107
5. New enzyme inhibitors
The most common therapeutic approach for many cancers is chemotherapy and radiation exposure. However, many patients relapse after treatment due to the development of chemoresistance. Recently, the development of novel approaches to destroy cancer cells or to improve their sensibility to the already existing therapies represent challenging tasks to achieve1.
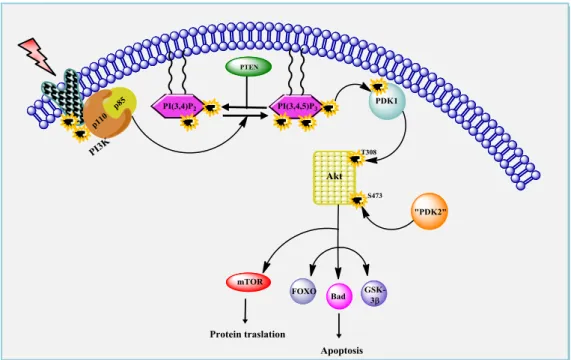
Many types of immune cellular stimulation or toxic insults activate the generalized systemic Akt/PDK1 pathway that regulates the basic cellular functions such as transcription, proliferation, growth and survival. Several studies report that alterations to this signaling pathway are frequent in a large percentage (30–60%) of common tumor types, including melanoma, glioblastoma and breast, lung, gastric, prostate, hematological and ovarian cancers1.
By stimulation with growth factors and cytokines, Akt is recruited from the cytosol to the plasma membrane and phosphorylated at two key regulatory sites, Thr308 and Ser473, by PDK1. The activated Akt stimulates cell growth and survival by phosphorilating numerous downstream substrates including caspase-9, glycogen synthase kinase-3 (GSK3), BCL antagonist of cell death (BAD) and forkhead box transcription factors (FOXO).
Figure 5.1:The PDK1/Akt signaling pathway. This allows the recruitment of PI3K (p85 and p110) to the receptor. Activated PI3K can generate PIP3 from PIP2. PIP3 subsequently recruits Akt to the cell membrane allowing it to be phosphorylated by PDK1
108 Serine/threonine kinase Akt/PKB is a crucial kinase in this pathway2. In mammals, there are three isoforms of Akt (Akt1, 2, 3). All three isoforms share a high degree of amino acid identity and are composed of three functionally distinct regions: an N-terminal pleckstrin homology (PH) domain, a central catalytic domain, and a C-terminal hydrophobic motif (HM).
Figure 5.2: Akt structure. Akt contain a PH (pleckstrin homology) domain in the N-terminus, a central catalytic domain with kinase activity, and a regulatory domain in the C-terminus. The PH domain binds inositol triphosphate. Akt also contains two main phosphorylation sites: one threonine in the kinase domain (Thr308) and one serine the regulatory domain (Ser473).
The members of Akt family share similar domain structure and are activated by various stimuli in a phosphatidylinositol 3-kinase (PI3K)-dependent manner.
Activation of Akt depends both on the integrity of the pleckstrin homology (PH) domain, which mediates its membrane translocation, and on the phosphorylation of Thr308 in the activation loop. Full activation of Akt is also associated with phosphorylation of Ser4733 within a C-terminal hydrophobic motif characteristic of kinases in the AGC kinase family.
The activity of Akt is negatively regulated by tumor suppressor PTEN (phophatase and tensin homolog delete on chromosome ten), which is frequently mutated in human malignancy. PTEN encodes a dual-specificity protein and lipid phosphatase that reduces intracellular levels of PtdIns(3,4,5)P3 by converting them to PtdIns(4,5)P2, thereby inhibiting the PI3K/Akt pathway.
5.1. Disrupting of Akt/PDK1 pathway
Promotion of cell survival is the most studied function of the Akt pathway. Akt develops its anti-apoptotic role through phosphorylation of downstream substrates that control the apoptotic machinery. Akt regulates cell growth through its effects on the mTOR and p70 S6 kinase pathways, as well as cell cycle and cell
109 proliferation through its direct action on the CDK inhibitors p21 and p27, and its indirect effect on the levels of cyclin D1 and p53. Akt is a major mediator of cell survival through direct inhibition of pro-apoptotic signals such as Bad and the Forkhead family of transcription factors. These findings make Akt an important therapeutic target for the treatment of cancer.
At present there are only few Akt inhibitors being evaluated in various stages of clinical trials. Progress in the development of Akt inhibitors appeared to be relatively slow and this might be hindered, at least in part, by the unexpected toxicity of Akt inhibitors due to their site of interaction.
