CHAPTER 3: MATERIALS AND METHODS
3.1 PLASMIDS USED FOR VECTOR PRODUCTION
Vector and packaging constructs were developed from p∆00, a replication-competent molecular clone of the Petaluma strain of FIV (FIV-Pet), derived from plasmid p34TF10 [GenBank:NC_001482] and produced in our laboratory by substituting a tryptophan codon for the stop codon in the accessory gene ORF-A (Pistello et al., 2002). Due to minimal activity of the FIV LTR in non feline cells packaging construct p∆env1 had the 5’ and 3’ LTRs replaced with CMVp and BGH poly A, respectively. Also, deletion of the 5’LTR was extended to nucleotide (nt) position 507 to remove most of the RNA ψ. Moreover it retains Rev/RRE system, whereas an internal 1,044 nt deletion within env (nt position 7246-8289) was carried out by digestion with the restriction enzymes BclI and SpeI (New England Biolabs, Celbio, Milan, Italy) (Fig. 3.1).
Vector construct LA34, was produced by sequential steps. The U3 region of the 5’ LTR (nt position 1-203, as referred to NC_001482) was replaced with CMVp amplified from pcDNA3.1 plasmid (Invitrogen Life Technologies, Milan, Italy) by digestion with the restriction enzymes PshAI and SacI (New England Biolabs, Celbio, Milan, Italy). To prevent LTR reconstruction during reverse transcription, an internal 120 bp segment of U3 in the 3’LTR (nt position 9201-9320), containing the cis-acting transcriptional elements AP-1, AP-4 and ATF-binding sites and TATA box, was also removed by PCR using overlapping primers. The same strategy was applied to delete the region from nt position 749 to 9045, encompassing most of the gag and the entire pol and env genes. Finally, the env segment containing RRE (nt position 8650-9038) was inserted downstream the gag stretch together with a multiple cloning site (MCS) containing AsuII, ClaI, SacII, BlpI, KpnI, and PacI restriction sites in the expression cassette CMV-GFP was inserted (Fig. 3.2).
Figure 3.2: LA34 vector construct.
FIV particles were pseudotyped VSV-G (495 aa) or the chimeric retrovirus GP RD114/TR (546 aa) (Sandrin et al., 2002). VSV-G was encoded by CMV-VSV-G derived from the pcDNA3.1 plasmid and RD114/TR by the phCMV-RD114/TR plasmid (kind gift of Dr. François-Loic Cosset, Ecole Normale Supérieure, Lyon, France).
3.2 CLONING STRATEGIES
Clones produced and described in this thesis were aimed to optimize heterologous cells transduction and maximize BRCA1 delivery into target cells. Correct cloning were verified by PCR or digestion screening and sequenced through ALF ExpressII DNA 20 sequencer (GE Healthcare, Cologno Monzese, Italy) using specific primers as reported in table 3.1. To reduce the risk of recombination both intermediate and final constructs were expanded in Stbl2 cells (Invitrogen Life Technologies, Milan, Italy), at 30 °C in Luria-Bertani broth and purified using either Midi or Maxi Plasmid Kits (Qiagen, Milan, Italy).
3.2.1 Vector optimization
LAW34 was obtained by the insertion of the woodchuck post-transcriptional regulatory element (WPRE) downstream the MCS (fig.3.3). WPRE region (kind gift of Dr. Stefano Indraccolo, University of Padua, Italy) was inserted in vector construct using MCS BlpI and PacI enzymes whose sequence was inserted at WPRE ends by specific primers used to amplify the element (tab. 3.1).
Figure 3.3: LAW34 vector construct.
To extend gag region from the original 120 nucleotides (nt) (nt position 628-747) to 310 nt (628-937) a two step PCR was carried out. In a first cycle, 2 fragments 420 nt and 650 nt long respectively were produced. These amplicons had overlapping regions that were jointed in a third PCR to produce the final fragment that was inserted in vector construct through AsuII and SacI enzymes. This cloning was carried out both in LA34 and LAW34 to produce LA34L (fig. 3.4 A) and LAW34L (fig 3.4 B)
A)
B)
Figure 3.4: A) LA34L and B) LAW34L vector constructs.
