73
4. Synthesis and Characterization of Poly(thiophene-b-acrylonitrile)
In current research the Huisgen’s 1,3-dipolar cycloaddition-based “click” chemistry is becoming popular due to the high yields, simple and versatile reaction conditions. “Click” chemistry was introduced in 2001 by Sharpless and co-workers,1 who revisited the Huisgen’s 1,3-dipolar cycloaddition2 between azido and alkynyl groups using copper salts as catalysts. Many papers have demonstrated its applicability to the preparation of block copolymers.3 In particular, this synthetic approach presents advantages of considerable interest for organic semiconductors and their applications in electronics. Firstly, it provides pure and stable materials, properties which are of great importance in electronics. Secondly, “click” reactions offer quantitative yields, generate easy to remove and harmless byproducts and require simple reaction conditions, qualities of interest when considering possible future industrialization. Finally, this technique enables a precise control of the resulting block copolymer from well-defined functionalized parent homopolymers. Such functionalized polymers can be easily prepared by controlled/living radical polymerization (CRP) techniques such as atom transfer radical polymerization (ATRP), nitroxide-mediated radical polymerization (NMRP), and reversible addition-fragmentation transfer (RAFT) polymerization.4Due to the mechanism of CRP, polymer chains prepared by CRP or living ionic polymerization techniques are typically end-capped by a ‘‘dormant” unit (a halogen atom in ATRP, an alkoxyamine moiety in NMP, a dithioester or trithiocarbonate or similar moiety in RAFT), which can be transformed into diverse functional groups after polymerization. ATRP is probably the most practical technique, because the terminal alkyl halide can be used for standard nucleophilic substitutions or elimination reactions.5 For example, the terminal halide can be easily transformed into azides, and a diverse range of functional alkynes suitable for halide replacement are commercially available. However, depending on the targeted functional group, transformations can vary in difficulty and reaction efficiency. Reported publications on click chemistry demonstrated that copper(I) catalyzed 1,3-dipolar cycloaddition (CuAAC) can be used to efficiently functionalize polymers prepared using ATRP.6 Furthermore, ATRP/CuAAC chain-end modification strategies may be further exploited for the synthesis of defined macromolecular architectures such as block copolymers, graft copolymers, macromolecular brushes, stars, miktoarm stars, macrocycles, and networks.7
In this work, azide−terminated polyacrylonitrile of differing chain lengths was initially synthesized by atom transfer radical polymerization (ATRP) from an azide-functional initiator. By
74
using the alkynyl terminated rr-P3HT described in chapter 3 and the azide terminated polyacrylonitrile, P3HT-b-PAN block copolymers could be synthesized by “Click” coupling. This block copolymer was expected to be an effective stabilizer for the nucleation and stabilization of CdSe nanocrystals since the CN group can coordinate with Cd2+.8 If used for the fabrication of the active layer in a PV device, the resulting P3HT-b-PAN/CdSe nanocomposites would thus possess a well-defined interface in which, due to the minimum distance of acceptor phase (CdSe nanocrystals) from any excitation point of donor phase (P3HT), charge transfer could occur before the radiation less decay of the photogenerated exciton.
4.1 ATRP polymerization of AN to obtain azide group at one chain end of PAN 4.1.1 Synthesis of 3-azido-1-propanol
Sodium azide (5.95 g, 91.5 mmol) was added to a solution of 3-bromopropanol (3.18 g, 22.9 mmol) in a mixture of 20 mL of water and 20 mL of ethanol. This reaction mixture was stirred at 70 o
C for 24 hr. The aqueous layer was then extracted with dichloromethane (DCM; 3x25 mL). The organic extracts were collected and washed with 25 mL of water, dried over NaSO4, filtered, and the solvent evaporated to dryness giving 3-azidopropanol as a pale-yellow liquid in a 85% yield.
1
H NMR (300 MHz, CDCl3): δ= 4.53 (2H, t, CH2-O), 3.47 (2H, t, CH2-N3), 3.38 (1H, s, -OH), 1.67 (2H, m, CH2-C-O) ppm.