Akt inhibitors might be classified as:
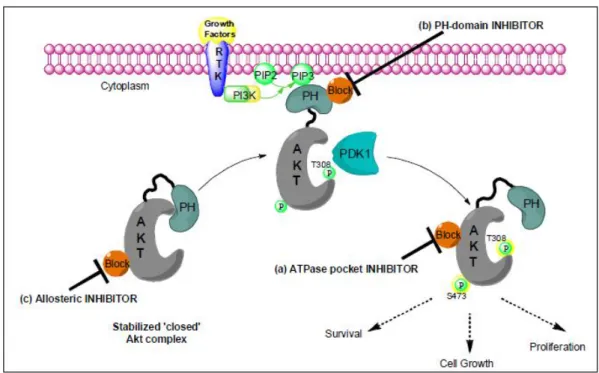
a) ATP-pocket binders which interact with the ATP-pocket of the enzyme, b) PH-domain inhibitors which devoid the Akt activation (phosphorylation at
Ser)
c) allosteric inhibitors which stabilized the ‘closed’ Akt complex in the cytoplasm
d) ‘hybrid-design’ inhibitors
Figure 5.3: Akt inhibition. It is thought that the N-terminal PH domain precludes kinase access to phosphorylation of the activation-loop at Thr 308 by PDK-1. Allosteric Inhibitors stabilized the ‘closed’ Akt complex. PH-domain inhibitors devoid the recruitment of Akt to the membrane by binding to its PH domain. ATP-binding pocket inhibitors interact with the activated Akt isoform by precluding its kinase activity (but not its activation).
110
a) ATPase pocket inhibitors
The architecture of the ATP binding site of all protein kinases is very similar, making it a difficult but challenging task to identify highly selective protein kinase inhibitors. The presence of an ATPase pocket in many other intracellular proteins (e.g. metabolic enzymes and ion pumps), represent the first potential source for off-target effects of Akt inhibitors rarely considered in screening programmes. Despite this important drawback the development of new ATPase inhibitors is clearly successful and numerous ATP-competitive inhibitors have been developed for Akt.
N H N O OH N N N N O NH2 N N N H O NH NH2 GSK690693 A-443654
Figure 5.4: The figure shows examples of compounds that are ATP-pocket binders.
b)PH-domain inhibitors
Theoretically, compounds that compete with PtdIns(3,4,5)P3 to bind to the PH domain of Akt or compete with PtdIns(4,5)P2 to bind to PI3K could serve as novel Akt inhibitors. This mode of inhibition would prevent Akt translocation to the plasma membrane and activation. The feasibility of this approach was suggested by the demonstration that D-3-deoxy-myo-inositol inhibited the growth of transformed cell.
Several phosphoinositide analogues, alkyl phospholipids (e.g. perifosine,) have been identified as competitive inhibitors of the binding of the Akt PH domain to PI(3,4,5)P3 and PI(3,4)P24.
N+ O P O C18H37 O H O Me Me Perifosine
111 While this approach obviates the selectivity issues associated with the ATP-binding domain homology among kinases, there is the potential to interfere with the function of other PH domain containing proteins. In addition, compounds in this class commonly have problems with solubility, aggregation, pharmacokinetics and potency that limit their utility as drugs.
c) Allosteric inhibitors
Merck & Co, Inc have developed a novel class of allosteric inhibitors of Akt. The initial lead compound was identified by highthroughput screening and further derivatised to compounds that specifically inhibit Akt1 (Akti-1), Akt2 (Akti-2) or both (Akti-1/2), with minimal activity against Akt35. These compounds do not bind to the ATP binding site or the PH domain, but inhibit Akt activity in a manner that requires the PH domain itself 6,7.
The allosteric Akt inhibitors should be more efficient than the ATP-pocket binders in attenuating Akt activity because the activation of Akt via the feedback mechanism is abolished. Although the potency and physiological properties are being optimized, the preliminary studies revealed that simultaneous inhibition of Akt1 and Akt2 is superior to a single isoform of either Akt1 or Akt2, as measured by apoptosis reduction. Greater induction of apoptosis by antisense inhibition of both Akt1 and Akt2 was also observed in comparison to a single antisense inhibition.
Figure 5.6: The figure shows example of compound that is inhibitors of activation of Akt
d) ‘hybrid-design’ inhibitors
The majority of kinase inhibitors that have been developed so far, known as type I inhibitors, target the ATP binding site of the kinase in its active conformation, in which the activation loop is phosphorylated. Extensive crystallographic and
N N H N N NH O Me O N N H N N NH O Me Me Me
112 associated molecular modeling efforts have allowed much kinase inhibitor potency and selective activity to be rationalized. For example, the binding modes of well-known inhibitors such as oxindole, quinazoline and phenyl amino pyrimidine can be predicted with high accuracy and used to guide lead optimization. Inhibitors such as imatinib (STI571), BIRB796 and sorafenib (BAY43-9006), known as type II inhibitors, have revealed a new binding mode that exploits an additional binding site immediately adjacent to the region occupied by ATP. This pocket is made accessible by an activation-loop rearrangement that is characteristic of kinases in an inactive conformation.
