Risultati
3. Risultati
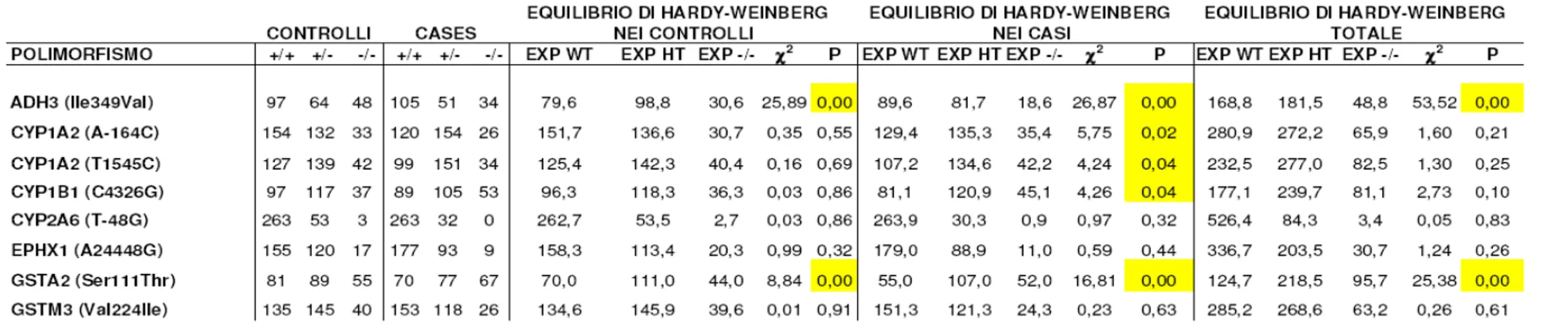
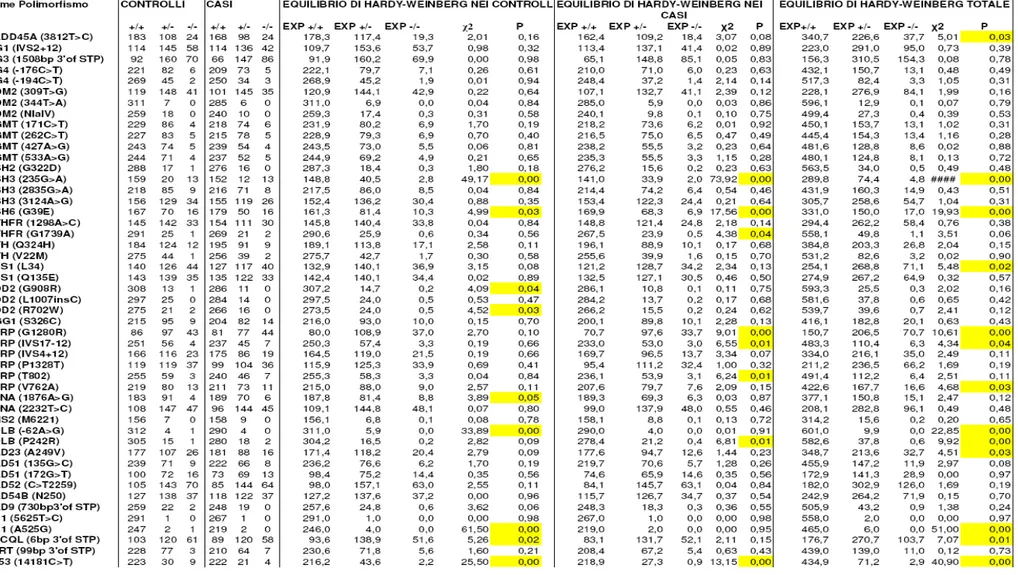
I polimorfismi nei geni ABCG2, ADH1B, ADH2, ALDH2, COMT, CYP1A1, CYP2C9, CYP2D6, CYP2C19, CYP2C18, CYP2E1, CYP3A4, CDA, DRD2, DRD4, GRPR, GSTA4, GSTM1, DIA4/NQO1, GSTP1, GSTT1, GSTT2, IRS2, MDR1, MNSOD2, NAT1, NAT2, NT5E, SULT1A1, SULT1A2, TERC, SLC6A3/DAT1, UGT1A7, non hanno mostrato associazioni statisticamente significative con il rischio di tumore polmonare (Tabella 1 a,b,c; Tabella 2 a,b,c). Abbiamo invece trovato associazioni per i geni: ADH3/ADH1C (polimorfismo Ile349Val), CYP1B1 (polimorfismo C4326G), CYP1A2 (polimorfismi A-164C; T1545C), CYP2A6 (polimorfismo T-48G), EPHX1 (polimorfismo A24448G), GSTA2 (polimorfismo Ser111Thr) e GSTM3 (polimorfismo Val1224Ile) (Tabella 3; Tabella 4).
