MATERIALI
CEPPO BATTERICO: DH5α, del batterio Escherichia coli, conservate in aliquote da 400 µl a –80°C.
Usato nel protocollo di trasformazione per amplificare vettori plasmidici per il loro utilizzo successivo. La competenza dei batteri, che in genere oscilla tra 108 e 109 colonie/µg di DNA plasmidico supercoiled, è prodotta con il metodo del Cloruro di Rubidio. Questo ceppo è difettivo per la restrizione e porta le mutazioni recA1 e relA1 per migliorare la stabilità e la qualità dei plasmidi ricombinanti preparati dalle “mini-“ e “midi- preps”. Contiene inoltre il marcatore φ80lacZ∆M15 che permette la α-complementazione del gene della β-galattosidasi e la selezione dei ricombinanti in un test bianco-blu.
VETTORI PLASMIDICI UTILIZZATI
Sono i vettori che portano la sequenze codificanti le proteine di fusione Xrx1-GR e GR-Xrx1. Entrambi sono stati sfruttati per la presenza del promotore SP6 per la RNA Polimerasi specifica, che ha permesso le trascrizioni dei costrutti codificanti le due proteine.
• pSP64 (Fig. 22, in alto). Può essere usato come vettore di clonaggio, e, grazie alla presenza del promotore SP6, anche per la trascrizione in vitro. Oltre alla presenza del promotore e del sito di inizio per la trascrizione della RNA Polimerasi SP6, possiede una regione per il clonaggio multiplo (Polylinker), una regione di Poly-A, una regione codificante la β-lattamasi per la resistenza all’antibiotico Ampicillina (permette la selezione delle colonie positive), e un sito di legame per il primer reverse per pUC/M13 (Promega). Il laboratorio di Sally A. Moody ci ha fornito questo vettore in cui è stato clonato il costrutto Xrx1-GR (Fig. 22, in basso) sotto forma di DNA plasmidico adsorbito su carta da filtro. Il DNA è stato poi amplificato, linearizzato (attraverso l’uso dell’enzima BamHI e trascritto in base alle indicazioni disponibili).
pSP64RIXrx1-GR
GR, LBD Xrx1
XhoI
KpnI BamHI
Figura 22 - In alto, mappa circolare del vettore pSP64. In basso, semplice rappresentazione schematica del costrutto generante la proteina di fusione GR-Xrx1
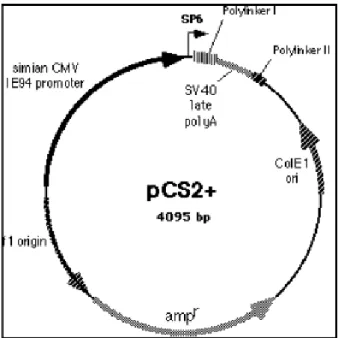
• pCS2+ (Fig. 23). E’ un vettore di espressione che consente vari usi. E’ progettato per l’espressione di proteine in embrioni di Xenopus, sia da RNA che da DNA microiniettati e si può usare in esperimenti di trascrizione/traduzione in vitro. Contiene un forte promotore virale (CMV IE94) seguito da un “polylinker” e dal sito di poliadenilazione del virus SV40. Un promotore SP6 consente la trascrizione di RNA da sequenze clonate nel “polylinker”; un promotore T7 inserito tra il “polylinker” e il sito polyA SV40 ad orientamento invertito consente la sintesi di sonde; un secondo polylinker successivo al sito polyA SV40 fornisce diversi siti di restrizione con cui linearizzare il vettore per la trascrizione SP6 (Rupp et al., 1994; Turner e Weintraub, 1994). Contiene il gene codificante per la resistenza all’antibiotico Ampicillina, sfruttato per la selezione delle colonie positive.
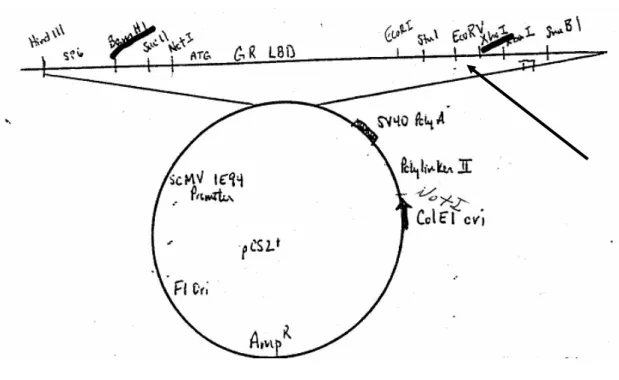
Sfruttando la presenza dei siti di restrizione presenti nel polylinker I, all’interno del vettore venne clonata la sequenza codificante il ‘ligand binding domain’ del recettore per glucocorticoidi (GR), originando così il vettore di espressione pCS2+GR (Fig. 24, A)
A
B
Figura 24 - (A) Rappresentazione schematica del vettore di espressione pCS2+GR. (B) Dettaglio sequenze al 5’ e al 3’ della LBD del GR. Le freccie nere indicano i siti di restrizione sfruttati per il clonaggio direzionale della sequenza codificante il fattore di trascrizione Xrx1
Durante il lavoro di tesi per la mia laurea triennale ho eseguito il clonaggio direzionale della sequenza codificante il fattore di trascrizione Xrx1, a valle della sequenza codificante la LBD del GR, sfruttando i siti di restrizione EcoRV e XhoI presenti all’interno del polylinker I (Fig. 24, B), in modo che le due sequenze fossero fuse ‘in frame’ (Fig. 25)
pCS2
+GR-Xrx1
GR, LBD Xrx1
EcoRV XhoI
Figura 25 - Semplice rappresentazione schematica del costrutto generante la proteina di fusione GR-Xrx1
CLONI
Utilizzati per la trascrizione di sonde:
• MC 19/11 (Xotx2): frammento di 860 pb clonato in pGem3 nei siti EcoRI e StuI, linearizzato con EcoRI, trascritto con SP6 RNA polimerasi (Pannese et al., 1995).
• fZIC2 (Zic2): frammento di 2230 bp clonato in pCDNA1-amp nei siti EcoRI,
linearizzato con BamHI, trascritto con SP6 RNA polimerasi.
OLIGO-PRIMER USATI NELLE REAZIONI DI PCR EFFETTUATE DURANTE I SAGGI SU ANIMAL CAPS
Primer progettati per essere specifici ciascuno del gene del quale ci interessa rilevare la presenza e il livello di espressione nei vari saggi su animal caps che sono stati effettuati durante il lavoro di tesi.
Zic2
Forward: 5’-CACCCCATTCAGCCTACTCT-3’ Tm: 63.0 °C Reverse: 5’-CGTCCTACCGAACACCTCTC-3’ Tm: 63.6 °C Lunghezza frammento amplificato: 299 bp
Odc
Forward: 5’-ACCCGAATGCAAAGCTTGTT-3’ Tm: 65.5 °C Reverse: 5’-TCCAGGGCTGGGTTTATCAC-3’ Tm: 65.9 °C Lunghezza frammento amplificato: 343 bp
Hairy2
Forward: 5’-GGCGTCAACACCGAAGTGAG-3’ Tm: 68.1 °C Reverse: 5’-ATCTTGTGGCCGAAGGTGGT-3’ Tm: 67.8 °C Lunghezza frammento amplificato: 377 bp
C-mycII
Forward: 5’-GCCAAGCTGGAGAAAGTGGT-3’ Tm: 65.5 °C Reverse: 5’-CTCCTCTTCCTCGTCGCAGT-3’ Tm: 65.9 °C Lunghezza frammento amplificato: 375 bp
EphB1
Forward: 5’-CCTCCAGAGCAACTCACGAC-3’ Tm: 65.1 °C Reverse: 5’-GACGGCAATTTTGGAGTTGA-3’ Tm: 64.8 °C Lunghezza frammento ampificato: 180 bp
Xl173
Forward: 5’-ATGCACTAAATCGCCCTCCT-3’ Tm: 64.6 °C Reverse: 5’-ATATTGTTTCCGGCCATTCC-3’ Tm: 64.4 °C Lunghezza frammento amplificato: 201 bp
Xl086
Forward: 5’-GCCCATAAGCCTGAATGTGA-3’ Tm: 64.8 °C Reverse: 5’-GTTTGCGCCGTGTCATTTAT-3’ Tm: 64.6 °C Lunghezza frammento amplificato: 164 bp
Xl443
Forward: 5’-CTTTTGGCCAATGGGAATCT-3’ Tm: 64.8 °C Reverse: 5’-GTTTGCTTGTTGGCAGGAAA-3’ Tm: 64.9 °C Lunghezza frammento amplificato: 206 bp
Xl520
Forward 5’-GAGAGTCGGCCATGTTTCTG-3’ Tm: 64.3 °C Reverse 5’-GAATCGTTTGCCGACAGGTA-3’ Tm: 64.8 °C Lunghezza frammento amplificato: 220 bp
Xbra
Forward 5’-TATATCCACCCAGACTCACCC-3’ Tm: 54.2 °C Reverse 5’-GATAGAGAGAGAGGTGCCCCG-3’ Tm: 58.4 °C Lunghezza frammento amplificato: 442 bp
TERRENI DI COLTURA E SOLUZIONI PER TRASFORMAZIONE BATTERICA
• Mezzo di coltura Luria-Bertani (L.B.), per Lt. di soluzione: Bacto Triptone 10 gr
Bacto-yeast extract 5 gr NaCl 10 gr
•
Terreno solido, Bottom Agar:agar sciolto in LB 1,5% (autoclavare)
• Antibiotici usati per terreni selettivi: Ampicillina 100 µg/µl
SOLUZIONI PER ‘MIDI-PREP’
Le soluzioni sono quelle contenute nel kit ‘NucleoBond Xtra Midi’ (Macherey-Nagel): ‘Resuspension Buffer + Rnasi’, ‘Lysis Buffer’, ‘Equilibration Buffer’, ‘Neutralization Buffer’, ‘Washing Buffer’, ‘Elution Buffer’.