3.2.2 BRCA1 and BRCA1-IRES-GFP cloning
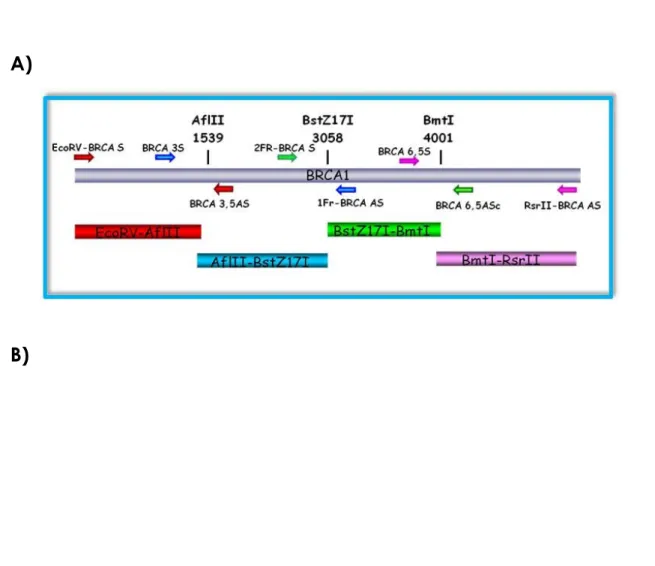
Due to its large size, BRCA1 cloning into LAW34 was carried out by subcloning the gene in four contiguous fragments. To this purpose, a preliminary computer restriction endonuclease digestion of BRCA1 gene and vector was carried out to find out the most suitable enzymes (Bioedit software, version 7.0.5.3; www.mbio.ncsu.edu/BioEdit/BioEdit.html). The main criterion used to select the enzymes was no restriction sites in the vector and only one into BRCA1. AflII, BstZ17I, BmtI enzymes cutting in position 1418, 2937, 3880 respectively were identified and their sequences were used to design specific oligonucleotides to produce a MCS to be inserted into vector construct known as LAW34III (tab. 3.1).
The two oligonucleotides were annealed by letting them at 95°C for 10 minutes and then at room temperature (RT) in the way that their temperature slowly decreased. Following this procedure, they were digested and inserted in LAW34III. The selected enzymes were used to digest the BRCA1 amplicons obtained by PCR amplification
with ad hoc designed oligonucleotides (fig 3.5 A) from pcDNA3-BRCA1 and were
inserted in LAW34III to obtain the vector expressing BRCA1 known as LAW34-BRCA1 (fig. 3.5 B).
A)
B)
Figure 3.5: A) BRCA1 cloning strategy B) LAW34-BRCA1 vector.
IRES-GFP cassette was inserted downstream BRCA1 in LAW34-BRCA1 to produce the bicistronic vector BRCA1-IRES-GFP (fig 3.6). Briefly, IRES-GFP cassette was amplified from a plasmid previously produced in our laboratory using primers containing RsrII restriction enzyme sequence. The PCR amplicon was purified, digested and inserted in LAW34-BRCA1 previously digested with RsrII.
