonammidi inverse di struttura 17, che hanno l'N solfonammidico dalla parte opposta del gruppo idrossamico e le solfonammidi dirette corrispondenti alla struttura 18 dove l'N solfonammidico è, invece, dalla stessa parte (tabella 11). È stato messo in evidenza che l'inserimento in posizione meta del gruppo solfonammidico rende i composti di struttura 17 (17a-b-c-d) più attivi degli analoghi inversi di struttura 18 (18a-b-c-d). Lo stesso andamento si riscontra
Figura 10: composto LAQ-824 e struttura generale di acidi idrossamici con linker
quando un metilene è introdotto tra il gruppo arilico e la porzione solfonammidica (17e e 18e). Nei composti para sostituiti la differenza tra solfonammidi dirette ed inverse è molto piccola (17f e 18f; 17g e 18g); infine la sostituzione in orto, rende inattive le molecole (17h-i). Di questi composti solo il 17a, identificato come PXD101 (vedi anche 1c tabella 1) è attualmente sperimentato in fase clinica II.
SK chemicals e In2Gen Co. hanno riportato una serie di derivati cinnamoilici, dove la CU è un gruppo benzilammidico, con struttura generale 19 (tabella 12). Questi composti hanno mostrato attività antiproliferativa nei confronti di cellule di cancro al polmone, alla mammella ed allo stomaco con risultati che hanno spinto i ricercatori alla sperimentazione preclinica dei composti 19a e 19b.
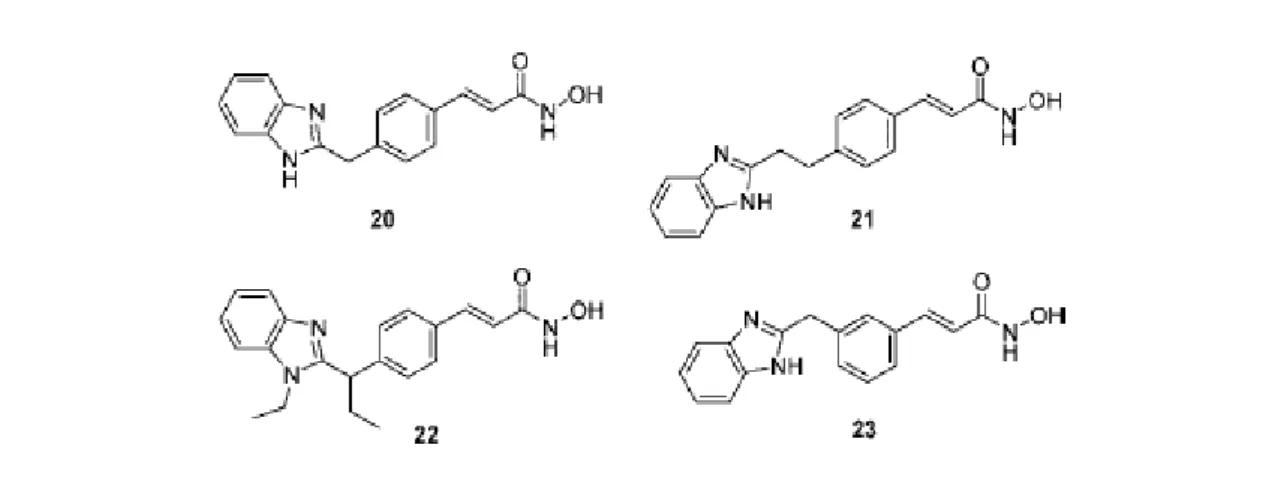
Un'altra serie di inibitori idrossamici con linker cinnamoilico è caratterizzata da un residuo benzimidazolico nella parte idrofobica terminale e da un gruppo metilenico come unità di connessione. Quattro di questi composti (20, 21, 22, e 23) sono stati testati come HDACi (tabella 13).
Un'ulteriore classe di inibitori, rappresentati dal composto 24 (tabella 14), che hanno come CAP un benzimidazolo sono stati studiati dalla Syrrx (ora facente parte della casa farmaceutica Takeda); un secondo studio ha portato ad una nuova classe di derivati, rappresentati dal composto 25, nei quali il CAP è un imidazolo sostituito; questi si sono dimostrati più efficaci dei primi (tabella 14).
Nel 2005 Miyachi [13] e collaboratori hanno pubblicato una nuova
classe di derivati ftalimmidici di formula generale 26 (tabella 15) con buona attività inibitoria dell'enzima. Studi più recenti hanno portato alla progettazione di composti in cui l'unità di connessione è un'ammide od un'immide ciclica, ottenuti per riduzione selettiva di uno dei gruppi carbonilici del composto 26. È interessante notare come dei due regioisomeri 27 e 28 (tabella 15), quello che ha il gruppo carbonilico in meta è molto più potente di quello in para. Ciò mette in evidenza che la regioselettività, e di conseguenza un diverso ingombro sterico, influisce anche sull'azione inibitrice dei composti, cambiando la loro modalità d'interazione con il sito attivo dell'enzima HDAC.
Acidi cinnamoil idrossamici che contengono una porzione eteroarilica sono stati proposti da vari gruppi di ricerca. I derivati benzimidazolici mostrano un'eccellente attività anticancro ed una capacità di promuovere la differenziazione e l'apoptosi. Uno di questi è il composto 29 (tabella 15), creato dalla S*BIO; tutte le strutture derivate dalla 29 sono generalmente ben tollerate in vivo con minimi segni di tossicità e mostrano una significativa riduzione del volume della massa tumorale.
Massa e Mai hanno riportato nelle loro pubblicazioni la progettazione e la sintesi di una serie di HDACi dove la porzione aromatica del linker cinnamoilico è un pirrolo. Partendo dal composto 30 (tabella 16) sono stati ottenuti numerosi derivati. Gli stessi autori hanno progettato una nuova classe di analoghi del composto 31 (tabella 17) dove la porzione aromatica è una
piridina o un fenile. Sei composti di questa classe hanno mostrato di bloccare in vivo il papilloma indotto sperimentalmente attraverso una miscela di DMPA e TPA, noti cancerogeni chimici.
Altana ha sintetizzato una nuova classe di derivati sulfonilpirrolici derivati del composto 32 (tabella 17) che non hanno portato a dei risultati specifici. Infine sono stati sintetizzati una serie di composti strutturalmente analoghi ai precedenti di formula generale 33 (tabella 17), nei quali la funzionalità α/β insatura dell'acido idrossamico è collegata ad un anello piridinico di una imidazo[1,2-a]piridina. Elaborazioni della struttura 33 hanno portato al composto 34 che si è dimostrato particolarmente attivo (tabella 17).
3.2.1.c HDACi CON LINKER AROMATICO OD ETEROAROMATICO [10].
Alcuni HDACi, di struttura generale 35 (Figura 11), contengono come linker una porzione aromatica o eteroaromatica.
Figura 11: struttura generale di acidi idrossamici con linker aromatico od
Uno dei primi esempi di HDACi appartenenti a questa classe è il composto 36 realizzato da Roche nel 2004 nel quale il nucleo tiofenico usato come linker (tabella 18) è la maggiore novità.
Sempre nel 2004 Argenta Discovery ha pubblicato studi su molecole con un tiofene come linker seguito da un altro eterociclo addizionale che può essere una piridina, una pirimidina, un imidazolo od un pirazolo. Di settantacinque composti testati dalla Argenta Discovery il composto 37 (tabella 18) è stato identificato come inibitore submicromolare degli HDAC.