Altre soluzioni impiegate:
• Isopropanolo al 100 % a temperatura ambiente • Etanolo al 70 % a temperatura ambiente
ENZIMI DI RESTRIZIONE
Usati per digestioni di diagnostica e per linearizzazione dei due costrutti Xrx1-GR e GR-Xrx1 per sottoporli alla reazione di trascrizione, tutti i seguenti enzimi di restrizione presentano attività massima a 37°C e una concentrazione negli stock in uso pari a 10 u/µl : Bam HI (Fermentas), KpnI (Fermentas), XhoI (Fermentas), EcoRI (Fermentas), EcoRV (Roche), NotI (Fermentas).
SOLUZIONI PER L’ ELETTROFORESI SU GEL DI AGAROSIO 10x TBE pH 8.0 (per la preparazione del gel è usato 1x)
Tris base 0.089 M
Acido borico 0.089 M
6x ‘Loading Buffer’ “Tipo III”: Glicerolo 5 % Blu di bromofenolo 0.05 % Xilene cianolo 0.05 % Et-Br 1:10000 peso/volume Agarosio 1-1.2 % (peso/vol.) 2x ‘Loading Buffer’ per RNA:
Formammide 95%
Xilene cianolo 0.025%
Blu di bromofenolo 0.025%
EDTA 18 mM
SDS 0.025%
Marcatori di quantità per RNA utilizzati:
tRNA, soluzione stock 7.26 µg/µl Marcatori di lunghezza usati:
• Ready-Load 1 Kb Plus DNA Ladder (Invitrogen) (Fig. 26, A) • GeneRuler 1 kb DNA Ladder (Fermentas) (Fig. 26, B)
• GeneRuler DNA Ladder mix (Fermentas) (Fig. 26, C)
(A) (B) (C)
Figura 26 - I marcatori dilunghezza usati nelle elettroforesi su gel d’agarosio durante questo lavoro di tesi
12.000 5.000 2.000 1.650 1.000 850 650 500 400 300 200 100 10.000 8.000 5.000 4.000 3.000 2.500 2.000 1.500 1.000 750 500 250
SOLUZIONI PER TRASCRIZIONE mRNA DA MICROINIETTARE
Le seguenti soluzioni provengono dal kit per la trascrizione dell’mRNA ‘capped’ “mMESSAGE mMACHINE” • H2O, ‘Nuclease-Free’ • Enzima SP6, in glicerolo al 50% • Tampone di reazione, 10x • 2x NTP/CAP: ATP 10 mM CTP 10 mM UTP 10 mM GTP 2 mM cap analog 8 mM • DNasi Turbo 2U/µl
• LiCl, soluzione di precipitazione: LiCl 7.5 mM
EDTA 50 mM
(Tutti i reagenti sono conservati a -20°C) Altre soluzioni impiegate:
• Etanolo R.F. al 70%
SOLUZIONI PER LA REAZIONE DI TRASCRIZIONE INVERSA
• Random Primer 3 µg/µl • dNTP mix ATP 10 mM GTP 10 mM CTP 10 mM TTP 10 mM
• 5x ‘First Strand Buffer’ (Invitrogen)
Tris-HCl, pH 8.3 250 mM
KCl 375 mM
• DTT (invitrogen) 0.1 M
• RNasi OUT (Invitrogen) 40 U/µl
• SuperScript III RT (Invitrogen) 200 U/µl
SOLUZIONI PER PCR
• Oligo-primer 10 µM
• dNTP mix 10 mM
• GoTaq DNA Polimerasi (Promega) 5 u/µl • 5x Green GoTaq Reaction Buffer (Promega)
SOLUZIONI PER EMBRIONI DI Xenopus laevis
• MMR NaCl 0.1 M KCl 2 mM MgSO4 1 mM CaCl2 2 mM HEPES 5 mM, pH 7.8 EDTA 0.1 mM • Soluzione degelificante DTT 3.2 mM Tris-HCl 0.2 M, pH 8.8 • Soluzione per il testicolo
MMR 1x
Siero di agnello inattivato al calore 10%
Gentamicina 20µg/ml • MEMFA MOPS 0.1 M, pH 7.4 EGTA 2 mM MgSO4 1 mM Formaldeide 3.7%
Generalmente si preparano “stocks” sterili di una soluzione 10x dei Sali (MEM) che viene diluita e addizionata di formaldeide al momento dell’uso.
SOLUZIONI DI USO COMUNE
• PBS NaCl 8 g Kcl 0.2 g Na2PO416H2O 1.44 g KH2PO4 0.24 g pH 7.4 • SSC 20x NaCl 3.0 M Na citrato 0.3 M
SOLUZIONI PER LA REAZIONE CROMOGENICA DELLA β -GALATTOSIDASI • Soluzione di ferri K3Fe(CN)6 30 mM K4Fe(CN)6.3H2O 30 mM PBS 1x • Stock Salmon-gal
Salmon-gal (Sigma) 5% in metanolo • Stock X-gal
X-gal 20 µg/µl • ‘Wash solution’ (conservare a 4°C)
PBS, pH 7.4 1x Sodio deossicolato 0.01% Nonidet P40 0.02%
SOLUZIONI PER IBRIDAZIONE IN SITU SU EMBRIONI INTERI • TBS NaCl 8 g KCl 0.2 g Tris Base 3 g pH 7.4 • PBTw 0.1% PBS 1x Tween 20 0.1% • TBSx TBS 1x NaCl 8 g KCl 0.2 g Tris Base 3 g pH 7.4 Triton X-100 0.1% • Paraformaldeide 20% 1. Sciogliere 5 g di paraformaldeide in 100 ml H2O R.F. a 60°C 2. Chiarificare con 10 µl NaOH 10 N, portare a volume e filtrare 3. Conservare a -20°C. Una volta scongelata usare entro un mese • Tampone di ibridazione (NIH)
Formammide 50%
SSC 5x
RNA di morula 1 mg/ml
Eparina 100 µg/ml
Roche Blocking reagent 1%
Tween 20 0.1%
CHAPS 0.1%
EDTA 10 mM
• APB (“Alcaline Phosphatase Buffer”) Tris HCl, ph 9.5 100 mM MgCl2 50 mM NaCl 100 mM Tween 20 0.1% Tetramisole 2 mM • “Egg extract”
1. Omogenizzare embrioni conservati a -20°C in 1 volume di PBS 1x 2. Centrifugare 10’ a 4°C a 12000 rpm, recuperare la fase acquosa 3. Centrifugare 5x10’ a 4°C a 12000 rpm, recuperando la fase acquosa 4. Denaturare a 56°C per 15’, centrifugare per 10’ a 12000 rpm a 4°C 5. Aliquotare e conservare a -20°C
• “Blocking buffer”
TBSx 1.6 ml
Siero di agnello inattivato al calore 300 µl
Egg extract 100 µl
Roche blocking reagent 400 µl
SOLUZIONI PER LA DEPIGMENTAZIONE DEGLI EMBRIONI (“BLEACHING”)
• “Bleaching solution”
H2O2 1%-3%
SSC 0.5x
Formammide 5%
SOLUZIONI PER L’ESPIANTO E LA COLTIVAZIONE DEGLI ANIMAL CAPS • MBS 10x, pH 7.6 NaCl 0.88 M KCl 0.01 M MgSO4*7H2O 8.0 mM HEPES 0.1 M NaHCO3 0.024 M
• CaCl2 100 mM • Gentamicina 1000x 50 µg/ml • Dexamethasone (Sigma) 500x 2 mg/ml • Cicloesimmide (Sigma) 1000x 10 mg/ml
SOLUZIONI PER L’ESTRAZIONE DELL’RNA DAGLI ANIMAL CAPS E SUCCESSIVA PURIFICAZIONE
• Estrazione
TRIzol Reagent (Invitrogen), conservato a 4°C Cloroformio
Isopropanolo R.F. al 100%, a temperatura ambiente Etanolo R.F. al 75%, a temperatura ambiente
DNasi RQ1 ‘RNase-Free’ 1 U/µl
10x DNasi RQ1 ‘Reaction Buffer’
RNasi OUT 40 U/µl
• Purificazione
I seguenti reagenti sono contenuti nel kit ‘Total RNA Isolation, Nucleospin RNA XS’ della ‘Macherey-Nagel’: ‘Lyisis Buffer RA1’, ‘Wash Buffer RA3’, H2O ‘RNase-Free’.
