1
UNIVERSITY OF PISA
BIOS- Research School in BIOmolecular Science
Course in Molecular Biotechnology XXIV cycle
Cellular Mechanisms in Niemann Pick type C disease
Dr.ssa Grazia Della Sala
Tutor
Dr.ssa Laura Colombaioni
Pisa, March 2012
2
Table of Contents
ABSTRACT _______________________________________________________________ 5 CHAPTER I _______________________________________________________________ 9 Cholesterol trafficking and Niemann-Pick disease ________________________________ 9
Introduction ___________________________________________________________________ 9 1.1 Cholesterol trafficking in cells _________________________________________________ 9 Cholesterol distribution and function in the brain ___________________________________ 10 Cholesterol removal from the brain _______________________________________________ 10 Intracellular cholesterol transport ________________________________________________ 11 1.2 Niemann Pick type C disease _________________________________________________ 14 Clinical description ____________________________________________________________ 14 Early forms of Niemann-Pick disease _____________________________________________ 16 Juvenile period (6-15 years) (classical form) ________________________________________ 18 Late onset forms (>15 years) _____________________________________________________ 18 1.3 NPC1 protein ______________________________________________________________ 19 1.4. Disease causing mutations and genotype-phenotype relationships __________________ 22 1.5 Consequences of NPC1-deficiency in the brain___________________________________ 23 Neuron death and pathological changes in NPC1-deficient brains ______________________ 24 Axonal transport and degeneration _______________________________________________ 27 1.6 Therapies for NPC disease ___________________________________________________ 28 1.7 Aim of the study ____________________________________________________________ 30
CHAPTER II _____________________________________________________________ 31 Experimental Procedures ___________________________________________________ 31
3 2.2 Cell culture ________________________________________________________________ 31 2.3 Process of freezing and thawing of the cellular line _______________________________ 31 2.4 Plasmids __________________________________________________________________ 32 2.5 Statistical Analysis __________________________________________________________ 32 BACTERIAL TRANSFORMATION _____________________________________________ 32 2.6 Competent cell preparation___________________________________________________ 32 2.7 Transformation of the vector into E. coli________________________________________ 32 2.8 Isolation of plasmid DNA from small amounts of bacteria (mini-prep) _______________ 33 2.9 Transient Transfection ______________________________________________________ 33
Image Analysis____________________________________________________________ 34
2.10 Confocal microscopy _______________________________________________________ 34 2.11 Confocal Microscopy of TMRM ______________________________________________ 34 2.12 Confocal Microscopy of Cameleons: new calcium probe _________________________ 34 2.13 Cell surface deduced from protein fluorescence levels ____________________________ 37 2.14 Vesicles Identification ______________________________________________________ 38 2.15 NPC1-bearing vesicles motility analysis _______________________________________ 38 2.16 Morphological Analysis of Endoplasmic Reticulum______________________________ 39 2.17 Colocalitation analysis ______________________________________________________ 44
CHAPTER III ____________________________________________________________ 48 Distribution and Motility of NPC1 I1061T________________________________________ 48
Introduction __________________________________________________________________ 48 3.1 Expression of NPC1wt and NPC1I1061T GFP in HN9.10e neuroblasts. _________________ 48 3.2 Dynamics of vesicles containing NPC1 protein ___________________________________ 55 3.3 NPC1 I1061T is mislocalized in HN9.10e cells______________________________________ 61
CHAPTER IV ____________________________________________________________ 65 Effect of NPC1 I1061T expression on morphology and function of Endoplasmic Reticulum 65
4 4.1 Distribution of wt and I1061T NPC1 on Endoplasmic Reticulum ___________________ 65 4.2 Expression of NPC1I1061T causes a reorganization of ER ___________________________ 66 4.3 How ER responds to stressing agents___________________________________________ 73 4.4 Effect of I1061T on ER [Ca] 2+________________________________________________ 76
CHAPTER V _____________________________________________________________ 80 Effect of I1061T missense mutation on Mitochondria. ____________________________ 80
Introduction __________________________________________________________________ 80 5.1 Distribution of NPC1 protein on Mitochondria __________________________________ 80 5.2 Effect of NPC1I1061T on membrane potential and mass of mitochondria ______________ 82
5.3 The effect of I1061T on mitochondrial Ca2+ _____________________________________ 86
DISCUSSION ____________________________________________________________ 88 SUMMARY AND CONCLUSIONS ___________________________________________ 93 Bibliography______________________________________________________________ 94 Acknowledgements _______________________________________________________ 115
5
Table of Figures
Figure 1.1- Intracellular cholesterol transport. ... 13
Figure 1.2- Age of onset of neurological disease vs. lifespan... 15
Figure 1.3 -Niemann Pick disease symptoms as function of age. ... 16
Figure 1.4- Hypothetical model of NPC1 dimerization to form a transmembrane channel operated by and for sterols... 20
Figure 1.5- Proposed pathway for transfer of cholesterol from LDL or β-VLDL to NPC2 to NPC1 to membranes. ... 21
Figure 1.6-Cholesterol accumulation in NPC1-deficient neurons... 27
Figure 2.1- Schematic of cameleon function... 36
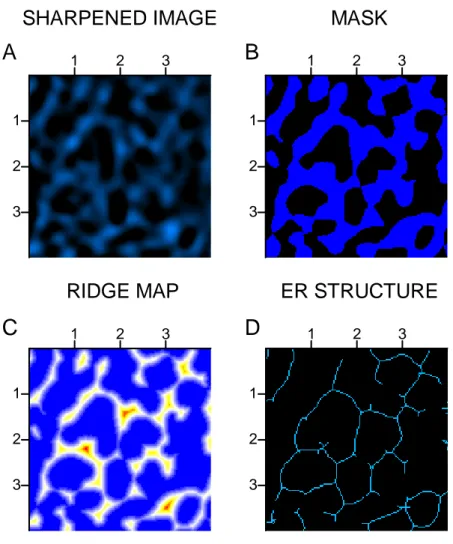
Figure 2.2 Processing stages for extracting the ER structure ... 41
Figure 2.3- Map of the distances from the ER ... 43
Figure 2.4 Classification of the skeleton in segments and branching points... 44
Figure 2.5- Schematic diagram of colocalization between two fluorophores. ... 45
Figure 3.1- Super resolution confocal images of cells transfected with wt (a, b) and I1061T (c, d) of NPC1 protein... 50
Figure 3.2- Fraction of NPC1 (wt and I1061T) proteins stored in vesicles. ... 51