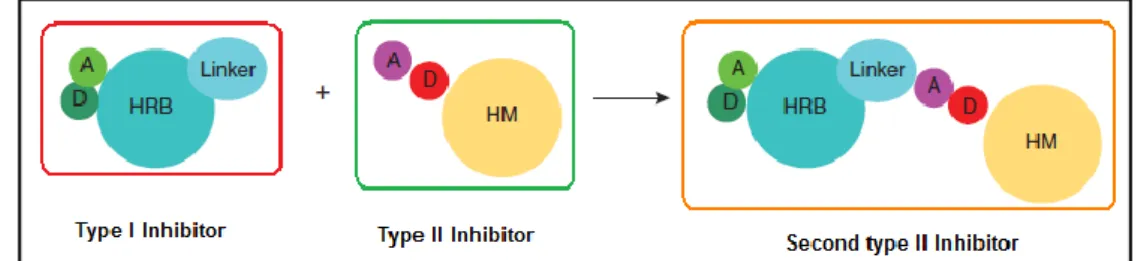
Second-generation type II inhibitor molecules can be broken down into two portions. The head portion is a normal type I inhibitor that has hydrogen bonding interactions with the kinase hinge residues and hydrophobic interactions in and around the adenine region of the ATP binding pocket. The tail portion bears a moiety similar to that seen on the known type II inhibitors that contains a pair of hydrogen bond donating and accepting groups. The ‘hybrid-design’ approach consists of appending a type II tail onto a type I scaffold (Figure 5.7). The first step is to select a type I scaffold based on its kinase activity and selectivity and based on the ease with which a linker can be installed to serve as anchor point to attach the type II tail. The second step is to attach the type II tail moiety, which consists of a hydrogen bond donor-acceptor pair and a hydrophobic motif. The resulting potential type II molecule can then be docked to an available DFG-out (the activation loop, which contains the conserved DFG motif, being in an ‘out’ conformation) kinase structure or to a homology model to verify that the type I head, the linker and the type II tail can fit into the ATP and allosteric pocket and form the expected hydrogen bonding and hydrophobic interactions8.
Figure 5.7: A general pharmacophore model for rational design of type II inhibitors. The second generation type II kinase inhibitors can be broken down into a type I head (black) attached to a type II tail. A, hydrogen bond acceptor; D, hydrogen bond donor; HRB, hinge-region binding; HM, hydrophobic motif.
113
5.2. State of Art
PDK1 belongs to the AGC kinase family, this enzyme has been characterized as a master kinase, due to its propensity to activate other important downstream AGC kinases such as Akt. Several nonselective inhibitors for PDK1 have already been reported in the literature9and have been shown to block survival of cancer cells. The understanding of the biological complexity of this pathway and the lack of effective therapies, led us to develop new molecules able to interact with one or more targets of PDK1/Akt pathway considered relevant to the genesis and progression of this disease.
Recently, new compounds designed to target some components of this pathway have been developed. In 2006 researchers at Bayer ScheringPharma AG patented a class of indolinone derivatives as PDK1 inhibitors.
BX-517 inhibits PDK1 in vitro with an IC50 value of 0.52M. It also blocks the
activation of Akt in tumor cells10.
N H N H O NH O N H2 BX-517
Figure 5.8: The figure shows BX-517 that is Inhibitor of PDK1/Akt
Despite its high enzymatic potency, the development of this compound was limited because of its poor solubility and unfavourable pharmacokinetic profile.
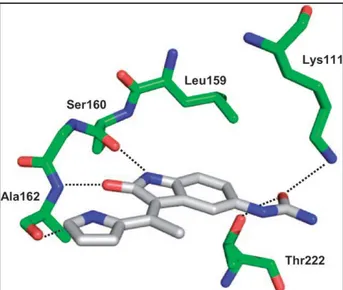
An X-ray crystallographic analysis of BX-517 in the ATP binding pocket of PDK1 disclosed the binding mode at the molecular level.
In particular the pyrrole–indolinone core is involved in three key H bonds. The indolinone nitrogen interacts with the Ser160; the indolinone oxygen accepts an H bond from Ala162. The pyrrole nitrogen addresses an H bond to the carbonyl group of Ala162.
The 5-urea group accepts an H bond directly from both the side chains of Lys111 and the hydroxyl function of Thr222.
114
Figure 5.9: Binding mode of BX-517 in the ATP binding pocket of PDK1
In 2008, the identification and initial medicinal chemistry optimization of an indoline-based series of PDK1 modulators has been published into a patent from Jiangsu Simcere Pharmaceutical. The compounds of the invention which had the chemical structure of Formula (I) showed inhibitory activity against kinases and the IC50 value was generally below 10-7 M. For these reasons the derivatives can be used
as scaffold to develop new anti-tumor agents11.
N H N H R2 R3 O NH R1 O
Figure 5.10: The figure shows structure of Formula (I) patented from Jiangsu Simcere Pharmaceutical
More recently, Vertex Pharmaceuticals (PCT April 2010) described a series of compounds as inhibitors of protein kinases, such as inhibitors of Akt or PDK1. Chemotherapeutic agents or other anti-proliferative agents may be combined with all compounds to treat proliferative diseases and cancer12.
115 N N H N N N S R1
Figure 5.11: The figure shows structure of VERTEX PHARMACEUTICALS INC., inhibitors of Akt or PDK1
Researchers at Merck Patent GmbH (PCT November 2010) recently patented a series of substituted pyridines derivatives as potent PDK1 inhibitors. The compounds of general formula 2 can be used for treating different type of tumors such us leukemia, breast, colon and lung cancers13.
