ABSTRACT
5’-Nucleotidases, which dephosphorylate non-cyclic (deoxy)ribonucleoside monophosphates to nucleosides and orthophosphate, constitute a heterogeneous family of widespread enzymes that vary in subcellular location, tissue-specific expression and substrate specificity.
Seven human 5’-nucleotidases have been isolated, five of which are located in the cytosol (cN-IA, cN-IB, cN-II, cN-II, cdN), one in the mitochondrial matrix (mdN) and one is anchorated to the outer plasma membrane by a glycosyl phosphatidylinositol anchor (eN). By opposing the phosphorylation of nucleosides by kinases, intracellular 5’-nucleotidases take part in substrate cycles thereby regulating the intracellular levels of (deoxy)ribonucleoside pools.
The focus of this research project is the cytosolic 5’-nucleotidase II (cN-II), a widespread enzyme with a remarkable sequence conservation through evolution. The Mg2+-dependent cN-II is a bifunctional enzyme, since it can transfer the orthophosphate from a nucleoside monophosphate (preferentially (d)IMP and (d)GMP) not only to water, but also to the 5’ position of a nucleoside acceptor, usually (d)inosine, by a ping-pong reaction mechanism; this is due to the formation of a pentacovalent phosphoester enzyme intermediate, a feature of all members of the HAD superfamily cN-II belongs to. CN-II is submitted to an intricate allosteric regulation, being activated by several phosphorylated compounds (Ap4A, dATP, ATP, ADP, 2,3-BPG) and inhibited by inorganic phosphate; moreover adenylate energy charge is able to affect its activity. In particular, at physiological values of adenylate energy charge, pH and Pi, if a suitable nucleoside acceptor is available, cN-II behaves mainly as a phosphotransferase. Nevertheless, it is unknown whether the transfer of orthophosphate normally occurs in vivo, as Km values for nucleoside substrates are quite high. In vivo, in fact, hydrolysis seems to be cN-II major activity.
CN-II has always been described as a homotetramer of 60 kDa subunits: the crystallized structure of cN-II highlighted the residues responsible for the interaction of two single subunits at interface A and those which hold the two dimers together at interface B. Besides, also the presence of two putative effector sites has been hypothesized. Moreover, it has been proposed by other authors that the effectors influence cN-II oligomerization state: the activators would promote subunit aggregation, while Pi woud act in the opposite way.
Clinical interest in this enzyme increases enormously in these last years, as it seems that cN-II is involved in anticancer and antiviral prodrug metabolism: in fact, not only it could be responsible for resistance to nucleoside analogues with its catabolic function, but it could also be able to activate them by its phosphotransferase activity. Besides mRNA level of cN-II probably has a prognostic value in adult acute myeloid leukemia.
In the light of this, any findings concerning how cN-II activity is regulated and its physiological role will offer new possible therapeutical approaches. Therefore, we designed this research project combining two experimental branches: one focused on the regulatory features of cN-II, seeking to ascertain if the effectors are able to affect cN-II subunit association and enzymatic activity; the other, instead, directed towards the clarification of the role of cN-II in purine metabolism.
In this way, the results obtained would help to improve existing therapeutic treatments leading to the design of personalized chemotherapy; moreover, the cellular systems we built could be very important for the determination not only of the role of cN-II in purine metabolism but also of the relationship between its altered activity and neurological and/or neoplastic pathologies.
INTRODUCTION
1. The mammalian 5’-Nucleotidases
5’-Nucleotidase activity was first described in heart and skeletal muscle about 80 years ago by M.J. Reis. Since then, nucleoside monophosphate phospho-hydrolases, or 5’-nucleotidases (EC 3.1.3.5.), has been described in all phyla. As inferable from the name, these enzymes catalyses the hydrolysis of phosphate esterified at carbon 5’ of the ribose and deoxyribose portions
of non-cyclic nucleotides (Zimmermann, 1992; Bianchi et Spychala, 2003).
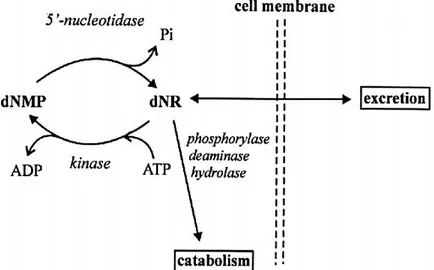
By opposing the phosphorylation of nucleosides by kinases, intracellular 5’-nucleotidases constitute the catabolic member of the substrate cycles postulated to be involved in the regulation of cellular levels of ribo- and deoxyribonucleoside monophosphates (figure 1.1). These cycles thus regulate the balance between anabolism and catabolism of (deoxy)ribonucleotides and are safeguards against the accumulation of dNTP pools. Impairment of their normal function, in fact, may result in severe immunodeficiency (Rampazzo et al., 1999; Camici et al., 2010).
Intracellular 5’-nucleotidases have relatively high Km values and operate on substrates generally present at very low concentrations. Thus they are extremely sensitive to oscillations of substrate concentration (Bianchi and Spychala, 2003). While intracellular nucleotidases catalyze the final step in nucleotide dephosphorylation leading to the export of nucleosides from the cell, the extracellular 5’-nucleotidase, being part of the ecto-nucleotidase cascade, participates in the regulation of extracellular nucleotides (Hunsucker et al., 2005).
In mammalians, seven human 5’-nucleotidases with different subcellular localization have been cloned, five of which are located in the cytosol (cN-IA, cN-IB, cN-II, cN-III, cdN), one in the mitochondrial matrix (mdN) and one is anchorated to the outer plasma membrane (eN). These Figure 1.1: Schematic representation of a substrate cycle involving a deoxyribonucleotide, dNMP, and a deoxyribonucleoside, dNR (from
enzymes are characterized by different subcellular location, substrate specificity and tissue-specific expression, as summarized in table 1.1.
Table 1.1: 5’-nucleotidases peculiarities (modified from Hunsucker et al., 2005).
1.1 Ecto-5’-Nucleotidase
Ecto-5’-nucleotidase (eN), also known as CD73, is the only - among mammalian 5’-nucleotidases - to be an ecto-enzyme and not to belong to HAD superfamily; crystallographic studies of the E. coli 5’-nucleotidase, in fact, confirmed the hypothesis, arose from data of sequence homology, which suggested that eN belongs to a superfamly of dinuclear metallophosphoesterases, which hydrolyse very different substrates, including phosphoproteins, nucleotides and nucleic acids (Sträter, 2006).
The eN gene is located on chromosome 6q14-q21 (Hunsucker et al., 2005). The coding region of human eN sequence is 1725 bp long and translates to a 574 amino acid protein (Misumi et al.,
1990). Allignment of the human eN sequence with that of other species reveals 86% to 89%
homology between mammalian eN proteins and 24% to 36% identity to proteins from bacteria (Hunsucker et al., 2005).
EN is a glycosylated protein bound to the outer surface of the plasma membrane by a glycosyl phosphatidylinositol anchor colocalizing with detergent-resistant and glycolipid-rich membrane subdomains called lipid rafts (Misumi et al., 1990; Bianchi et Spychala, 2003). Like other ecto-enzymes, eN recycles through the endosomal compartment during internalization of the plasma membrane; for this reason, immunohistochemistry shows eN localized to the plasma membrane, multivesicular endosomes and, to a lesser extent, the Golgi complex (Van den Bosch et al., 1988).