Figure 3.3-Distribution of NPC1 (wt and I1061T) in the vesicles ... 52
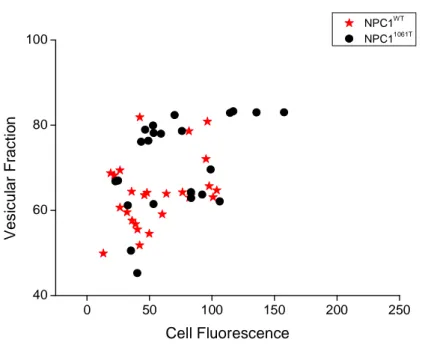
Figure 3.4- Vesicular fraction as function of cell fluorescence. ... 53
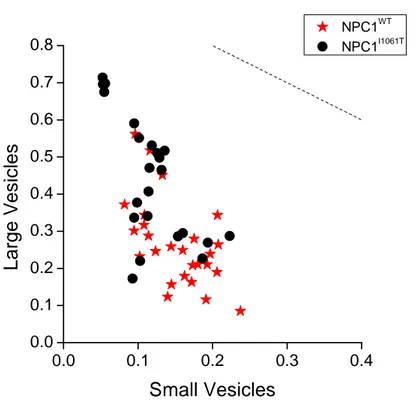
Figure 3.5- Distribution in small and large vesicles. ... 54
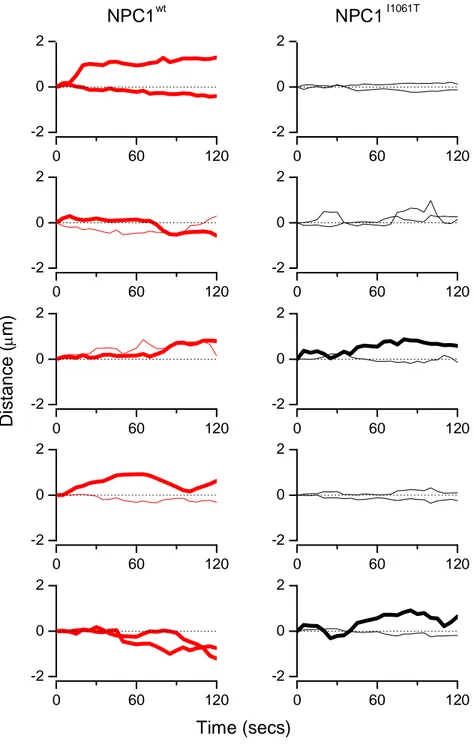
Figure 3.6- Dynamics of vesicles positive for NPC1wt and NPC1I1061T proteins ... 56
Figure 3.7 - Vesicle Trajectory along the main axis of drift is plotted as function of time ... 57
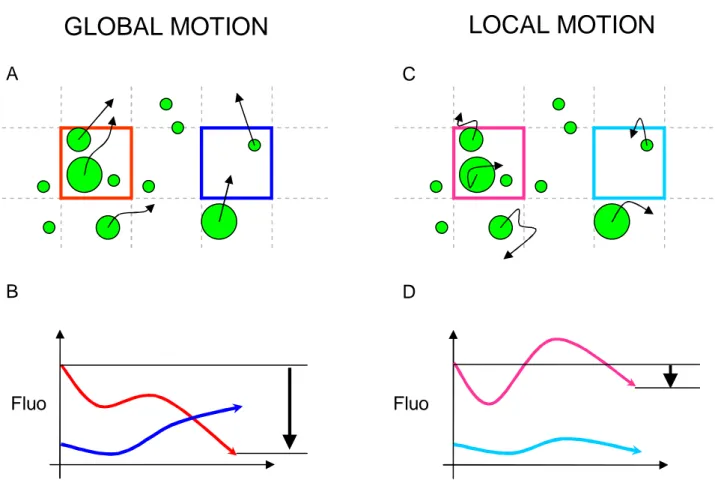
Figure 3.8- Temporal evolution of fluorescence in the case of large scale and small scale motion... 58
6
Figure 3.9-Fluorescence change is plotted as function of sampling interval ... 59
Figure 3.10- Superesolution representation of fluorescence change in the cytoplasm of two sample cells. ... 60
Figure 3.11-Confocal images of HN9.10e cells transfected with NPC1 (wt and mutant) and labeled with LysoTracker ... 62
Figure 3.12- Colocalitation analysis of NPC1 and endosomal compartments... 63
Figure 3.13- Distribution histograms for two population cells as function of late endosomes radius... 64
Figure 4.1- Colocalitation analysis in HN9.10e cotransfected with a fluorescent protein targeted to ER (blue) and NPC1 protein, wt or I1061T, (green). ... 67
Figure 4.2- Colocalitation analysis of NPC1 (wt and mutant) with ER. ... 68
Figure 4.3 -Confocal images of HN9.10e transfected with wt and I1061T protein. ... 69
Figure 4.4- Amount of OSERs in HN9.10e with wt and or I1061T mutation of NPC1 ... 70
Figure 4.5- Analysis of ER morphology... 71
Figure 4.6- Average Morphological parameters in cells cotransfected with Cerulean -ER (white) and the wild type protein (red) or the I1061T mutant form (black)... 72
Figure 4.7- Segment length plotted either as function of NPC1 protein on ER or as function of ER coverage ... 71
Figure 4.8- Confocal images of HN9.10e transfected with the ER-tagged cerulean protein for 48 h and treated with typical ER stressors... 71
Figure 4.9- Amount of OSERs in HN9.10e after treatments with two ER stressors... 75
Figure 4.10- DTT’s effect on ER Ca2+.... 77
Figure 4.11-Time course of DTT effect on HN9.10e cells. ... 78
7
Figure 5.1- High resolution confocal images of HN9.10e cells transfected with NPC1wt (top), and NPC1 I1061T (bottom) and labelled with TMRM a specific probe for mitochondria ... 81 Figure 5.2- Colocalitation analysis of NPC1 (wt and mutant) with ER and Mitochondria .... 82 Figure 5.3- Comparison of mitochondrial membrane potential between cells transfected with wt and I1061T protein and control cells ... 85 Figure 5.4-Measure of mitochondrial mass in HN9.10e cells transfected with wt and mutant forms of NPC1 protein ... 85 Figure 5.5-Measure of Ca2+ in mitochondria after expression of normal and mutant protein of NPC1 ... 87
8
ABSTRACT
Over 200 disease-causing mutations have been identified in the NPC1 gene. NPC1 is a 1278 amino acid protein with 13 transmembrane domains that is crucial for normal cholesterol homeostasis. The most prevalent mutation, NPC1I1061T, is predicted to lie within the cysteine-rich luminal domain and is associated with the classic juvenile-onset phenotype of Niemann-Pick type C disease.
To gain some insights on how loss of NPC1 function leads to neurodegeneration we have examined the global pattern of expression the protein in HN9.10e a neuronal cell line. In this research we studied first the distribution of the protein in the cell and motility and then we focused on the cell suborganelles seeking evidence for mislocalization of the protein as well as structural damage.
Our research shows that the NPC1I1061T protein fails to advance in the secretory pathway and remains trapped into the endoplasmic reticulum with consequent alteration in the reticulum structure and function. Further analyses reveal that the presence of missense mutation of NPC1 protein affects also mitochondria functions.
Understanding the behaviour and properties of organelles containing NPC1 will help to address the substantial mysteries of NPC1 function. This work could be useful to understand the basis of Niemann-Pick disease and devise prevention or treatment strategies.
9
CHAPTER I
Cholesterol trafficking and Niemann-Pick disease
Introduction
Niemann-Pick Type C (NPC) disease is associated with accumulation of cholesterol and other lipids in late endosomal/lysosomal compartment. Despite cholesterol accumulation occurs in several cell types, neurodegeneration represents the fatal cause for the disease. Genetic analysis has identified loss-of-function mutations in NPC1 and NPC2 genes as the molecular triggers for the disease. Although the precise function of these proteins has not yet been clarified, recent research suggests that they regulate cholesterol efflux from late endosomes/lysosomes. NPC protein deficits result in impairment in intracellular cholesterol trafficking and deregulations of cholesterol biosynthesis.
The first chapter will focus on the importance of cholesterol in the nervous system; in particular how it is distributed and transported through the neuronal cells.
The second part will take in consideration the Niemann Pick disease with particular focus to the consequences of NPC1 deficiency in the brain.
1.1 Cholesterol trafficking in cells
Cholesterol plays a crucial role in cell structure and function. It is an essential component of the cell membranes required for membrane lipid organization. The nervous system is the part of the body that contains the highest levels of cholesterol and normal brain function is guaranteed by trafficking of cholesterol both between nerve cells and between intracellular organelles. The intracellular organelles endosomes and lysosomes receive and distribute cholesterol through the endocytic and retrograde transport pathways. Deregulated cholesterol trafficking appears to be involved in the pathogenesis of Alzheimer’s disease(AD), Parkinson’s disease (PD) and Niemann-Pick disease type C (NPC) diseases.
10
Cholesterol distribution and function in the brain
Outside the brain, cholesterol requirement is covered either by de novo synthesis or by cellular uptake of dietary cholesterol under the form of lipoprotein cholesterol complexes. In the brain, however, the blood-brain barrier prevents the uptake of this lipid from the circulation. Thus, practically all the cholesterol present in this organ is provided by de novo synthesis with nerve cells themselves that have to synthesize all the cholesterol the brain needs (Dietschy, 2009).