Altre soluzioni impiegate:
METODI
PREPARAZIONE DI CELLULE COMPETENTI
Mediante il seguente protocollo le cellule di E. coli (ceppo DH5α) sono state rese competenti per la successiva trasformazione con vettori plasmidici.
• Prelevare una colonia cresciuta ON su terreno solido e inocularla in 50 ml di terreno liquido.
• Far procedere la crescita a 37°C in agitazione fino a che la densità ottica (OD) misurata a 600 nm raggiunge il valore di 0,2.
• Interrompere la crescita mantenendo 3’ in ghiaccio. • Centrifugare in tubo sterile per 10’ a 5000 rpm a 4°C.
• Eliminare il sopranatante e risospendere in ½ del volume iniziale (circa 25 ml) di RbCl 50 mM freddo.
• Mantenere 30’ in ghiaccio.
• Centrifugare in tubo sterile per 10’ a 5000 rpm a 4°C.
• Eliminare il sopranatante e risospendere in 1/50 del volume iniziale (circa 0,8 ml) di RbCl 50 mM freddo.
• Conservare in aliquote a –80°C.
TRASFORMAZIONE BATTERICA
Il seguente protocollo è utilizzato per trasformare cellule batteriche competenti con DNA plasmidico.
• Si aggiungono 5 ng di DNA plasmidico a 100 µl di cellule batteriche competenti (DH5α, E. Coli), si mescola con cura ma non si vortexa, per non danneggiare irreversibilmente le cellule.
• Si pone la provetta contenente le cellule e il DNA plasmidico in ghiaccio per circa mezz’ora.
• Si sottopone la provetta a shock termico per 45 sec a 42°C e si pone nuovamente la provetta in ghiaccio per circa 5 minuti. Lo shock termico dovrebbe permettere al DNA palsmidico di entrare nelle cellule.
• Si aggiunge ai 100 µl di cellule e al DNA plasmidico 800 µl di brodo L.B. si pone la provetta in incubatore sulla ruota a 37°C per circa 1 ora. Questo passaggio consentirà alle cellule che sono state trasformate con il DNA plasmidico di esprimere il gene di resistenza agli antibiotici in esso contenuto.
• Si recupera la provetta dall’incubatore e si centrifuga a 12.000 rpm per 2 minuti. A questo punto si ha un pellet di cellule e un sovranatante. Si elimina quest’ultimo lasciandone però un minimo volume per risospendere le cellule.
• Si risospende le cellule.
• Si piastra la sospensione ottenuta su piastre di L.B. agar contenenti ampicillina e porre le piastre stesse in incubatore a 37°C O.N..
CONSERVAZIONE CELLULE BATTERICHE TRASFORMATE
E’ importante, ai fini di un successivo utilizzo, porre in glicerolo una parte delle cellule trasformate con DNA plasmidico e cresciute in terreno liquido. Dalla coltura si prelevano in genere 500 µl di sospensione batterica e si pongono in altrettanti µl di glicerolo. Tale sostanza protegge dal congelamento le cellule a -80°C, temperatura alla quale in genere vengono conservati gli stock in glicerolo. Per poter utilizzare nuovamente tali cellule, si preleva dallo stock una piccola quantità di sospensione con un’apposita ansa sterile e si piastra su piastre di L.B. agar contenenti Ampicillina. Poi la piastra viene posta in incubatore O.N. a 37°C.
ESTRAZIONE DI DNA PLASMIDICO SU MEDIA SCALA MEDIANTE COLONNINE NUCLEOBOND A SCAMBIO ANIONICO (“MIDI-PREP”)
Le colonne a scambio anionico e le soluzioni impiegate sono fornite dal kit ‘NucleoBond Xtra Midi’ (Macherey-Nagel). Queste colonne sono dotate di una resina basata sulla silice, sviluppata per separare differenti classi di acidi nucleici quali oligonucleotidi, RNA e DNA plasmidico. Con questa tecnica è possibile estrarre in media da 3 µg/µl a 12 µg/µl di DNA plasmidico a partire da 100 ml di sospensione batterica satura.
• A partire da colonie cresciute su piastra o da stock in glicerolo di cellule batteriche, si effettuano inoculi su terreno liquido (3-4 ml) in cui in precedenza è stato posto l’antibiotico Ampicillina (concentrazione 100 mg/ml, 1 µl di antibiotico ogni ml di terreno).
• I tubi da batteri in cui sono stati eseguiti gli inoculi vengono posti nell’incubatore sulla ruota a 37°C per circa 8 ore (pre-crescita).
• Da un tubo da batterio viene prelevato 1 ml di pre-crescita, che viene successivamente inoculato in 100 ml di terreno contenente Ampicillina. La coltura viene incubata O.N. a 37°C con agitazione.
• Si risospende completamente il pellet di cellule formatosi in 8 ml di ‘resuspension buffer RES + RNase A’, fino a quando la soluzione non diventa omogenea.
• Si aggiungono 8 ml di ‘lysis buffer LYS’ alla sospensione, si agita delicatamente invertendo il contenitore 5 volte. Non si vortexa la soluzione, poiché ciò potrebbe causare il distacco del DNA genomico dalla parete cellulare batterica. Si incuba la soluzione 5 minuti a R.T.
• Si aggiungono alla colonna già assemblata con un filtro 12 ml di ‘equilibration buffer EQU’. Si applica la soluzione entro il margine del filtro, in modo da bagnarlo completamente. Il liquido passa attraverso la colonna per gravità. Ad ogni passaggio si attende che il liquido fluisca completamente attraverso la resina della colonna.
• Si aggiungono 8 ml di ‘neutralization buffer NEU’ alla sospensione e si mescola delicatamente invertendo 10-15 volte il contenitore.
• Si pone tutto il lisato nella colonnina con il filtro, e si attende che questa si svuoti per gravità.
• Si aggiungono 5 ml di ‘equilibration buffer EQU’ alla colonna, per lavarla insieme al filtro in essa contenuto. Si applica la soluzione entro il margine del filtro, bagnandolo completamente. Si attende che la colonna si svuoti per gravità.
• Si toglie il filtro dalla colonna e si elimina.
• Si lava la colonna aggiungendo 8 ml della ‘washing buffer WASH’. Si attende che la colonna si svuoti per gravità. Si elimina il liquido raccolto.
• Si aggiunge alla colonnina 5 ml di ‘elution buffer ELU’ per eluire il DNA plasmidico. Si raccoglie l’eluato in un nuovo contenitore.
• Si aggiunge alla soluzione raccolta 3.5 ml di Isopropanolo a R.T. per permettere la precipitazione del DNA plasmidico eluito. Si vortexa la soluzione e si attende almeno 2 minuti prima di procedere al passaggio successivo.