Per cercare un'ottimizzazione di questo composto, sono stati introdotti differenti gruppi lipofili su uno degli N pirazolici sintetizzando le molecole di struttura generale 38 (tabella 19). Queste hanno dato notevoli risultati in vitro dimostrando anche una significativa inibizione del CIT P450, in particolare il composto 38h è risultato efficace.
È stata anche provata la sostituzione dell'anello pirazolico del composto 37 con altri anelli aromatici od eteroaromatici. In particolare la sostituzione con la piridina ha generato tre possibili isomeri di struttura 39 (tabella 20) dei quali l'orto derivato 39a ha dato i migliori risultati.
L'ottimizzazione del nuovo lead 39a è stata effettuata inserendo sostituenti addizionali sull'anello piridinico (tabella 21). Sulla base dei risultati farmacologici ottenuti e della semplificazione strutturale è stato selezionato il composto di formula 40 come prototipo per ulteriori strutture. Questo è risultato più attivo del SAHA nei test di inibizione enzimatica e di proliferazione cellulare.
Studi della S*BIO nel 2005 hanno proposto l'introduzione di nuovi linker eterobiarilici, in particolare, molte di queste molecole hanno come linker un sistema biarilico costituito da un tiazolo-tiofene. Numerosi composti, tra i quali quelli di struttura generale 41 e 42 (tabella 22), hanno mostrato una certa selettività per l'HDAC8.
Molti ricercatori hanno sintetizzato molecole nelle quali il linker è semplicemente un fenile. Osaka Industrial Promotion ha proposto nel 2003 una classe di composti il cui maggior rappresentante è il composto 43 (tabella 23); questo in vivo mostra marcati effetti antitumorali contro il tumore HT-29 dei topi.
La Merk nel 2006 ha pubblicato i derivati di struttura 44 e 45 (tabella 23) dove l'unità di connessione è rispettivamente un'ammide o una solfonammide posta in para all'anello aromatico. Il CAP idrofobico è scelto tra una larga varietà di sistemi monociclici e biciclici aromatici ed eteroaromatici. Altri ricercatori hanno introdotto CU alternative costituite da benzilcarbammati, fenileteri, alchini o alcheni.
La Mikana Terapeutics nel 2004 ha utilizzato come linker il 2-aminotiazolo sintetizzando il composto 46 (tabella 24). Successivamente lo stesso gruppo di ricerca ha sintetizzato una serie di composti contenenti la 2-amminopirimidina come linker, e corrispondenti ai composti di formula generale
47 (tabella 24); questi, testati su cellule cancerose umane, hanno mostrato una
Utilizzando la struttura di 47 nel 2003 la Janness Pharmaceutica ha sviluppato nuove molecole, fra cui il composto 1m (tabella 1), entrato nella sperimentazione clinica, ed i composti 48, 49 e 50 (tabella 25).
Nel 2005 sono stati testati dalla Aton Pharma HDACi, di struttura generale 51, che contengono come linker un benzotiofene (tabella 26). L'acido idrossamico 2-benzotiofenico 51a non sostituito, ha una buona attività; la sostituzione delle posizioni 5 e 6 dell'anello tiofenico con una fenilammide (51b-c), una benzilammide (51d-e), o una naftilammide (51g-f) provoca un forte aumento della potenza inibitoria. Sostituzioni con una fenilammide inversa (51h) ha evidenziato un piccolo incremento dell'efficacia rispetto al composto 51a; la sostituzione rispettivamente in posizione 5 e 6 con una benzilammide inversa (51i e 51j) invece è ben tollerata portando di nuovo un aumento dell'attività inibitoria. La sostituzione con una naftilammide inversa (51k) si è dimostrata deleteria per l'attività. L'introduzione in posizione 6 di una solfonammide come unità di connessione (51l, 51m e 51n) ha dato, invece, buoni risultati.
La casa farmaceutica Syrrx ha sperimentato molecole nelle quali il linker è un benzofurano o un benzimidazolo, corrispondenti ai composti di struttura rispettivamente 52 e 53 (tabella 27). Questi sono stati testati sugli HDAC 1, 2, 6 e 8, ma ad oggi non sono stati pubblicati i risultati della loro attività biologica.
3.2.2. HDACi CON “ZBG” DIVERSO DALL'ACIDO IDROSSAMICO [10]
Come accennato precedentemente, sono stati sintetizzati composti con azione inibitrice sugli HDAC che, come porzione di chelazione allo Zn, presente nel sito attivo dell'enzima HDAC, hanno un gruppo funzionale diverso da quello idrossamico.
3.2.2.a TIOLI E TIOLO DERIVATI [10].
I tioli sono gli inibitori meglio conosciuti degli enzimi Zn2+-dipendenti,
come gli ACE, le metallo proteinasi di matrice, e chiaramente gli HDAC. La loro struttura generale è riassunta in Figura 12.
A questa classe di inibitori appartiene il composto 1b (tabella 1), un ditiolo ridotto, entrato nella sperimentazione clinica di fase II.
La prima modifica strutturale del gruppo idrossamico del SAHA con
un tiolo è stata riportata da Suzuki e collaboratori con buoni risultati. L'analogo tiolico del SAHA, 54a (tabella 28), ha mostrato un'attività simile al SAHA negli studi sull'enzima isolato. Modificazioni ad hoc della struttura sono state la base di studi che hanno quindi analizzato l'importanza del gruppo tiolico libero e gli effetti del linker sull'attività inibitoria. La conversione del gruppo tiolico in tioacetato (54b) o in tiometile (54c), rispettivamente, riduce l'attività del composto o lo rende inattivo, suggerendo che il tiolo libero è fondamentale per l'interazione con lo Zn presente nel sito attivo. L'attività inibitoria dipende anche dalla lunghezza della catena: i valori ottimali sono di cinque o sei unità di C. Le similitudini nelle relazioni struttura-attività tra gli acidi idrossamici ed i tioli hanno messo in evidenza che entrambi hanno modalità di legame simili con il sito attivo dell'enzima HDAC. Assumendo come valido questo meccanismo è stato sintetizzato il composto 54g (tabella 28). La molecola progettata si basa sul concetto che, in accordo con il meccanismo proposto, la chelazione con lo Zn attiva una molecola d'acqua che instaura un attacco nucleofilo sul carbonio carbonilico di una lisina acetilata provocandone la deacetilazione. Se la molecola di acqua è rimossa dal sito attivo questo porta ad un aumento dell'energia di legame ed a una conseguente mancata deacetilazione. Composti con un gruppo α-mercaptoacetammidico come gruppo di chelazione allo Zn2+ possono entrare
nel sito attivo dell'enzima e rimuovere la molecola di acqua dallo ione Zn stesso come, appunto, nel caso del composto 54g.
introducendo sul gruppo aromatico un secondo fenile, in posizione meta, o una chinolina ottenendo rispettivamente i composti 55 e 56 (tabella 29). Questo ha portato ad un aumento dell'attività.