ADH3/ADH1C. Il polimorfismo Ile349Val nel gene ADH3/ADH1C mostra un OR statisticamente significativo di 0,56 per gli omozigoti per l’allele più raro nel modello codominante, con un intervallo di confidenza compreso tra 0,31 ed 1. Il fatto che gli eterozigoti appaiano con un rischio intermedio (OR=0.74) (ma statisticamente non significativo) e il trend test risulti “borderline” suggerisce la possibilità di una codominanza.
CYP1A2. Abbiamo osservato un valore statisticamente significativo per due polimorfismi del gene CYP1A2 (A-164C; T1545C). Il polimorfismo A-164C di CYP1A2 ha mostrato OR significativi sia utilizzando un modello codominante sia secondo un modello dominante. L’OR per l’eterozigote nel modello codominante è risultato essere di 1,61 con un intervallo di confidenza tra 1,12 e 2,32. L’OR secondo il modello dominante invece è di 1,40 in un intervallo di confidenza compreso tra 1,02 e 1,93. Il polimorfismo T1545C del gene CYP1A2 ha mostrato invece nel solo modello codominante un OR di 1,54 per l’eterozigote, con un intervallo di confidenza tra 1,05 e 2,26.
CYP1B1. CYP1B1 (polimorfismo C4326G O Val432Leu), ha mostrato un valore significativo di OR di 1,74, secondo il modello codominante, nell’omozigote per l’allele raro, con un intervallo di confidenza compreso tra 1,3 e 0,3.
CYP2A6. Per il polimorfismo T-48G dI CYP2A6 osserviamo un OR statisticamente significativo di 0,56 per gli eterozigoti nel modello codominante, con un intervallo di
Risultati
confidenza compreso tra 0,34 e 0.94. Inoltre con il modello dominante è risultato un OR altrettanto significativo di 0,57 in un intervallo di 0,36-0,91.
EPHX1. Per il polimorfismo A24448G dell’ EPHX1 abbiamo ottenuto valori significativi di OR sia nell’omozigote per l’allele raro che nell’eterozigote seguendo il modello codominante. Infatti per l’omozigote l’OR è risultato 0,46 con un intervallo di confidenza compreso tra 0,19 e 1,12; per l’eterozigote invece l’OR è risultato di 0,66 in un intervallo di confidenza di 0.45-0.97. Il trend test significativo suggerisce un effetto dose-dipendente del rischio in funzione della quantità di alleli varianti. Il modello dominante fornisce un OR significativo uguale a 0,65 (intervallo di confidenza compreso tra 0,47 e 0,91).
GSTA2. Per il polimorfismo Thr111Ser di GSTA2 abbiamo ottenuto un OR di 1,78 (intervallo di confidenza di 1,05-3,02) (secondo il modello codominante) nell’omozigote per l’allele raro.
GSTM3. Il polimorfismo Val224Ile del gene GSTM3 ha mostrato, secondo il modello codominante, un OR per l’eterozigote di 0,69 (intervallo di confidenza di 0,48-1); mentre l’OR per l’omozigote raro ugualmente significativo è di 0,55 (intervallo di confidenza di 0,31-0.99). Secondo il modello dominante inoltre l’OR di 0,69 con un intervallo di confidenza compreso tra 0,5 e 0,94, si è dimostrato statisticamente significativo mostrando anche il questo caso una tendenza per questo enzima ad essere protettivo sul rischio di tumore al polmone.
Risultati
Risultati
Tabella 3. Varianti alleliche associate. Calcolo degli OR secondo il modello codominante, dominante e recessivo.