• Si centrifuga la soluzione a 15.000 x g per 30 minuti a 4°C. Successivamente, rimuove con attenzione il sovranatante e si elimina.
• Si lava il pellet con 2 ml di etanolo al 70 % a R.T. e si centrifuga la soluzione a 15.000 x g per 5 minuti a 20-25 °C.
• Si rimuove completamente l’etanolo dal tubo e si lascia asciugare il pellet a temperatura ambiente per 5 minuti.
• Si sospende il pellet in 100 µl di H2O. Se non è previsto un immediato utilizzo del DNA plasmidico estratto, conservarlo a –20°C.
DETERMINAZIONE SPETTROFOTOMETRICA DELLA CONCENTRAZIONE DEL DNA.
Per la valutazione della concentrazione di DNA plasmidico estratto tramite ‘midi-prep’, si utilizzano opportune cuvette di quarzo che consentono la lettura nell’ultravioletto. In questo lavoro di tesi è stata in genere scelta una diluizione 1:200 del campione di DNA plasmidico, per la lettura dell’assorbanza di tale campione sottoposto ad un fascio di 260 nm di lunghezza d’onda.
Per la valutazione della concentrazione di DNA proveniente ad esempio da una digestione preparativa e purificato utilizzando l’apposito kit, o si scelgono diluizioni minori (1:50), oppure si utilizzano capillari di quarzo, in cui vengono caricati 4 µl di materiale diluito 3 volte (1 µl DNA + 3 µl di H2O).
Per valutare la pulizia dei campioni, in entrambi i casi si è tenuto conto del rapporto A260/A280, che deve mantenersi tra 1.8 e 2.0. Campioni che presentano tale rapporto inferiore a 1.8 dovrebbero essere scartati in quanto l’elevata proporzione della concentrazione di proteine in soluzione (A280), potrebbe inibire eventuali reazioni enzimatiche effettuate usando tale campione.
DIGESTIONE DI DNA CON ENZIMI DI RESTRIZIONE
L’uso di enzimi di restrizione in questo lavoro di tesi è stato necessario per effettuare digestioni diagnostiche, verificare la presenza e l’orientamento di un inserto all’interno di un vettore, linearizzare plasmidi per usarli come DNA stampo lineare in esperimenti di trascrizione in vitro (digestione preparativa).
Seguono le linee fondamentali da seguire nell’assemblaggio di una reazione di digestione enzimatica :
• Per ogni µg di DNA si impiegano 3 unità di enzima.
• Per impedire il congelamento e quindi l’inattivazione degli enzimi, tali enzimi sono conservati in glicerolo a –20°C. Il glicerolo inibisce l’attività dell’enzima, quindi la quantità di enzima che si deve porre nella reazione non deve superare il 10 % del volume totale di reazione.
• Si pone la mix di reazione in incubatore alla temperatura alla quale tali enzimi hanno massima attività. La durata dell’incubazione dipende dalla quantità di DNA da digerire, dalla quantità dell’enzima e dal tampone di reazione usato. Di solito è limitata a 2-3 ore.
PURIFICAZIONE DEL DNA DOPO DIGESTIONE PREPARATIVA
Viene utilizzato a questo scopo il “GenElute PCR Clean-Up kit” della Sigma, progettato per purificare prodotti di PCR ma anche prodotti di digestioni enzimatiche da eccessi di nucleotidi, enzima e sali, sfruttando colonnine dotate di una resina a base di silice.
• Si assembla la colonnina al tubo collettore. Si aggiungono 0.5 ml della ‘Column Preparation Solution’ a ciascuna colonna e si centrifuga a 12.000 x g da 30 secondi a 1 minuto. Si elimina il liquido raccolto.
• Si aggiungono 5 volumi di ‘Binding Solution’ a 1 volume di DNA e si mescola. Ad esempio, si aggiungono 500 µl di ‘Binding Solution’ a 100 µl di soluzione della reazione enzimatica.
• Si trasferisce la soluzione nella colonnina e si centrifuga alla massima velocità (12.000-16.000 x g) per 1 minuto. Si elimina il liquido raccolto ma si conserva il tubo collettore.
• Si riposiziona la colonnina nel tubo collettore. Si aggiungono 0.5 ml della ‘Wash Solution’ diluita e si centrifuga alla velocità massima per 1 minuto. Si elimina raccolto ma si conserva il tubo collettore.
• Si riposiziona la colonnina nel tubo collettore. Si centrifuga la colonnina alla velocità massima per 2 minuti, senza aggiungere altra ‘Wash Solution’, per rimuovere l’eccesso di etanolo. Si elimina l’eluato così come il tubo collettore
• Si trasferisce la colonnina in nuovo tubo collettore da 2 ml. Si aggiungono 50 µl della ‘Elution Solution’ o H2O al centro di ciascuna colonnina e si lascia ad incubare a temperatura ambiente per 1 minuto.
• Per eluire il DNA, si centrifuga la colonnina alla velocità massima per 1 minuto. Il prodotto purificato è ora presente nell’eluato e pronto per un uso immediato, altrimenti può essere conservato a -20°C.
CORSA ELETTROFORETICA SU GEL DI AGAROSIO Viene eseguita in questo lavoro di tesi per i seguenti scopi:
• Per valutare l’avvenuta amplificazione o meno di un gene a seguito di una reazione di PCR, prelevando dalla mix una piccola aliquota e correndola su gel a ~ 90 mV. Dopo la corsa viene verificata l’assenza di bande nel controllo negativo e la presenza della banda attesa nel controllo positivo. In seguito si passa all’analisi della banda corrispondente al prodotto amplificato:
1- Si osserva se la banda migra all’altezza attesa, utilizzando l’apposito marcatore di lunghezza (Ladder).
2- Si valuta la pulizia della banda, ovvero se la banda è netta. Si riscontra la presenza o meno di bande aspecifiche e ‘smear’. La presenza di bande aspecifiche è significativa del fatto che il prodotto amplificato non è rappresentato solo dal gene che ci interessa amplificare, ma anche da prodotti aspecifici. Per evitare ciò, probabilmente devono essere modificate le condizioni della reazione di PCR, rendendola più stringente e quindi più specifica.
3- Si cerca, ad occhio, di valutare l’intensità delle bande, perché l’intensità è proporzionale alla quantità di DNA specifico di quel determinato gene nel campione di partenza. Se le condizioni di PCR sono le stesse per tutte le reazioni caricate sul gel, può essere fatto un confronto tra le intensità delle bande, indicativo di un confronto tra i livelli di espressione di geni diversi, o dello stesso gene in condizioni sperimentali imposte diverse.
• Per valutare l’esito di una digestione enzimatica.
Una digestione enzimatica può avere due scopi: analitici o preparativi. La digestione a scopi analitici viene effettuata in questo lavoro come diagnostica su prodotti di trasformazione. Per tali scopi si carica su gel una piccola parte del prodotto digerito e si confronta con il non digerito, utilizzando il marcatore di lunghezza (Ladder) per valutare differenze di migrazione tra non digerito e digerito, e se quest’ultimo migra all’altezza attesa, oppure si eseguono digestioni doppie, che solitamente rimuovono una sequenza di lunghezza conosciuta, e si valuta se tale sequenza migra all’altezza attesa. Le digestioni preparative in questo lavoro vengono eseguite per linearizzare un DNA plasmidico, che poi rappresenterà il templato per la successiva reazione di trascrizione.
• Per valutazioni quantitative, utilizzando marcatori di quantità. Sono utilizzate in questo lavoro per valutare la quantità e la qualità di RNA estratto da Animal Caps. Si caricano insieme su gel i marcatori di quantità e campioni di volume noto del materiale che deve essere valutato. Si effettua una breve corsa e si confronta l’intensità delle bande dei campioni con quella dei marcatori a peso noto. Invece per una valutazione positiva della qualità del campione di RNA, si devono riscontrare su gel almeno la presenza di due bande, che rappresentano l’rRNA 28s e l’rRNA 18s. Le bande devono essere abbastanza nette e definite e si deve riscontrare l’assenza di smear, segno evidente di parziale degradazione dell’RNA.
RNA degradato può compromettere la riuscita e la valutazione di esperimenti successivi che utilizzino proprio tale RNA.
• Per valutare la bontà dell’avvenuta trascrizione in vitro di un determinato mRNA e anche la sua concentrazione. Essenzialmente si deve valutare l’intensità della banda di mRNA e confrontarla con quella di marcatori a peso noto e non si deve riscontrare la presenza di DNA nel prodotto finale.