13
C NMR (300 MHz, CDCl3): δ = 57.7 (CH2-OH), 47.8 (CH2-N3), 31.5 (CH2-CH2-CH2) ppm.
4.1.2 Synthesis of 3-azidopropyl 2-bromoisobutyrate
A solution of α-bromoisobutyryl bromide (1.28 mL, 10.4 mmol) in THF (10mL) was added dropwise to a solution of 3-azido-1-propanol (1 g, 9.9 mmol) and triethylamine (TEA) (1.80 mL, 12.88 mmol) at 0 °C. After stirring the mixture for 1 hr at 0 °C, and another 3 hr at room temperature followed by addition of degassed methanol (10 mL), the precipitated triethylammonium bromide salt was filtered off and the solution was concentrated by removing the solvent at the rotary evaporator. The crude product was dissolved in dichloromethane and washed 3 times with a saturated NaCl solution, then 3 times with de-ionized water. The organic layer was dried on NaSO4, filtered and solvent was evaporated by rotary evaporator to yield a pale yellow oil. The last traces of solvent were removed under vacuum to yield 81% of product.
75 1 H NMR (300 MHz, CDCl3): δ = 1.92 (6 H, s, (CH3)2C), 1.96 (2 H, m, CH2-CH2-O), 3.44 (2 H, t, CH2-N3), 4.27 (2 H, t, CH2-O-C=O) ppm. 13 C {1H} NMR (300 MHz, CDCl3): δ = 27.97 (CH2-N3), 30.70 ((CH3)2-C), 48.03 (CH2-CH2-COR), 55.66 (C-Br), 62.74(CH2-O), 171.53 (C=O) ppm.
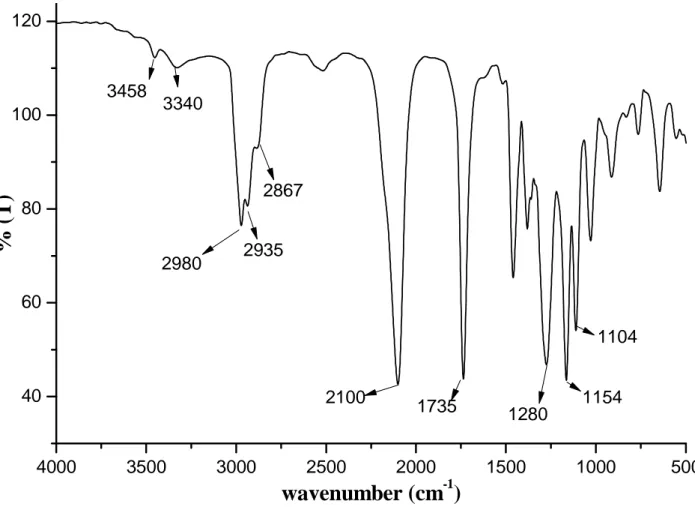
FT-IR (KBr window): ν = 2980, 2935, 2867 (aliphatic C-H stretching), 2100 (C≡N stretching), 1735 (C=O stretching), 1280, 1154, 1104 ( C-O stretching) cm-1.
4.1.3 Polymerization of acrylonitrile by ATRP
A 25 mL schlenk flask was loaded with 4 mL (3.20 g, 60.3 mmol) of acrylonitrile (AN) and 5 mL of ethylene carbonate, and the mixture was degassed by three freeze-pump-thaw cycles. The flask was filled with nitrogen while the mixture was kept frozen, then 0.143 g (1 mmol) of CuBr and 0.468 g (3 mmol) of 2,2’-bipyridine were added. The flask was then evacuated and back-filled with nitrogen several times, and the mixture was left to thaw. The obtained homogeneous brown reaction mixture was heated to 60 °C, and 0.25 g (1 mmol) of 3-azidopropyl-2-bromoisobutyrate previously deoxygenated by purging with nitrogen was injected. Samples of the reaction mixture were collected after 20, 40, and 60 min reaction time. After 2 hr the viscosity had reached its maximum and the reaction was stopped. The obtained acrylonitrile homopolymer (PAN) solution was precipitated in methanol, filtered, redissolved in DMF and reprecipitated in methanol in order to remove unreacted monomer and catalyst. The purified PAN was filtered and dried under high vacuum at 55 °C. PANs of different molecular weights were prepared in the same manner by using different polymerization times. The PAN samples were characterized by 1H NMR and GPC.