Cholesterol removal from the brain
Cholesterol in the adult brain is considered metabolically inert; however, a small fraction of the pool (about 0.02% in humans and 0.4% in mouse) turns over each day (Dietschy and Turley, 2004). On the other hand, cholesterol synthesis in the developing CNS is relatively high (Dietschy et al., 1983). To maintain the steady state, a small amount of cholesterol has to be continuously exported from the brain into the circulation. At the moment, two different mechanisms for cholesterol elimination from the brain are known. The major mechanism by which cholesterol is metabolized in the brain is by conversion to 24(S)-hydroxycholesterol (Bjorkhem and Diczfalusy, 2004) that diffuses out from the cells, crosses the blood–brain barrier, and is cleared by the liver (Li-Hawkins et al., 2000;Bjorkhem et al., 2001). This reaction is catalyzed by a cytochrome P450 enzyme, Cholesterol 24-hydroxylase (CYP46), which is selectively expressed in the brain (Lund et al., 1999). This enzyme seems particularly crucial for the disposal of cholesterol in neuronal cells in particular in pyramidal neurons of the hippocampus and cortex, in Purkinje cells of the cerebellum, and in hippocampal and cerebellar interneurons (Lund et al., 1999; Ramirez et al., 2008). Interestingly, Bogdanovic et al. (2001) have also reported CYP46 immunoreactivity in glial cells from brain of AD patients. The other mechanism for cholesterol elimination, called reverse cholesterol transport pathway, involves the cholesterol transport across the plasma membrane to apolipoprotein acceptors in peripheral tissues (Jessup et al., 2006; Kim et al., 2007) by ATP binding cassette (ABC) transporters.
At neuronal level, evidence suggests that a major role is played by members of the ABC-A sub-family (Pohl et al., 2005; Kim et al., 2006) and members of the ABCG sub-family (Nakamura et al., 2004; Tachikawa et al., 2005). In particular evidence suggests that ABC-A1 and ABC-G1 mediate the cholesterol efflux from astrocytes and microglia (Xu et al.,
11 2000;Wahrle et al., 2004). All the above supports the current concept that the synthesis and elimination of cholesterol in the adult brain is compartmentalized: astrocytes are believed to be responsible for the majority of synthesis in the adult brain while they contribute relatively little to brain elimination.
On the other hand, the neuron specific cholesterol 24-hydroxylase, CYP46A1, is responsible for the elimination of about two thirds of the cholesterol synthesised by the brain (Bjorkhem et al., 2001; Xie et al., 2003). Indeed, knockout (KO) mice lacking CYP46 have a ≈50% reduction in brain cholesterol excretion (Lund et al., 2003).This decrease is compensated by the reduction in de novo synthesis, resulting in steady state levels of cholesterol in the brains of KO mice that are similar to those of wild type (WT) mice. This reduced synthesis is likely to be mediated by a decrease in the activity of HMG CoA reductase.
Intracellular cholesterol transport
Regardless of whether cells acquire cholesterol by synthesis or uptake, the molecule must be distributed to the different cellular compartments. So far, little is known about these processes, even in non neuronal cells (Schmitz and Orso, 2001). One of the trafficking routes of cholesterol is the endocytic transport of LDL particles to lysosomes (see fig 1.1). Endosomes constitute a membrane transport pathway starting at the plasma membrane and ending in lysosomes, where cargo is degraded (Saftig and Klumperman, 2009). Endocytic cargo is first transported to early/sorting endosomes, then further into late endosomes and finally to lysosomes. In this endosomal progression the pH of the organelles gradually decreases and the outer endosomal membrane buds inwards forming internal membranes (Saftig and Klumperman, 2009). LDL particle bound to the LDL receptor at the plasma membrane is transported along this pathway to endolysosomes where the LDL-derived cholesteryl esters are hydrolyzed, followed by efflux of cholesterol to other cellular membranes (Ikonen and Jansen, 2008). The transport of cholesterol out of endolysosomes appears to be slow (Schoer et al., 2000) and to some extents depend on membrane trafficking (Holtta-Vuori et al., 2000). In contrast, the transport of cholesterol from the ER to the plasma membrane and from the plasma membrane to ER and lipid droplets has been shown to be mostly independent of vesicular trafficking (Urbani and Simoni, 1990;Jansen et al., 2010). Many disorders of endosomal function such as Niemann-Pick diseases, Gaucher disease (Vaccaro et al., 2010) and Mucolipidosis type IV (Soyombo et al., 2006) lead to accumulation of free cholesterol in endosomes. Reasons why sterol transport in/from endolysosomes is so
12 different from other cellular membranes may include the highly glycosylated limiting membrane of lysosomes. This rigid membrane functions to protect the lysosomal outer membrane from degradation, isolating the outer membrane from the lysosomal lumen and may play a role also in cholesterol mobility.
The multivesicular late endosomes harbour two proteins, NPC1 and NPC2, which appear to be crucial for moving cholesterol out of the endosomal system. Deficiency of either protein leads to the accumulation of LDL-derived unesterified cholesterol in late endocytic organelles (Sturley et al., 2004). Genetic and phenotypic evidence in mutant mice suggest that the NPC proteins participate in different steps of the same pathway and that neither can compensate for the other (Sleat et al., 2004). Both proteins can bind sterol (Ohgami et al., 2004; Xu et al., 2007) but recent evidence suggests that they may also be implicated in sphingolipid binding or mobilization (Koivusalo et al., 2007; Schrantz et al., 2007). Thus, the question about the primary functions of the NPC proteins — whether it is cholesterol or sphingolipid mobilization, or something else — still awaits resolution. On release from the endolysosomal system, cholesterol is delivered to other membranes, such as the plasma membrane, ER, recycling endosomes and mitochondria. Two proteins in late endosomes have been identified with sterol-binding domains that are exposed to the cytosol. These proteins, MLN64 and ORP1, bind cholesterol and 25-hydroxycholesterol, respectively (Tsujishita and Hurley, 2000) (Suchanek et al., 2007). Another site in the endocytic pathway for active sterol exchange is the recycling compartment. Recycling endosomes are sterol- and sphingolipid-enriched (Gagescu et al., 2000), and can serve as acceptors for non-vesicular sterol flux through the cytoplasm. Vesicular trafficking, in particular membrane transport regulated by the Rab11 GTPase, has been implicated in recycling membrane cholesterol to the plasma membrane (Holtta-Vuori et al., 2002). LDL does not intersect with the endosomal recycling circuits en route to late endosomes (Holtta-Vuori et al., 2002).
13 Figure 1.1- Intracellular cholesterol transport. Cholesterol is salvaged through the low-density
lipoprotein receptor (LDLR) pathway at the plasma membrane. After endocytosis of the LDL particles in clathrin-coated pits, cholesterol is released into the endosomal–lysosomal system. The products of three genes in this system —Niemann–Pick C 1 (NPC1), NPC2 and MLN64 — are thought to mediate cholesterol exit from the endosomal system and it is eventual transport to the plasma membrane. Caveolins are thought to facilitate cholesterol movement to the plasma membrane where an ABC-type plasma membrane transporter (ABCA1) can facilitate its efflux onto high-density lipoprotein (HDL) particles. Excess cholesterol can be transported from the plasma membrane to the endoplasmic reticulum (ER) where acyl-coenzyme A:cholesterol acetyltransferase (ACAT) facilitates esterification of cholesterol for storage in lipid droplets in the cytosol. (ABCA1, ATP-binding cassette transporter; MLN64, malignancy antigen 64; TGN, trans-Golgi network.) (Ioannou, 2001).
14
1.2 Niemann Pick type C disease
Clinical description
The clinical presentation of NPC is extremely heterogeneous, with an age of onset ranging from the perinatal period until well into adult age.