35 Figure 3.6: Bicistronic vector BRCA1-IRES-GFP.
3.3 ASSESSMENT FOR VECTOR SAFETY
Genomic DNA and RNA from 6 x 104 transfected or transduced 293T cells were
extracted with QIAamp DNA Blood Kit and RNeasy kit (Qiagen), respectively. Viral and genomic RNAs were treated with Rnase-free Dnase (Qiagen) to eliminate residual DNA. Presence of p∆env1 plasmid and RNA transcripts in supernatants and transduced cells was investigated by PCR using 295s-296as primers (Tab 3.1) targeting FIV p25 capsid protein. Translocation of the U3 deletion from the 3’ to the 5’LTR in the vector provirus was examined by amplifying genomic DNA from transduced cells with U3s-R3as primers. Inactivation of LTR mediated transcription was ascertained by amplifying cDNA of transduced cells with In s and RRE as primers. Amplification profiles were as follows: initial denaturation 94°C 2 minutes; cycling 94 °C 30 seconds, 60 °C (54°C for 295s-296as) 30 seconds, 72°C 30 seconds (40 seconds for 295s-296as), 35 cycles; extension 72°C 10 minutes. Results were also confermed by sequencing. The possibility that infectious virions were released from transduced 293T cells was investigated by measuring for FIV p25 content the supernatant harvested 3 days post transfection and passing 0.5 ml of it serially twice in fresh cells, which 3 days later were monitored for presence of GFP-positive cells by flow cytometry and p25 content. RRE gag ΔU3 CMV R U5 CMV SD GFP WPRE R U5 BRCA1 ires
36
3.4 CELL LINES AND PRIMARY CELLS
The cell lines used for this thesis are feline Crandell cells (CrFK) and murine fibroblast NIH-3T3 (used as control) and two human cell lines. Human 293T cells aside, used for vector production and in vitro tests as described in chapter 1, vector performances were evaluated on human HCC1937 cells. 293T and NIH-3T3 were propagated in Dulbecco’s modified Eagle medium (D-MEM, Sigma-Aldrich, Milan, Italy) whereas HCC1937 in RPMI-1640 (Sigma-Aldrich). Epithelial and primary cells were isolated from a breast cancer patient, partially separated by tripsinization and cultivated in D-MEM/F12. All medium were supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 µg/ml streptomycin and 2 mM L-glutamine (Sigma-Aldrich) and all the cell lines were mantained at 37°C in 5% CO2.
3.5 VECTOR PRODUCTION AND TITRATION
Vectors were generated into 293T cells. Briefly, 3 X 106 cells were seeded in 10 cm
Petri dishes 24 hours before transfection and one day later co-transfected with packaging construct p∆env1, vector construct and either the VSV-G or the RD114/TR Env (4:5:1; 20 µg total DNA) using a modified calcium phosphate method (Pistello et al., 2006). Vector-containing supernatant was collected 48 hours later and transfection efficiency was evaluated by measuring capsid protein p25, RT activity through ELISA test and, when possible, by counting GFP-positive cells by flow cytometry with a FACScan and CELLQuest Version 2 software (BD Biosciences, Milan, Italy). Then, vectors were aliquoted in 1 ml volume and stored at -80°C until use following clarification at 1,500 rpm for 10 min and 0,45 µm filtration.
Vector titre was determined by evaluating RNA copies in transfected cells supernatant through real-time PCR. Previously RNA was extracted with QIAamp Viral RNA Mini Kit (Qiagen gmbH, Hilden, Germany), treated with Rnase-free Dnase (Ambion, foster city, USA) to remove possible plasmid residuals and retrotranscribed through avian myeloblastosis virus (AMV) RT (AMV reverse transcriptase Finnzymes Oy, Finland).
Real Time PCR was carried out through TaqMan probe; primers were designed in the WPRE region that, being a heterologous element reduces contamination risk. Reaction mix was carried out by TaqMan universal PCR Master Mix (Applied
37 Biosystem, Roche, New jersey USA) and amplification carried out in BioRad IQ5 cycler (Bio-Rad Laboratories S.r.l., Milan, Italy)
3.6 TRANSDUCTION PROTOCOLS
The day before transduction, 24-well plates were seeded with 7 X 104 293T cells, 5 X
104 NIH3T3 cells, 3 X 104 HCC1937, 2 X 104 epithelial primary cells or 3 X 104 primary
fibroblasts per well in 1 ml complete medium. Eighteen h later, the medium was replaced with the same volume of vector suspension. For Polybrene (PB) transduction 8 µg/ml of PB were added at vector suspension whereas for double transduction (DT) cells were treated with the vector twice, four h apart. Independently from the protocol used, the medium was changed 6 hours post transduction. Transduction efficiency was evaluated 2 days post-transduction by flow cytometry (FACS) or Western Blot (WB).