Soluble forms of the enzyme exist, which are derived from the membrane-bound form by hydrolysis of the GPI anchor by phosphatidylinositol-specific phospholipase or by proteolytic cleavage (Sträter, 2006). Human ecto-5’-nucleotidase consists of two 71 kDa subunits linked by non-covalent bonds
(Martinez-Martinez et al., 2000;
except the enzyme from bull seminal plasma in which eight cysteines are all involved in intramolecular
disulfide bridges: Cys 51–Cys 57, Cys 353– Cys 358, Cys 365–Cys 387 and Cys 476–Cys 479; Fini et al., 2000) - while the calculated molecular mass from the human cDNA is 63 kDa, as eN undergoes
extensive co- and post-translational processing into the mature form. In fact, not only the final 25 amino acids of the C-terminus are cleaved and a specie-tissue specific GPI anchor is added to Ser
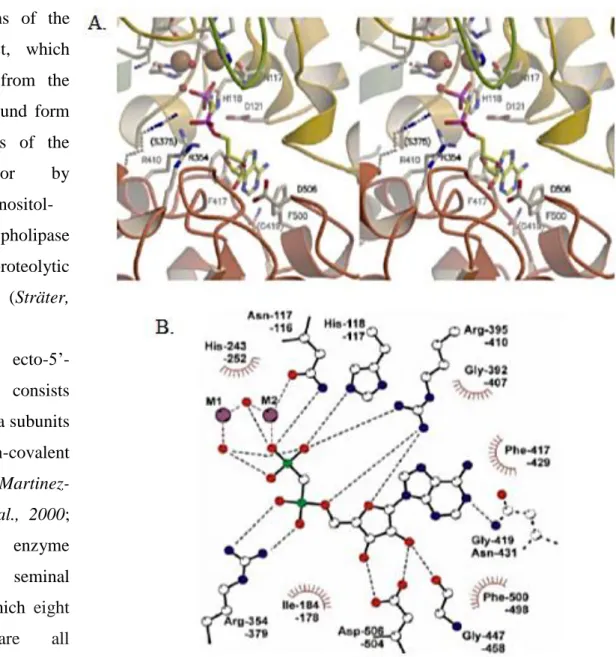
Figure 1.1.1: Binding mode of the substrate analogue inhibitor -methylene-ADP. A. Stereo view of the active site. The residues which are not conserved in eN are depicted in broken lines. The labels refer to the human enzyme. B. Scheme of the interaction between the protein and the inhibitor. The upper label for each residue refers to human eN and the bottom label to E. coli 5’-NT. All residues are conserved with the exception of N431 of E. coli (shown in broken lines), which is a glycine in the mammalian enzymes. One oxygen atom of the terminal phosphate group is coordinated to the site 2 metal ion (M2). In addition, the catalytic histidine and an arginine bind and polarize the phosphate group for attack of the nucleophile. The nucleophilic water is supposed to be the terminal water coordinated to the site 1 metal ion (M1). This water molecule is in a perfect position for an in-line attack on the phosphorus atom. It is located at a distance of 3.2 Å and the angle between this water, the phosphorus atom and the leaving group is 155°. However, the metal bridging water ligand is also a possible candidate for the nucleophile. The two metal ions, the arginine and the catalytic histidine stabilize the transition state. No protein residue is positioned to protonate the leaving group. Therefore, a water molecule might provide a proton for the leaving group (from Sträter 2006).
523, but also 26 amino acids of the N-terminus are cleaved, cotranslationally (Misumi et al, 1990;
Zimmermann, 1992).
EN is expressed in all tissues; however, in most cases, it is specifically located only in certain cell types, as – for example – in the thymus, where is mainly located on epithelial cells and reticular fibroblasts (Resta et al., 1993; Resta et al., 1998).
Ecto-5’-nucleotidase has a broad substrate specificity, being able to hydrolyse both ribo- and deoxyribonucleoside monophosphates, even if it has the highest affinity for AMP; for this reason, eN is known also as “low Km-nucleotidase” (Km value for AMP are in the low micromolar range;
Hunsucker et al, 2005). Hydrolysis of AMP is stereoselective, as the L-enantiomer is not
hydrolysed (Zimmermann, 1992). By the Vmax/Km ratio, AMP seems to be the most efficient substrate and ribonucleotides are far better substrates than deoxy-ribonucleotides (in order of increasing activity UMP, IMP, GMP, AMP).
Cations are not necessary for enzyme activity; anyway, both Mg2+ and Mn2+ increase enzyme activity, while Pb2+ and Hg2+ act as inhibitors. It seems that the natural metal ligand of eN is Zn2+ since the purified enzyme from chicken gizzard contains tightly associated zinc at a molar ratio zinc/protein of 2 (Zimmermann, 1992). These results were confirmed by X-ray crystallographic data and site-mutagenesis studies performed on the E. coli 5’-nucleotidase which suggested the presence of two metal-ion-binding sites in the catalytic cleft, designated M1 and M2, respectively the high- and the low-affinity metal-ion binding sites (Knofel and Sträter, 1999; McMillen et al.,
2003).
ADP, ATP and adenosine 5’-[, - methylene]diphosphate are highly effective competitive inhibitors for human eN (instead, ADP and ATP are substrate of E. coli 5’-nucleotidase), whereas inhibition by concanavalin A is non-competitive (Zimmermann, 1992).
The pH optimum of eN depends on the source of the enzyme (between 6.8 and 9; Hunsucker et al.,
2005).
Even if there is low overall sequence homology between E. coli 5’-N and mammalian e-N, the crystal structure of the latter enzyme provided insight into the catalytic mechanism of mammalian eN.
The N-terminal domain (residues 25-342) coordinates the dimetal cluster and the conserved Asp-His dyad, forming the catalytic core structure, whereas the C-terminal domain (residues 362-550) provides the substrate specificity pocket for the nucleotides (Sträter, 2006). Thus, the active site is located between these two domains. A long -helix (residues 343–361) connects the two domains (for more details see figure 1.1.1; Sträter, 2006).
Catalysis proceeds by nucleophilic attack on the phosphate group of the substrate by a water molecule bound to one of the metal ions (M1); after the hydrolysis of the phosphate bond, a phosphoenzyme intermediate is formed before orthophosphate is released (figure 1.1.2; Knofel and
Sträter 1999; Knofel and Sträter, 2001; Sträter 2006).
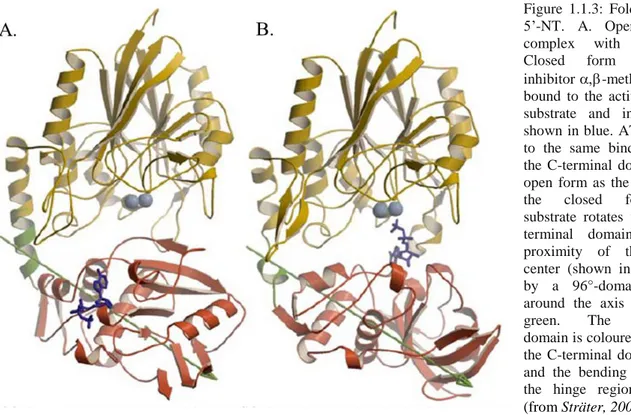
In E. coli 5’-NT, a domain rotation is necessary for catalytic activity, presumably allowing substrate binding and product release. In the crystal structures, in fact, the protein has been characterised in two different conformations differing in the relative orientation of the two domains. The domain movement can be described as a rotation of the C-terminal domain around the axis shown in figure 1.1.3, which passes through the center of the C-terminal domain. A rotation of up to 96° is necessary for the change between
the inactive open and the active closed conformation (Sträter 2006). A sequence alignment of E. coli 5’-NT and the mammalian ecto-enzymes shows that both domains are conserved and a homology model can be built for eN (figure 1.1.4 A). In this model the loop from residue 319 to 337 in the E.
coli enzyme, whose function is
unknown and which is stabilized by two interacting -strands, is missing in eN (L1 in figure 1.1.4 B). The nearby loop around residue 50 is longer in the mammalian enzyme (L2): it is stabilized by a cystine bridge (51–57) and carries a glycosylation on Asn 53. On the other side of the protein, three loops
are predicted to differ significantly between the mammalian and bacterial nucleotidases. The loop near residue 146 in E. coli (L3) is longer in the mammalian enzymes, whereas the short helix at residue 183 in E. coli 5’-NT is missing (L4). More, in the C-terminal domain two insertions are predicted to be present in eN (at residue 482 (L5) and 398 (L6) of the bacterial sequence). The loop
Fig 1.1.2: Scheme of the dimetal center and proposed structure of the Michaelis complex for 5’-NT catalysis. Residue labels refer to human eN. All residues except for N245 are conserved in the E. coli enzyme. Therefore the coordination of N245 via a water molecule to metal ion 1 is hypothetical. In the bacterial enzyme N245 is replaced by a glutamine, which is directly coordinated to the metal ion (from Sträter, 2006).
around residue 228 in E. coli 5’-NT is significantly shorter in ecto-5’-nucleotidase (L7; Sträter 2006).
Further, as the mammalian enzymes form dimers, whereas the bacterial enzymes are monomers, it is not possible to predict the interaction site between the monomers with confidence; however, it seems improbable that the left side of the monomer, as depicted in figure 1.1.4 B, forms the dimer interface since it carries several glycosylation sites. It is more likely that the other side of the protein near loops L3 and L4 is part of the dimer interface. If a domain rotation is also part of the catalytic cycle of the mammalian enzymes, it might be speculated that the two monomers interact only with the larger N-terminal domains and not at the smaller C-terminal domain, which rotates around its center in the E. coli enzyme (Sträter, 2006).
As ecto-5’-nucleotidase hydrolyses AMP to adenosine, there are several proposed functions for eN, including local generation of adenosine and activation of G-protein coupled seven transmembrane spanning adenosine receptors (A1, A2A, A2B, A3) or internalization through dipyridamole-sensitive carriers, salvaging of extracellular nucleotides, mediating cell–cell adhesion and possibly acting as a coreceptor for T-cell activation; thus ecto-5’-nucleotidase would be fundamental not only as enzyme, but also as protein per se (Airas et al., 1995; Hunsucker et al., 2005; Colgan et al., 2006;
Deaglio et al., 2011).