1
H NMR (300 MHz, CDCl3): δ = 1.2 (s, 6H, CH3), 2.07 (d, methylene of AN units), 2.38 (m, 2H, N3-C-CH2), 3.17 (m, methane of AN units), 3.42 (t, 2H, CH2-N3), 4.09 (t, 2H, CH2-O) ppm.
13
C {1H} NMR (300 MHz, CDCl3): δ = 16.60 (CH2-C(CN)-CH2), 28.17 (CH2-N3), 29.70 ((CH3)2 -C), 30.05 (CH2-C(CN)-Br), 34.20 (C(CN)-CH2-C(CN)), 48.03 (CH2-CH2-CH2), 55.66 (C-Br), 62.74 (CH2-O), 119.20 (C≡N), 171.53 (C=O) ppm.
4.2 Click reaction between alkynyl-terminated P3HT and azide-terminated poly(acrylonitrile)
In a typical experiment, alkynyl-terminated P3HT (65 mg, 6.27×10-6 mol) and PMDETA (4.1 mg, 5 µL, 2.38×10-5 mol) were dissolved in 6 mL of THF in a schlenk tube that was then evacuated and back filled with nitrogen thrice. In another schlenk tube, azide-terminated PAN (93
76
mg, 6.27×10-6 mol) and CuBr (3.80 mg, 2.38×10-5 mol) were dissolved in 2 mL DMF, evacuated and back filled with nitrogen, then transferred in the first schlenk. This reaction mixture was stirred for 5 min and degassed three times by freeze-pump-thaw cycles to remove any residual oxygen. The schlenk tube was placed in a constant temperature oil bath at 45 °C for 6 days. The reaction was monitored by FTIR by checking the decrease of the intensity of azide absorption at 2100 cm-1. At the end of the reaction the solution was concentrated at the rotary evaporator and the product was recovered by precipitation in methanol and was dried under vacuum. The copolymer was characterized by 1H NMR, FT-IR, TGA and DSC.
1
H NMR (300 MHz, CDCl3): δ = 0.91 (s, 1H, CH2-CH2-CH3), 1.25 ( hexyl proton and C(CH3)2-CH2-CH(CN)), 1.341 (hexyl chain), 1.55 (CBr-CH2-CH(CN), 1.82 (C(CH3)2), 2.05 (CH2-CH2-O), 2.82 (hexyl chain), 3.5, 3.78 (allyl chain end), 6.98 (thiophene proton), 7.61 (triazole proton) ppm. FT-IR (KBr window): ν = 3100 (triazole C–H stretching), 2932, 2857 (aliphatic C-H stretching), 2242, (PAN C≡N stretching), 1710 (C=O stretching), 1554 (triazole C=N stretching), 1124 (triazole C–N stretching), 665 (allyl CH2 twisting vibration) cm-1.