3.7 ANALYSIS OF TRANSGENE EXPRESSION
GFP expression from LAW34 and BRCA1-IRES-GFP was evaluated through FACS analysis. BRCA1 expression was evaluated by both FACS and WB. Due to the large size of BRCA1 in the case of WB the transfer was carried out overnight (O.N.) at 4 °C. Cells were then labeled with a specific monoclonal antibody (mAb) anti-BRCA1 (Calbiochem, Birmigham, UK) and then with an anti-mouse secondary Ab (Sigma-Aldrich). Development was carried out with Horseradish Peroxidase Conjugate Substrate Kit (Bio-Rad). For FACS analysis, 2-4 days post transduction cells were trypsinized washed with FACS buffer solution and permeabilized with saponin 5% for 20 minutes. Following permabilization cells were incubated first with 2 µg/ml mouse monoclonal anti-BRCA1 antibody (Calbiochem) for 1 hour at room temperature. Cells were then washed and stained with the secondary antibody goat anti mouse IgG1: R-Phycoerythrin (RPE) (Serotec) at a dilution of 1:100 for 1 hour in the Dark. Following washing, cells were resuspended in an appropriate volume and subjected to FACS analysis.
38
3.8 BRCA1 FUNCTIONAL TEST
Functional activity of vector expressing BRCA1 was evaluated in HCC1937 by rescue of radiation resistance test (RRR). Briefly 24 hours before the experiment 1,8 x 105
HCC1937 were seeded in 6 cm Petri dishes. The following day cell were irradiated with 0,8; 1,1; 2,3; 4,6 and 8,8 Grey (Gy) performed with 137Cs-source (3,94 Gy/min)
ionizing irradiation and placed into complete medium at 37 °C to perform DSB repair. To choose the most correct irradiation dose not transduced cells were trypsinized at 6 hours and 1, 2, 3, and 5 days post irradiation and analysed for cell viability through tripan blue staining and relative growth calculated with the following formula:
Relative growth = Cell number following irradiation
Initial cell number
For LAW34-GFP and BRCA1-IRES-GFP transduced HCC1937, following irradiation with 2,3 Gy cells were incubated at 37°C, trypsinized at day 1, 3, 5 and 7 post irradiation and monitored for GFP expression through FACS analysis.
39 Table 3.1: primers used for cloning and screening.
Primer Name Sequence (5’-3’)
SeqWPRE as Cy5-cac ata gcg taa aag gag c SeqEnd CMV
s Cy5-agc tct ctg gct aac tag ScrEC as Cy-5ttg att gtc gac act aga tat tc
BlpIW s tta cca gct aag cag gaa tat cta gtg tcg aca atc aac ctc tgg att ac PacIW as atc ctt aat taa ggt cca ggc ggg gag gcg gcc caa agg g
Long gag s tct act gct gct tga gtt aac gta tag gag tgt tat tat tga ttt tat gt Long gag as tac gtt aac tca agc agc agt aga cac cgt cat att taa aag Lpcr as cga ctt cta caa cgg gag aca gca c
In s ttt acc tgt gag gtc tcg gaa tcc ggg ccg aga act tcg cag ttg gcg ccc gaa cag gac ttg att gag agt gat tga g
MCSIII s atc ctt aag cta tac gct agc cgg acc g MCSIII as gtc cgg cta gcg tat acc tta agg at EcoRV BRCA s ttt gat atc atg gat tta tct gc RsrII BRCA as tcc cgg tcc gtc agt agt ggc tgt gg IIFr BRCA s gta tca aag gag gct cta gg IFr BRCA as ccc att tct ctt tca ggt gac BRCA 6,5 s tga ctg cag taa cca gg BRCA 6,5 as cca atc aag aaa gga tcc 295 s ggc ata tcc tat tca aac agt 296 as cac cag gat ata aaa tgc aac tct t U3R s tgg gat gag tat tgg aac cct ga R3 as tgc gaa gtt ctc ggc ccg gat tcc RRE as gtg tat cct agt atc ttg tgg a