N N H R1 N N N R2
Figure 5.12: The figure shows structure of Formula (II) patented from Merck Patent GmbH
5.3. Aim of the thesis
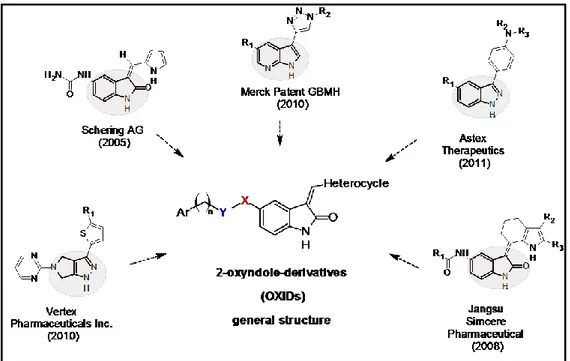
The principal aim of this work is to design and synthesise new ligands able to disrupt the PDK1/Akt pathway: this could be achieved through the development of molecules that act directly on Akt enzyme or hindering its activation by the simultaneous inhibition of PDK1 and Akt enzymes. Starting from an analysis of recent patented Akt/PDK1 pathway inhibitors chosen as lead structures, my PhD project aimed at the design and synthesis of new collection of 2-oxyindole derivatives (OXIDs). Although the 2-oxyindole nucleus has been widely investigated, and in particular as regards the effects induced by different type of substitutions in 3-position, only few cases regarded the insertion of electron reach groups in 5-position. On this bases, we synthesised OXIDs in which the C5 carbon
116 was substituted by different aromatic moieties anchored to the 2-oxyindole nucleus through different amidoalkyl chains.
Figure 5.13: Design of OXIDs starting from the analysis of recent patented Akt pathway inhibitors.
The initial optimization efforts focused on modifications at the 3 position by the insertion of small heteroaromatic residues. Moreover the 5 position of the oxindole was substituted with different amidoalkyl moieties. On this basis I synthesized the final products 1a-d and its intermediate 12.
N H N O O H N MeO MeO R N H X O H Heterocycle Ar [ ]n OXIDs N H N O O H N MeO MeO
Figure 5.14: Structure of OXIDs derivates
Compd R Compd R Z-1a S Z-1c N H N E-1b N N Me E-1d N 12 1a-d
117
5.3.1. Synthesis
The desired compounds 1a-d were synthesized according to the procedure reported in Scheme 5.1.
The amino derivative 10 was obtained by catalytic hydrogenation of the commercial 5-nitro-1,3-dihydro-2H-indol-one. The subsequent reaction of 10 with 2-chloroacetyl chloride afforded the 2-chloro-N-(2-oxo-2,3-dihydro-1H-indol-5-yl)acetamide 11, which was submitted to the reaction with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride in presence of K2CO3 to give the derivative 12.
Final compounds 1a-d were obtained by reaction of derivative 12 with 2-thiophencarboxaldehyde, 1-methyl-5-imidazolecarboxaldehyde, 1H-imidazole-4-carbaldehyde and 2-pyridinecarboxaldehyde, respectively, in the presence of a catalytic amount of pirrolidine.
Scheme 5.1 N H O2N O N H H2N O N H N O O Cl H N H N O O H N MeO MeO N H N O O H N MeO MeO R 10 1a-d I II III IV 12 11
Scheme 5.1: Reagents and conditions: I: H2, Pd/C, EtOH, r.t.; II: ClCOCH2Cl, Acetone, DMF, r.t.;
III: K2CO3, DMF, CH3CN, 82°C; IV: Appropriate carboxaldehyde, EtOH, pirrolidine, 110°C.
NMR analysis of these compounds indicated differences in E and Z-isomer populations. The compounds 1a and 1c exist as Z-isomers, while 1b and 1d adopts the E-isomeric form.
118 Z-Isomers E-Isomers 1a 1c 1b 1d N H O H R' R H C2 C3 C4 C5 C6 C7 N H O NH H O N MeO MeO S N H O NH H O N MeO MeO N H N N H O NH O N MeO MeO H N N Me N H O NH O N MeO MeO H N
Figure 5.15: Structure of E and Z isomer populations.
Compound H-C4 H-vinyl Compound H-C4 H-vinyl
1a(Z) 8.09 7.65 1b(E) 9.27 7.47
1c(Z) 7.93 7.60 1d(E) 8.88 7.42
The chemical shifts for the protons at the C4 were around 7.90-8.10 ppm for the Z-isomer but 8.80-9.30 ppm for the E-Z-isomer. The vinyl proton presents chemical shift value around 7.60-7.70 ppm for the Z-isomer but 7.40-7.50 ppm for the E-isomer. The Z-isomerism may be the result of the electrostatic interaction between the C-2 carbonyl oxygen atom of the indolin-2-one ring and the sulfur of the thiophene ring (1a) or the proton on the N-1of imidazole ring (1c). On the contrary, the electrostatic repulsion between the C-2 carbonyl oxygen atom and the methyl group on N-1-imidazole (1b) or with the long pair of pyridine nitrogen (1d) could stabilize the form of the E-isomer. The configurations of the compounds were assigned by comparison of their 1H NMR spectrum with those of analogues previously synthesized14.