4.3 Results and discussion
Here, our strategy was to obtain a poly(3-hexylthiophene)-block-poly(acrylonitrile)
(P3HT-b-PAN) by Huisgen’s 1,3 dipolar cycloaddition, “click” reaction of the separately prepared
azide-terminated PAN and alkynyl-azide-terminated P3HT. The copper(I) catalyzed azide-alkyne (3+2) cycloaddition based “click” reaction is used due to high yields and no byproducts.9 The alkynyl/allyl terminated rr-P3HT was synthesized by Grignard metathesis polymerization (GRIM) and Sonogoshira coupling reaction as discussed in chapter 3. The azide terminated poly(acrylonitrile) was synthesized by atom transfer radical polymerization (ATRP) by using the azide-functional initiator . 3-azidopropyl 2-bromoisobutyrate was prepared in two steps as shown in scheme 4.1.10
77 Br OH N3 OH NaN3 EtOH/H2O Br Br O N3 O Br O THF/TEA
Scheme 4.1 Synthesis of 3-azidopropyl 2-bromoisobutyrate
After purification, the structure of the reaction product was confirmed by 1H NMR. In figure 4.1, the proton resonances of the initiator occur at 4.30, 3.47 and 2.02 ppm. The azide group absorption occurs at 2100 cm-1 in the FTIR spectrum of figure 4.2. The presence of additional absorptions at 3458 and 3340 cm-1 can be ascribed to the presence of primary amine deriving from unwanted photocleavage of azide group caused by exposure to sunlight during the purification procedures.
78
Figure 4.1 1HNMR of 3-azidopropyl 2-bromoisobutyrate
4000 3500 3000 2500 2000 1500 1000 500 40 60 80 100 120
%
(
T
)
wavenumber (cm
-1)
2980 2935 2867 2100 1735 1280 1154 1104 3458 3340Figure 4.2 FTIR spectrum of 3-azidopropyl 2-bromoisobutyrate
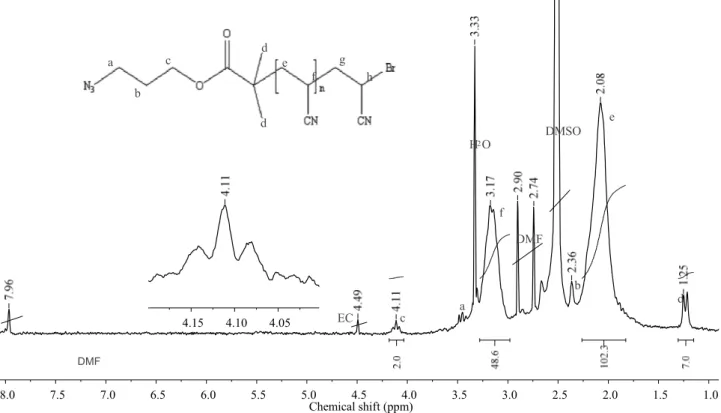
Azide-terminated PAN was obtained by ATRP with 3-aziodopropyl 2-bromoisobutyrate as the initiator and CuBr/bipyridine as the catalytic system. Several PANs with different molecular weights were prepared by varying the reaction time, and thus conversion when the reaction times were 20, 40, 60 and 120 minutes, polymer products with molecular weights 26000, 31000, 35000 and 50000 g/mol, respectively were obtained. The molecular weights were determined by SEC analysis in DMF by using a poly(styrene) (PS) standards calibration curve, and they are therefore expected to be overestimated due to the reported phenomenon of association11 and to the stiff structure generated from the polar character of the –CN groups of PAN in any solvent, including DMF. In fact figure 4.3 shows the SEC traces of the various samples of PAN, in which the presence of a shoulder at lower retention time is indicative of either poor control of the ATRP process or, more likely, of the occurrence of some aggregation in solution. The azide terminated PAN structure was confirmed by the 1H NMR spectrum, which was recorded from the polymer obtained using a polymerization time of 60 min. The resonance signals at 3.17 and 2.07 ppm are observed due to the backbone CH2 and CH protons, while the N3 terminated chain end from the initiator is responsible for the weak resonances at 4.10, 3.42, 2.27 and 1.20 ppm, respectively, as shown in figure 4.4. By
79