A number of studies have implicated eN in the control of tissue barrier function. Lennon and colleagues examined the influence of polymorphonuclear leukocyte (PMN) on endothelial para-
Figure 1.1.3: Fold of E. coli 5’-NT. A. Open form in complex with ATP; B. Closed form with the inhibitor -methylene-ADP bound to the active site. The substrate and inhibitor are shown in blue. ATP is bound to the same binding site of the C-terminal domain in the open form as the inhibitor in the closed form. The substrate rotates with the C-terminal domain into the proximity of the dimetal center (shown in light blue) by a 96°-domain rotation around the axis depicted in green. The N-terminal domain is coloured in yellow, the C-terminal domain in red and the bending residues of the hinge region in green
interact with the substrate by hydrogen bonding are marked with a capital ‘S’ whereas residues involved in non-polar interactions are indicated by a lower-case ‘s’. Cystine bridges of the bovine enzyme are marked by lines connecting the cysteines. B. Superposition of the C traces of the experimental X-ray structure of E. coli 5’-NT (grey) and the
homology model for human eN. Larger insertions and deletions of the ecto-5’-NT are coloured in magenta and labeled L1 to L7. The closed conformation of E. coli 5’-NT has been used as template for the homology modeling according to the sequence alignment listed in A. The exact loop conformations of longer insertions (L2, L3, L5 and L6) in ecto-5’-nucleotidase cannot be modeled with confidence. The experimentally determined disulfide bridges of bovine eN are shown in yellow and the glycosylated asparagines are shown in red. Depicted in dark blue is the substrate analogue inhibitor -methylene-ADP (from Sträter, 2006).
Fig. 1.1.4: A. Sequence alignment of E. coli, bovine, human and electric ray 5’-nucleotidases. The secondary structure and the residue numbering shown above the alignment are from the E. coli structure. Functionally important residues are marked below the alignment with the following labels: ‘1’ and ‘2’ mark ligands to metal ions 1 and 2, respectively; the catalytic Asp–His dyad is labeled ‘C’; ‘R’ marks the three arginine side chains in the active site of the E. coli enzyme; ‘F’ are the phenylalanines that bind the adenine moiety; residues that
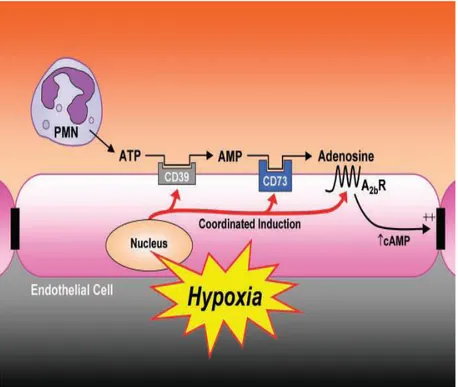
cellular permeability, whose increase is a measure of PMN-mediate injury and disfunction during inflammation. In fact, during severe inflammation, PMN transendothelial migration has the potential to disturb vascular barrier. Lennon et al. showed that, in order to dampen fluid loss during PMN-endothelial interactions, activated PMNs release 5’-AMP. This undergoes conversion to adenosine through the action of eN at the endothelial cell surface, where adenosine activates A2B receptors, thus increasing intracellular cAMP and hence resulting in increased endothelial barrier function (for more details, figure 1.1.5; Lennon et al., 1998).
It is well-documented that during hypoxia multiple cell types release adenine nucleotides also in the form of AMP, which is converted into adenosine, a vasodilation regulator, by eN. It has been demonstrated that, during limited oxygen availability, eN is transcriptionally up-regulated. EN gene promoter, in fact, contains a cAMP response element (CRE) and during hypoxia the activation of the adenosine receptors A2A or A2B leads to the increase of intracellular level of cAMP. More, it has been shown that in the transcriptional regulation of eN the hypoxia-inducible factor-1 (HIF-1) is fundamental (Synnestvedt et al., 2002; Colgan et al., 2006). In a similar way, cells of the cardiovascular system generate and release adenosine during stress conditions: after ischemic preconditioning, ecto-5’-nucleotidase activity increases; eN, in fact, seems to be primarily responsible for adenosine production under these circumstances (Hunsucker et al., 2005; Colgan et
al., 2006). Similarly, it has been shown the protective role of ecto-5’-nucleotidase in renal and in
hepatic ischemia; furthermore, the significant attenuation of renal injury after ischemia by soluble 5’-nucleotidase treatment suggests possible new strategies to ameliorate the consequences of renal hypoxia and hepatic ischemia (Grenz et al., 2007; Hart et al., 2008).
EN also plays a role in cell adhesion and acts as a coreceptor for T-cell activation under certain conditions. In fact, in addition to its catalytic activity, eN has adhesive properties being involved in lymphocyte adhesion to vascular endothelial cells, an important step in the migration of lymphocytes through the blood vessel wall and into sites of inflammation. It seems that the properties of eN adhesion depends strongly on the cell type in which it is expressed (Hunsucker et
al., 2005).
It has been appreciated that eN may also contribute in host responses to microbial infection. Ecto-5’-nucleotidase, in fact, seems to be responsible for symptomology related to epithelial infection by enteropathogenic E. coli and to contribute to responses following virus infection (Colgan et al.,
2006).
EN have recently been attributed an intracellular function in the regulation of lipid metabolism in rat adipocytes: Müller et al. showed that in response to certain physiological and pharmacological signals – as the absence/presence of H2O2 palmitate and glimepiride – microvesicles are released
from donor adipocytes that transfer their constituent GPI- protein eN to the plasma membrane DIGs (detergent-insoluble glycolipid-enriched plasma membrane microdomains) and cytoplasmic LDs (lipid droplets) of acceptor adipocytes, resulting in the stimulation of esterification of fatty acids into lipids (Müller et al., 2010).
Very recently, it was shown that eN is expressed in peptidergic and nonpeptidergic nociceptive neurons. Moreover, experiments performed on eN-/- mice established that eN accounts for about 50% of all AMP hydrolytic activity in nociceptive neurons, where eN colocalized with prostatic acid phospahtase (PAP), another ectonucleotidase hydrolyzing AMP to adenosine. Besides, both pap-/- and eN-/- mice showed enhanced thermal hyperalgesia in animal models of inflammatory pain. Two years ago, Sowa and colleagues demonstrated that recombinant eN is involved in the long lasting antinociceptive circuits activated by adenosine A1 receptor: these findings suggest that ectonucleotidases represent a new class of antinociceptive tools (Sowa et al., 2010).
In 1997, Page and colleagues described a neurological and developmental syndrome characterized by onset within the first few years of life of the patients, seizures, ataxia, recurrent infections, developmental delay, hyperactivity, short attention and poor interaction. Investigation of the enzymatic set involved in purine and pyrimidine metabolism showed an increase of 6- to 10- fold of a AMP hydrolysing activity. The identity of this nucleotidase remained unknown until few years ago, when Pesi and colleagues demonstrated that ecto-5’-nucleotidase is responsible for the AMP hydrolysing hyperactivity described by Page et al. (Page et al., 1997; Pesi et al., 2008). This syndrome has been called “Nucleotidase Associated Pervasive Developmental Disorder” (NAPDD). Figure 1.1.5: Proposed model of
coordinated nucleotide metabolism and nucleoside signaling in posthypoxic endothelial cells: in areas of ongoing inflammation, diminished oxygen supply coordinates the induction of CD39, CD73 and AdoRA2B. At such sites,
activated PMNs provide a readily available extracellular source of ATP that through two enzymatic steps results in the liberation of extracellular adenosine, available for activation of surface endothelial adenosine receptors, particularly the AdoRA2B.
Postreceptor increase in intracellular cAMP results in enhanced barrier function: this protective mechanism may provide an innate mechanism to preserve vascular integrity and prevent intravascular fluid loss (from
The mechanism through which ecto 5’-nucleotidase hyperactivity causes NAPDD is still unknown (Camici et al., 2010).