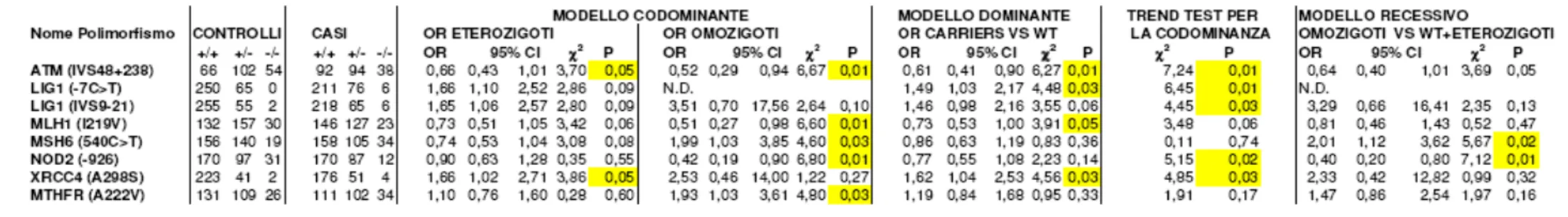
Risultati I risultati dell’associazione tra cancro polmonare e polimorfismi dei geni del controllo del ciclo cellulare e della stabilita’ genomca sono riportati nelle tabelle 5, 6a, 6b, 6c, 3 A, 3 B e 3 C. In particolare, abbiamo osservato associazione con i seguenti polimorfismi: ATM (C→G, introne 48), LIG1 (C→T, 5’UTR e A→G, introne 9), MSH6 (C→T, esone 3), MLH1 (A→G, codone 219), NOD2 (G→A, promotore), XRCC4 (G→A, codone 298) e MTHFR (C→T, esone 4).
Mediante l’analisi statistica si può notare che il polimorfismo C→G nell’introne 48 del gene ATM e’ associato ad un diminuito rischio di cancro al polmone (tabella 5). La riduzione del rischio appare seguire un modello di tipo codominante (trend test per la codominanza, statisticamente significativo), dove il genotipo eterozigote ha un OR di 0,66 (intervallo di confidenza al 95% tra 0,43 e 1.01) e quello omozigote di 0.52 (0,29-0.94). Per quanto riguarda LIG1, si osserva un rischio di 1.49 (intervallo 1.03-2.17) per i portatori dell’allele variante, nel modello dominante. La mancanza di omozigoti tra i controlli impedisce di calcolare il rischio per questo genotipo. E’ probabile che la variante intronica IVS9-21 entro lo stesso gene, risulti associata alla malattia in maniera statisticamente “borderline”, poiche’ i polimorfismi C→T (5’UTR) e A→G (introne 9) di LIG1 risultano avere un elevato livello di Linkage Disequilibrium. Il calcolo dei rischi effettuato per quest’ultima variante indica la possibilita’ di un effetto di codominanza (trend test statisticamente significativo). Il polimorfismo Isoleucina>Valina del codone 219 del gene MLH1, e’ associato ad un diminuito rischio della neoplasia (OR: 0.51 e 95%CI: 0.27, 0.98 per il genotipo omozigote nel modello codominante). Il valore del trend test non raggiunge formalmente la significativita’ statistica (p=0.06), nonostante il rischio degli eterozigoti (OR=0.73) risulti intermedio (tabella 5). Il genotipo omozigote per l’allele variante nel gene MSH6 risulta associato ad un aumento del rischio del tumore con un valore di OR di 1.99 e un intervallo di confidenza tra 1.03 e 3.85. Per questo gene il modello di recessivita’ sembrerebbe il migliore, in quanto gli eterozigoti appaiono avere un rischio non dissimile al riferimento. Quando si utilizzano questi ultimi due gruppi come riferimento, il rischio per gli omozigoti varianti risulta di 2.01 con un’alta significativita’ statistica (intervallo di confidenza al 95%: 1.12-3.62, tabella 1).
Il polimorfismo G→A nel promotore di NOD2/ CARD15 è associato ad una diminuzione del rischio del cancro con un OR: 0.42 e 95%CI compreso tra 0.19 e 0.9 per quanto riguarda il genotipo omozigote. Il valore del trend test risulta significativo, tuttavia i nostri dati potrebbero essere spiegati anche attraverso il modello recessivo in quanto gli
Risultati riferiti agli altri due genotipi raggruppati. Per il polimorfismo A298S del gene XRCC4, i nostri risultati indicano un’associazione con il tumore nel genotipo eterozigote (OR: 1.66 e 95%CI compreso tra 1.02 e 2.71). Individui omozigoti sembrano aver un rischio maggiore per la malattia ed anche il valore del trend test risulta significativo, indicando una possibile codominanza. Tuttavia il basso numero di campioni omozigoti per l’allele variante non permette di stabilire con fiducia il rischio per questa categoria. Infine, il polimorfismo Ala222Val di MTHFR è associato ad un aumento del rischio di cancro al polmone nei soli individui omozigoti (OR: 1.93 e 95%CI: 1.03, 3.61, tabella1).