considering the integrals of the carbalkoxy methylene proton resonance from the initiator moiety at 4.11 ppm and of the main chain methyne resonance at 3.17 ppm, the number average molecular weight of the various PAN samples obtained using reaction times of 20, 40, 60 and 120 min were estimated as 2302, 2567, 2885 and 4316 g.mol-1, respectively (chain end groups included). These values should be considered as more reliable than those determined by SEC suggesting that indeed significant aggregation of PAN chains occurs in DMF solution, resulting in SEC, molecular weights nearly ten times larger than those determined by NMR. The PAN sample obtained by polymerization for 60 minutes, with a DPn=49 as calculated from the 1H NMR was chosen for additional characterizations and further used in the “click” reaction with the alkynyl terminated P3HT. In the FTIR spectrum, the N3 group at the chain end of PAN and the CN stretching absorption of acrylonitrile units can be observed at 2242 and 2100 cm-1, respectively, as shown in figure 4.5a. 1 5 1 6 17 18 1 9 2 0 21 R e te n tio n tim e (m in )
2 0 M in
4 0 M in
6 0 M in
1 2 0 M in
Figure 4.3 SEC traces of PAN samples obtained at different reaction times from the same reaction mixture composition elution in DMF
80 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 Chemical shift (ppm) DMF EC DMSO H2O a b a b c d d d e e f f g h c DMF 4.05 4.10 4.15
Figure 4.4 1H NMR spectrum of azide terminated polyacrylonitrile
The of block copolymer synthesis by the “click” reaction is shown in scheme 4.2. The alkynyl terminated regioregular poly(3-hexylthiophene) (rr-P3HT) was synthesized by Grignard Metathesis polymerization (GRIM) followed by Sonogoshira coupling reaction to introduce the alkynyl end group, as already described in chapter 3.
Scheme 4.2 “Click” reaction for P3HT-b-PAN
O
O
Br
NC
CN
n
N
3S
m
N
N
N
S
O
O
Br
NC
CN
n
m
CuBr, PMDETA
DMF:THF, 40
oC
81
A Huisgen’s “click” condensation was then carried out between azide terminated PAN and alkynyl terminated rr-P3HT. This copper(I) catalyzed 1,3-dipolar cycloaddition was performed in a 3:1 mixture of THF and DMF solvents using CuBr/PMDETA as the catalyst. The main problem faced to perform the “click” reaction between PAN and alkynyl-P3HT, was the poor solubility of the two polymers in a common solvent. While DMF is generally suggested as the best solvent for such cycloaddition reactions,12,13 simple test tube solubility assessments showed that the alkynyl terminated P3HT is soluble in THF but has a poor solubility in DMF, whereas the azide terminated PAN behaves just in the opposite way. To overcome this limitation, and given the generally poor solubility of PAN in most solvents, a 3:1 THF:DMF solvent mixture was found to be the best compromise for carrying out the reaction in homogenous phase. Thus P3HT was solubilized in THF, PAN was separately solubilized in DMF, and then they were transferred slowly into the same schlenk tube and added with the CuBr/PMDETA catalyst, which is soluble in both solvents as well as in their mixture. The reaction was carried out at 45 °C for 5 days, and the reacting mixture analyzed periodically by FTIR to follow the decrease in the azide peak absorption with the reaction time. After 4 days reaction no further decrease in the still present azide signal could be observed, possibly because a fraction of the azide groups, located inside the coil shape of PAN, were unable to react with alkynyl groups. The obtained mixture of P3HT-b-PAN and unreacted P3HT and PAN was precipitated in methanol, filtered and redispersed sequentially in DMF and THF, in order to remove the unreacted PAN and P3HT homopolymers. This purification cycle was repeated twice and after drying the final product was characterized by 1H NMR and FTIR, to confirm that the diblock structure of the poly(P3HT-b-PAN) had been actually achieved. In the FTIR spectrum of the azide terminated