1.2 Intracellular 5’-nucleotidases and HAD superfamily
The six intracellular 5’-nucleotidases belong to the haloacid dehalogenase (HAD) superfamily, which was first described by Koonin and Tatusov in 1994 and named after the structural characterization of the first family member, 2-haloacid dehalogenase precisely (Koonin and
Tatusov, 1994; Koonin et al., 1994). HAD superfamily consists of more than 3000 members and is
represented in the proteomes of organisms from all three superkingdoms of life, colonizing numerous very disparate biological functions (the vaste majority of which are unknown), which vary in their degree of essentiality to the cell. Members with known function fall into one of five subfamilies:
1. Haloalkanoate dehalogenases (C-Cl bond hydrolysis); 2. Phosphonoacetaldehyde hydrolases (P-C bond hydrolysis); 3. Phosphate monoesterases (P-OC bond hydrolysis);
4. ATPases (POP bond hydrolysis);
5. Phosphomutases (P-OC cleavage with intramolecular phosphoryl group transfer).
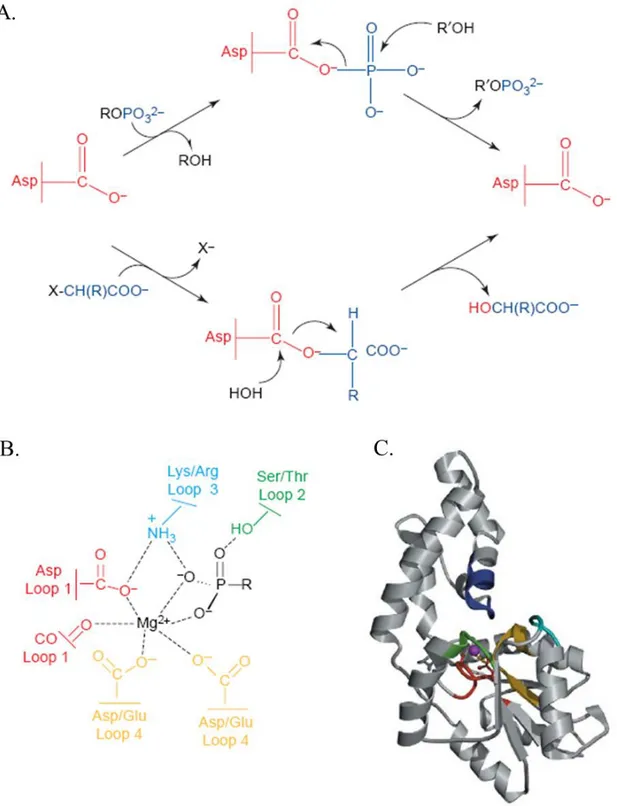
The core chemistry catalyzed by this superfamily is the "group transfer", mediated by a nucleophilic aspartate: using this catalytic strategy, a covalent bond is made to the nucleophile with the formation of a phosphoenzyme intermediate (figure 1.2.1 A and B for more details; Liu et al., 1995;
Li et a., 1998; Lahiri et al., 2004; Allen and Dunaway-Mariano, 2004; Burroughs et al., 2006).
Even if the sequence homology of these enzymes is less than 10%, they contain a highly conserved / core domain, in which the active site is formed by four loops conserved residues that
orchestrate the core chemistry (Figure 1.2.1 C; we have to specify that some authors refer to three, and not four, conserved motifs; in this case motifs III is composed not only by the conserved lysine, but also by the two conserved aspartates; Hisano et al., 1996; Rinaldo-Matthis et al., 2002; Allen
and Dunaway-Mariano, 2004; for a detailed description of the HAD superfamily "evolutionary
exploration of substrate space" read Burroughs et al., 2006). This common structural feature is the "HAD domain", phylogenetically correlated with other superfamilies containing the / Rossmann fold (DHH, CheY-Like, von Willebrand A, TOPRIM, classical hystone deacetylases, PIN/FLAP nuclease domain): the HAD domain is distinguished from these related Rossamannoid folds by two key structural signatures, a "squiggle" (a single helical turn) and a "flap" (a beta hairpin motif) located immediately downstream of the first -strand of their core Rossmannoid fold (Burroughs et
al., 2006). These structures probably provide the necessary mobility to alternate between the "open"
and the "closed" conformations (Burroughs et al., 2006). The core catalytic domain of the HAD
Figure 1.2.1: The catalytic scaffold in the HAD superfamily of phosphotransferases. A. In HAD enzymes, aspartate mediates carbon-group transfer to water and phosphoryl-group transfer to a variety of acceptors. Thus, the HAD superfamily is unique in catalyzing both phosphoryl-group transfer (top) and carbon-group transfer (bottom). B. Schematic of the roles of the four loops that comprise the catalytic scaffold. The activity ‘switch’ is located on loop 4 of the catalytic scaffold (yellow) which positions one carboxylate residue to function as a general base for the dehalogenases and either two or three carboxylates to bind the Mg2+ cofactor, essential for the phosphotransferases. CO represents the backbone carbonyl oxygen of the moiety that is two residues downstream from the loop 1 (red). The
side-chain at this position is also used as an acid-base catalyst by phosphatase and phosphomutase HAD members. Loop 2 (green) and loop 3 (cyan) serve to position the nucleophile and substrate phosphoryl moiety. C. Ribbon diagram (core domain: loop 1, red; loop 2, cyan; loop 3, green; loop 4, yellow; cap domain: specificity loop, blue) of the fold supporting the catalytic scaffold of phosphonatase (from Allen and Dunaway-Mariano, 2004).
superfamily contains a three-layered / sandwich comprised of repeating units which adopt the topology typical of the Rossmannoid class / folds. The central sheet is parallel and is typically comprised of at least five strands in a 54123 strand order (these strands are hereinafter referred to as S1-S5; for intracellular 5’-nucleotidases whose structure is known the strand order is 321456; figure 1.2.2; Burroughs et al., 2006).
As mentioned, all members of HAD superfamily contain four highly conserved sequence motifs. Sequence motif I corresponds to strand S1 and the DXD signature is present at the end of this strand (figures 1.2.1 B and 1.2.3): the carboxylate group of the first aspartate and the backbone C=O of the second aspartate coordinate the Mg2+ cofactor, which provides orientation and charge shielding for nucleophilic attack performed by the first aspartate, thus forming the abovementioned aspartyl-intermediate (Ridder and Dijkstra, 1999; Morais et al., 2000; Burroughs et al., 2006). The presence of the divalent ion magnesium is an absolute catalytic requirement for HAD superfamily (Allen and
Dunaway-Mariano 2004; Burroughs et al., 2006). Replacement of either aspartate of this motif by
both a conservative and a non-conservative residue resulted in, at least, partial inactivation of the enzyme (Collet et al., 1998). Motif II corresponds to the S2 strand, which is characterized by a highly conserved threonine or serine at the end, while Motif III is centered on a conserved lysine that occurs around the N-terminus of the helix located upstream of S4: these two motifs contribute to the stability of the reaction intermediates of the hydrolysis reaction (figures 1.2.2 and 1.2.3). Motif IV maps to strand S4: the conserved acidic residues located at its end (typically one of these three basic signatures: DD, GDXXXD, GDXXXXD) coordinate Mg2+ in the active site together with those of motif I (Burroughs et al., 2006). Motifs I-IV are spatially arranged around a single “binding cleft” at the C-terminal end of the strands of the central sheet that forms the active site of the HAD superfamily (figure 1.2.2). This binding cleft is partly covered by the -hairpin flap Figure 1.2.2: Classic HAD Rossmannoid domain. Strands are shown as arrows with the arrowhead on the C-terminal end and are labeled from S1 to S6. The strand containing the catalytic D residue is rendered in yellow; other core strands conserved across all members of HAD superfamily are in blue; non-conserved elements are in grey-broken lines. The pink loop represents the conserved squiggle. For more details read the text (from Burroughs et al., 2006).
occurring after S1. Additional inserts (if present) occurring between the two strands of the flap or in the region immediately after S3 provide extensive shielding for the catalytic cavity. These inserts, called “cap domains” (or loop 5), often contribute for specificity acting as a dynamic lid over the core domain active site: a tight -turn in the helix-loop-helix motif of the cap domain, in fact, contains a stringently conserved glycine located at the N-terminal end of the hairpin turn (i.e., helix2-(Gly)turn-helix3) which affords flexibility to the enzyme structures (Zhang et al., 2002;
Lahiri et al., 2004).
Figure 1.2.3: Multiple alignment of mammalian 5’-nucleotidases and members of the P-type ATPase-L-2-haloacid dehalogenase (ATPase-HAD) superfamily. Proteins are listed under their SWISS-PROT codes. Bb, Bos bovis; Ec,
Escherichia coli; Eh, Enterococcus hirae; Hs, Homo sapiens; Mg, Mycoplasma genitalium; Psp, Pseudomonas sp.; Sa, Staphylococcus aureus; Sc, Saccharomyces cerevisiae; Sp, Schizosaccharomyces pombe. Only the four common
sequence motifs are reported. The numbers indicate the distances to the N-terminus of each protein and the sizes of the gaps between aligned segments. The upper, middle and lower block of sequences include mammalian 5’-nucleotidases, members of the HAD superfamily and P-type ATPases, respectively. Blue shading indicates conserved amino acid residues required for catalytic activity. Red shading indicates conserved amino acids alternatively present in motif I. Yellow shading indicates uncharged amino acid residues. Common secondary structure elements are indicated as -helices, -strands and l (loop) regions (from Allegrini et al., 2004).