Similarly, the lifespan of the patients varies between a few days until over 60 years of age, although a majority of cases die between 10 and 25 years of age (Vanier and Millat, 2003;Trendelenburg et al., 2006; Wraith et al., 2009). NPC is classically a neurovisceral condition. Importantly, visceral involvement (of liver, spleen, and sometimes lung) and neurologic or psychiatric manifestations arise at different times, and follow completely independent courses. Apart from a small subset of patients who die at birth or in the first 6 months of life from hepatic or respiratory failure, and some exceptional adult cases, all patients ultimately will develop a progressive and fatal neurological disease. Systemic symptoms, when present, always precedes onset of neurological symptoms, however the systemic component may be absent or minimal in approximately 15% of all patients, and in the case of adult-onset patients the percentage of them who does not present systemic symptoms is even higher, about 50%. In typical patients, the neurologic disorder consists mainly of cerebellar ataxia, dysarthria, dysphagia, and progressive dementia, and the majority of cases show characteristic vertical supranuclear gaze palsy (VSGP) (Kandt et al., 1982). Cataplexy, seizures, and dystonia are other quite common features, and psychiatric disturbances are frequent in late-onset patients. The proper recognition of VSGP is essential but this sign is often overlooked at an early stage, because slow pursuit in some cases if maintained whilst other oculomotor parameters such as saccadic peak velocity are already impaired. Cataplexy (with or without narcolepsy), usually laughter-induced, is another more specific symptom (Kandt et al., 1982; Oyama et al., 2006). Except for the perinatal period, the systemic disease is usually not very severe and is well tolerated. The splenomegaly has been described to fluctuate and to decrease with time. Severe lung involvement has been reported in a few patients but is not frequent.
15 Figure 1.2- Age of onset of neurological disease vs. lifespan. Data from a metaanalysis by Vanier
(2010). Each horizontal bar depicts one patient. Cases are sorted by the age of onset of Neurological symptoms (green lines). The subsequent part of the segment shows remaining lifetime (blue if patient was alive, red otherwise). Data are from 97 cases showing neurological symptoms out of a pool of 181 patients.
A description of the various clinical forms by age categories has been used in recent reviews (Vanier and Millat, 2003;Wraith et al., 2009). Of essential importance is to note that the age of onset of the systemic symptoms is not related with that of the neurological disease (the latter can occur many years or even decades later), while there is a correlation between the age of onset of the neurological symptoms and the general further course of the disease and lifespan (fig. 1.2) (Vanier, 2010). With an exception for the severe early infantile neurological form which is quite significantly distinct, recent large studies have however demonstrated an overlap between the neurological forms, and thus a continuum (Wraith et al., 2009). A schematic representation is illustrated in figure 1.3.
16 Figure 1.3 -Niemann Pick disease symptoms as function of age. Schematic representation of the
main forms of the disease, dividing between systemic and neurological symptoms showing the age range at which the symptoms appear (Vanier, 2010).
Early forms of Niemann-Pick disease
Perinatal presentationNiemann-Pick C disease is now recognized as a relatively common cause of liver disease in early life. Fetal hydrops or fetal ascites can be observed (Spiegel et al., 2009). Above all, a prolonged neonatal cholestatic icterus, appearing in the first days or weeks of life and usually associated with progressive hepatosplenomegaly is present in close to half of patients, although with very variable intensity (Vanier et al., 1988; Kelly et al., 1993; Yerushalmi et al., 2002). In most cases, the icterus resolves spontaneously by 2 to 4 months of age, and only hepatosplenomegaly remains for a highly variable period, preceding onset of neurologic symptoms (see below). In about 10% of these patients, however, the icterus quickly worsens and leads to liver failure. Children with this dramatic "acute" neonatal cholestatic rapidly fatal form usually die before the age of 6 months (Vanier et al., 1988) .Some other infants, especially (but not exclusively) those having mutations in the NPC2 gene, present with a severe respiratory insufficiency (together with hepatosplenomegaly or more severe liver disease) that may also be fatal. Patients with NPC do not show neurological manifestations
17 during the neonatal period (important for differential diagnosis). But there are many examples of patients dying from a severe perinatal form having siblings with a neurologic infantile or juvenile onset form (Vanier et al., 1988;Vanier and Millat, 2003).
Early infantile onset (2 months – 2 years)
Between 2 months and 2 years of age isolated hepatosplenomegaly can appear but it can well stay isolated for many years, in spite of the early onset. In these infants, hepatosplenomegaly has almost invariably been present since birth or the first months of life. Delay of developmental motor milestones from the age of 8-9 months and central hypotonia constitute the first neurologic symptoms, which become evident between the age of 1 and 2 years. Subsequent clinical course includes a loss of acquired motor skills, proportionally less marked mental regression, followed by pronounced spasticity with pyramidal tract involvement. Many of these children never learn to walk. Intention tremor is frequently present; supranuclear gaze palsy is usually not recognized. Seizures are uncommon. Brain imaging (MRI and MRS) shows signs of leukodystrophy and cerebral atrophy. Survival rarely exceeds 5 years. This form seems to be more frequent in Southern Europe (where it constitutes >20% of the cases) and the Middle East (Vanier et al., 1988; Vanier and Millat, 2003; Imrie et al., 2007).
Late infantile period (2-6 years)
Many patients start their disease by discovery of an isolated hepatosplenomegaly or splenomegaly during this period. However hepatosplenomegaly in these patients often has been present for a varying length of time. On the neurological side, language delay is frequent. The child often presents with gait problems, frequent falls and clumsiness between 3 and 5 years of age, due to ataxia. VSGP is usually present but may not be recognized at an early stage. Hearing loss has also been described. Cataplexy develops relatively frequently and may occasionally be the presenting symptom. The motor problems worsen, and impairment in mental development becomes more obvious. A significant proportion of patients develop seizures which may be partial, generalized, or both. They generally respond to standard treatment but refractory cases may occur, with some patients dying from status epilepticus or complications of seizures. Severe epilepsy has a bad prognosis and significantly shortens the lifespan of the patients. As ataxia progresses, dysphagia, dysarthria, and dementia develop. At later stages, the patients develop pyramidal signs and spasticity, and pronounced swallowing problems. Death most often occurs between 7 to 12 years in this form.
18
Juvenile period (6-15 years) (classical form)
Juvenile (or classical) form of Niemann-Pick disease appears between 6 and 15 years of age. On the systemic side diagnosis sometimes is made on the basis of an isolated splenomegaly (or, rarely, of a hepatosplenomegaly). Despite diagnosis through the systemic symptoms is often crucial, in most countries the most common form of the disease is its neurological form. A moderate splenomegaly (or hepatosplenomegaly) is frequent, and may have been detected at any earlier time, including the neonatal period. However, cases in which a splenomegaly had been noted in early childhood but is hardly detectable at the time first neurological symptoms arise are not rare; and absence of a detectable organomegaly has been reported to occur in at least 10% of cases. School problems with difficulties in writing and impaired attention are very common and may lead to misdiagnosis. The disease may also mimic dyspraxia. VSGP is almost invariably present and often the initial sign. The child becomes clumsier and shows more learning disabilities. Cataplexy, with or without narcolepsy, typically laughter-induced, is another common symptom. Ataxia soon becomes obvious, with frequent falls and difficulties to run, and progresses at a variable rate. Dysarthria develops, as well as dysphagia. Action dystonia is also frequent, Motor impairment is major and intellectual decline may be variable. About half of the patients with the classic form develop seizures of variable type and severity (see above). At a later stage, dysarthria worsens and patients often stop talking. At a late stage, patients develop pyramidal signs and spasticity, and pronounced swallowing problems, requiring gastrostomy. The lifespan is quite variable, some patients being still alive by age 30 or later (Wraith et al., 2009).
Late onset forms (>15 years)
Even though Niemann-Pick with systemic symptoms is generally an early onset disease, the finding of three patients aged 53-63 years with isolated splenomegaly and a biochemical and molecular diagnosis of NP-C (Fensom et al., 1999;Dvorakova et al., 2006) suggests the existence of a rare non-neuronopathic form of the disease (possibly corresponding to the ill-described historical "type E"). Nevertheless, apart from these exceptional cases and from infants with early death, as stated above, all NP-C patients develop neurologic symptoms. More patients with a neurologic adult onset form of the disease (often in the second or third decade, but as late as 50 years or older) have been described in recent years (Trendelenburg et al., 2006; Sevin et al., 2007; Shulman et al., 1995; Klarner et al., 2007) .This diagnosis is
19 probably underestimated. Absence of clinically detectable splenomegaly has been reported in a significant proportion of patients but abdominal sonography may reveal a slightly enlarged spleen. VSGP is usually present but may also be missing. The most common symptomatology is that of an attenuated juvenile form with an insidious onset, with in at least one third of cases, a psychiatric presentation that may be isolated for several years before the onset of motor and cognitive signs. Psychiatric signs are most often consistent with psychosis, including paranoid delusions, auditory or visual hallucinations, and interpretative thoughts. Onset may be acute or progressive, eventually with relapses. At this stage the neurologic examination may be normal. Other types of psychiatric disturbances are depressive syndrome, behavioral problems with aggressiveness, or social isolation. Cases have also been reported with bipolar disorders, obsessive-compulsive disorders, or transient visual hallucinations. From compilation of the literature (Sevin et al., 2007) the most common features are: cerebellar ataxia (76%), vertical supranuclear ophthalmoplegia (75%), dysarthria (63%), cognitive troubles (61%), movement disorders (58%), splenomegaly (54%), psychiatric disorders (45%) and dysphagia (37%). Movement disorders (dystonia, Parkinsonism, chorea) are more frequent than in the juvenile form. Some patients show severe ataxia, dystonia, and dysarthria with variable cognitive dysfunction, whereas psychiatric symptoms and dementia dominate in others. Epilepsy is rare in adult onset NPC (15%). Later course is similar to that in the juvenile form.