Il composto 54a è stato testato su cellule di cancro polmonare umano senza grandi successi quindi, nel tentativo di migliorarne l'efficienza, è stata studiata la possibilità di sintetizzare disolfuri e S-acil derivati da utilizzare come profarmaci. Mentre i disolfuri hanno dato risultati deludenti, i secondi sono più promettenti ed in particolare i tioesteri. L'S-isobutirril derivato 57 (tabella 29) ha mostrato una certa attività nei test su cellule tumorali polmonari, questa attività migliora se si ottimizza il CAP sostituendo il semplice fenile con un feniltiazolo, ottenendo il derivato 58 (tabella 29), che ha mostrato un'attività paragonabile a quella del SAHA.
Per cercare di ottenere nuove strutture più attive, sono state apportate varie modifiche strutturali alla formula generale dei derivati tiolici variando il CAP, la CU e gli HS ed ottenendo i composti di formula generale 59 (tabella 30). Per quanto riguarda la lunghezza della catena i migliori composti sono quelli portanti tre o quattro carboni, per quanto riguarda l'unità di connessione un gruppo ammidico sembra migliore dell'urea. I composti 59g e
59h, contenenti un gruppo ammidico inverso, si sono dimostrati molto attivi in
vitro, mentre la loro attività diminuisce notevolmente nei saggi su linee cellulari tumorali.
SAHA, di formula generale 60, dove l'acido idrossamico è sostituito da gruppi funzionali contenenti lo S quali tioammide o mercaptotioacetammide (tabella 31). Inaspettatamente l'α-aceto mercapto chetone (60f) è risultato più potente del corrispondente tiolo libero (60e) e del SAHA. Poiché lo stesso risultato non si ottiene con i derivati mercaptoacetammidici (60c e 60g), può essere messo in evidenza che il chetone carbonilico sinergizza con il gruppo α-tioacetilico. La S-metilazione del composto 60c per creare l'α-metiltiochetone (60h) ha invece portato ad un calo dell'attività.
Infine l'Argenta Discovery ha introdotto una porzione mercaptochetonica su un composto caratterizzato da un linker erteroaromatico, del quale non sono stati pubblicati studi sull'attività farmacologica.
3.2.2.b AMMINOFENILAMMIDI [10].
I composti appartenenti alla classe delle 2-amminofenilammidi hanno la formula generale mostrata in Figura 13. Si ritiene che porzione la 2-amminofenilammidica sia il gruppo di chelazione per lo zinco.
Suzuki e collaboratori hanno sintetizzato il composto 1g (tabella 1) che, avendo dimostrato ottima attività antiproliferativa su numerose linee cellulari tumorali umane, è attualmente in fase II di sperimentazione clinica.
Ipotizzando che il gruppo diamminobenzenico si lega al sito attivo dell'enzima HDAC, Vaisburg ha progettato varie 2-amminofenilammidi di acidi alcanoici ω-sostituiti, ibridi cioè tra 1g ed il SAHA, corrispondenti alla struttura
61 (tabella 32). Il composto 61b è stato valutato in diversi modelli xenografici
di tumore con buoni risultati [15].
I composti 1g e 1c sono stati quindi utilizzati come punto di partenza per la sintesi di nuove 2-amminofenilammidi con struttura generale 62, 63 e
64 (tabella 33).
Nonostante la modesta potenza in vitro, i composti 62g e 63d hanno dimostrato in vivo una buona efficacia antitumorale se somministrati per iniezione intraperitoneale o orale; una buona biodisponibilità è stata evidenziata anche dopo somministrazione per via endovenosa.
Presso la Hoffmann-La Roche sono stati studiati derivati 2-amminofenilammidici con un linker eterociclico (tabella 34); mentre la AstraZeneca ha pubblicato un ampio studio sulle relazioni struttura attività di derivati 2-amminofenilammidici con un linker aromatico (tabella 35); ma di nessuno di questi è stata resa nota l'attività inibitrice degli HDAC.
Ricercatori della MethylGene nello sforzo di analizzare la tasca adiacente al sito catalitico dell'enzima HDAC, hanno evidenziato che inibitori
o-amminobenzammidici con sostituenti aromatici od eteroaromatici, fenile, furano o tienile, in posizione para all'anello dell'anilina possono avere una significativa attività HDAC inibitoria. I composti 65 e 66 (tabella 36) mostrano una certa attività, che è pero' limitata dalla loro scarsa solubilità in acqua, per cui sono state preparate molecole più idrofobiche, analoghe del composto 1g (tabella 1) e rappresentate dalla struttura generale 67 (tabella 37). In questa serie di composti, tutte le molecole hanno capacità inibitrice analoga a quella del composto 1g, ad eccezione del composto 67a che non risulta attivo. I composti
67b-f hanno mostrato una certa azione sulle cellule fibroblastiche con attività
paragonabile al composto 1g.
Altre strutture 2-amminofenilammidiche sono state studiate da varie ditte farmaceutiche; le loro formule generali 68, 69, 70, 71, e 72 sono riportate nella tabella 38. Inoltre recenti studi hanno portato allo sviluppo di un gruppo di derivati N,N-diarilmalonammidici di formula generale 73 (tabella 39), fra i quali il composto 73a ha mostrato una certa attività inibitoria nei confronti dell'HDAC3.
3.2.2.c CHETONI [10].
Chetoni elettrofili o semplici metilchetoni sono stati proposti spesso come inibitori di varie proteasi tra cui gli HDAC. È da sottolineare che il meccanismo d'azione espletato dai chetoni è analogo a quello degli acidi
idrossamici. Sono stati testati come HDACi gruppi trifluorometilchetonici, α-chetoammidi, α-chetoesteri, e α-chetoeterocicli.
Studi sistematici sulle molecole di struttura generale 74 (tabella 40) con linker costituito da una catena lineare, un etere come CU ed un gruppo p-bifenilico come CAP, hanno mostrato che gli α-chetoesteri (74d-e-f), e l'α-chetoammide (74h) sono potenti HDACi. Questi composti, come anche i trifluorometilchetoni, sono inattivati in vivo, in quanto trasformati nei corrispondenti alcool.
Sono state studiate α-chetoammidi di struttura generale 75 (tabella 41), in cui è stata modificata la geometria della porzione aromatica; di queste, il composto
75a si è dimostrato molto attivo sull'enzima isolato, ma scarsamente attivo in
vivo. L'aumento della polarità e della solubilità in acqua ottenuti con l'introduzione di un eteroatomo polare come nei derivati feniltiazolici o 4-piridiltiazolici (75b-c-d), ha portato ad un aumento dell'attività cellulare, soprattutto su linee cellulari di cancro al seno. In particolare, il composto 75b ha mostrato un significativo effetto antitumorale su modelli animali, mentre nelle colture cellulari e nel sangue, ha un'emivita breve che ne diminuisce l'attività; questo è dovuto alla sua rapida conversione nell'α-idrossiammide inattiva. Inoltre il composto 75b ha evidenziato anche segni di tossicità come mortalità e perdita di peso, se somministrato ad alte dosi.
infatti, gli α-chetoossazoli, inibitori eterociclici di struttura generale 76 si sono rilevati estremamente potenti (tabella 42). I composti più potenti in vitro hanno attività antiproliferativa nel fibrosarcoma e nel carcinoma al seno, ma la loro attività sulle cellule in vivo, è compromessa dalla rapida riduzione ad alcool, che risulta inattivo.