5.3.2. Results and discussion of the derivatives 5.3.2.1. Cytotoxicity
Compounds 1a-d and 12, as well as the reference drug perifosine (a Akt inhibitor in phase II of clinical trials), were submitted to a preliminary
119 pharmacological evaluation on A549 cell line, a non-small-cell lung cancer (NSCLC) characterised by aberrant Akt expression15.
A dose-dependent inhibition of cell growth was observed with perifosine and OXIDs derivatives (1a-d, 12) after 24, 48, 72h in A549 cell line (Fig. 5.16A). The exposure to 72h is the best one to highlight the cytotoxic effect. Compounds 1a-d and 12 (Fig.
5.16B) exhibit potency level in the micromolar/submicromolar range. In particular
compound 1a showed a potency (IC50= 0.66±0.07µM) 6-fold greater than the
reference drug (Perifosine, IC50 values of 4.17±0.83µM). Moreover, also compounds
1b and 1c showed appreciable cytotoxicity with IC50 value of 2.75 and 2.19 µM,
respectively.
Figure 5.16: A) Concentration-dependent cytotoxicity of perifosine and Akt inhibitors in A549 cancer cells. Each data point represents the percentage of proliferating cells with respect to untreated control and is the average of three independent experiments. Bars, s.d. B) Values of IC50 of the compounds
120
5.3.2.2. Induction of apoptosis
The new compounds were also characterized for their ability to induce apoptosis, on A549 cell line. Upon exposure at IC50 values to perifosine and OXIDs
derivatives (1a-d, 12), lung cancer cells presented typical apoptotic morphology with cell shrinkage, nuclear fragmentation and cellular rupture into debris. The occurrence of apoptosis was significantly higher in cells treated with new compounds than control. In particular, the OXIDs induced cell death with apoptotic index values ranging from 12% (12) to 24% (1a), in all cases lesser than perifosine (32%).
Figure 5.17: Percentage of cells with damaged DNA after perifosine and Akt inhibitors exposures in A549 cell line. Upper panels, morphological appearance of control and treated cells
5.3.2.3. Effects of treatments on cell cycle
Besides Akt signaling pathway plays a crucial role for cell survival, it modulates cell cycle progression16.
Taking this into account, also the cell-cycle distribution was analyzed. Data were collected by flow cytometry in A549 cells exposed to OXIDs 1a-d and 12 and perifosine. The results are reported as percentage variation on cells population in G1, S and G2 phases (Fig. 5.18). Perifosine, enhanced cellular population in the G1 phase with respect to control of 18.56% of cells. Treatment with 1a and 12 caused
a significant increase in the population of cells in the G1 phase (1a 21.04%, 12
C Perifosine 1a 1b 1c 1d 12 0 5 10 15 20 25 30 35 A po pt ot ic Ind e x ( % ) * * * * * * Drugs Control Perifosine 1a
121 19.24%, respectively) associated to a reduction of G2/M phase and a relative loss of S-phase population, as a result of the inhibition of DNA synthesis. This preliminary data indicate that OXIDs 1a and 12 are able to affect both cell survival and cell cycle progression confirming a possible disruption of Akt pathway. On the contrary, compounds 1b and 1d resulted to be unable to have a significant effect on cell cycle progression.
Figure 5.18: Cell cycle distribution analysis by DNA flow cytometry. (A) A549 cells were treated with Control, Perifosine and 1a. The cell cycle distribution was analyzed by flow cytometry. (B) The percentage of cells in G1, S, G2/M phase.Values (%) are means from three independent experiments and differences (Δ) are calculated with respect to control.
5.3.2.4. Inhibition of Akt phosphorylation
To prove that the ability of OXIDs to inhibit cells growth and induce apoptosis should be related with the inhibition of Akt phosphorylation, the % of P-Ser473 and P-Thr308 in A459 cell lines were evaluated through ELISA technique. As reported in Figure 5.19 the OXIDs derivatives (1a-d, 12). determined a reduction of phosphorylation on both the aminoacids thus indicating their ability to affect the Akt activation. This result has been observed also for the reference drug perifosine (-20.0% pSer473, -25.0% pThr308). The results in Figure 5.19 show that the most
122 interesting compounds able to reduce the phosphorylation of both Ser and Thr are 1a and 1d.
Figure 5.19: Reduction of pSer473 and pThr308 Akt by perifosine, 1a-d and 12 in A549 cell line.
Starting from successful results obtained from the OXIDs compound, we carried out a systematic medicinal chemistry optimization in order to increase the drug-like properties of this class of derivatives. In particular we afforded the following investigation:
the importance of the bioisosteric replacement of the tetrahydroisoquinoline moiety with benzyl structure, and the substitution of the amido group with the amidosulfonyl ones;
the influence of the replacement of the amido group of 1a with 5-sulfonylamido moiety;
the insertion on elettron-rich linker such as a ureidic alkyl chain or ureidosulfonyl alkyl group
123
5.3.3. Substitution of the tetrahydroisoquinoline moiety
On the basis of biological results and in order to investigate the role played by
the 5-position substituent we decided to study the effect of the replacement of the
tetrahydroisoquinoline moiety with the methoxybenzylamine derivative endowed of fewer steric constrains.