PREPARAZIONE DEL GEL E CARICAMENTO DEI CAMPIONI
In questo lavoro è stato usato gel all’1% di agarosio. La percentuale si riferisce al rapporto peso del gel/volume di TBE 1x usato. Si pesa 1g di Agarosio per porlo in 100 ml di TBE 1x. Si mescola la soluzione in modo da renderla omogenea, e si porta ad ebollizione. Si aggiunge alla soluzione il Bromuro di Etidio (un agente che si intercala tra le basi del DNA, rendendolo in questo modo visibile ai raggi UV. Si usa diluito 1:10000). Si lascia raffreddare per qualche minuto e poi si cola negli appositi stampi, in cui vengono appoggiati dei pettini, e si attende il tempo necessario affinché il gel solidifichi. I pettini servono a formare i pozzetti necessari per il caricamento dei campioni. Pettini di diversa grandezza determinano pozzetti di diversa grandezza. Quelli utilizzati in questo lavoro sono quelli per corse a scopi analitici, che possono contenere un volume massimo di circa 15 µl.
I campioni prima di essere caricati devono essere appesantiti con il ‘Loading Buffer 6x’, se si tratta di DNA (1x finale in soluzione), o con il ‘Loading Buffer 2x’ se si tratta di RNA. Tale soluzione non serve solo ad appesantire il campione (per la presenza di glicerolo in soluzione) per farlo scendere sul fondo del pozzetto, ma anche a renderlo visibile durante il caricamento e la corsa. Il range di quantità di DNA per una buona visibilità dello stesso su gel previa illuminazione con raggi UV e per una migliore discriminazione delle bande varia da 100 ng e 500 ng di materiale caricato.
TRASCRIZIONE IN VITRO DI mRNA PER MICROINIEZIONI
Per trascrivere mRNA senso da iniettare in embrioni di Xenopus laevis a partire da plasmidi linearizzati contenenti la sequenza di interesse clonata a valle del promotore SP6 è stato utilizzato il kit ‘mMESSAGE mMACHINE’. Con questo kit viene trascritto un RNA che presenta una ‘terminal cap structure’ (‘cap’) all’estremità 5’, che ne aumenta l’efficienza di traduzione.
E’ importante prima di procedere all’assemblaggio della reazione, vortexare il tampone di reazione 10x e la soluzione contenente 2x NTP/CAP, e, in particolare, mantenere il
tampone di reazione a temperatura ambiente durante l’assemblaggio. La Spermidina presente nel tampone di reazione può far precipitare il templato (DNA) se la reazione viene assemblata in ghiaccio.
La reazione è stata assemblata come segue, importante è aggiungere il tampone di reazione dopo l’H2O e i ribonucleotidi:
2x NTP/CAP 5 mM
Tampone di reazione 10x 1x DNA, templato linearizzato 1 µg
RNA Polimerasi SP6 2 µl
H2O ‘Nuclease-Free’ fino ad un volume totale di 20 µl • Si mescola delicatamente e si pone la reazione a 37°C per 2 ore
• Al termine, prelevare dalla mix di reazione 1 µl, e conservarlo per una successiva analisi
• Si aggiunge alla reazione 1 µl di DNasi TURBO (2 U) e si pone la reazione nuovamente a 37°C, per 15 minuti
• Si ferma la reazione e si precipita l’RNA aggiungendo 30 µl di H2O ‘Nuclease-Free’ e 30 µl di LiCl (soluzione di precipitazione)
• Si mescola abbondantemente e si pone la reazione a -20°C per un tempo non inferiore ai 30 minuti
• Si centrifuga la mix a 4°C per 15 minuti alla velocità massima per raccogliere l’RNA in un pellet
• Si rimuove con cautela il sovranatante, si lava il pellet una volta con Etanolo al 70% e si centrifuga nuovamente per massimizzare la rimozione dei nucleotidi non incorporati.
• Si rimuove con cautela l’etanolo al 70% e si rispospende l’RNA in 20 µl, se ne determina la concentrazione allo spettrofotometro, se ne valuta la bontà e la eventuale presenza di DNA tramite corsa elettroforetica su gel di agarosio, si aliquota e si conservano le aliquote in attesa dell’uso a -80°C.
Per quanto riguarda la corsa elettroforetica, si caricano dei marcatori di peso, 1 µl della reazione di trascrizione prelevato prima dell’aggiunta in soluzione della DNasi e 1 µl dell’RNA finale rispospeso in 20 µl di H2O ‘Nuclease-Free’. Dopo la corsa elettroforetica (~ 150 mV per 3-4 minuti), nella ‘line’ dove è stato caricato l’RNA pre-DNasi si dovrebbero vedere 2 bande: una superiore corrispondente al DNA e una inferiore corrispondente all’RNA. Mentre, se la DNasi avrà funzionato correttamente, nella ‘line’ corrispondente
all’RNA finale si dovrebbe vedere solo la banda inferiore, corrispondente all’mRNA neo-sintetizzato
TRASCRIZIONE IN VITRO DI UNA SONDA ANTISENSO E PURIFICAZIONE
Questo protocollo è stato usato nella trascrizione di RNA antisenso marcato con DIG-UTP (digossigenina-DIG-UTP) da usare come sonda negli esperimenti di ibridazione in situ: l’RNA Polimerasi necessaria (SP6) è stata scelta in base alla direzione di clonaggio della sequenza di interesse nel vettore, che è stato linearizzato di conseguenza.
• Si assembla la reazione con 1-1.5 µg di DNA linearizzato, 2 µl DTT 100 mM, 4 µl transcription buffer 5x, 2 µl DIG-labeling mix (Roche), 1 µl RNase OUT (Invitrogen), 2 µl RNA polimerasi SP6. Si porta ad un volume finale di 20 µl con H2O mQ.
• Si pone la reazione 2h a 37°C.
• Si preleva 1µl del volume di reazione e si conserva in ghiaccio per la successiva corsa elettroforetica di controllo.
• Si aggiungono 2µl di DNase/RNase free (Invitrogen) e si pone la mix di reazione per 15’ a 37°C.
• Si aggiungono, per la precipitazione dell’RNA, 1 µl EDTA 0.5 M, 1/10 Vol. di NH4Ac, 1 Vol. isopropanolo. Si rimescola e si lascia 20-40’ a -20°C.
• Si centrifuga per 15’ a 13000 rpm a 4°C e si rimuove il sovranatante.
• Si lava il pellet di RNA prodotto con EtOH al 75% (non meno di 100 µl) e si centrifuga 3’ a 13000 rpm RT.
• Si elimina il sovranantante e si risospende in 22 µl H2O mQ.
• Si prelevare 1 µl del campione e si carica in un gel di agarosio 1% con “loading buffer” 2x per RNA assieme al campione prelevato in precedenza e a soluzioni di marcatori di RNA.
• L’assenza della banda del DNA stampo successiva al trattamento con DNasi indica il buon fine della reazione; il confronto con i marcatori dà una stima della concentrazione dell’mRNA prodotto, che deve comunque essere valutata allo spettrofotometro.
SINTESI DI cDNA A PARTIRE DA RNA (TRASCRIZIONE INVERSA)
Passaggio che si è reso necessario in questo lavoro di tesi durante i saggi su animal caps, successivamente all’estrazione e alla purificazione dell’RNA, per fornire il templato alle specifiche reazioni di PCR.
1. Si aggiungono ad una mini-eppendorf (una per ogni reazione) ‘Nuclease-Free’, i seguenti componenti:
• Random Primers 250 ng
• Total RNA 10 pg - 5 µ g
• dNTP mix 10 mM 1 µl H2O mQ, fino a un volume totale di 13 µl
2. Si pone la mix a 65°C per 5 minuti e successivamente in ghiaccio per almeno 1 minuto.
3. Si centrifuga brevemente per raccogliere la reazione e si aggiunge: • 5x ‘First Strand Buffer’ 4 µl
• DTT 0.1 M 1 µl
• RNase OUT 1 µl
• SuperScript III RT 1 µl
4. Si mescola la reazione delicatamente e si pone 5 minuti a 25°C 5. Si pone a 55°C per 60 minuti (fase di retrotrascrizione)
6. Si inattiva la reazione ponendola a 70°C per 15 minuti
Ora il cDNA prodotto può essere usato come templato per l’amplificazione tramite PCR.