Recentemente alcuni derivati metilchetonici semplici sono stati riportati come inibitori dell'HDAC (tabella 43). Jones e collaboratori hanno selezionato il prodotto naturale Apicidin, un peptide ciclico contenente il gruppo etilchetonico, come punto di partenza per la progettazione di nuovi inibitori HDAC. Di questi sono stati scelti composti che contengono l'amminoacido L-AODA (acido 2-amino-8-oxodecanoico), e fra questi, il composto 77 (tabella 43) si è mostrato un promettente punto di inizio. Approfonditi studi SAR sono stati fatti sui due gruppi ammidici ed è stato ipotizzato che: il legame di una porzione ammidica con la superficie del sito attivo influenza anche il legame del secondo gruppo ammidico. Dal composto 77 sono stati poi sintetizzati i composti di struttura generale 78 (tabella 43). La molecola 78m è stata testata come inibitore HDAC ed ha mostrato una certa stabilità.
3.2.3. COMPOSTI A STRUTTURA VARIA [10].
Alcune molecole che non sono comprese nelle classi fino a qui descritte sono state scoperte attraverso lo studio di composti naturali o
attraverso studi biologici mirati.
Nel 2003 Hu e collaboratori hanno identificato, i composti 79a e 79b (tabella 44) come HDACi atipici; il primo selettivo per l'HDAC1 ed il secondo sull'HDAC8.
Il gruppo di Fusetani, ha studiato alcune specie di invertebrati marini del Giappone identificando due classi di molecole: le azumammidi (da A a E) e le ciclostellettammine (tabella 45). L'azumammide A, mostra attività nell'ordine del nanomolare, mentre le ciclostellettammine sono meno potenti. Un esempio di questa classe è la molecola 80b.
Altri composti studiati sono i silanodioli rappresentati dalla struttura generale
81 (tabella 46) che mimano lo stato di transizione dell'enzima e potrebbero
essere interessanti sostituti degli acidi idrossamici.
3.3. SELETTIVITÀ DELLE ISOFORME [10].
Come già detto, le molecole attualmente utilizzate negli studi clinici sono in genere pan-inibitori di tutti gli HDAC, in rari casi distinguono tra gli HDAC di classe I e II (es le amminofenilammidi).
L'introduzione di nuove classi di composti e la disponibilità di alcune isoforme di HDAC, ha portato alla ricerca di inibitori selettivi con alcuni successi.
I composti 19a e 19b (tabella 12) appartenenti alla classe degli acidi idrossamici con linker cinnamoilico sono selettivi per gli HDAC 1 e 2. La stessa
selettività si trova nei composti della classe delle amminofenilammidi sostituite in para al gruppo amminico con un anello eteroaromatico come ad esempio le molecole 82a e 82b (tabella 47). Questi interagiscono con la cavità vicino al sito di legame con lo Zn e mostrano alta attività per gli HDAC 1 e 2, mentre sono inattive nei confronti degli HDAC 3 e 8. I composti di struttura 83a e 83b (tabella 47) appartengono alla classe degli acidi idrossamici con linker cinnamoil-pirrolico e sono selettivi per gli HDAC di classe II umani.
3.4. SPERIMENTAZIONE CLINICA [10].
L'evidenza del crescente interesse per questo tipo di inibitori è messa in luce dalle numerose sperimentazioni effettuate che hanno portato a diversi inibitori con buona selettività antitumorale in vitro e marcatamente bassa tossicità negli studi preclinici, suggerendo così alta selettività nei confronti delle cellule neoplastiche.
In questo paragrafo verranno dettagliate le caratteristiche di alcuni composti entrati nella sperimentazione clinica ed i risultati ottenuti da tali studi.
Il farmaco Vorinostat o SAHA (acido suberoil-anilide idrossammico, 1a tabella 1) è il primo inibitore HDAC ad essere stato messo sul mercato. Il suo nome commerciale è Zolinza® ed è distribuito dalla Merck & Co. Inc. È un acido
idrossamico che funziona come pan-inibitore degli HDAC. È stato approvato negli USA dalla FDA per il trattamento del CTCL, in quanto dagli studi di fase I e
II è emerso che pazienti trattati con il farmaco hanno risposto positivamente nel 30% dei casi [16]. Vorinostat è stato anche valutato recentemente nei confronti
di altre tipologie di cancro come i linfomi Hodgkin e le leucemie, dove è stato utilizzato come monoterapia o in combinazione ad altri chemioterapici. Tuttavia è emerso che il SAHA non ha azione su alcuni tumori refrattari come il mieloma, il cancro al cervello, al seno ed alla tiroide [17].
Romidepsin o depsipeptide (FK 228) (1b tabella 1) [18] è un peptide ciclico che
inibisce selettivamente gli HDAC di tipo 1, 2, 4 e 6. Gli studi di fase I hanno fornito diversi dati riguardanti le dosi terapeutiche, che sono stati poi approfonditi negli studi di fase II. Gli effetti collaterali dose-limitanti più evidenti sono affaticamento, nausea, vomito, trombocitopenia e neutropenia transitorie. L'uso del Romidepsin come monoterapia si è dimostrato efficace nel trattamento della AML (leucemia mieloide acuta) e del CTCL, purtroppo come per il Vorinostat, non ha azione su alcuni tumori refrattari. Sono stati sollevati tuttavia dubbi sulla sua sicurezza, a causa di una possibile tossicità cardiaca [17].
Altri composti sono in fase di sperimentazione clinica, con risultati che non evidenziano marcate differenze tra di essi.
Il derivato denominato Belinostat (1c tabella 1) potente inibitore un vitro, ha completato la fase I di sperimentazione clinica ed attualmente è in fase II [17].
Il Panobinostat (1d tabella 1) è uno dei più potenti HDACi nelle sperimentazioni in vitro, e terminata la fase I della sperimentazione, viene valutata la sua attività nei confronti del CTCL e dei tumori Hodgkin [17].
Il derivato N-idrossibenzammidico CRA-024781 (1e tabella 1) è coinvolto in studi di fase I. È stato dimostrato che inibisce gli HDAC ricombinanti 1, 2, 3, 6, 8 e 10 sia in vivo che in vitro. Inoltre ha attività in vitro su linee cellulari di cancro del colon, della prostata, del seno e delle ovaie. In vivo ha azione su diversi tumori a dosaggi ben tollerati se somministrato per via intraperitoneale od endovenosa [19].
Il composto MGCD-0103 (1f tabella 1) è appartenente alla classe dei derivati 2-amminofenilammidici con buona attività inibitoria di alcuni HDAC di classe I (HDAC 1, 2 e 3), di classe II (HDAC 4, 5, 6, 7, 9 e 10) e di classe IV (HDAC11). Può essere somministrato per via orale ed ha dimostrato di avere una potenza antiproliferativa maggiore di quella del SAHA ed effetti sinergici e/o additivi se somministrato in associazione con gemcitabina, azacitidina, doxorubicina, cisplatino e bortezomib. Nella fase I di sperimentazione sono state testate la tossicità di base, la farmacocinetica ed i parametri di dosaggio su pazienti affetti da tumori del sangue, leucemia o MDS (Sindrome Mielodisplastica), o tumori solidi; dimostrando come segni di tossicità, affaticamento, nausea, vomito e diarrea. Dati i risultati promettenti della sperimentazione di fase I, il composto è passato alla sperimentazione di fase II, dove è stato saggiato in pazienti affetti da linfoma di Hodgkin mostrando buoni risultati e tollerabilità. Un'altra sperimentazione è stata fatta come monoterapia in pazienti recidivi o affetti da linfomi refrattari come il linfoma follicolare (FL) con buoni risultati, dimostrando che l'MGCD-0103 può essere utilizzato con successo alle dosi saggiate [19].