Figure 5.21: Structure of compound 2.
5.3.3.1. Synthesis
The analogue 2 was synthesized following the synthetic procedure illustrate in Scheme 5.2. The chloroacetamide 11 was subjected to alkylation reaction with 3,4-dimethoxybenzylamine to give derivative 13. The subsequent condensation of compound 13 and thiophencarboxaldehyde afforded compound 2 as Z-isomer.
Scheme 5.2 N H NH O Cl O N H NH O NH O MeO MeO N H NH O NH O MeO MeO S 11 13 2 I II
Reagents and conditions: I: 3,4-Dimethoxybenzylamine, K2CO3, DMF, CH3CN, 82°C; II:
124
5.3.3.2. Results and discussion
The acetamido derivatives (1a-d, 2 and 12) were evaluated for their ability to inhibit the protein kinases: AKT1 (PKB alpha) and PDK1 Direct. The compounds were tested in duplicate up to 10µM. The inhibitory activity was evaluated by Invitrogen Corporation provided by a Z’-LYTE kit assay17.
Results are depicted in table 5.1.
Table 5.1: AKT1 (PKB alpha) and PDK1 inhibition data of synthesised compounds a
Tested in duplicate.
Data reported in table 5.1 indicate that the % inhibition at 10µM against Akt of compounds synthesize is appreciable only for compound 1a (37%). All the other seem to be inactive.
The compounds displaying no effect on PDK1 with the exception of the derivative 1c that exhibited a good inhibitory selectivity with a % value of 92% in a concentration of 10µM. To evaluate the potency of 1c has been submitted to further study to determine of IC50 value. (Data not yet available)
N H NH O R O N MeO MeO NH NH O O N MeO MeO S 2 1a-c, 12 Compound R %Inhibition (10µM )a Akt1 PDK1 Z-1a S 37 0 E-1b N N Me 14 - 23 Z-1c N H N 13 92 E-1d N 8 - 2 Z-2 / 8 - 3 12 H 3 - 29
125
5.3.4. Substitution of the acetamido linker with a 5-aminosulfonyl chain
In order to improve Akt/PDK1 inhibition activity of OXIDs derivatives and better understand the SAR of these class of compounds we decided to afford the amido group replacement with its bioisosteric amidosulfonyl moiety. Sulfonamide is well-known pharmacofore commonly notourius a key element to confer anticancer property18.
Figure 5.22: General structure of compounds of series B.
The compounds synthesized were 5-mesyl substituted (3a-c) or 5-tosyl substituted
(4a-c); moreover the C3 position of oxindole has been replaced with small heteroaromatic residues such as thiophene, 1-methyl-midazole or imidazole.
N H O NH SO2 Me R N H O NH SO2 Me R 4 a-c 3 a-c N H O NH SO2 R' R SERIE B
Compd R Compd R Compd R
a S b N N Me c N H N
126
5.3.4.1. Synthesis
The compounds 3a-c and 4a-c were obtained following the synthetic procedure illustrated in Scheme 5.3. The 5-amino-2-oxindole 10 reacted with p-toluenesulfonyl chloride or methanesulfonyl chloride to give derivatives 14 or 15, respectively. Then the reaction of 14 and 15 with appropriate carboxaldehyde afforded the final compounds 3a-c and 4a-c, respectively. The compounds 3a,4a and
3c,4c exist as Z-isomers, while 3b,4b adopts the E-isomeric form.
Scheme 5.3 N H O N H2 N H O NH SO2 Me N H O NH SO2 Me R N H O NH SO2 Me N H O NH SO2 Me R 10 15 4 a-c 14 3 a-c I II IV III
Compd R Compd R Compd R
a S b N N Me c N H N
Reagents and conditions: I: Methanesulfonyl chloride, H2O, r.t.; II: Appropriate carboxaldehyde,
EtOH, Pirrolidine, 110°C; III: p-Toluenesulfonyl chloride, H2O, r.t.; IV: Appropriate carboxaldehyde,
127
5.3.4.2. Results and discussion of the 5-amidosulfonyl chain
The 5-amidosulfonyl derivatives were evaluated for their ability to inhibit the protein kinases: AKT1 (PKB alpha) and PDK1 Direct. The compounds were tested by Invitrogen (see experimental part). Results are depicted in table 5.2.
Table 5.2: AKT1 (PKB alpha) and PDK1 inhibition data of synthesised compounds a
Tested in duplicate.
The results listed in table 5.2 displayed that methanesulfonamide derivatives showed higher % inhibition value than p-toluensulfonamide compounds (4a,b).
None of the compounds synthesize, showed to have an appreciable activity against Akt.
The compound 3a, substituted in 3-position with thiophene showed appreciable activity against PDK1 with a % of inhibition of 50%; the replacement with a methyl-imidazole, in the same position (3b) induced a decrease in the inhibitory activity. The compound 3c, that possess a imidazole group, showed the best inhibitory activity with a % value of 97% in a concentration of 10 µM.