PAN (Figure 4.5a) the N3 and CN stretching can be observed at 2100, and 2242 cm-1, respectively. The weaker C≡C stretching absorption cannot be observed in the IR spectrum of the mixed reagents (Figure 4.5b) because it lies underneath the CN stretching at 2100 cm-1. As the “click” reaction was performed, the intensity ratio of the N3 to CN stretching absorptions decreased progressively from time zero (Figure 4.5b) till day five (Figure 4.5c) due to the progressive consumption of the azide group. In the spectrum of the initial reaction mixture (Figure 4.5b) the strong and broad absorption at 3400 cm-1 is due to the presence of residual reaction solvents (THF/DMF mixture). The spectrum of final product (Figure 4.5c) shows a significant decrease of the intensity of the N=N=N stretching band and a new absorption at 1554 cm-1 from the 1,2,3-triazole,14 indicating the successful although not complete conversion of the reagents according to the “Click” reaction, In the 1H NMR spectra a new resonance at 7.61 ppm due to the newly formed triazole was observed, allowing to follow the cycloaddition between azide and
82
alkynyl groups. The 1H NMR spectrum of the final copolymer shown in figure 4.6 presents the additional P3HT proton resonances at 6.98, 3.49, 2.82, 1.25 and 0.91 ppm, and those from the PAN chain and the bridging moiety at 3.75, 2.82, 2.03, 1.85 and 1.44 ppm. The PAN resonance peaks intensity is very low, due to the poor solubility of PAN in chloroform-d. But the chain end of PAN can be recognized clearly. SEC measurements could not be carried out due to the inadequate solubility in common organic solvents, including the mixture used for the click reaction.
Figure 4.5 FTIR monitoring of the “Click” reaction steps: a) azide−PAN; b) mixture of alkynyl−P3HT and azide−PAN at time zero; c) same mixture as in (b) after 5 days reaction.
83
Figure 4.6 1H NMR spectrum of block copolymer of P3HT-b-PAN
The thermal behavior of P3HT-b-PAN was studied by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). DSC scans were performed in the range between -20 °C and 350 °C with nitrogen purging gas at heating and cooling rates of -20°C/min. During the first heating scan, two deflections of the DSC trace (Figure 4.7) were observed at 42 °C and 80 °C, respectively. According to the literature,15 , 16 the glass transition temperature (Tg) for P3HT is indeed observed below 50 °C, whereas for PAN at about 87 °C. The melting temperature at 210– 220 °C is consistent with the values given by Malik et al.15 for regioregular P3HT. The endotherm observed at 170 °C could be due to evaporation of residual DMF solvent or, more probably, to the melting endotherm of PAN plasticized by DMF. In fact it is reported in the literature the occurrence of a melting transition in the same temperature range for PAN plasticized with water17 and ethylene carbonate, 18 although the authors used a fast heating scan or sealed pans to prevent degradation prior to the onset of the melting transition; a possible melting transition of unplasticized PAN was only observed above 300 °C.19 In the cooling scan a crystallization exotherm at 215 °C from the P3HT domain indicates that this polymer block structure was carefully preserved even if some degradation must have occurred during the heating scan, as shown by the following TGA analysis. On the other hand, no crystallization exotherm of PAN was observed in the cooling scan, as one would expect as the result of evaporation of the plasticizing solvent.