Il composto MS-275 (1g tabella 1), appartenente alla classe delle amminobenzanilidi, ha mostrato selettività per alcuni HDAC di classe I (HDAC 1, 2 e 3); test preclinici hanno inoltre mostrato un'attività sia in vivo che in vitro nei confronti di diversi tipi di neoplasie, tra cui quella alla prostata, all'endometrio ed alla glia; inoltre ha mostrato azione antiproliferativa in alcuni tumori solidi dei bambini. La sua attività è sinergica se associato ad analoghi dei nucleotidi o usato contemporaneamente alla terapia con radiazioni ionizzanti. La farmacocinetica e la farmacodinamica sono state studiate nella sperimentazione di fase I dove è stato utilizzato in monoterapia su pazienti affetti da tumori solidi o da linfomi refrattari e per la leucemia acuta. Essendo caratterizzato da un'emivita particolarmente lunga (80h) anche a basse dosi ha manifestato una certa tossicità (inclusa la comparsa di infezione, neurotossicità, nausea, vomito, sonnolenza ed affaticamento). Sono stati effettuati anche esperimenti in fase II in pazienti affetti da melanoma metastatico avanzato. È stato inoltre analizzato in terapia combinata con la 5-azacitidina in pazienti affetti da MDS, con risultati meno apprezzabili di quelli ottenuti dal MGCD-0103 [19].
L'acido valproico (1h tabella 1) è un acido grasso a catena corta che è risultato essere un debole inibitore degli HDAC, tuttavia la sua lunga biodisponibilità come anticonvulsivo hanno portato gli studiosi a valorizzarlo come farmaco anticancro di tipo epigenetico. Utilizzato come monoterapia non ha dato grossi risultati, mentre sono buoni quelli registrati per l'utilizzo in associazione con altre terapie. Sono esempi positivi gli studi di fase I e II sulla combinazione con
inibitori della metilazione del DNA: acido decitabina e acido valproico-azacitabina-acido retinoico (ATRA). In tutti i casi si ha un aumento dell'attività, ma anche della tossicità [17].
Il composto SB-639 [19] (1l tabella 1), un acido idrossamico derivato
benzimidazolico, si sta dimostrando uno dei più promettenti candidati agli studi clinici, con attività antiproliferativa in vitro nell'ordine del nanomolare nei confronti del cancro al polmone, alla prostata, al seno ed al fegato. L'agente è ben tollerato dopo somministrazione per via intraperitoneale nei ratti; inoltre promuove l'acetilazione dell'istone H3 in tessuti tumorali, per tempi prolungati.
3.5. TABELLE
1a: Vorinostat (SAHA) – In commercio (Merk) 1b: Romidepsin (FK228) – Fase II (Fujisawa)
1c: Belinostat (PXD101) – Fase II
(Topotarget) 1d(Novartis): Panobinostat (LBH-589) – Fase III
1e: CRA-024781 – Fase I (Celera Genomics) 1f: MGCD-0103 – Fase II (Methylgene)
1g: MS-275 – Fase II (Bayer/Syndax) 1h: acido valproico – Fase II (G2M)
1i: ITF-2357 – Fase II (Italfarmaco)
1l: SB-939 – Fase I (S*BIO)
1m: JNJ-16241199 – Fase I (J&J) 1n: LAQ-824 – Fase II (Novartis)
1o: Tacedinaline (CI-994) – Fase II (Pfizer)
Composto R1 R2 2a C6H5 C6H5 2b C6H5 -CH2 C6H5 -CH2O 2c C6H5 -CH2 2d C6H5 -CH2 C6H5 2e C6H5 -CH2 2f C6H5 -CH2O
Composto R1 R2 2g C6H5 2h 2i 2j 2k 2l C6H5
Composto
n
R1 R2 3a 6 C6H5 C6H5 4a 5 4b 5 4c 5 C6H5 4d 5 5a 6 6a 6 C6H5 -CH2 C6H5 -(CH2)2Composto R Composto R 7a H 9a H 7b p-CH3O 9b p-CH3O 7c P-Br 9c P-Br 7d P-ClC6H4 9d p-Ph 8a H 9e p-CF3 8b P-Br 9f p-CF3O 8c p-CF3O 9g m-Br 8d p-Ph 9h 2,6-(CH3O) 8e P-F 9i β-naftile
Composto n R 10a 4 10b 5 10c 6 10d 7 10e 6
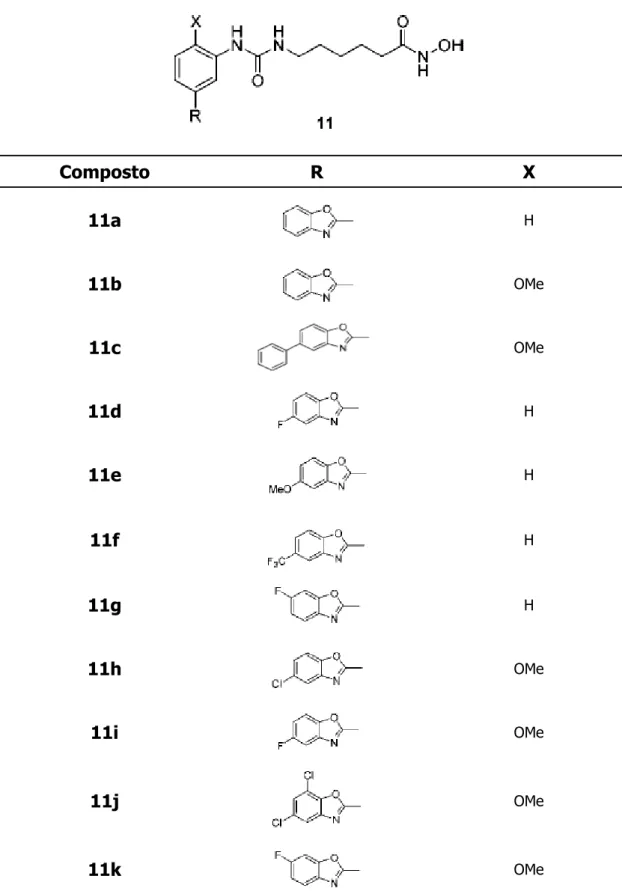
Composto R X 11a H 11b OMe 11c OMe 11d H 11e H 11f H 11g H 11h OMe 11i OMe 11j OMe 11k OMe
Composto X n R 12a S 5 H 12b SO 5 H 12c SO2 5 H 12d S 5 Cl 12e SO 5 Cl 12f SO 5 Me2N 12g SO 6 Cl 12h S 5 4Cl-C6H4SO2NH
Tabella 7: acidi idrossamici con uno S, un gruppo solfossido o un solfone nella CU
Composto R X
13a C6H5 (CH2)4
13b C6H5 (CH2)5
13c C6H5CH2 (CH2)5
13d C6H5CH(CH3)2 (CH2)5
Composto R 14a 14b 14c 14d 14e 14f
Tabella 10: acidi idrossamici aventi come gruppo linker una piperidina 4-alchil sostituita ed
Composto Posizione R R1 17a meta C6H5 H 18a meta C6H5 -17b meta 4-MeC6H4 H 18b meta 4-MeC6H4 -17c meta 3,4-dimetossifenile H 18c meta 3,4-dimetossifenile -17d meta [1,1'-bifenil]-4-yl H 18d meta [1,1'-bifenil]-4-yl -17e meta C6H5 -CH2 H 18e meta C6H5 -CH2 -17f para C6H5 H 18f para C6H5 -17g para [1,1'-bifenil]-4-yl H 18g para [1,1'-bifenil]-4-yl -17h orto C6H5 H
17i orto naftalen-2-yl H
17k meta C6H5 CH3
Tabella 11: acidi idrossamici con linker cinnamoilico ed una CU costituita da una
Composto R
19a 4-(dimetilammino)-fenile
19b 4-(pirrolidin-1-yl)-fenile
19c 3-piridil
19d 2-piridil
Tabella 12: acidi idrossamici con linker cinnamoilico ed un gruppo benzilammidico come CU
Tabella 13: acidi idrossamici con linker cinnamoilico, CAP benzimidazolico e CU metilenica.