To evaluate the potency of 3c, has been submitted to further study to determine of IC50 value. (Date not yet avaible)
N H NH O SO2 R1 R2 Compound R1 R2 %Inhibition (10µM )a Akt1 PDK1 Z-3a Me S 9 50 E-3b Me N N Me 0 - 13 Z-3c Me N H N 5 97 Z-4a Me S 6 -19 E-4b Me N N Me 2 - 15
128
5.3.5. The replacement of the 5-amido moiety with 5-sulfonylamido group
To better understand the role played by 5-position substituent in of 2-oxindole, we replaced the amido function of 1a with a 5-sulfonylamido ones to obtain derivatives of type C.
Figure 5.23: General structure of compounds of series C.
5.3.5.1. Synthesis
Compounds 5a-c and 6a-c were synthesized following the synthetic procedure illustrated in Scheme 5.4. The reductive cyclization of 2-nitrophenylacetic acid with Fe and acetic acid gave indolin-2-one 16. Then the reaction with chlorosulfonic acid afforded the 5-(chlorosulfonyl)indolin-2-one 17. The subsequence reaction with methoxybenzylamine (23a,b), yielded methoxybenzyl-2-oxoindoline-5-sulfonamide 18 or 19, wich reacted with the appropriate carboxaldehydes affording compounds 5a-c and 6a-c. The reaction afforded exclusively Z-isomers for 5a,6a and 5c,6c and E -isomers for 5b,6b.
129 Scheme 5.4 O NO2 OH N H O N H O S O O Cl + R' OMe N H2 N H O SO2 NH R' MeO R N H O SO2 NH R' MeO R'= H 5a-c R'=OCH3 6a-c I II III IV 17 16 R'= H 18 R'=OCH3 19
Compd R Compd R Compd R
a S b N N Me c N H N R'= H 23a R'=OCH3 23b
Reagents and conditions: I: Fe, AcOH, reflux, 2h.; II: ClSO2OH, r.t., 1.5h, 68°C, 1h; III:
Appropriate methoxybenzylamine, EtOH, r.t., 4 h; IV: Appropriate carboxaldehyde, EtOH, pirrolidine, 110°C.
5.3.5.2. Results and discussion of the 5-sulfonylamido chain
The 5-sulfonylamido derivatives were evaluated for their ability to inhibit the protein kinases: AKT1 (PKB alpha) and PDK1 Direct. The compounds were tested by Invitrogen (see experimental part). Results are depicted in table 5.3.
130
Table 5.3: AKT1 (PKB alpha) and PDK1 inhibition data of synthesised compounds a
Tested in duplicate.
Table 5.3, report the activity against both Akt and PDK1 expressed as % inhibition
of the compounds synthesize. None of the compounds synthesize, showed to have an appreciable activity against Akt.
The substitution of thiophene in 3-position 5a, with other heteroaromatic residue (methyl-imidazole, imidazole) not proved to effect on the activity (5b, 5c).
The replacement of the methoxy substituent 5a with the analog di-substituted 6a induced an increased of % of inhibition against PDK1 with a value of 107% in a concentration of 10µM. To evaluate the potency of 6a, has been submitted to further study to determine of IC50 value. (Date not yet avaible)
5.3.6. Substitution of the acetamido chain with a elettron-rich linker
A further investigation on 5-position of 2-oxindole carried out through replacement of an acetamido chain with an elettron-rich moiety. This chemical manipulation was aiming at increase the possible interaction.
N H SO2 O NH R2 MeO R1 Compound R1 R2 %Inhibition (10µM )a Akt1 PDK1 Z-5a H S -3 -11 E-5b H N N Me -8 - 26 Z-5c H N H N 0 6 Z-6a OMe S 9 107 E-6b OMe N N Me -1 - 47 Z-6c OMe N H N -4 -34
131 On this basis compounds of type D and derivatives of series E were synthesized. These two series differ each other for the linker in 5-position: a ureido alkyl moiety or (series D) or a ureidosolfonyl ones (serie E).
Actually, no data about the activity against Akt and PDK1 is available for derivatives.
Figure 5.24: General structure of compounds of series D and E
5.3.6.1. Synthesis of urea-derivatives of serie D
The compounds 7a-c and 8a-c were obtained following the synthetic procedure illustrated in scheme 5.5. The 5-amino-2-oxindole 10 reacted with 4-(methylthio)phenyl-isocyanate to give derivative 20; then the reaction with the appropriate carboxaldehyde afforded the final compounds 7a-c. Similarly, at the previously synthesize compound 7a and 7c as exist Z-isomers, while 7b exist as E-isomer. Oxidation with (1.2eq) oxone in MeOH-THF yielded final products 8a-c.
132 Scheme 5.5 I II N H O N H2 SMe OCN
+
N H O NH NH O MeS N H O NH NH O MeS R N H O NH NH O MeO2S R III 10 20 7 a-c 8 a-cCompd R Compd R Compd R
a S b N N Me c N H N
Reagents and conditions: I: Ar-NCO, DCM/methanol, 10°C–r.t., 3–4h; II: Appropriate carboxaldehyde, EtOH, pirrolidine, 110°C; III: Oxone, MeOH/THF, r.t., 12h.