84
Figure 4.7 DSC curve of P3HT-b-PAN. (Inset the magnified region of heating scan between 30 and 95 °C with marks for the two Tg)
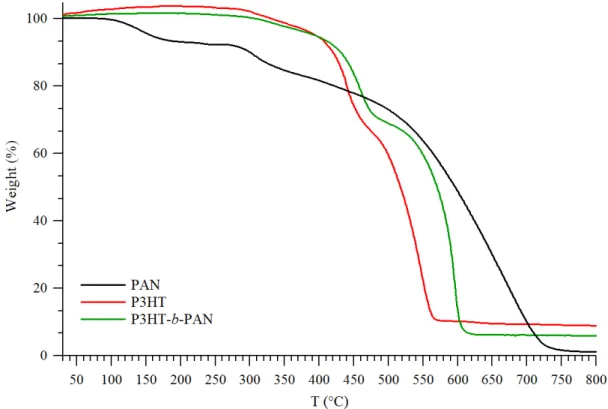
To estimate the thermal stability of P3HT-b-PAN, TGA analysis was performed in air at a heating scan of 10 °C/min and the obtained weight loss and weight loss rate thermograms are shown in figure 4.8. It is observed that the weight loss takes place in a three-step mechanism. Incipient decomposition appears to start at around 250 °C and leads to about 7% weight loss at 380 °C (although the presence of residual DMF evaporating at that temperature could not be excluded), followed by a first main decomposition step with maximum weight loss rate at 459 °C leading to a total (1st and 2nd stage) 33% weight loss, possibly due to the loss of hexyl side groups attached to the poly thiophene backbone and of some additional weight loss from the initial graphitization of PAN. The observed two-stage decomposition behavior in air is consistent with the results of a study by A. R. Adhikari et al.20 on the thermal properties of rr-P3HT/CNT composites and one by R. Ramani et al.21 on the thermal decomposition of pure P3HT showing a single-step decomposition in
nitrogen but a two stage decomposition in air. The first decomposition was attributed to cleavage of the alkyl chains while the second one was explained as due to thermo-oxidative decomposition of the polymer backbone. As the temperature increases above 500 °C, the oxidation is accelerated and both the PAN and P3HT structures are decomposed, leaving a negligible residue above 650 °C.
85
According to the literature,22 the mass loss of polyacrylonitrile starts from 350 °C in air and proceeds to complete decomposition at above 650 °C, so in figure 4.8 the PAN degradation overlaps with that of the hexyl chains of P3HT, not allowing to attempt the estimation of copolymer composition. In figure 4.8, the block copolymer thermal stability increased with respect to that of pure P3HT and PAN in the temperature range from 350 to 550 °C. On the other hand the thermal stability of the PAN block is decreased with respect to that of pure PAN in the high temperature range (above 550 °C). These changes in the thermal properties with respect to the homopolymers confirms that the product of the click reaction is in fact a block copolymer, and suggests that the PAN block exerts a protective effect towards the P3HT one, whereas the onset of degradation of P3HT promotes a faster oxidative decomposition of the residual PAN blocks.
Figure 4.8 TGA thermogram of PAN (−−), P3HT (−−) and P3HT-b-PAN (−−) (air, at 10 °C/min heating rate). The PAN-N3 and allyl/alkynyl-terminated P3HT were used as the reference homopolymers.
In conclusion, an azide-terminated poly(acrylonitrile) was synthesized by using an azido−functional ATRP initiator, and the Huisgen’s 1,3-dipolar cycloaddition-based “click” coupling of this azide-PAN with an alkynyl terminated poly(3-hexylthiophene) was also performed successfully resulting in a block copolymer. In chapter 6, the preparation of P3HT-b-PAN /CdSe hybrids starting from this block copolymer will be discussed.
86
1
Kolb, H. C., Finn, M. G., Sharpless, K. B., Angew. Chem. Int. Ed., 2001, 40, 2004. 2
Huisgen, R. Angew. Chem., Int. Ed. Engl., 1963, 2, 633. 3
(a) Opsteen, J. A.; Hest, J. C. M. v. Chem. Commun., 2005, 57–59 (b) Quémener, D., Davis, T. P.; Barner-Kowollik, C.; Stenzel, M. H. Chem. Commun., 2006, 5051 (c) Agut, W.; Taton, D.; Lecommandoux, S. Macromolecules, 2007, 40, 5653.
4
K. Matyjaszewski, J. Xia, Chem. Rev., 2001, 101, 2921, (b) Y.P. Wang, Y.Q. Shen, X.W. Pei, S.C. Zhang, H.G. Liu, J.M. Ren, React. Funct. Polym., 2008, 68, 1225, (c) J.S. Wang, K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614.
5
V. Coessens, T. Pintauer, K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337. 6
(a) Q.C. Liu, Y.M. Chen, J. Polym. Sci. Part A: Polym. Chem., 2006, 44, 6103, (b) J.F. Lutz, H.G. Borner, K. Weicnhehan, Macromol. Rapid Commun., 2005, 26, 514.