Tabella 14: acidi idrossamici con linker cinnamoilico, CAP benzimidazolico od imidazolico
Tabella 15: acidi idrossamici che contengono una molecola ciclica come CU
Tabella 17: acidi idrossamici recentemente sintetizzati.
Composto R 38a 38b 38c 38d 38e 38f 38g 38h
Tabella 19 (segue): acidi idrossamici che contengono come linker un nucleo tiofenico
Composto R 38i 38l 38k 38j 38m 38n 38o 38p
Tabella 19: acidi idrossamici che contengono come linker un nucleo tiofenico seguito da un
Composto R 39a
39b 39c
Tabella 20: altri acidi idrossamici che contengono come linker un nucleo tiofenico
Tabella 21: acidi idrossamici sintetizzati dall'ottimizzazione del composto 39a; il composto 40
Tabella 22: acidi idrossamici con linker biarilico costituito da un tiazolo-tiofene
Tabella 24: acidi idrossamici con linker 2-amminotiazolico o 2-amminopirimidinico
Tabella 25: acidi idrossamici sintetizzati dalla Janness Pharmaceutica nel tentativo di
R'= Ph, Arile, Bifenile, Naftile n=1-4 Composto Posizione R 51a - H 51b 5 51c 6 51d 5 51e 6 51f 5 51g 6
R'= Ph, Arile, Bifenile, Naftile n=1-4 Composto Posizione R 51h 5 51i 5 51j 6 51k 5 51l 6 51m 6 51n 6 51o 5
Tabella 27: acidi idrossamici che contengono come linker un benzofurano o un benzimidazolo Composto X n R 54a -NHCO- 6 -SH 54b -NHCO- 6 -SAc 54c -NHCO- 6 -SMe 54d -NHCO- 4 -SH 54e -NHCO- 5 -SH 54f -NHCO- 7 -SH 54g -NHCO- 5 -NHCOCH2SH
Tabella 29: inibitori derivati dall'ottimizzazione del composto 54a. Composto R X n 59a p-(CH3N)C6H5 -CONH- 3 59b p-(CH3N)C6H5 -CONH- 4 59c p-(CH3N)C6H5 -CONH- (CH2)n=P-C6H5 59d 8-chinolina -CONH- 3 59e C6H5 -NHCONH- 3 59f C6H5 -NHCONH- 4 59g 8-chinolina -NHCO- 3 59h 3-chinolina -NHCO- 3
Composto X Y 60a -CH2CSNH2- -OH 60b -NHCOCH2- -OH 60c -NHCOCH2- -OH 60d -NHCSCH2- -SH 60e -CH2COCH2- -SH 60f -CH2COCH2- -SH 60g -NHCOCH2- -SAc 60h -CH2COCH2- -SCH3
Tabella 31: derivati tioammidici o mercaptotioacetammidici analoghi del SAHA
Composto Ar
61a
61b
61c
Composto X Y R 62a N - C6H5CH2NH 62b N -62c N -62d N -62e CH -62f CH -62g CH
-Tabella 33 (segue): nuove 2-amminofenilammidi derivate dall'ottimizzazione dei composti
Composto X Y R 63a N -63b N -63c CH -63d CH -63e CH -63f CH -63g CH -63h CH
-Tabella 33 (segue): nuove 2-amminofenilammidi derivate dall'ottimizzazione dei composti
Composto X Y R
64a NH CO
64b NH CO
64c NH CH2
64d NH CH2
Tabella 34: derivati 2-amminofenilammidici con un linker eterociclico studiati da Hoffmann-La
Roche.
Tabella 36: inibitori 2-amminofenilammidici p-eteroaril sostituiti. Composto Ar 67a 67b 67c 67d 67e 67f
Composto Ar 73a 73b 73c 73d 73e 73f 73g 73h
Composto n x 74a 1 -CONHOH 74b 1 -COCF3 74c 1 -COCO2H 74d 1 -COCO2CH3 74e 0 -COCO2CH3 74f 2 -COCO2CH3 74g 1 -COCONH2 74h 1 -COCONHCH3 74i 1 -COCONHEt 74j 1 -COCON(CH3)2 74k 1 -COCONH(CH2)2N(CH3)2 74l 1 -COCONH(CH2)CO2H
Tabella 40: chetoni elettrofili con linker costituito da una catena lineare, un etere come CU
Composto R 75a 75b 75c 75d Tabella 41: α-chetoammidi
Composto n X R 76a 5 -O-76b 5 -CONH-76c 4 - CONH-76d 5 -NHCO-76e 5 -NHCO-Tabella 42: α-chetoossazoli
Composto R R' 78a 78b -CH3 78c 78d 78e 78f 78g
Composto R R' 78h 78i 78j 78k 78l 78m
Tabella 44: inibitori HDAC studiati da Hu e collaboratori.
Tabella 46: silanodioli
3.6. ATTIVITÀ IN VIVO DEGLI INIBITORI DEGLI HDAC DI CLASSE I E II [10].
Gli HDACi hanno mostrato attività antitumorale come farmaci singoli con buona efficacia; tuttavia un aspetto significativo delle loro potenzialità risiede nell'associazione con altri agenti citotossici.
La capacità degli HDACi di aprire la struttura della cromatina, facilitando così l'accesso al DNA, può essere utilizzata per cercare di incrementare gli effetti di farmaci che hanno come bersaglio il DNA. Un aumento di citotossicità è stato mostrato negli studi di combinazione con docetaxel, adriamicina, VP-16 e cisplatino in diversi modelli di tumore. Questa interessante azione sinergica con gli inibitori della topoisomerasi II è stata mostrata in vitro e confermata in vivo, in modelli preclinici.
Molto spesso proteine oncogene incorporano gli HDAC e il caso più studiato è quello della leucemia acuta promielocitica (APL). Normalmente questa malattia risponde al trattamento con acido retinoico (RA) che induce differenziazione ed arresto della crescita cellulare. Tuttavia, quando il recettore per il RA (RAR) è espresso come una proteina di fusione con il “leucemia promielocitica zinc finger” (PLZF), le cellule diventano resistenti al RA. La nuova proteina RAR-PLZF si associa agli HDAC provocando la repressione di geni normalmente indotti dal RA che regolano la differenziazione. In questo modo le cellule diventano resistenti al RA. Il trattamento di questi pazienti con un'associazione RA e HDACi ristabilisce la sensibilità cellulare al RA. In generale quando un tumore è
caratterizzato dalla comparsa di queste proteine di fusione, un'associazione del farmaco originale con un HDACi può essere di grande beneficio.