133
5.3.6.2. Synthesis of sulfonilureido-derivatives of serie E
Compounds 9a-c were synthesised following the synthetic procedure illustrated in Scheme 5.6. The 5-nitro-2-oxindole was condensing with the appropriate carboxaldehyde afford the analogous 21a-c; 21a and 21c as Z-isomers, and 21b as E-isomer. The nitro derivative was then reduced to the corresponding amine 22a-c with hydrazine hydrate in the presence of a catalytic amount of ferric chloride and activated carbon. Treatment of 22a-c with commercial p-Toluenesulfonyl isocyanate afforded the final compounds 9a-c.
Scheme 5.6 N H O R O2N N H O N H2 R + Me OCNO2S I II III N H O O2N N H O NH R NH SO2 O Me 21a-c 22a-c 9 a-c
Compd R Compd R Compd R
a S b N N Me c N H N
Reagents and conditions: I: Appropriate carboxaldehyde, EtOH, pirrolidine, 110°C; II: H2, Pd/C,
134
5.3.7. Conclusion
In conclusion, taking together, these pharmacological results on A549 cell line seems to confirm that the choice of 2-oxindole nucleus as central core to develop new ligands able to disrupt PDK1/Akt pathway has been successful.
Concerning the compounds synthesized, the results show that the substituent in 3-posizion on the 2-oxindole nucleus is able to effect their activity. In particular in tetrahydroisoquinoline compounds, derivatives in a Z-configuration is the most active ligand for its ability to inhibit the cell growth; as concern the derivatives of E-configation show a reduced activity with respect to Z-isomer.
The substitution in 5-position seems to be detrimental for the activity against Akt. Moreover, the results of this study showed that the replacement of amido moiety with solfonamido or amidosulfonil group increases the selectivity against PDK1. Unfortunately, the effects produced by the replacement of the amido groups with ureido alkyl or ureidosolfonyl moiety is not avaible.
Futher investigation to assign the observed pharmacological effects to a direct inhibition of Akt, PDK1 or both enzymes are in progress.
135
Reference chapter 5
1. Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, McCubrey JA, Travali S. Int J Oncol. 2011, 4(5), 771-7.
2. Vivanco I, Sawyers CL. National Review Cancer. 2002, 2, 489-501.
3. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings
BA. EMBO J. 1996, 15(23), 6541-51.
4. T. Maffucci, E. Piccolo, A. Cumashi, M. Iezzi, A.M. Riley, A. Saiardi, H.Y.
Godage, C. Rossi, M. Broggini, S. Iacobelli, B.V. Potter, P. Innocenti, M. Falasca,
Cancer Res. 2005, 65 (18), 8339–8349.
5. C.W. Lindsley, Z. Zhao, W.H. Leister, R.G. Robinson, S.F. Barnett, D.
Defeo-Jones, R.E. Defeo-Jones, G.D. Hartman, J.R. Huff, H.E. Huber, M.E. Duggan, Bioorg. Med.
Chem. Lett. 2005, 15 (3), 761–764.
6. S.F. Barnett, M.T. Bilodeau, C.W. Lindsley, Curr Top Med Chem. 2005, 5 (2),
109–125.
7. J.C. Hartnett, S.F. Barnett, M.T. Bilodeau, D. Defeo-Jones, G.D. Hartman, H.E. Huber, R.E. Jones, A.M. Kral, R.G. Robinson, Z. Wu, Bioorg. Med. Chem. Lett.
2008, 18 (6), 2194–2197.
8. Liu Y, Gray NS. Nat. Chem. Biol. 2006, 2 (7), 358-64.
9. Alexandra Hofler et al. Analytical Biochemistry, 2011, 414 (2), 179–186.
10. Islam, Imadul; Bryant, Judi; Chou, Yuo-Ling; Kochanny, Monica J.; Lee,
Wheeseong; Phillips, Gary B.; Yu, Hongyi; Adler, Marc; Whitlow, Marc; Ho, Elena; et al . Bioorganic & Medicinal Chemistry Letters. 2007, 17(14), 3814-3818.
11. Tang, Feng; Shen, Han; Jin, Qiu; Ding, Lei; Yang, Jie; Yin, Xiaojin; Lu, Shiyue.
PCT Int. Appl: WO 2008067756 A1 20080612, 2008.
12. Binch, Hayley; Everitt, Simon; Mazzei, Francesca; Robinson, Daniel. PCT Int.
Appl: US 20100087467 A1, 2010.
13. Dorsch, Dieter; Wucherer-Plietker, Margarita; Mueller, Thomas J. J.; Merkul,
Eugen. PCT Int. Appl: WO 2010127754 A1 20101111, 2010.
14. Li Sun, Ngoc Tran, Flora Tang, Harald App, Peter Hirth, Gerald McMahon, and
Cho Tang. J. Med. Chem. 1998, 41, 2588-2603
15. Tsurutani J, Castillo SS, Brognard J, et al. Carcinogenesis. 2005, 26(7),
136
16. H Sun et al. Proc Natl Acad Sci U S A. 1999, 96(11), 6199–6204.
17. Toral-Barza, L., et al. Molecular Cancer Therapeutics. 2007, 6 (11), 3028-3038.