1.3 NPC1 protein
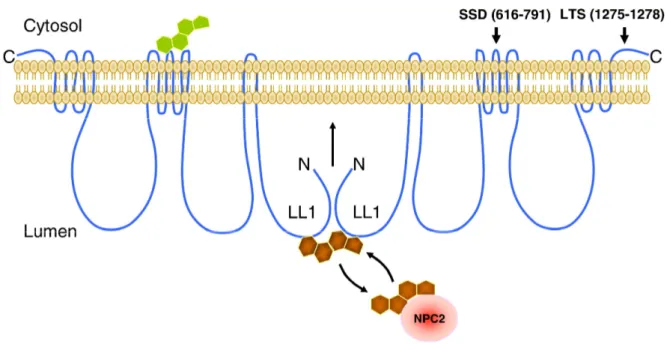
The majority (95%) of NPC cases arise from the mutation and the remainder by mutation of the protein NPC2 (Carstea et al., 1993; Carstea et al., 1997; Naureckiene et al., 2000). Mutations of NPC1 in patients result in cholesterol aggregation in lysosomes. NPC1 is a large transmembrane protein (1278 amino acid residues) localized to the late endosomal membrane where it transiently interacts with lysosomes and the Golgi apparatus to mediate cholesterol trafficking (Higgins et al., 1999; Cruz et al., 2000; Wiegand et al., 2003). The protein possesses 13 hydrophobic helices possibly spanning the membrane with three loops (each of ~240 amino acids) between the transmembrane segments (Davies and Ioannou, 2000). NPC1 possesses a sterol-sensing domain (SSD, 616–791) on the cytoplasmic side between the two large loops in the lysosomal lumen SSD shares homology with the SSD of SCAP (SREBP cleavage-activating protein) and HMG-CoA (3-hydroxy-3-methyl-glutaryl
20 coenzyme A) that are involved in cholesterol homeostasis (Davies and Ioannou, 2000). Although the mechanism by which NPC1 regulates cholesterol remains unknown, recent studies suggest that NPC1 binds cholesterol directly in vitro (Infante et al., 2008a; Infante et al., 2008b). Purified N-terminal domain (NTD, amino acids 25–264) of NPC1 binds cholesterol. Binding shows increased affinity if cholesterol is oxidized at the hydroxyl group of the 24, 25, or 27 positions. The findings that 25-hydroxyl cholesterol competitively inhibits cholesterol binding to NPC1 suggest that cholesterol and oxysterols regulate each other's binding to NPC1 for transport. Moreover, the recombinant protein of the large loop-1 (LL1) of 240 amino acid residues of NPC1 forms a homodimer that interacts with a single molecule of cholesterol. It is then tempting to speculate that the two monomers of LL1 operate cooperatively as gateway to regulate cholesterol access to NPC1 for transport. NPC1 homodimer might form a cholesterol transport channel with the luminal loops regulating cholesterol transport from the lysosomal lumen whereby the two LL1 domains might function as entry gate recruiting cholesterol and facilitating cholesterol transport across the flycocalyx of the lysosome (figure 1.4).
Figure 1.4- Hypothetical model of NPC1 dimerization to form a transmembrane channel operated by and for sterols. NPC1 in lysosomal membrane might recruit and gate cholesterol for
transmembrane transport via intra-molecular interaction between the luminal loop-1(LL1) or N-terminal domains (NTDs). Oxysterol (green) interacts with NPC1 sterol sensing domain (SSD) to
21
regulate the dimerization and thereby the close (or open) of the LL1 domains between the two NPC1 molecules. Cholesterol bound to and release from NPC1 is facilitated by soluble cholesterol binding protein NPC2 (colored red).
In support of NPC1 transport of cholesterol include the findings from both genetic analysis and biochemical data. In vitro analysis of [3H]-cholesterol interaction between NPC1, NPC2 and phosphatidylcholine liposomes shows that NPC2 rapidly donates or accepts cholesterol from liposomes, whereas the NTD of NPC1 acts slowly (Infante et al., 2008c). A bidirectional transfer of cholesterol between NPC1 NTD and liposomes is accelerated N100-fold by NPC2. Transfer of cholesterol to NPC1 requires NPC2 binding to cholesterol, as a naturally occurring mutant of NPC2 (Pro120Ser) that does not bind cholesterol fails to stimulate cholesterol transfer from NPC1 NTD to liposomes (figure 1.5). The observations that both NPC1 and NPC2 interact with cholesterol suggest a common pathway by which both proteins cooperate in the cholesterol egress process from lysosomes and that mutation in either protein produces a similar cellular phenotype of cholesterol segregation in the lysosomal compartment.
Figure 1.5- Proposed pathway for transfer of cholesterol from LDL or β-VLDL to NPC2 to NPC1 to membranes. This model suggest that NPC2 delivers LDL-C to the N-terminal domain of
22
1.4. Disease causing mutations and genotype-phenotype relationships
The Niemann-Pick type C disease variation database (Runz et al., 2008) listed by January 2010 reports 244 NPC1 and 18 NPC2 gene sequence variants. The current number for identified NPC1 disease-causing mutations is most likely close to 300. More than 60 polymorphisms of NPC1 have also been described, some of them very common. In early genetic complementation studies, it was stated that about 95% of the families had mutations in the NPC1 gene (Vanier et al., 1996). In France, among the 132 families genotyped so far, 9 had NPC2 mutations. To date, only c:a 30 families have been identified worldwide with mutations in the NPC2 gene.The NPC1 gene, mapped to chromosome 18q11-q12, spans 56 kb and contains 25 exons. One mutant allele, p.I1061T, is particularly frequent (Millat et al., 1999) (approximately 20-25% of alleles in patients diagnosed in France or the United Kingdom). This mutation is also highly prevalent in patients from a Spanish-American isolate from the upper Rio Grande valley, but much less frequent in Portugal, Spain or Italy (Ribeiro et al., 2001; Fernandez-Valero et al., 2005; Fancello et al., 2009). In the homoallelic state, it is associated with prominent cellular cholesterol trafficking abnormalities in fibroblasts of patients, and it correlates with a juvenile neurologic onset form of the disease (Millat et al., 1999). In the heteroallelic state, so far it has never been found associated with the most severe infantile neurologic onset form. The I1061T mutant was shown to be a functional protein selected for endoplasmic reticulum-associated degradation due to protein misfolding and thus a potential target for chaperone therapy (Gelsthorpe et al., 2008).The second most recurrent NPC1 mutation in Europe, p.P1007A, is the prototype of a "biochemical variant" mutation (Greer et al., 1999; Millat et al., 2001a; Millat et al., 2005) .In the homozygous state, it has been described in a family with two adult onset siblings (Ribeiro et al., 2001).A number of other recurrent NPC1 mutations seem to be associated with adult neurological onset of the disease (Millat et al., 2005; Imrie et al., 2007) . The mutation p.G992W, typical of Nova-Scotian patients (Greer et al., 1998) is sporadically (but rarely) found in patients of other origin.