Un certo numero di strategie di associazione sono state proposte in diverse tipologie di leucemia; sono stati proposti trattamenti con SAHA e imatinib nelle fasi avanzate di leucemia mieloide cronica; con SAHA e dasatinib per la leucemia mieloide cronica resistente all'imatinib. Inoltre sono state sperimentate in vitro associazioni con fludarabina (antimetabolita); e con il bortezomib (inibitore proteosomico). Per quanto riguarda i tumori solidi è stata dimostrata una certa sinergia d'azione con tamoxifene e 5-fluorouracile.
Infine è stato dimostrato che tutti gli HDACi sinergizzano con le radiazioni ionizzanti (radiazioni γ), anche in vivo, contribuendo alla morte cellulare. Questo effetto può essere dovuto a varie ragioni, tra cui una down-regulation dei geni e delle proteine coinvolte nella risposta al danno al DNA indotta dagli HDACi. La riduzione di proteine di riparazione e sopravvivenza è anche parzialmente responsabile degli effetti sinergici con composti chemioterapici classici.
3.7. TOSSICITÀ E RESISTENZA [17]
Gli HDACi sono in genere ben tollerati e mostrano un profilo di tossicità accettabile. Durante gli esperimenti clinici è emerso un quadro comune di effetti collaterali indice di un meccanismo generale alla base della loro
comparsa, tuttavia ancora sconosciuto. Gli effetti collaterali più comuni sono senso di affaticamento e debolezza. Un altro segno di tossicità è la comparsa di eventi gastrointestinali in particolare diarrea e disturbi elettrolitici connessi; questa tuttavia, è di modesta entità e tollerata dalla maggior parte dei pazienti. Manifestazioni neurocorticali sono state evidenziate principalmente con la somministrazione di acido valproico.
Effetti cardiaci come cambiamenti non specifici nell'ECG sono emersi negli esperimenti di fase I con il romidepsin, mentre nella sperimentazione di fase II si sono riscontrati casi di morte improvvisa di alcuni soggetti dovuta a complicanze cardiache; tuttavia, in questi pazienti si riscontravano altri fattori di rischio che possono aver causato il decesso. È stato rilevato anche un prolungamento dell'intervallo QT dopo somministrazione di entrambe le formulazioni del Vorinostat, intravenosa e orale, ed in modelli dose-dipendenti del panobinostat.
In generale, per gli HDACi, è emersa, in via sperimentale su cavie, trombocitopenia dose-limitante che nell'uomo alle dosi terapeutiche, è di modesta entità e transitoria. Anemia e neutropenia compaiono sporadicamente e le complicazioni settiche sono rare.
Tuttavia spesso si riscontra, nel soggetto trattato con HDACi, la precoce comparsa della resistenza che limita in parte i benefici della terapia. Diversi esperimenti hanno dimostrato che esistono numerosi meccanismi coinvolti nella biologia della resistenza; in particolare quelli associati all'iperespressione di
proteine antiapoptotiche (Bcl-2) e quelli relativi alla regolazione della traduzione genica.
3.8. UNO SGUARDO AL CAPOSTIPITE: ZOLINZA®
Zolinza® è indicato per il trattamento delle manifestazioni cutanee nei
pazienti affetti da CTCL che hanno manifestato progressione o persistenza della malattia, tendenza alla recidiva o che seguono terapie binarie senza grandi risultati. Come noto il principio attivo è il Vorinostat o N-idrossi-N'-fenilottandiammide, inibitore degli HDAC1, HDAC2, HDAC3 e HDAC6, che è formulato in capsule rigide, da 100 mg ciascuna (vedi foto)
Le capsule non devono essere aperte o schiacciate in quanto il contatto diretto della polvere con la pelle o le mucose può rendere inattivo il farmaco. Zolinza®
ha il vantaggio di venire somministrato oralmente in un'unica dose giornaliera di 400 mg assunta al momento del pasto. La dose può essere ridotta a 300 mg il giorno o, in casi estremi, a 300mg per cinque giorni nell'arco di una settimana
se compaiono segni di tossicità od intolleranza. Il trattamento può essere prolungato fino alla comparsa di tossicità inaccettabile da parte del paziente o fino all'evidente mancanza di segni di guarigione.
Per quanto riguarda la farmacocinetica il Vorinostat viene assorbito dal tratto gastrointestinale e successivamente si distribuisce nel sangue dove si lega alla proteine plasmatiche per circa il 71%. Il farmaco viene metabolizzato nel fegato, attraverso reazioni di demolizione che comportano l'idrolisi seguita da β-ossidazione e principalmente, tramite la O-glucuronidazione, reazione metabolica di fase II dove il principio attivo viene reso idoneo all'escrezione biliare per mezzo di una sua combinazione con l'acido glucuronico. Queste reazioni portano nel complesso alla trasformazione del SAHA nei suoi metaboliti inattivi: l'acido 4-anilino-4-oxobutanoico e l'O-glucuronide. In vitro, studi sui microsomi del fegato, indicano che il SAHA subisce anche una trascurabile biotrasformazione da parte del citocromo P450. L'emivita per il Vorinostat è di 2 ore come anche per l'O-glucuronide, mentre per l'acido 4-anilino-4-oxobutanoico è di 11 ore. L'escrezione renale non è la principale via di eliminazione del farmaco infatti solo l'1% si trova inalterato nelle urine; come già accennato, infatti la principale via è la sua inattivazione metabolica.
È stata riscontrata la comparsa di alcuni effetti collaterali, fra i quali si notano affaticamento e comparsa di brividi, alterazioni del gusto e secchezza delle fauci; infatti anche la disidratazione rientra tra gli effetti collaterali, pertanto i pazienti devono bere almeno due litri di liquidi al giorno per
mantenere una buona idratazione. Gli effetti a livello gastrointestinale possono manifestarsi con comparsa di nausea, vomito e diarrea contrastati con la concomitante somministrazione di farmaci antiemetici, antidiarroici e con flebo contenenti elettroliti. Per quanto riguarda il quadro ematologico il più comune e severo sintomo di tossicità è la possibilità del verificarsi di tromboembolie, in particolare embolie polmonari e trombosi delle vene profonde sono state riportate come reazioni avverse. Inoltre l'assunzione del Vorinostat può causare anemia e trombocitopenia entrambe correlate alla dose somministrata. Talvolta può comparire iperglicemia, pertanto si deve porre particolare attenzione nel trattamento di pazienti diabetici. Sono emersi anche effetti avversi a livello cardiaco: studi definitivi sul prolungamento dell'intervallo QT non sono stati ancora terminati, ma dagli studi condotti in fase I e II è evidente la comparsa di questo segno di tossicità in un buon numero di pazienti.
In pazienti che non sono affetti da CTCL si sono riscontrate manifestazioni avverse più serie rispetto ai pazienti che manifestano la malattia. Si sono evidenziate visione sfuocata, astenia, iponatriemia, emorragie tumorali, sindrome di Guillain-Barre, insufficienza renale, ritenzione urinaria, tosse, emottisi, ipertensione e vasculiti.