7
(a) J.A. Johnson, D.R. Lewis, D.D. Diaz, M.G. Finn, J.T. Koberstein, N.J. Turro, J. Am. Chem.
Soc., 2006, 128, 6564, (b) L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, O. Coulembier,
P. Degeé, R. Flammang, P. Dubois, Macromol. Rapid Commun., 2007, 28, 2151 (c) J.F. Lutz, H.T. Schlaad, Polymer, 2008, 49, 817, (d) O. Altintas, G. Hizal, U. Tunca, J. Polym. Sci. Part A:
Polym. Chem., 2008, 46, 1218, (e) J. Xu, J. Ye, S.Y. Liu, Macromolecules, 2007, 40, 9103, (f)
W. Van Camp, V. Germonpre, L. Mespouille, P. Dubois, E.J. Goethals, F.E. Du Prez, React.
Funct. Polym., 2007, 67, 1168 (g) M.F. Xie, Y. Kong, H.J. Han, J.X. Shi, L. Ding, C.M. Song,
Y.Q. Zhang, React. Funct. Polym., 2008, 68, 1601. 8
Zhongli Lei, Xiangyu Wei, Youhua Fan, Yalan Liu, Shuxian Bi, Journal of Colloid and Interface
Science, 2006, 304, 402–407.
9
(a) Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem., Int. Ed., 2005, 44, 2210-2215. (b) Narayan, S.; Muldoon, J.; Finn, M. G.; Fokin, V. V.; Kolb, H. C.; Sharpless, K. B. Angew. Chem.,
Int. Ed. 2005, 44, 3275-3279. (c) Li, Z. M.; Seo, T. S.; Ju, J. Tetrahedron Lett. 2004,
45,3143-3146. (d) Kolb, H. C.; Sharpless, K. B.; Drug Discovery Today 2003, 8, 1128-1137. (e) Kolb, H. C.; Finn, M. G.; Sharpless, K. B.; Angew. Chem., Int. Ed. 2001, 40, 2004-2021.
10
Quemener, D., Davis, T. P., Barner-Kowollik, C., Stenzel, M. H., Chem.Commun., 2006, 5051. 11
Valentina Pitto, Brigitte I. Voit, Ton J. A. Loontjens, Rolf A. T. M. van Benthem, Macromol.
Chem. Phys., 2004, 205, 2346–235.
12
Binder, W. H.; Sachsenhofer, R. Macromol. Rapid Commun,. 2007, 28, 15. 13
Ornelas, C.; Aranzaes, J. R.; Cloutet, E.; Alves, S.; Astruc, D. Angew. Chem., Int. Ed., 2007, 46, 872.
87
14
Meghann A. White, Jeremiah A. Johnson, Jeffrey T. Koberstein, Nicholas J. Turro , J. Am. Chem.
Soc,. 2006, 128, 11356-11357.
15
Malik S, Nandi AK, J Polym Sci: Part B, Polym Phys, 2002, 40, 2073. 16
Yue Zhao, Guoxiong Yuan, Philippe Roche, Polymer, 1994, 36, 2211. 17
G. Frushour, Bruce, Polym. Bull. 1982, 7, 1-8. 18
B. G. Min, T. W. Son, B. C. Kim, W. H. Jo, Polym. J. 1992, 24, 841-8. 19
A. K. Gupta, D. K. Paliwal, P. Bajaj, J. Appl. Polym. Sci. 1998, 70, 2703-2709. 20
A R Adhikari1, M. Huang1, H Bakhru1, M Chipara, C Yryu, P M Ajayan, Nanotechnology,
2006, 17, 5947–5953.
21
R. Ramani, J. Srivastava and S. Alam, Thermochimica Acta, 2010, 499, 34–39. 22
(a) Vincent Crook, John Ebdon, Barry Hunt, Paul Joseph, Paul Wyman, Polymer Degradation
and Stability, 2010, 1-9, (b) Michael S. Silversteina, Youval Najarya, Yulia Lumelskya, Irene