The few genotype-phenotype studies published so far in NPC1 patients generally showed good correlation between nonsense or frame shift mutations and the most severe neurologic course. Missense mutations have emphasized the functional significance of two particular domains of the NPC1 protein. Homozygous mutations in the sterol-sensing domain were found to be very deleterious, corresponding to a lack of mature NPC1 protein and to a very
23 severe disease phenotype, biochemically and clinically (Millat et al., 2001b) The cysteine-rich luminal loop contains approximately one third of all described mutations, with a variable cellular and clinical impact (Greer et al., 1998;Millat et al., 2001b;Park et al., 2003;Millat et al., 2005). Interestingly, mutations leading to a less severe impairment of cellular trafficking ("variant" phenotype) are typically located on this loop.
The NPC2 gene (initially known as HE1), mapped to chromosome 14q24.3, spans 13.5 Kb and contains 5 exons (Naureckiene et al., 2000). One nonsense mutation (E20X) appears relatively frequent (Millat et al., 2001a;Verot et al., 2007), and many other mutations also lead to a truncated protein. They have so far been associated with very severe clinical phenotypes. Described missense mutations have corresponded to more varied phenotypes, including juvenile and adult onset patients (Millat et al., 2001a; Chikh et al., 2005; Verot et al., 2007) Finally, for both NPC1 and NPC2, the study of a large number of multiplex families has clearly shown that mutations correlate with the neurological form of the disease, but not with the systemic manifestations (Vanier and Millat, 2003).
1.5 Consequences of NPC1-deficiency in the brain
Most studies on NPC1-deficient cells have utilized human fibroblasts or CHO cell mutants. Studies on primary neurons from Npc1−/−mice are technically challenging and only a few groups have investigated NPC1 functions in these cells. In one of the first such studies, Henderson et al. (Henderson et al., 2000) reported that striatal neurons isolated from NPC1-deficient mice had reduced axon length and a reduced number of branch points, as well as impaired responsiveness to brain-derived neurotrophic factor (BDNF); BDNF failed to elicit normal autophosphorylation of its receptor, TrkB (Henderson et al., 2000). Since extraction of cholesterol from Npc1+/+ neurons also reduced the phosphorylation of TrkB upon BDNF binding, it was proposed that NPC1-deficiency reduced the cholesterol-rich domains of the plasma membrane where the TrkB receptor resides (Henderson et al., 2000). In contrast, growth and morphology of NPC1-deficient mouse sympathetic neurons, as well as TrkA activation by nerve growth factor (Karten et al., 2002), are normal.
Functional defects are less obvious in the peripheral nervous system than in the CNS. However, the ability of NPC1-deficient mice to reutilize cholesterol liberated from myelin during peripheral nerve injury is impaired, leading to reduced growth and myelination of the regenerating axons (Goodrum and Pentchev, 1997).
24 Some symptoms of NPC disease, such as the occurrence of seizures, suggest that synaptic transmission is altered. In NPC1-deficient mice, brain area-specific changes in levels of neurotransmitters were observed. For example, GABA and glutamate were decreased in the cerebral cortex, whereas the cerebellum showed an increased level of glycine but no alteration in GABA or glutamate (Yadid et al., 1998; Byun et al., 2006). This reciprocal change in GABAergic and glutamatergic systems indicates that the balance of inhibitory (GABA) and excitatory (glutamate) synaptic transmission is disturbed which might contribute to some of the observed neurological deficiencies (Phillips et al., 2008).
The NPC1 protein is present in axons of sympathetic neurons and in isolated synaptosomes from mouse brains suggesting that NPC1 has a function in axons (Karten et al., 2006). In NPC1-deficient synaptosomes some enlarged vesicular structures, either aberrant synaptic vesicles or endosomal structures, were observed. In addition, the protein composition of synaptic vesicles isolated from NPC1-deficient synaptosomes was altered (Karten et al., 2006) Several electrophysiological parameters (resting membrane potential, neuronal input resistance and action potential amplitude) were measured in isolated cortical neurons from embryonic mice, and in cortical brain slices from older mice, but no significant differences were detected between Npc1+/+ and Npc1−/− neurons in synaptic activity or growth morphology (Deisz et al., 2005;Wasser et al., 2007).
Most neurodegenerative diseases are characterized by impaired mitochondrial function, which in turn can contribute to neuronal dysfunction and death. Neurons have a particularly high energy requirement for maintenance of membrane potential and seem to be highly dependent on mitochondrial oxidative phosphorylation. Mitochondria isolated from NPC1-deficient brains had a lower rate of ATP generation than did mitochondria from wild-type brains (Yu et al., 2005) .Moreover, ATP levels in primary cortical neurons isolated from NPC1-deficient mice were lower than in control neurons, as were neurite length and number; the defects were normalized by addition of exogenous ATP (Yu et al., 2005). These changes were ascribed to a possible build-up of cholesterol in mitochondrial outer and inner membranes.
Neuron death and pathological changes in NPC1-deficient brains
In the brain, the consequence of NPC1-deficiency is the progressive loss of Purkinje cells in the cerebellum, which correlates with gait ataxia, dysarthria and dysphagia. In the BALB/c mouse model, which closely resembles late infantile human NPC disease , Purkinje cell death begins around 3 weeks of age and follows a consistent pattern wherein zebrin-II negative25 Purkinje cells are lost first, causing a “striped” pattern of neurodegeneration (Sarna et al., 2003). Neuronal loss also occurs in the substantia nigra of the midbrain, the pons, certain areas of the brainstem and, to a lesser extent, in the thalamus and pre-frontal cortex (German et al., 2001a; Ong et al., 2001 ;Li et al., 2005; Luan et al., 2008). The reason for the selective death of defined populations of neurons in NPC1-deficient brains is not clear. However, changes in activities and distribution of lysosomal enzyme are more pronounced in areas of the brain that undergo extensive neuron loss (cerebellum) than in relatively protected areas (hippocampus) .
The precise mode of neuronal death in NPC1-deficiency is not yet clear. The widespread occurrence of apoptosis in NPC1-deficient brains is indicated by an increased number of TUNEL-positive nuclei (Wu et al., 2005), increases in caspase 1 and 3 mRNA (Wu et al., 2005)and marked increases in tumor necrosis factor-α (TNFα) and its downstream signaling molecules caspase 8, FADD, TNFRp55, TRADD, and RIP (Li et al., 2005;Wu et al., 2005). Consistent with these observations, TNFα signaling was identified as a key pathway leading to apoptosis of hepatocytes from mice in which NPC1 deficiency was induced in the liver (Rimkunas et al., 2009). It is likely that several pro-apoptotic factors contribute to neuronal death in NPC1-deficiency, including lysosomal dysfunction, increased oxidative stress, ER stress and/or energy deprivation.
Neuronal death in NPC disease might also involve autophagy as well as apoptosis. Autophagy is the mechanism by which long-lived proteins and organelles are degraded by lysosomes. Cytoplasmic constituents are enclosed by a limiting membrane thereby forming autophagosomes that subsequently fuse with lysosomes, allowing the cargo to be digested by lysosomal enzymes (Abeliovich et al., 2000; Klionsky and Emr, 2000; Chu, 2006). A basal level of autophagy is crucial for neuron viability (Kuma et al., 2004; Hara et al., 2006; Komatsu et al., 2006).
Autophagy has been observed in a range of neurodegenerative disorders including Alzheimer disease, Parkinson disease and Huntington disease. Electron microscopic examination of degenerating Purkinje cells in NPC1-null mice showed increased numbers of autophagic vacuoles (Ko et al., 2005; Liao et al., 2007; Pacheco et al., 2007).The autophagy marker, microtubule-associated protein 1 light chain 3-II (LC3-II) (Kabeya et al., 2000), is increased in cerebellar and liver isolates from NPC1-null mice, as well as in fibroblasts from NPC patients (Cheng et al., 2006; Komatsu et al., 2007; Liao et al., 2007; Pacheco et al., 2007).