In generale, per la comparsa di tutti questi effetti collaterali, anche se di lieve entità, è necessario un monitoraggio continuo del paziente attraverso una serie di analisi di laboratorio. Sono necessari frequenti elettrocardiogrammi per verificare la tossicità cardiaca ed esami del sangue, in particolare volti a
monitorare la comparsa di danni ematici e a controllare i livelli degli elettroliti K e Mg.
Per quanto riguarda le interazioni con altri farmaci è stato dimostrato che il Vorinostat non è un inibitore del citocromo P450 del fegato, quindi non interagisce con molecole che provocano l'inibizione o l'induzione del citocromo P450 stesso. Associazioni con il Coumadin ed i suoi derivati anticoagulanti, hanno rilevato un allungamento del tempo di Quick e del Rapporto Internazionale Normalizzato (INR). È stato verificato inoltre, che l'assunzione con altri inibitori HDAC, come ad esempio l'acido valproico, ha portato ad una severa trombocitopenia e ad emorragie intestinali.
Particolare attenzione deve essere posta nella somministrazione del farmaco a specifiche popolazioni. Nelle donne in gravidanza potrebbe causare morte fetale. In studi di laboratorio condotti su cavie e conigli è stata messa in evidenza la sua teratogenicità: il farmaco attraversa la placenta e passa nel circolo sanguigno fetale dove si trova in concentrazione superiore al 50% provocando perdita di peso, malformazioni della cistifellea, incompleta ossificazione del cranio e delle vertebre toraciche, e variazioni nello scheletro, come la comparsa di costole cervicali o costole soprannumerarie. Non è noto se il principio attivo è escreto nel latte materno, pertanto si sconsiglia l'allattamento. Inoltre non è stata stabilita l'effettiva sicurezza per l'uso pediatrico, l'uso geriatrico invece non ha mostrato particolari eccezioni. Infine pazienti con danni epatici devono essere trattati con cautela, come anche quelli
con danni renali.
Non ci sono specifiche informazioni circa il sovradosaggio; nel caso si verificasse un tale evento si deve rimuovere il materiale non ancora assorbito dal tratto gastrointestinale, ed istituire un'adeguata terapia di supporto; non è noto se il Vorinostat è eliminabile tramite dialisi.
Prima di essere immesso sul mercato Zolinza® è stato accuratamente
testato in due distinte sperimentazioni open-label, dove cioè sia i ricercatori che i malati conoscono la terapia seguita, scegliendo come campione un gruppo di pazienti affetti da CTCL refrattario in stato avanzato; in entrambi gli studi i pazienti sono stati trattati fino alla progressione della malattia o alla comparsa di tossicità intollerabile. I pazienti totali sono stati 107: 47,7% femmine, 52,3% maschi dei quali l'81,4% bianchi, il 16,3% neri, l'1,2% asiatici e il restante 1,1% appartenente ad altre razze.
Lo Studio 1 comprendeva 74 pazienti, con età media di 61 anni, in prevalenza affetti da CTCL in stato avanzato, dallo stadio IIB agli stadi superiori; ed in minima parte affetti dalla sindrome di Sézary; in media erano tutti soggetti che erano stati prima sottoposti ad altre due terapie sistemiche di cui almeno una con Bexarotene. Per inciso, questo farmaco è un retinoide sintetico che viene utilizzato come trattamento del CTCL di grado avanzato, refrattario ad almeno un trattamento sistemico precedente, viene inoltre utilizzato in alcune forme di carcinoma come quello renale e nel trattamento delle metastasi da carcinoma mammario, la sua azione si esplica anche come
terapia contro la dermatite delle mani e la parapsoriasi.
Il gruppo di pazienti dello studio 1 è stato trattato con una dose unica giornaliera di 400 mg.
Sono state valutate la diminuzione dell'estensione della malattia e la sua regressione. L'estensione della malattia riscontrabile da segni visibili sulla pelle è stata quantitativamente misurata usando una modifica della SWAT, acronimo di “Severity Weighted Assessment Tool” o “Strumento per la valutazione della gravità ponderata”. Gli studiosi hanno misurato la percentuale totale della superficie corporea (%TBSA) coinvolta in tre differenti tipi di lesioni: macchie, placche e tumefazioni od ulcere, che si sono presentate su dodici distinte regioni corporee, usando il palmo del paziente come unità di riferimento. La %TSBA totale per ogni tipo di lesione è moltiplicata per un “Fattore di gravità ponderata” o “Severity weighting factor” corrispondente a 1 per le macchie, a 2 per le placche ed a 4 per le tumefazioni e/o le ulcere. Questi risultati sono sommati tra loro per avere il risultato definitivo della SWAT.
L'efficacia è stata invece misurata considerando due distinte risposte cliniche alla malattia: una risposta clinica completa (CCR), definita come la totale regressione della malattia, ed una risposta parziale (PR) definita come diminuzione ≥ del 50% nella stima della SWAT rispetto ai valori standard; CCR e PR dovevano rimanere stabili per almeno quattro settimane. I criteri di selezione sono stati piuttosto restrittivi: pazienti con diminuzione nei risultati SWAT da 0% a 49%, sono stati considerati come non rispondenti ed esclusi dal
gruppo che ha risposto positivamente alla terapia.
Dalla valutazione generale degli studi condotti è emerso che, il 29,7% (22 casi su 74) dei pazienti trattati ha avuto un miglioramento dello stato patologico (Grafico 1).
I pazienti affetti da CTCL nello stadio IIB o negli stadi più avanzati della malattia hanno avuto un miglioramento nel 29,5% dei casi (18 pazienti su 61) ed uno è completamente guarito. Il tempo medio di risposta è stato di 55-56 giorni; in pratica pero', la terapia si è protratta da un minimo di 28 giorni ad un massimo di 171 giorni; tuttavia, raramente sono stati necessari tempi lunghi, fino a sei mesi, per avere una risposta terapeutica (Grafico 2).
0 10 20 30 40 50 60 70 80 90 100 Grafico 1
analisi della risposta positiva relativa allo Studio 1
% casi positivi casi esaminati pe rc en tu al e di r isp os ta 27,9%
Lo Studio 2 comprendeva invece 33 soggetti: pazienti con età media di 67 anni dei quali il 55% maschi, refrattari ad almeno quattro precedenti terapie sistemiche, ed affetti da CTCL. Questo campione è stato suddiviso in tre coorti: alla prima coorte di 13 pazienti sono stati somministrati 400 mg una volta al giorno; alla seconda, che comprendeva 11 pazienti, 300 mg due volte al giorno per tre giorni consecutivi in una settimana; ed infine, alla terza di 9 pazienti, 300mg due volte al giorno per quattordici giorni consecutivi, seguiti da una settimana nella quale veniva somministrata una dose di mantenimento di 200 mg al giorno. Il tempo medio di risposta è stato di 83,5 giorni. Le risposte generali relative a questo studio sono state del 30,8% nella prima categoria, del 9,1% nella seconda e del 33,33% nella terza, quest'ultimo può sembrare un risultato promettente, tuttavia, nel complesso non ci sono stati miglioramenti rilevanti rispetto ai pazienti trattati nello studio 1; inoltre è stato messo in
0 1 2 3 4 5 6
Grafico 2
tempi di risposta positiva nello Studio 1
mesi per risposta positiva
mesi di terapia
evidenza che la dose di 300 mg somministrata alla coorte due non è adatta alla terapia in quanto ha dimostrato un'alta tossicità senza mostrare miglioramenti clinici [20].