REAZIONE DI PCR
La PCR (Polymerase Chain Reaction) è una reazione utilizzata per amplificare in modo esponenziale un determinato frammento di DNA, chiamato templato della reazione. In questo lavoro tale tecnica viene utilizzata per amplificare sequenze appartenenti a geni specifici presenti o meno in un pool di cDNA. Vengono impiegate coppie di primer specifiche per ogni gene.
La reazione, una volta assemblata, viene posta in un termo-ciclizzatore, dove viene sottoposta ad un numero variabile di cicli di amplificazione, ciascuno composto da più fasi a temperature diverse. Il ciclo consiste nel fornire inizialmente una temperatura molto elevata, che permette la denaturazione del filamento del cDNA. A questa fase iniziale seguono i cicli di amplificazione, che consistono di 3 fasi che si ripetono tante volte quante ne stabilisce lo sperimentatore secondo le proprie esigenze: una prima fase di denaturazione, una seconda fase di annealing, in cui si ha un abbassamento della temperatura per permettere l’ibridazione
dei primers (in genere tra 45°C-65°C), una terza fase di allungamento, in cui la temperatura si alza fino a raggiungere quella di funzionamento della DNA Polimerasi utilizzata. L’ultima fase di questo processo è l’allungamento dei frammenti amplificati per circa 10 minuti. La reazione prevede l’utilizzo di apposite DNA Polimeasi termostabili.
In questo lavoro è stata utilizzata una sola DNA Polimerasi, la GoTaq DNA Polymerase (Promega), che agisce a 72°C. La reazione è stata assemblata come segue:
5X Green GoTaq Reaction Buffer 1X dNTP mix (10mM ciascuno) 200 µM
Primer ‘Forward’ (10 µM) 250 nM
Primer ‘Reverse’ (10µM) 250 nM
GoTaq DNA Polymerase (5U/µl) 0.025 U/µl
Templato 20 ng
H2O fino ad un volume totale di 50 µl
Per ogni reazione di amplificazione, è stato eseguito anche un controllo negativo, assemblando una normale reazione di PCR, eliminando il templato, e sostituendolo con H2O. Questo controllo viene eseguito per verificare che non esistano contaminazioni da DNA a livello degli oligo-primer, dei dNTP, dell’H2O utilizzata. Sono stati eseguiti anche controlli positivi, assemblando una reazione di PCR usando gli stessi oligo-primer su un templato del quale si ha la certezza che venga amplificato. Questo controllo viene eseguito per valutare se la reazione procede regolarmente o se ci sono difetti a livello della Master Mix o dei primer, che potrebbero non risultare efficienti.
EMBRIONI DI Xenopus laevis
Gli embrioni di Xenopus laevis per gli esperimenti sono stati ottenuti mediante fecondazione in vitro. Prima di essere operato per la rimozione dei testicoli, il maschio e stato anestetizzato immergendolo in una soluzione 0.1% di MSS (metansulfonato dell’estere etilico dell’acido 3-amminobenzoico) disposta in ghiaccio per abbassare rapidamente il metabolismo dell’animale. I testicoli si possono conservare per 3-5 giorni a 4°C immersi nella soluzione per testicoli. Dopo l’operazione il maschio viene sacrificato. Lo stoccaggio avviene secondo la vigente normativa veterinaria. Le femmine di Xenopus laevis sono state pre-stimolate con 100 UI di Folligon Intervet per uso veterinario da 4 a 11 giorni prima della deposizione e con 1000 UI di Profasi HP 5000 Serono (gonadotropina corionica) 10-12 h prima della deposizione: entrambi gli ormoni sono stati somministrati mediante iniezione nel sacco
perilinfatico. Le uova da fecondare sono state ottenute esercitando una leggera pressione sull’addome degli animali e raccolte in piastre Petri: questa operazione puo essere ripetuta a intervalli di 1 ora. La fecondazione e stata effettuata bagnando le uova con una sospensione ottenuta sminuzzando un frammento di testicolo in MMR 1x e lasciandole in poco MMR 0.1x. Dopo almeno 30 minuti o quando comunque fosse ben rilevabile la rotazione corticale degli embrioni, effetto dell’avvenuta fecondazione, gli embrioni sono stati privati del loro rivestimento gelatinoso ricoprendoli di soluzione degelificante e lasciandoveli 5-10 minuti e comunque fino a che non fosse evidente la perdita dl suddetto rivestimento. La soluzione degelificante e stata eliminata sciacquando gli embrioni in MMR 0,1x.
In questo lavoro di tesi la fecondazione in vitro viene effettuata per raccogliere gli embrioni necessari per la microiniezione, che è stata effettuata agli stadi 2 e 4 di sviluppo. Gli stadi sono classificati secondo i criteri di Nieuwkoop e Faber (Nieuwkoop et al.,1967a; Nieuwkoop et al.,1967b). Gli embrioni sono stati fatti sviluppare fino allo stadio desiderato in un ambiente a temperatura controllata (14°C o 18°C) e poi sottoposti a microiniezione.
MICROINIEZIONE DI EMBRIONI DI Xenopus laevis
Per gli esperimenti di microiniezione sono stati usati embrioni pigmentati in cui e possibile distinguere il polo animale da quello vegetativo e i blastomeri dorsali da quelli ventrali. Al momento della microiniezione gli embrioni degelificati vengono trasferiti in una piastra Petri del diametro di 5 cm sul fondo della quale e stata fissata una reticella di plastica, con maglie di 1 mm, che ne limita gli spostamenti. Inoltre gli embrioni sono immersi in una soluzione di Ficoll al 4% (peso/volume) sciolto in MMR 0.1x: il Ficoll e uno zucchero viscoso, permette agli embrioni di mantenere la forma sferica durante la microiniezione e ne limita la perdita di citoplasma successivamente. Gli embrioni microiniettati sono stati lasciati sviluppare ON in MMR 0.1x-ficoll 4%, la prima notte a 14°C e poi trasferiti in MMR 0.1x a 18°C, finche gli embrioni di controllo non inietttati non avessero raggiunto lo stadio desiderato e quindi fissati. Alcuni embrioni iniettati, durante questo lavoro di tesi sono stati trattati con l’ormone Dexamethasone, aggiungendolo al mezzo di coltura, ad una concentrazione finale di 4 µg/ml. Le microiniezioni sono state eseguite con un microiniettore Drummond Nanoject, che consente l’iniezione di volumi compresi tra 4.6 nl e 73.6 nl ad incrementi discreti. Gli aghi sono stati preparati per tiratura a caldo a partire da capillari forniti da Drummond: la loro bonta e stata controllata allo stereoscopio. Prima di essere montati sul microiniettore gli aghi sono stati riempiti di olio minerale con una siringa. Il caricamento degli aghi con la soluzione da iniettare e eseguito dal microiniettore stesso.
Quando giungono allo stadio desiderato gli embrioni microiniettati e tutti i relativi embrioni di controllo vengono fissati in MEMFA in ‘vials’ di vetro da 4 ml e lasciati in agitazione orizzontale per circa 45 minuti. Gli embrioni vengono trasferiti in Etanolo assoluto, per la conservazione a -20°C, per una immediata analisi fenotipica degli eventuali effetti imposti dalla microiniezione, oppure per sottoporli all’esperimento di ibridazione in situ su embrioni interi. Gli embrioni, quando co-iniettati con il marcatore β-Gal, prima di una qualsiasi analisi o precedura sperimentale, dopo la fissazione, vengono sottoposti alla reazione cromogenica della β-Galattosidasi.
REAZIONE CROMOGENICA DELLA Β-GALATTOSIDASI
Negli esperimenti di microiniezione in genere è stato co-iniettato mRNA di β-galattosidasi nucleare come marcatore: ciò ha permesso di valutare la bontà della microiniezione attraverso la reazione catalizzata dall’enzima, che in presenza di un substrato cromogeno produce un precipitato colorato. Come substrato cromogeno è stato utilizzato il Salmon-gal, che produce un precipitato di colore rosso, oppure in alternativa la X-Gal, che invece produce un precipitato di colore azzurro, seguendo il seguente protocollo:
• Dopo la fissazione in MEMFA, senza porre gli embrioni in etanolo, si effettuano 2 lavaggi da 5 minuti in PBS 1x (si puo lasciare a 4°C fino a 48 h).