1.2.1 Cytosolic 5’-nucleotidases IA and IB
cN-IA was first described as an AMP preferring enzyme (it is known also as AMP-specific 5’-nucleotidase) partially purified from pigeon ventricle by Gibson & Drummond in 1972 and was later purified from rabbit, rat, pigeon and dog hearts and from pigeon breast muscle (Skladanowski
and Newby, 1990; Tkacz-Stachowska et al., 2005; Hunsucker et al., 2005). It is expressed at a high
level in skeletal and heart muscle, at an intermediate level in pancreas and brain and at a low level in kidney, testis and uterus (Hunsucker et al., 2001).
The human cN-IA gene is located on chromosome 1p34.3–p33: while the open reading frame of the cloned human cDNA consists of 6 exons and is 1107 bp, northern blot analysis showed that the mRNA migrates at nearly 10 kb (Hunsucker et al., 2001). The pigeon cN-IA has also been cloned; the mRNA transcript in this case is 2.3 kb (Sala-Newby et al., 1999).
Electron microscopy using immunogold staining of dog heart tissues showed association of cN-IA with myofilaments in ventricular cells; these findings was confirmed by immunocytochemistry on pigeon heart demonstrating a striated pattern of cN-IA in the cytosol that may be due to association with contractile elements (Darvish et al., 1993a; Sala-Newby et al., 1999).
The subunit size of purified cN-IA is 41 kDa in human heart, 43 kDa in dog heart and 40 kDa in pigeon heart (Darvish et al., 1993b; Sala-Newby et al., 1999; Hunsucker et al., 2001). Gel filtration experiments of the purified dog heart cN-IA indicated that the enzyme is a tetramer (Darvish et al,
1993b).
Alignment studies of amino acid sequence demonstrated not only that the human cN-IA is related to pigeon cN-IA, bu also that it is significantly related to proteobacteria Xylella fastidiosa 5’-nucleotidase F82601.
In figure 1.2.1.1 we report the alignment of the three enzymes: all three cN-I sequences contain a conserved classic Walker B motif (R/K)X1–4GX2–4X2(D/E) ( is a hydrophobic amino acid) at amino acids 220 to 233 found in many ATP-binding proteins; thus, it could serve as part of the nucleotide binding pocket. All three cN-I sequences also contain a conserved kinase-2 motif near the C terminus, IFFDD (at amino acids 338 to 342 on the alignment). As the sequences of kinase-2 domains - although highly variable - usually consist of four hydrophobic residues followed by an aspartate that interacts with Mg2+ or other divalent metal ions, this conserved domain may be essential for enzyme function as Mg2+ is necessary for cN-I activity
(Hunsucker et al., 2001; Skladanowski and Newby, 1990; Darvish et al., 1993b). There are several additional putative nucleotide binding domains that are conserved only in the
human and pigeon cN-IA enzymes: two kinase-2 domains are located at amino acids 31–35 and 313–317 and a putative kinase 3a domain is located at amino acids 299–304. Moreover, three putative nucleoside monophosphate binding motifs (NMP-2), which may bind the pentose mojety of the substrate, are located at amino acids 68–73, 177–182 and 313–318 on the alignment (Hunsucker et al., 2001).
Figure 1.2.1.1 Alignment of human, pigeon and X. fastidiosa cN-I amino acid sequences. Similar conserved amino acids are shaded. The conserved Walker B and kinase 2 motifs are indicated below the alignment (from Hunsucker et
al., 2001).
As already countered, cN-IA has a preference for AMP (Km for AMP ranges from 1.2 to 8.3 mM using cN-IA purified from rat, pigeon and rabbit hearts; Hunsucker et al., 2005). Although the Km values for IMP (0.8 to 8.3 mM) are in the same range, the Vmax with this substrate is lower, making the reaction less efficient (Skladanowski and Newby, 1990; Hunsucker et al., 2001). Even if cN-IA has maximal relative activity using AMP as a substrate, the rabbit heart enzyme is most efficient with pyrimidine deoxyribonucleoside monophosphates, with Km values of 61 M for dCMP and 20 M for dTMP (Garvey et al., 1998). In fact, when enzyme efficiency is calculated as Vmax/Km, cN-IA is 8.6-fold more efficient with TMP and 3.6-fold more efficient with dCMP than with AMP (Garvey et al., 1998). Although they are less efficient substrates than dCMP evaluated by the Vmax/Km ratio, dAMP (Km 130 M), dGMP (Km 120 M), CMP (Km 110 M) and GMP (Km 320 M) all have Km values in the micromolar range using the purified rabbit enzyme (Garvey et al.,
1998). Similar results were obtained with the recombinant human enzyme, which has high affinity
for dCMP (Km 12 M) and dTMP (Km 20 M; Hunsucker et al., 2001; Hunsucker et al., 2005). The low Km values for the deoxyribonucleoside monophosphates suggest that they may be physiological substrates of cN-IA. dUMP inhibits cN-IA activity with a Ki of 40 M, suggesting that it could also be an excellent substrate (Garvey et al., 1998).
Being a member of HAD superfamily, cN-IA is Mg2+-dependent, with maximum activation at 3.5 to 10 mM, depending on the species; Mn2+ and Co2+ activate the enzyme to a lesser extent (Darvish
et al., 1993b; Hunsucker et al., 2005).
The pH optimum of pigeon and human heart cN-IA is 7.0, while that of the rabbit enzyme is between pH 6.5 to 6.7 (Hunsucker et al., 2005).
IA is activated by ADP (K50 value between 10 M and 89 M, depending on the source of cN-IA and the substrate concentration ) and, to a lesser extent, by GTP (Skladanowski and Newby,
1990; Hunsucker et al., 2001). cN-IA is also activated by dNDPs, with maximal activation in the
low micromolar range similar to that of GTP for dADP, dTDP, dCDP, dGDP and dUDP (Hunsucker et al., 2005). In product inhibition studies, thymidine inhibited the enzyme non-competitively while inorganic phosphate non-competitively, consistent with an ordered release of nucleoside prior to phosphate (Garvey et al., 1998). Mirroring nucleotide substrate specificities, pyrimidine nucleosides were more potent product inhibitors than purine nucleosides: 5-ethynyl-2‘,3‘-dideoxyuridine (5-EddU) has been developed as a selective inhibitor of cN-IA based on inhibition of the enzyme by the pyrimidine dideoxynucleotides ddT, ddU and ddC (Garvey et al.,
1998).
cN-I might play a role not only in dephosphorylation of deoxyribonucleoside monophosphates, as indicated by their low Km values, but also in inactivation of nucleoside analogs. In order to unravel this point, Hunsucker and colleagues expressed human cN-I in Jurkat and HEK 293 cells: this conferred resistance to 2-chloro-2’-deoxyadenosine, with a 49-fold increase in the IC50 in HEK 293 and a greater than 400-fold increase in the IC50 in Jurkat cells. Expression of cN-I also conferred a 22-fold increase in the IC50 to 2’,3’-difluorodeoxycytidine in HEK 293 cells and an 82-fold increase in the IC50 to 2’,3’-dideoxycytidine in Jurkat cells. These data indicate that cN-I could be responsible for resistance to specific nucleoside analogs (Hunsucker et al., 2001).
There are several evidences that cN-IA is responsible for the production of adenosine during the imbalance of energy supply and demand such as under ischemic or hypoxic conditions. In fact, overexpression of the cloned pigeon cN-IA in COS-7 cells, H9c2 rat cardiomyocytes and neonatal rat cardiomyocytes showed that the enzyme is capable of producing adenosine under conditions of ATP breakdown, despite the fact that the Km for AMP is in the millimolar range (Sala-Newby et al.,
1999, 2000, 2003).
IA mRNA levels varies among different pigeon muscles; in particular, 5–10 times more of cN-IA transcript is present in red, oxidative muscles (breast muscle and gastrocnemius) than in white ones, composed of glycolytic fibers (biceps brachii; Lechward and Tkacz-Stachowska, 2009). On the contrary, human muscle cN-I does not reflect the ratio of oxidative fibers to the total mass of the
muscle sample in humans. That difference indicates that there are certain mechanisms that differentially regulate the expression of cN-IA in muscle tissues of mammals and lower vertebrates (Lechward and Tkacz-Stachowska, 2009).
A cN-IA homologous sequence, related to human autoimmune infertility gene (AIRP), has been cloned and designated cN-IB (figure 1.2.1.2; Sala-Newby and Newby, 2001). It was identified on chromosome 2p24.3 (Sala-Newby and Newby, 2001).
Human cN-IB is ubiquitously expressed, as determined by RT-PCR analysis, with highest mRNA expression in testis and lowest expression in brain and skeletal muscle (Sala-Newby and Newby,
2001). The cloned murine and human cN-IB cDNAs have several putative translation start sites: it
is unclear which site(s) is (are) used in vivo (Sala-Newbyand and Newby, 2001). It is also unknown whether the cN-IB subunits function as a tetramer like cN-IA or exist as a monomer or dimer like other intracellular nucleotidases.