26 The late endosomal/lysosomal sequestration of cholesterol and GSLs, including glucosylceramide, lactosylceramide and complex gangliosides (mainly GM2 and GM3), in NPC1-deficient brains (Elleder et al., 1984; Vanier, 1999; Sugimoto et al., 2001; Taniguchi et al., 2001; Zervas et al., 2001) is thought to be responsible not only for neuron death and axonal degeneration but also for some of the profound morphological changes that occur in human, feline and murine NPC1-deficient brains (reviewed in (Walkley and Suzuki, 2004)). Swollen neuronal cell bodies, containing lamellar and multi-vesicular structures, have been detected throughout the CNS in NPC1-deficient mice and cats (Yamada et al., 2001;Walkley and Suzuki, 2004). Moreover, abnormal lipid storage in NPC1-deficient brains is thought to induce the formation of “meganeurites” which are enlargements of axonal hillocks in neurons (Walkley and Suzuki, 2004) and are probably caused by the aberrant movement of lipid-filled storage vesicles from cell bodies into proximal axons (Zervas et al., 2001). Similarly, in cultured NPC1-deficient neurons LAMP1-immunoreactive lysosomal structures, which normally are largely restricted to cell bodies, were detected in proximal neurites. The presence of these large storage vesicles in proximal axons might create a “roadblock” that would impair axonal trafficking.
Although much insight has been provided in the last few years into the links between impaired lipid homeostasis and morphological/functional changes in the brain in NPC disease, the mechanistic pathways that connect the loss of NPC1 function to neuronal dysfunction and the symptoms of NPC disease remain to be elucidated.
27 Figure 1.6-Cholesterol accumulation in NPC1-deficient neurons. Hippocampal neurons isolated
from embryonic wild-type (Npc1+/+) and NPC1-deficient (Npc1−/−) mice were cultured for 20 days in serum-free medium, then fixed and stained with filipin (blue, cholesterol) and rat anti-LAMP1 antibodies followed by Texas Red-conjugated anti-rat IgG (red, LE/L) (Karten et al., 2006).
Axonal transport and degeneration
In addition to classical apoptosis, where cell death is initiated in the cell body, axonal degeneration that can lead to neuronal dysfunction and death is also observed in NPC disease (Walkley and Suzuki, 2004). Degenerating axonal termini have been observed histochemically in many areas of NPC1-deficient brains, even in areas that do not show prominent cell death, such as the hippocampus (Patel et al., 1999). Widespread formation of axonal spheroids in NPC1-deficient brains correlates with neuroaxonal dystrophy and degeneration of axons (March et al., 1997; Zervas et al., 2001; Bu et al., 2002; Ohara et al., 2004; Walkley and Suzuki, 2004).
In addition to axonal spheroids, neurofibrillary tangles (NFT) that are ultrastructurally identical to those seen in Alzheimer disease, were observed in human post-mortem brains from NPC patients as young as 10 years of age (Auer et al., 1995;Love et al., 1995;Suzuki et al., 1995;Distl et al., 2003); NPC1 disease is the only known condition in which tangle formation occurs at such an early age. The presence of NFT has been linked to impaired axonal transport, and often follows axonal spheroid formation (Leroy et al., 2007). The exact mechanism of tangle formation and the role of lipid/cholesterol homeostasis in this process are not clear although several observations indicate that defective cholesterol homeostasis is
28 involved in NFT formation. First, neurons that have high cholesterol content are more likely to contain NFT than are neurons with little cholesterol storage (Distl et al., 2003). Second, cholesterol deficiency in cultured murine neurons leads to hyperphosphorylation of tau and microtubule degeneration (Fan et al., 2002;Meske et al., 2003). Although NPC1-deficient mice do not form NFT (German et al., 2001b), these mice show a tau phosphorylation pattern that is characteristic of the NFT that are seen in human Alzheimer disease (Sawamura et al., 2001; Bu et al., 2002; Treiber-Held et al., 2003). It should be noted that the lack of tangle formation by hyperphosphorylated tau is common to most rodents and has been ascribed to species differences in tau (Dodart et al., 2002).
1.6 Therapies for NPC disease
No treatments are currently available for NPC disease other than those used to alleviate the symptoms caused by disease progression. Recently, miglustat was approved in Europe for the treatment of adult and paediatric patients with NPC disease (Cox et al., 2000). Administration of miglustat to animal models of NPC disease, including mice and cats, reduced ganglioside accumulation in the brain, delayed neurological symptoms, and increased life-span (Zervas et al., 2001). Data from initial clinical trials suggest that long-term treatment of NPC patients with miglustat is well tolerated in adult and juvenile patients, and can stabilize some of the neurological symptoms (Patterson et al.; Pineda et al., 2009)
Another therapeutic strategy that was proposed for NPC disease was the administration of neurosteroids(Griffin et al., 2004). Cholesterol is a precursor of the neurosteroids (i.e. steroids that are synthesized in the brain) that are important for neuron growth and development. In brains of NPC1-deficient mice, levels of pregnenolone and the neurosteroid allopregnanolone (ALLO), as well as the activities of the enzymes involved in ALLO synthesis, are lower than in wild-type mice . Remarkably, a single injection of ALLO in 7-day-old NPC1-deficient mice delayed the onset of neurological symptoms and increased life-span. Purkinje cell survival was also increased, while levels of GM1 and GM2 gangliosides were reduced (Griffin et al., 2004).
Recently, the reported beneficial effects of ALLO have come under intense scrutiny due to effects caused by the vehicle, cyclodextrin (CD) that was used in the experiments with ALLO described above. CD is a cyclic oligosaccharide capable of sequestering lipids, especially cholesterol, within its hydrophobic core (Kilsdonk et al., 1995).Interestingly, recent work of Liu et al. found that treatment of NPC1-deficient mice with CD delayed the
29 neurodegeneration and prolonged the life of the mice independent of ALLO administration (Liu et al., 2008). A single dose of CD given to 7-day-old NPC1-deficient mice reduced the accumulation of unesterified cholesterol, increased the amount of CE, and decreased cholesterol synthesis in the liver, spleen and, to a lesser extent, the brain(Liu et al., 2009).
Although the mechanism underlying this striking effect of CD has not been completely elucidated, the current model is that CD enters the endocytic pathway through bulk-phase endocytosis and releases cholesterol and other lipids that are trapped in the LE/L so that the cholesterol can reach the ER (Liu et al., 2009).
Other experiments by Davidson et al. suggest that the administration of ALLO alone to NPC1-deficient mice, without CD has no beneficial effects, whereas the administration of CD with ALLO is more beneficial than CD alone (Davidson et al., 2009), although these conclusions remain controversial.
Thus, further investigations are needed to clarify the role of ALLO in the treatment of NPC disease.
30
1.7 Aim of the study
The aim of the present study was to develop an in vitro neuronal model to study the NPC1 disease. We used the hippocampal neuroblast line HN9.10e transfected with the fusion proteins NPC1 wt-GFP and I1061T-GFP. I1061T is the most common mutation, responsible of the classic juvenile form of NPC disease. We investigated the subcellular localization of NPC1 in neurons to gain insight into the mechanism of action of NPC1 in neuronal metabolism.
Confocal imaging was used to evaluate the impact of the I1061T mutation, in specific compartments such as lysosomes, endoplasmic reticulum and mitochondria. Quantitative analyses were assessed to determine important parameters to evaluate how the presence of the mutant NPC1 protein affects the organization of these compartments.
The results reveal a novel mechanism whereby the expression of mutant protein adversely affects the endoplasmic reticulum and mitochondria.
31
CHAPTER II
Experimental Procedures
2.1 Chemicals
F12, DMSO Dimethyl sulfoxide, FBS, DMEM, L-glutamine, penicillin, streptomycin, EDTA, bacto-tryptone, bacto-yeast extract, agarose, MidiPrep forniti da Sigma; TMRM Tetramethyl rhodamine Methyl Ester is provided by Molecular Probes Inc, Bacteria DH5α, lipofectamin and Optimem provided by Invitrogen.
2.2 Cell culture
The HN9.10e cell line is the cellular model of study. It was generated by fusion of embryonic mouse hippocampus and N18TG2 neuroblastoma cells(Lee et al., 1990).HN9.10e hippocampal neuroblasts were grown in F12 medium supplemented with 2 mM -glutamine and 10% heat-inactivated fetal bovine serum (Sigma). Cells were maintained at 37 °C in humidified atmosphere containing 5% CO2. Sterility of culture medium was maintained
without antibiotics to avoid toxic effects on susceptible subcellular compartments. For imaging experiments, cells were plated on 13 mm diameter glass cover slips 2–3 days before use to reach 50–60% of confluence and to obtain a stable substrate adhesion.