• 2 lavaggi da 15’ in “Wash solution” (o ON a 4°C). • Lasciare 5’ in soluzione di ferri.
• Rivelare in 0,5 ml totali di soluzione di ferri contente 5 µl di stock Salmon-gal, incubando a 37°C. Controllare periodicamente la rivelazione.
• In alternativa, in luogo della Salmon Gal, possono essere posti 37 µl di X-Gal in 0.5 ml totali di soluzione di ferri.
• 2 lavaggi da 5 minuti in “Wash solution”.
• Si fissano gli embrioni in MEMFA per 30’-45’ e si trasferiscono in etanolo 100%.
IBRIDAZIONE IN SITU SU EMBRIONI INTERI (WHOLE MOUNT)
L’ibridazione in situ su embrioni interi è stata effettuata in questo lavoro di tesi per studiare il “pattern” di espressione di due geni allo stadio 13 di sviluppo di Xenopus laevis, su embrioni iniettati ed embrioni di controllo, mediante l’ibridazione di sonde marcate con DIG-UTP ai trascritti presenti nei tessuti; il protocollo seguito e sostanzialmente quello descritto da Niehrs (Gawantka et al., 1995):
Gli embrioni vengono posti in ‘vials da 4 ml RF (RNase free)
• Si reidratano gli embrioni conservati in etanolo a -20°C, con passaggi di 5’, ciascuno in: Etanolo 75 % PBTw 25 %
Etanolo 50 % PBTw 50 % Etanolo 25 % PBTw 75 % PBTw 100%
• Si lava ancora una volta per 5’ in PBTw con agitazione orizzontale.
• Si aggiungono 0,5 ml/vial di soluzione di proteinasi K in PBTw 1:2000; si incuba 5’ esatti senza agitazione (omettere questo passaggio e successivi lavaggi in PBTw con embrioni emisezionati o con espianti).
• Si effettuano due brevi lavaggi con PBTw con agitazione manuale.
• Si sostituisce il PBTw con paraformaldeide al 4% in PBS, 1 ml per vial; si incuba 20’ a RT con agitazione manuale ogni 5’ (omettere questo passaggio e successivi lavaggi in PBTw con embrioni emisezionati o con espianti).
• Si effettuano due brevi lavaggi con PBTw con agitazione manuale, quindi si effettuano 4 lavaggio x 5’ in PBTw con agitazione orizzontale.
• Si sostituisce il PBTw con 0,5 ml/vial di tampone di ibridazione al 50% in PBS, si incuba 3’, quindi si sostituisce con tampone di ibridazione 100% e si incuba 3’.
• Si sostituisce con 0,5 ml/vial di tampone di ibridazione 100% e si preibrida 2-3 h a 60°C.
• Nel frattempo si denaturano 50 ng di sonda di RNA marcato in 10 µl di H2O mQ per 2’ a 95°C, si passa rapidamente in ghiaccio e si aggiungono 600 µl di tampone di ibridazione (concentrazione finale della sonda: 83.3 pg/µl). Si aggiunge la miscela alle vials e si ibrida ON a 60°C.
• Si prepara per il giorno successivo una soluzione SSC 2x/0.1% CHAPS e si lascia ON a 37°C e una soluzione SSC 0.2x/0.1% CHAPS, che si lascia ON a 60°C.
• Il giorno seguente si recuperare la miscela con la sonda e si sostituisce con 1 ml/vial 50% tampone di ibridazione/50% (SSC 2x/0.1% CHAPS); si incuba per 10’.
• Si incuba 2 volte x 30’ con 1ml/vial di SSC 2x/0.1% CHAPS preequilibrato a 37°C.
• Si incuba 2 volte x 30’ con 1 ml/vial SSC 0.2x/0.1% CHAPS preequilibrato a 60°C.
• Si sostituisce con 50% (SSC 0.2x/0.1% CHAPS)/50% TBS 1x a RT per 5’, e quindi con TBS 1x per 5’.
• Si sostituisce con 0,5 ml/vial di blocking buffer, si incuba 2h a 4°C con agitazione orizzontale.
• Contemporaneamente, si diluisce l’anticorpo AP Fab Anti-DiG (Roche) 1:2500 in blocking buffer e si incuba anch’esso 2h a 4°C con agitazione orizzontale.
• Si sostituisce il “blocking buffer” con 0,5 ml/vial di soluzione di anticorpo diluito e si incuba 4 h a RT con agitazione verticale.
• Si elimina la soluzione di anticorpo e si effettuano 5 lavaggi da 1h ciascuno in TBSx a RT (il primo ON a 4°C, gli altri 4 il giorno successivo) con agitazione orizzontale.
• Si effettuano 2 lavaggi x 5’ con 1 ml/vial di APB.
• Si sostituisce l’APB con 0,5 ml/vial di “BM Purple AP substrate precipitaing” (Roche) e si incuba a RT al buio (incubando a 14°C il segnale risulta più pulito, ma è necessario un tempo piu lungo; a 4°C la reazione si blocca): si sostituisce il substrato quando è necessario e quando la marcatura è adeguata.
• Si sostituisce il “BM Purple” con TBSx per 10’ per bloccare la reazione. • Si fissano gli embrioni in MEMFA 1 h a RT oppure ON a 4°C e si conservano in etanolo a -20°C. Gli embrioni adesso sono pronti per essere analizzati allo stereomicroscopio.
La fosfatasi alcalina coniugata all’anticorpo scinde il substrato cromogenico (“BM Purple”) generando un precipitato colorato che evidenzia le zone in cui la sonda si e ibridata, dove il gene in esame è stato trascritto.
DEPIGMENTAZIONE DEGLI EMBRIONI (“BLEACHING”)
Per evidenziare meglio il segnale dell’ibridazione gli embrioni sono stati sottoposti a depigmentazione:
• Si sostituisce l’Etanolo in cui sono conservati gli embrioni con Etanolo 70% in H2O e si pongono per 5’ a RT in agitazione orizzontale.
• Si sostituisce con 50% Etanolo/50% SSC 1x e si pongono gli embrioni per 5’ a RT in agitazione orizzontale.
• Si sostituisce con “bleaching solution” e si pongono le vials in agitazione orizzontale sotto una lampada ad incandescenza, controllando costantemente la depigmentazione.
• Si ferma la depigmentazione passando gli embrioni in etanolo 70% per 5’ e successivamente in Etanolo al 100%. Conservare gli embrioni a -20°C.
‘ANIMAL CAPS’ DI Xenopus laevis. ESPIANTO, COLTIVAZIONE E TRATTAMENTO.
Gli animal caps (calotte animali) di embrioni di Xenopus laevis, vengono prelevate allo stadio di sviluppo 8-9, attraverso l’uso di apposite pinzette e con l’ausilio di uno stereomicroscopio. Gli espianti vengono eseguiti in apposite piastre Petri di 6 cm di diametro, in cui è stato posto un sottile strato di gel di Agarosio all’1% in H2O, e una soluzione contenente:
MBS 1x
Gentamicina 50 ng/ml
CaCl2 0.7 mM
In alternativa all’uso dell’MBS, può essere usato MMR 0.7x, omettendo l’aggiunta di CaCl2 in soluzione. La presenza di una concentrazione salina più elevata rispetto al mezzo in cui vengono fatti crescere o iniettati embrioni interi, è per permettere al cap spiantato di non disgregarsi e di richiudersi in oco tempo. Gli espianti vengono effettuati prima togliendo con le pinzette la membrana che ricopre l’embrione, successivamente andando a prelevare la porzione centrale del polo animale (cellule pigmentate) dell’embrione (ectoderma presuntivo), attraverso dei piccoli tagli effettuati con le pinzette. E’ necessario mantenersi ad una certa distanza dal confine tra cellule pigmentate e non pigmentate durante l’operazione di espianto, per non rischiare di prelevare cellule del mesoderma presuntivo. A volte, per permettere che gli animal caps si richiudano più rapidamente, se ne fondono due insieme, facendoli aderire l’uno con l’altro con la loro superficie interna. Si attende che i caps si richiudano e delicatamente si trasferiscono in un'altra piastra contenente la stessa soluzione e si pongono in un incubatore a temperatura controllata e costante (14°C o 18°C), assieme gli embrioni di controllo, che appartengono alla stessa fecondazione degli embrioni da cui abbiamo prelevato i caps, e che servono per identificare lo stadio di sviluppo degli animal caps stessi. La bontà degli animal caps può essere valutata qualche ora dopo l’espianto, osservandoli allo stereomicroscopio. Per essere accettabili devono essere di forma simil-sferica e comunque non allungata e devono presentare una superficie liscia.