Overexpression of the cloned murine cN-IB in COS-7 cells showed that the enzyme, like cN-IA, hydrolyzes AMP and is activated by ADP (Sala-Newby and Newby, 2001).
1.2.2 Cytosolic 5’-nucleotidase III
CN-III is also known as Pyrimidine 5’-nucleotidase (P5’-NT), as it catalyzes the dephosphorylation of the pyrimidine 5’-monophosphates UMP and CMP to the corresponding nucleosides (Amici et
al., 1997).
The gene encoding cN-III is located on chromosome 7p15-p14 and spans approximately 50 kb (Marinaki et al., 2001; Kanno et al., 2004). The codifying region is split into 11 exons (1, 2, R, 3-10) leading to the production of three mRNA forms by alternative splicing of exons 2 and R (Marinaki et al.,2001; Kanno et a.l, 2004). The transcript lacking both exons 2 and R is expressed ubiquitously, whereas transcripts including exon 2 alone or both exons 2 and R are almost
Figure 1.2.1.2. Protein structure of cN-IA and cN-IB aligned by catalytic motif I. Dark shaded boxes indicated catalytic motifs and light shading indicates sequences of high homology. Three possible translation start sites are indicated on cN-IB with open triangles (from Hunscker et
specifically expressed in reticulocytes (Kanno et al., 2004; Hunsucker et al., 2005; Zanella et al.,
2006). The biological significance of these various mRNAs found in reticulocytes is at present
unknown; among the three expected proteins (285, 286 and 297 amino acids long) only the protein consisting of 286 amino acid residues, identical to the previously identified lupus inclusion protein p36, has been isolated from erythrocytes (Amici et al., 2000; Lu et al., 2000; Zanella et al., 2006). It is produced using the initiation codon at the 3’-end of exon 1 and sequentially lengthened from exon 3, both exons 2 and R being spliced out (Marinaki et al, 2001; Kanno et al., 2004). The 286 amino acid form of cN-III has been cloned and studied in detail and the characteristics of this enzyme are the same as purified cN-III (Rees et al., 2003).
cN-III has mainly been studied in erythrocytes and is often thought of as an erythrocyte-specific enzyme; actually, it is also found in other tissues. Searches of the expressed sequence tag database show cN-III expression in multiple human tissues, including lymphoid cells, spleen, heart, liver, testis, colon, uterus, lung, pancreas, kidney and brain (Lu et al., 2000; Marinaki et al., 2001). The mouse homologue of p36, lupin, shows ubiquitous mRNA expression, with highest levels in spleen, brain, heart, blood and kidney (Lu et al., 2000). It seems that this protein has a high degree of evolutionary conservation (figure 1.2.2.1).
cN-III hydrolyzes only pyrimidine monophosphates and has no activity with purine substrates (Amici and Magni, 2002). The Km ranges from low micromolar to millimolar (CMP (10-150 M), UMP (330 M), dUMP (400 M), dCMP (580 M) and dTMP (1.0 mM); Amici and Magni, 2002;
Hunsucker et al., 2005). The Vmax/Km ratio shows that CMP is the most efficient substrate of cN-III, followed by UMP (Amici and Magni, 2002). Amici and colleagues, in 1997, showed that cN-III has also phosphotransferase activity, like cN-II (Amici et al., 1997). Because the transferred phosphate originates from a monophosphate substrate of the nucleotidase reaction, all pyrimidine substrates of cN-III can serve as phosphate donors, while cytidine (Km is 0.64 mM) and uridine (Km is 0.79 mM) appear to be the best phosphate acceptors (Amici et al., 1997). Besides, it seems that this activity could be important also in the activation pathway of some prodrugs, as cN-III is able to phosphorylate 3’-azido-3’-deoxy-thymidine (AZT), cytosine--D-arabinofuranoside (AraC) and 5-fluoro-2’-deoxy-uridine (5FdUrd; Amici et al., 1997; Amici and Magni, 2002).
Moreover, nucleotidase activity of cN-III could be responsible for the resistance to nucleoside analogs, as cN-III is able to use both AZTMP and araCMP as substrates (Amici and Magni, 2002). Any attempt to correlate cN-III expression and genetic variants, in order to explain the significantly association between gemcitabine and AraC citotoxicity and cN-III expression, gave unclear results (Jordheim et al., 2008; Aksoy et al., 2009).
Figure 1.2.2.1: Deduced amino acid sequences of Lupin proteins in various species are aligned to demonstrate evolutionary conservation. Identical residues are shown in bold and shaded; similar residues are lightly shaded. Gaps (–) are inserted to improve sequence alignment (from Lu et al, 2000).
cN-III is a Mg2+-dependent enzyme, a feature of HAD superfamily, and has a pH optimum of 7.5 (Amici and Magni, 2002). While nucleosides and phosphate inhibit cN-III activity, the enzyme is not affected by ADP, ATP or 2,3-BPG (Amici and Magni, 2002).
CN-III is the only nucleotidase known to function as a monomer (Amici et al., 1997; Marinaki et
al., 2001). Few years ago, Bitto and colleagues obtained the crystal structure of murine cN-III,
which has 92% sequence identity to its human counterpart, refined to 2.35 Å resolution (Bitto et al.,
2006). The structure showed that the enzyme adopts a fold similar to enzymes of the HAD
belongs to the /-class of proteins with a three-layer sandwich architecture and Rossmann fold topology in the core domain (cyan and red structure in figure 1.2.2.2). Extended loops of the fold spanning residues 55–142 and 190–211 form an auxiliary domain (yellow and green in figure 1.2.2.2): 4 and 5 helices of the auxiliary domain form an extended “lid” located over the core domain (Bitto et al., 2006). Structural homology search, using the DALI and VAST servers, identified a range of structural homologs of murine cN-III, many of which were indeed established members of the HAD superfamily; the closest structural homologs are the phosphoserine phosphatases
(PSPs) from different species. Specifically the top homolog found by DALI was a PSP from
Methanococcus jannaschii (MjPSP; figure 1.2.2.3; Bitto et al., 2006).
Fig. 1.2.2.3: Comparison of murine cN-III and its closest structural homolog. A. Stereo representation of structural superposition of murine cN-III (red) and human PSP (cyan). Every 10th C carbon of murine cN-III is highlighted by a red sphere and some are labeled by residue numbers for better orientation. B. Structural superposition of the active sites of murine cN-III (red) and human PSP (cyan; from Bitto et al., 2006).
Figure 1.2.2.2: A ribbon diagram of the murine cN-III structure. The core domain is coded in cyan and red. The lid domain is color-coded in yellow and green. The highly conserved residues of the four motifs peculiar of members of the HAD superfamily are depicted as blue sticks (D49, D51, T53, K213, G237, D238 and D242; from Bitto
The catalytic mechanism of MjPSP and hPSP has been studied in considerable structural detail by x-ray crystallography; because the active site of murine cN-III is nearly identical to that of PSPs, the authors hypothesized that the catalytic mechanism of cN-III might be analogous to that of PSPs; for more details read the caption of figure 1.2.2.4 (Bitto et al., 2006).
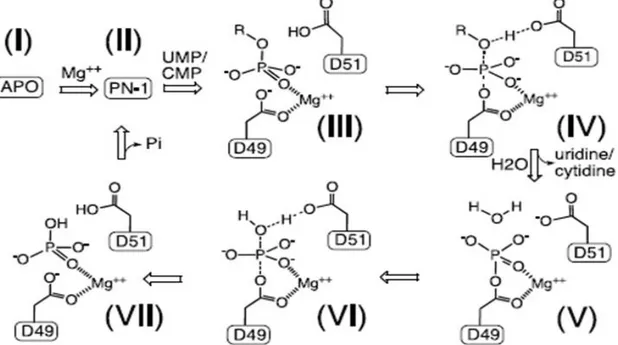
Figure 1.2.2.4: The general scheme of the proposed reaction cycle of murine cN-III, based on that of MjPSP, for nucleotidase activity. “R” represents ribonucleoside. Individual states of the reaction mechanism include apoenzyme (I), active enzyme (II), the substrate complex (III), the substrate-transition complex (IV), the phosphoenzyme intermediate (V), the product-transition complex (VI) and the product complex (VII; from Bitto et al., 2006).
In order to provide experimental evidence for this hypothesis, Bitto and colleagues set out to structurally characterize accessible states of the murine cN-III reaction cycle. In this way, they confirmed their hypothesis (for more details figures 1.2.2.4 and 1.2.2.5; Bitto et al., 2006).