2.3 Process of freezing and thawing of the cellular line
To freeze the cells is useful to prepare a medium of freezing consists of: 50% FBS 42,5% DMEM 7,5% DMSO
Cells to 80% of confluence, detached from the plate, are centrifuged at 1000 rpm for 6 minutes; the supernatant is removed and resuspended in the new freezing medium. The
32 suspension is aliquoted into freezing vials and then are placed at -80° and after 24h placed in liquid nitrogen.
To thaw cells vials are removed from liquid nitrogen and placed in a water bath at 37° C so that cells thaw quickly and evenly. Once thawed the suspension is transferred in an sterile tube with 5 ml of DMEM and centrifuged at 1000 rpm for 6 minutes at room temperature. The supernatant is removed from freezing medium with DMSO and the cells are ready to be plated.
2.4 Plasmids
The ΔU3mnpc1-I1061T-GFP construct was generated using the ΔU3mnpc1-WT-GFP construct as a template. ΔU3mnpc1-I1061T was generated by deletion of the COOH-terminal GFP Tag (plasmids were provided by Prof. Daniel Ory).
2.5 Statistical Analysis
The data obtained from the experiments are reported as a mean percentage of controls +- standard error of mean (SEM) The significance was obtained with ANOVA’s test: *p<0.5;**p<0.1;***p<0.01.
BACTERIAL TRANSFORMATION
2.6 Competent cell preparation
The competent cells were prepared as described by (Inoue et al., 1990). Competent cells were frozen in liquid nitrogen and stored in -80ºC for future use.
2.7 Transformation of the vector into E. coli
For transformation, 40 µl DH5α competent cells and 30 ng of vector DNA were added into a sterile tube, which was then incubated on ice for 20 minutes, followed by a heat shock at 43 ºC for 30 seconds and further incubation on ice for 5 minutes. Then 0.5 ml of LB Medium was added and the mixture was incubated at 37 ºC for 2 h. The bacterial solution was then poured on the solid LB medium with antibiotics for selection of transformed cells at 37ºC until single colonies appeared. Randomly chosen colonies were then cultured on solid LB medium and stored at 4°C for later use.
33
2.8 Isolation of plasmid DNA from small amounts of bacteria
(mini-prep)
Mini-preps were performed in accordance to the manufacturer’s instructions using the Sigma Plasmid Mini Prep. The final DNA pellet was dissolved in 50 µl H2O or TE buffer with the concentration set to 1µg/µl and stored at 4°C.2.9 DNA precipitation
To precipitate DNA is added 1/10 of the total volume of the sample of NaAc 3M pH 5, 2 and 2 volumes of pure and cold EtOH and then put the sample at -80° C for 20 minutes. Then, the samples are centrifuged at 14000 rpm for 30 minutes at 4 °C and the supernatant was removed.
Pellets are added 500 µl of 70% EtOH and centrifuged at 14000 rpm for 10 minutes, the supernatant is discarded and the washing is repeated again.
The EtOH is evaporated and the DNA resuspended in 50 µl of MilliQ H2O.
2.9 Transient Transfection
HN9.10e cells were transfected using the lipofectamine system as described in the manufacturer’s instructions. Cells were cultured were plated on 13 mm diameter glass coverslips overnight until they reached 70-90% confluence. Serum-free transfection medium (OPTIMEM) containing 1µg plasmid DNA/ml medium was mixed with 1,5 µl lipofectamine in OPTIMEM medium. The transfection mixture was incubated at RT for 30min. Cells were rinsed twice in OPTIMEM medium. The transfection mixture was diluted 1:10 in OPTIMEM medium and applied onto the cells for 2 hours under normal cell culturing conditions before the transfection medium was exchanged with HN9.10e medium. Cells were grown for 24 to 48h at 37°C.
34
Image Analysis
2.10 Confocal microscopy
Images digitized with the confocal microscope from TCS-NT Leica laser scanning confocal microscope (Leica Inc) using a 40x/NA 1.25 oil objective, were analyzed with custom routines implemented in Matlab (version 7.4, Mathworks Inc). Coverslips were assembled in a perfusion chamber and the images were collected at 37°C with a resolution of 1024x1024. The mutant and wild type NPC1-GFP protein were excited with the 488 laser line and the GFP emission was recorded at 509 nm.
The cerulean- ER (kindly supported from Nica Borgese) was excited with 435 nm and the emission was recorded at 488 nm.
The main computational steps of the each routine are outlined below together with some samples of analysis. Full code of the analysis program is available on request.
2.11 Confocal Microscopy of TMRM
Routinely, HN9.10e cells were incubated in medium containing 500 nM tetramethyl rhodamine methyl ester (TMRM; Invitrogen) for 25 min at 37°C. After loading, cells were washed with fresh medium, and coverslips were mounted in a glass chamber for imaging. A TCS-NT Leica laser scanning confocal microscope (Nussloch, Germany) was used. Laser power was kept at 10% of the maximal power to avoid both photo damage of cells and photo bleaching of fluorescent probes. Images were acquired at 1024x1024 pixel resolution and averaged 4 times to improve the signal/noise ratio. Oil immersion objectives 100x (1.4NA), 63x (1.4NA) or 40x (1.25NA) (Leica, Nussloch, Germany) were used. The TMRM probe was excited with the 540nm laser line and the emission was recorded at 599 nm.
2.12 Confocal Microscopy of Cameleons: new calcium probe
Dr. Tsien and colleagues have developed genetically encoded calcium sensitive indicators based on Fluorescence Resonance Energy Transfer (FRET) (Miyawaki et al., 1999). Briefly, FRET involves the non-radioactive energy transfer between two fluorophores. This process is highly dependent on the distance and relative orientation of fluorophores, with the biologically relevant interaction distance on the order of 5-10 nm. The calcium sensors were called cameleons because they are based on calmodulin (CaM) and sense calcium changes by
35 changing the relative emission of two different colour wavelengths. CaM is a small, 150 amino acid proteins with 4 calcium binding sites, organized in two domains with different calcium binding affinities. The protein has no catalytic action itself, but works by changing conformation in response to calcium concentration and thereby activates a long list of kinase. The cameleon proteins were engineered to take advantage of the conformational change in response to calcium concentration. The sensor consists of two fluorophores, cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP), linked by CaM and the calmodulin binding peptide M13. CFP can be selectively excited with appropriate filters and in low calcium surroundings cameleon will be in its most open conformation, positioning the two fluorophores far from each other and resulting in low FRET efficiency and a high CFP emission. In high calcium, CaM binds to M13 and the two fluorophores are brought closer together, enabling FRET and resulting in an increased YFP emission at the expense of CFP emission (figure 2.1).
36 Figure 2.1- Schematic of cameleon function. Upper part shows the cameleon protein in low calcium
surroundings with the two fluorophore positioned far apart. Lower part shows conformational change induced in high calcium surroundings. (Fiala and Spall, 2003).
We use these reciprocal changes in fluorescence as a measure of changes in calcium concentration. The ratio between YFP and CFP is a better measure of calcium concentration changes than the individual intensities, since the ratio reduces signal dependence on indicator concentration.
The particularity of the cameleons is that is possible to add specific sequences of different cellular compartments for example Endoplasmic Reticulum (ERD1CPV) and Mitochondria (MTCD2CPV).
The MTCD2CPV and ERD1CPV (kindly supplied from Roger Tsien and Tullio Pozzan) calcium probes were excited with the 458 laser line of a TCS-NT Leica laser scanning confocal microscope (Nussloch, Germany) and the emissions of CFP and YFP were recorded at 485 and 535 nm, respectively. The quantification was made offline with the MetaMorph 5.0 software (Universal Imaging, West Chester, PA) expressing the calcium concentration as ratio of emissions CFP/YFP (535/485 nm). The images were elaborated with the plug-in “FRET analyzer” of Image J the public domain, Java-based, image processing program developed at the National Institutes of Health.