Gli animal caps durante questo lavoro di tesi sono stati a volte trattati con l’ormone Dexamethasone, alla concentrazione finale in soluzione di 4 µg/ml e con l’inibitore di sintesi proteica Cicloesimmide alla concentrazione finale in soluzione di 10 µg/ml.
Una volta arrivati allo stadio di sviluppo desiderato, gli animal caps vengono fermati semplicemente ponendoli in una eppendorf e togliendoli quasi tutto il liquido di coltura. A
questo punto, o vengono utilizzati subito, ad esempio per l’estrazione di RNA, oppure devono essere immediatamente congelati ponendo le eppendorf in un bagnetto di Etanolo ghiacciato a -80°C per 30 minuti (sarebbe meglio porli in ghiaccio secco, se è a disposizione), e poi conservandoli a -80°C per un successivo utilizzo.
ESTRAZIONE E PURIFICAZIONE DI RNA DA ‘ANIMAL CAPS’
Per l’estrazione di RNA da ‘animal caps’ viene utilizzato un protocollo della Invitrogen che utilizza il reagente TRIzol, una soluzione mono-fasica di fenolo e guanidina isotiocianato.
• Gli animal caps vengono omogenizzati tramite l’uso di appositi pestelli (conservati in H2O mQ con NaOH 1N) in 1 ml del reagente TRizol. Il volume del campione non dovrebbe superare il 10% del volume di TRIzol impiegato.
• Si incuba il campione omogenizzato per 5 minuti ad una temperatura compresa tra 15°C e 30°C. Il TRIzol mantiene l’integrità dell’RNA, mentre distrugge le cellule e dissolve le componenti cellulari.
• Si aggiungono 0.2 ml di Cloroformio e si mescola la soluzione con vigore per 15 secondi. Si incuba il tutto per 2-3 minuti ad una temperatura compresa tra 15°C e 30°C.
• Si centrifuga il campione a non più di 12.000 x g per 15 minuti a 4°C. A seguito della centrifugazione, la soluzione si separa in una fase di colore rosso, più bassa, di fenolo-cloroformio, un interfase, e una fase superiore acquosa. L’RNA resta presente esclusivamente nella fase acquosa
• Si trasferisce la fase acquosa in una nuova eppendorf e si precipita l’RNA mescolando la soluzione con 0.5 ml di Isopropanolo. Si incuba la mix ad una temperatura compresa tra 15°C e 30°C per 10 minuti.
• Si centrifuga la soluzione a non più di 12.000 x g per 10 minuti a 4°C. L’RNA precipitato forma un pellet visibile sul fondo della eppendorf.
• Si elimina il sovranatante e di lava il pellet con 1 ml di Etanolo RF al 75%. Si mescola la soluzione usando il vortex e si centrifuga ad una velocità non inferiore a 7.500 x g per 5 minuti a 4°C.
• Si toglie tutto l’etanolo e si lascia asciugare il pellet per 5-10 minuti
• Si risospende il pellet in 20 µl di H2O ‘RNase Free’ delicatamente e si pone la soluzione per 10 minuti ad una temperatura compresa tra 55°C e 60°C.
A questo punto l’RNA viene posto in ghiaccio ed è pronto per la successiva reazione, quella di degradazione del DNA:
RNA 20 µl 10x DNasi RQ1 ‘Reaction Buffer’ 1x
DNasi RQ1 ‘RNase-Free’ 2 U
RNasi OUT 40 U
H2O ‘RNase Free’ fino ad un volume totale di 50 µl Si incuba la reazione per 30 minuti a 37°C
La successiva fase di purificazione dell’RNA viene eseguita utilizzando il kit ‘Total RNA Isolation, Nucleospin RNA XS’ della ‘Macherey-Nagel’ che sfrutta delle particolari colonnine cromatografiche, che contengono una resina a base di silice:
• Prima di tutto, si porta il volume della soluzione a 100 µl con H2O ‘RNase Free’.
• Si prepara una soluzione ideata allo scopo di permettere il legame dell’RNA alla resina della colonnina con alta efficienza, mescolando per ogni campione 25 µl di ‘Buffer RA1’ con 75 µl di Etanolo assoluto.
• Si aggiungono i 100 µ l della soluzione appena preparata alla soluzione contenente il campione, si mescola 2 volte per 5 secondi e si esegue un breve ‘spin’ del campione con una microcentrifuga.
• Si carica il campione sulla colonnina, già assemblata con il tubo collettore e si centrifuga per 30 secondi a 11.000 x g. Si scarta l’eluato e si assembla la colonnina con un nuovo tubo collettore.
• Si aggiungono 400 µl di ‘Buffer RA3’ alla colonnina e si centrifuga per 30 secondi a 11.000 x g. Si scarta l’eluato e si riposiziona la colonnina nel tubo collettore.
• Si aggiungono 200 µl alla colonnia e si centrifuga per 2 minuti a 11.000 x g. Si scarta il tubo collettore con l’eluato e si assembla la colonnina con un tubo collettore ‘Nuclease-Free’.
• Si aggiungono 10 µl di H2O ‘RNase-Free’ alla colonnina e si centrifuga per 30 secondi a 11.000 x g per l’eluizione dell’RNA.
A questo punto l’RNA purificato è pronto per la determinazione spettrofotometrica della sua concentrazione e per un successivo utilizzo, oppure per essere conservato a -80°C se l’utilizzo non è immediato.
DETERMINAZIONE SPETTROFOTOMETRICA DELLA CONCENTRAZIONE DELL’RNA
A questo scopo vengono utilizzati speciali capillari di quarzo che consentono la lettura nell’ultravioletto, in cui vengono caricati 4 µl del campione diluito 3 volte (1 µl RNA + 3 µl di H2O). Il capillare di quarzo con il campione all’interno dello spettrofotometro viene sottoposto ad un fascio di 260 nm di lunghezza d’onda. In base al valore dell’assorbanza e considerando la diluizione del campione, viene calcolata la sua concentrazione in soluzione. Per la valutazione della pulizia del campione si è tenuto conto di due rapporti tra assorbanze: A260/A280 e A260/A230. Il primo deve essere compreso tra 1.8 e 2, per le motivazione già elencate precedentemente, mentre il secondo dovrebbe presentare un valore non inferiore a 2.2. Valori inferiori a 2.2 indicano una eccessiva presenza di Sali in soluzione (Fenolo, ad esempio) che potrebbero anche avere effetti negativi e imprevedibili sull’esito delle successive reazioni enzimatiche.
FOTOGRAFIE
Le fotografie degli embrioni interi e degli animal caps sono state realizzate mediante una fotocamera digitale CoolSNAP-cf montata in asse focale ad uno stereoscopio Nikon SMZ1500 con l’ausilio di una coppia di fibre ottiche.
Le fotografie delle elettroforesi su gel di Agarosio sono state realizzate mediante una fotocamera montata sull’apparato Gel Doc 2000 Bio Rad in grado di emettere raggi ultravioletti, consentendo la visione delle bande del DNA o dell’RNA.
Il trattamento delle immagini è stato realizzato con l’ausilio del software Adobe Photoshop CS3.
SEQUENZIAMENTO
I risultati del sequenziamento visionati durante questo lavoro di tesi sono stati forniti dal Servizio Sequenziamento della M-Medical GENENCO-Laboratori di Roma.
STRUMENTI BIOINFORMATICI
Per le analisi bioinformatiche è stato utilizzato l’algoritmo ‘align2seq’, disponibile presso l’NCBI (National Center for Biotechnology Information) che solitamente viene utilizzato per confrontare due sequenze immesse direttamente dall’utente. Indirizzo dell’algoritmo:
&BLAST_PROG_DEF=megaBlast&SHOW_DEFAULTS=on&BLAST_SPEC=blast2se
q&LINK_LOC=align2seq.
Durante questo lavoro di tesi è stato utilizzato per localizzare all’interno della sequenza codificante di un gene, in quale punto si appaiavano i primer progettati per la sua amplificazione, per valutarne sia la correttezza e la specificità del legame, sia la lunghezza del frammento compreso tra i due primer, che è quello che verrà amplificato durante la reazione di PCR