Eukaryotic pyrimidine nucleotidases are enzymes involved in catabolism of RNA and in the pyrimidine salvage pathway. During erythrocyte maturation, the nuclei and mitochondria are expelled and the energy needed for cellular functions is subsequently produced by glycolysis. cN-III is up-regulated to catabolize uridine and cytidine ribonucleotides produced by RNA degradation (Rees et al., 2003; Hunsucker et al., 2005). Because the mature erythrocyte has lost the ability to synthesize nucleosides, it is important that the adenine nucleotide pools are not depleted during breakdown of RNA. Thus, the restriction of cN-III activity to pyrimidines ensures that purines are not lost (Hunsucker et al., 2005).
Two types of cN-III deficiency are known: hereditary and acquired. Hereditary deficiency is associated with nonspherocytic hemolytic anemia (NHA), while acquired deficiency results from inhibition of cN-III by Pb2+ during lead poisoning and its severity is related to the blood levels of lead (Rees et al., 2003; Hunsucker et al., 2005). Both NHA and severe lead poisoning (>80 g/dl
Pb2+ in blood) have the same clinical manifestation: erythrocytes in blood smears of affected patients show a distinct basophilic stippling upon Wright’s staining; at the molecular level, NHA is characterized by significantly increased levels of nucleotides in red blood cells (3–6 times),
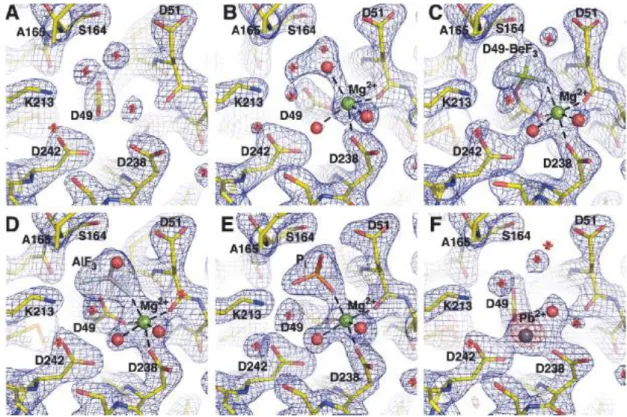
Figure 1.2.2.5: Individual states of the reaction cycle of cN-III include apoenzyme (I), active enzyme (II), the substrate complex (III), the substrate-transition complex (IV), the phosphoenzyme intermediate (V), the product-transition complex (VI) and the product complex (VII; figure 1.2.2.4). Bitto et al. obtained crystals of murine cN-III trapped in different states of the reaction mechanism: I, i.e. A; II, i.e. B: Mg2+ is octahedrally coordinated by the carboxyl oxygens of D49, D51, D238, three waters molecules; V, i.e. C: intermediate with the covalently modified catalytic D49; BeF3
-acts as a phosphate analog in proteins that are phosphorylated on aspartate residues; VI, i.e. D: D51 is likely involved in the activation of the nucleophile (water or nucleoside) as a general base; AlF3 acts as the transition state analog of the
phosphoryl group; VII, i.e. E. F represents murine cN-III inhibited by Pb2+ (from Bitto et al., 2006).
especially pyrimidine nucleotides that can account for up to 80% nucleotide content. Pyrimidine nucleotides are present only in minute amounts in normal erythrocytes (Hunsucker et al., 2005;
Bitto et al., 2006). Similar, although less pronounced, accumulation of pyrimidine nucleotides may
occur during severe lead poisoning. Bitto and colleagues elucidated the molecular mechanism of cN-III inhibition by Pb2+ (figure 1.2.2.5 F): Pb2+ binds specifically within the active site (but in a different position compared with Mg2+) in a way that compromises function of the cationic cavity, which is required for the recognition and binding of the phosphate group of nucleotides (Bitto et al.,
2006). Several studies have been made in order to identify the cause of hereditary cN-III
deficiency. Various distinct missense mutations in cN-III have been found in patients with NHA, but cN-III activity may not be considered a prognostic indicator in patient affected by the enzymopathy; some mutations, in fact, allow red cell to maintain a rather high activity, up to about
60% (Marinaki et al., 2001; Rees et al., 2003; Kanno et al., 2004; Chiarelli et al., 2005; Chiarelli
et al., 2008).
Few years ago, it was shown that low levels of cN-III mRNA were related to a worse overall survival in patient affected by acute myeloid leukemia and treated with araC, but there was no relationship between enzyme mRNA expression and response to induction treatment or disease-free survival (Galmarini et al., 2005). These findings means that cN-III is unlikely to be involved in araC metabolism; instead, this enzyme may reflect disease aggressiveness (Galmarini et al., 2005). In the attempt to clarify this aspect, Jordheim and colleagues studied if interindividual differences in cN-III expression is correlated with differences of sensitivity to araC therapy, but the results they obtained did not underlined a strong correlation (Jordheim et al., 2008).
1.2.3 Cytosolic 5’ (3’)-deoxyribonucleotidase
Cytosolic 5’(3’)-deoxyribonucleotidase (cdN), also known as dNT-1, was first purified from human placenta by Höglund and Reichard in 1990 (Höglund and Reichard, 1990). The cdN gene is located on chromosome 17q25.321–23 and consists of 5 exons and 4 introns; its mRNA is expressed ubiquitously in human tissues, with the highest levels in pancreas, skeletal muscle and heart (Höglund and Reichard, 1990; Rampazzo et al., 2002).
Human placenta and human erythrocytes cdN catalyzes the dephosphorylation of the nucleoside monophosphates containing the pyrimidine bases uracil or thymine preferring 3’-dUMP, 3’-dTMP, 3’-UMP and 2’-UMP (Km values in the millimolar range; Höglund and Reichard, 1990). In contrast to human cN-III, human cdN is not strictly pyrimidine-specific and works efficiently with dIMP and dGMP (Höglund and Reichard, 1990). CdN purified from rat, instead, uses all 5’-dNMPs although dAMP is a poor substrate (Höglund and Reichard, 1990; Hunsucker et al., 2005). We have to underline that the dNT-1 purified from human erythrocytes, but not that derived from placenta, seems to possess also pyrimidine-specific phosphotransferase activity (Amici et al., 1997). Anyway, further studies have to be performed in order to confirm this point as phosphotransferase activity was not found using recombinant murine cdN or purified cdN from human placenta (Höglund and Reichard, 1990; Rampazzo et al, 2000a).
As a member of the HAD superfamily, also cdN is a Mg2+-dependent enzyme and has a pH optimum of 6.0 to 6.5 for dUMP and 5.5 to 7.5 for 3’-UMP (Amici and Magni, 2002).
CdN activity is inhibited by dIno, dUrd and Pi when dUMP is used as a substrate, while its activity with 3’-nucleotides is increased in the presence of dGMP, dTMP, dUMP, dIMP, dIno, dGuo and to
a lesser extent dThd (Höglund and Reichard, 1990; Rampazzo et al., 2000a; Hunsucker et al.,
2005).
As depicted in the reported crystal structure of human cdN obtained by Walldén and colleagues in 2007, the enzyme is a homodimer in agreement with previous results from gel filtration chromatographies; each subunit is 23.4 kDa (figure 1.2.3.1; Höglund and Reichard, 1990;
Rampazzo et al., 2000a; Walldén et al, 2007a).
In particular, cdN has two domains, an Rossmann-like fold (core domain which covers residues
1-17 and 77-201), containing six antiparallel -strands that are sandwiched between -helixes, and a 4-helix bundle (cap domain which covers residue 18-76), forming an aromatic/hydrophobic pocket that coordinates the base of the nucleotide substrate by residues Phe 18, Phe 44, Leu 45 and Tyr 65.
The active site is situated in a cleft between the core domain and the cap domain. This structure is very similar to that of human mitochondrial 5’(3’)-deoxyribonucleotidase (mdN) described in next paragraph (average rmsd of 0.65 Å; Walldén et al., 2007a). These data and the 52% sequence identity between cdN and mdN support the hypothesis that these two separate genes arose from gene duplication (Rampazzo et al., 2000b).
In 2001, Gazziola and colleagues demonstrated that cytosolic 5’(3’)-deoxyribonucleotidase constitutes the catabolic counterpart of deoxycitidine kinase in the substrate cycle which regulates pyrimidine nucleotide pools (Gazziola et al., 2001). In fact, overproduction of murine cdN in human 293 cells led to excretion of deoxyuridine, deoxycytidine and deoxythymidine from cells,
Figure 1.2.3.1: Superposition of the homodimeric human cdN (yellow) and one protomer of dUMP-bound D12N variant of murine cdN (blue; this point mutant was built in order to capture substrates/products in cdN). Numbers are residue numbers of human cdN. One-letter codes of the following residues of the D12N variant of murine cdN are shown: N12 and D14 of Motif I, T101 of Motif II, K136 of Motif III and L47 and Y67 of the base recognition site. Nitrogens are colored in blue and oxygens in red. For more details read the text (from Walldén et al,
while overexpression of the enzyme in hamster V79 cells - that have higher dCK activity than 293 cells - led to a much smaller increase in deoxycytidine and deoxyuridine excretion (Gazziola et al.,
2001). Moreover, the reduced excretion of deoxynucleosides in cdN expressing cells in the presence
of increased dCK activity further suggests that the two enzymes form a substrate cycle (Gazziola et
al., 2001). Besides, the ribonucleotide pools in 293 cells expressing cdN were also affected, with
increased excretion of uridine and cytidine, although CMP is not a substrate for the recombinant mouse enzyme (Rampazzo et al., 2000b; Gazziola et al., 2001). Moreover, from results of isotope flow experiments it was suggested that cdN, together with the cytosolic thymidine kinase (TK1), participates in a substrate cycle regulating the intracellular concentration of deoxyuridine and thymidine nucleotides (Gazziola et al., 2001). The substrate specificity of cdN and data from cells overexpressing cdN implicates the enzyme in protecting cells from expansion of pyrimidine nucleotide pools, particularly dUTP and dTTP pools (Gazziola et al., 2001).
It seems that cytosolic 5’(3’)-deoxyribonucleotidase is involved not only in the regulation of intracellular pyrimidine levels, but also in pyrimidine nucleoside analog metabolism. In 2003, in fact, Mazzon and colleagues demonstrated that cdN is able to dephosphorylate the 5’-phosphates of various prodrugs used in antiviral and anti-cancer therapy; in particular FdUMP, AZTMP, BVdUMP and d4TMP seem to be quite good substrates of cdN (Km values of all substrates tested are in the millimolar range; Mazzon et al., 2003).
Recent RT-PCR studies performed on leukemic blasts from 114 patients with acute myeloid leukemia and treated with araC showed that low cdN mRNA expression is correlated with a worse clinical outcome, suggesting that cdN may play a role in sensitivity to araC in AML patients; anyway, Mazzon and colleagues registered no activity with araCMP (Mazzon et al, 2003;
Galmarini et al., 2004). Further studied have to be carried out to clarify this burning point.
1.2.4 Mitochondrial 5’(3’)-deoxyribonucleotidase
The human mitochondrial 5’(3’)-deoxyribonucleotidase is the only mammalian 5’-nucleotidase found so far in mitochondria; it is encoded by a nuclear gene located on
chromosome 17 p11.2 in the critical region deleted in the Smith–Magenis syndrome, a genetic disease of unknown etiology (Rampazzo et al., 2002). The mdN gene has 5 exons and 4 introns, with identical gene structure and intron/exon boundaries as cdN (Rampazzo et al., 2002). Excluding the mitochondrial leader sequence of mdN that is cleaved during import into the mitochondrial matrix, the two sequences have 52% homology, suggesting that the separate genes derived from a
gene duplication event (figures 1.2.4.1 and 1.2.4.2 A; Rampazzo et al., 2000b; Rampazzo et al.,
2002). This hypotesis is supported also by the strong similarity of the human cdN and mdN
structure (average rmsd of 0.65 Å; Walldén et al., 2007a).
In human tissues, mdN mRNA expression is highest in heart, brain and skeletal muscle, with much lower expression in pancreas and kidney (Rampazzo et al., 2000b). The cDNA of human mdN codes for a 25.9-kDa polypeptide with a typical mitochondrial leader peptide, providing the structural basis for two-step processing during import into the mitochondrial matrix.
Mitochondrial localization of mdN was demonstrated by the mitochondrial fluorescence of 293 cells expressing a mdN-GFP fusion protein (Rampazzo et al., 2002; Gallinaro et al., 2002).
Recombinant human mdN shows a narrower substrate specificity than cdN, in fact prefers 5’-dUMP, 5’- and 3’-dTMP and 5’-, 3’- and 2’-UMP as substrates (Km values for all substrates are between 0.1 and 0.15 mM; Mazzon and colleagues demonstrated that the Km values for dUMP and dTMP are, respectively, 100 M and 200 M; Rampazzo et al., 2000b; Gallinaro et al., 2002;
Mazzon et al., 2003).
The enzyme has an acid pH-optimum between 5.0 and 5.5 and shows an absolute requirement for Mg2+ for catalysis (Rampazzo et al., 2000b).
Two inhibitors of mdN have been developed: PMcP-U, a competitive inhibitor with a Ki of 40 M, and (S)-1-[2’deoxy-3’,5’-O-(1-phosphono)benzylidene--D-threo-pentofuranosyl]-thymine (DPB-T), an inhibitor with mixed-type inhibition and a Ki of 70 M (Mazzon et al., 2003). PMcP-U also inhibits cdN, while DPB-T does not. Resolution of the crystal structure of human mdN complexed with either PMcP-U or DPB-T demonstrated that the 2 inhibitors are coordinated in the active site by different amino acids (for more details, figure 1.2.4.2; Rinaldo-Matthis et al., 2004).
Figure 1.2.4.1: Comparison of mdN (i.e dNT-2) with cdN (i.e. dNT-1). A. Amino acid sequences of the two human enzymes, deduced from their cDNA sequences. Amino acid identities are indicated by shading. The peptide motif for mitochondrial two-step processing is underlined. The common exon/intron borders are indicated by arrow heads. B. Organization of the genes for the two enzymes. Exons are shaded. Notice the difference in scales (from
A.
B.
Figure 1.2.4.2: A. Sequence alignment of the human mdN (here named dNT-2) and cdN (here named dNT-1). The above numbering follows the amino acid sequence in dNT-2. Blue fields represent conserved residues, red fields represent residues with conserved mutations, yellow fields mark less conserved mutations than red fields and white marks indicate no conservation at all. ○, amino acids involved in catalysis; ▲, residues interacting with the base; ∆, residues coordinating the pentofuranosyl part of DPB-T; ●, residues coordinating the phosphonate of DPB-T in mdN. The sequence alignment was done using ALSCRIPT to clarify why dNT-1 is not inhibited by DBP-T inhibitor. B. Detailed view of the active site of dNT-2 with bound DPB-T inhibitor. In light blue R163. Structural comparison between cdN and mdN shows that when DPB-T binds mdN, only the thymine base is in the same position as cdN; the rest of the molecule is oriented toward the solvent; the amino acids that coordinate the phosphate and pentofuranosyl groups of DPB-T are not conserved between mdN and cdN, explaining why DPB-T does not inhibit cdN. PMcP-U, instead, is positioned in the conserved binding pocket similarly to the natural substrate and thus can inhibit both mdN and cdN (from Rinaldo-Matthis et al., 2004).
In 2004, Rinaldo-Matthis and colleagues published the crystal structure of mdN in complex with bound phosphate and beryllium trifluoride plus thymidine as model for a phosphoenzyme-product complex: in this way, they were able not only to outline determinants for substrate specificity recognition – which have been confirmed in later studies – but also to assess the membership of mdN to HAD superfamily (Rinaldo-Matthis et al., 2004). The crystal structure of mdN reveals a dimeric structure consistent with gel filtration data (figure 1.2.4.3; Rampazzo et al., 2000b;
comprising amino acids 33-42 and 111-227, forms a Rossmann fold, except for two loops, residues 175-187 and 194-203, which are helices in the canonical Rossmann fold (Rinaldo-Matthis et al., 2002). The small domain, instead, forms a truncated four-helix bundle inserted between strand 1 and helix 5 of the large domain. Two loops, residues 42-49 and residues 97-110, connect the two domains. The dimer interaction is formed primarily by 6, 3 and the loop
between 7 and 4 from the core domain. The interaction surface is relatively hydrofobic.
The active site of mdN is found in a cleft between the two domains and is accessible to solvent; in the active site of the native structure distinct nonprotein density was detected due to a bound phosphate ion and a Mg2+ ion, with the phosphate coordinating directly to the Mg2+ (figure 1.2.4.4;
Rinaldo-Matthis et al., 2002). The main components of the active site are constituted by loop 1, the
C-terminal part of strands 2 and 3, the N-terminal part of 4 and the loop between helices 2 and 3.
Figure 1.2.4.4: Stereo view of the final 2Fo – Fc map contoured at 1σ around the phosphate and the Mg2+ in the active
site. The Mg2+ ion is coordinated to the carboxylate side chains of D41 and D176 and the main chain carbonyl of D43. In addition, Mg2+ is coordinated by three exogenous ligands, the phosphate ion and two densities modeled as water molecules, completing an octahedral coordination (from Rinaldo-Matthis et al., 2002).
Figure 1.2.4.3: The mdN dimeric structure, with residues from 6, 3 and the loop between 7 and 4 forming the dimer interface. In the active site, the bound thymidine product is found. The grey ball represents the Mg2+ (from Rinaldo-Matthis et al., 2002).