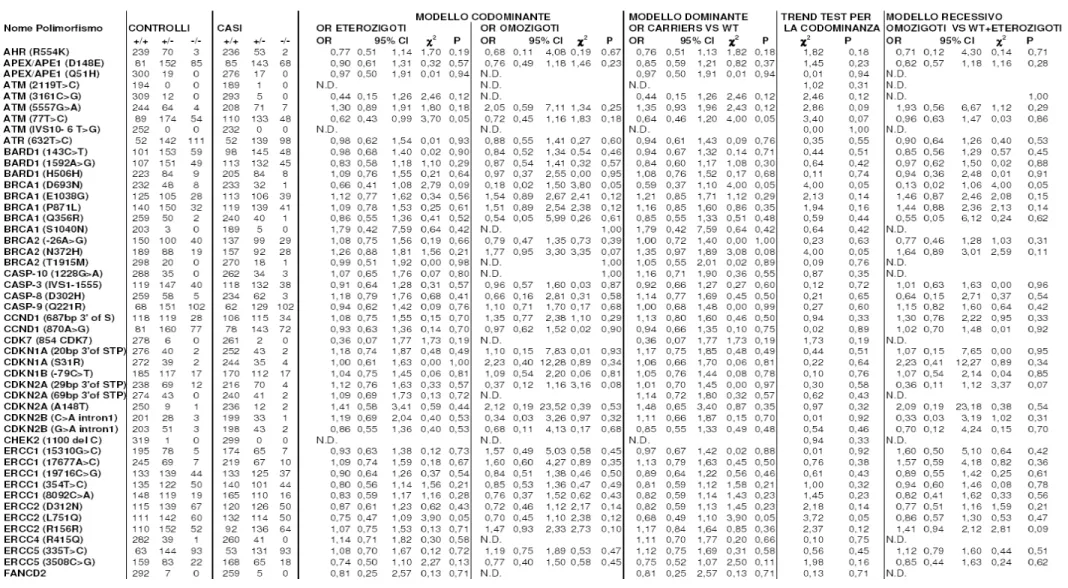
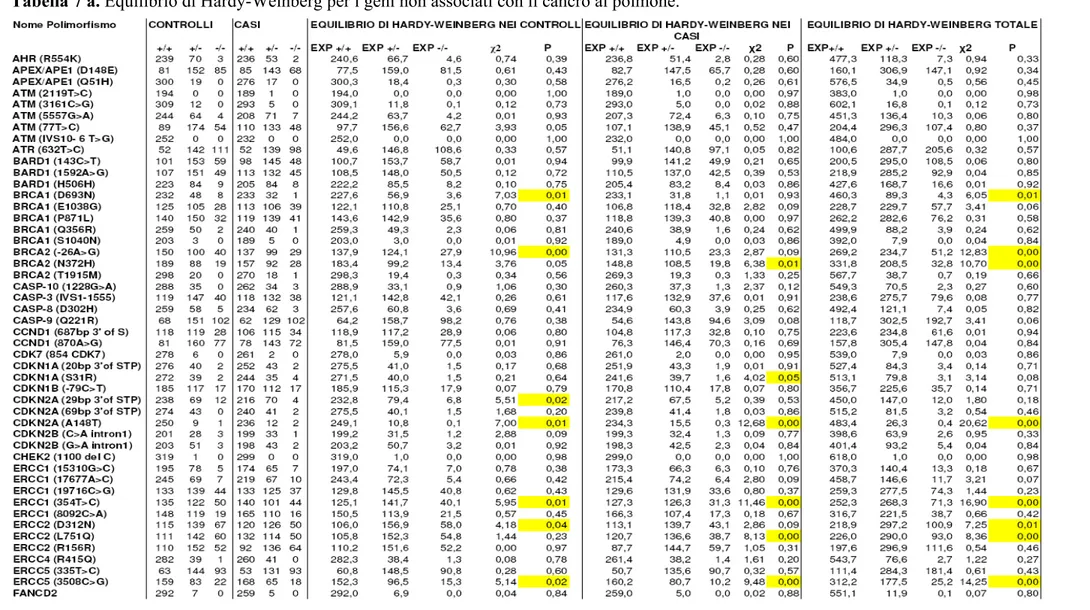
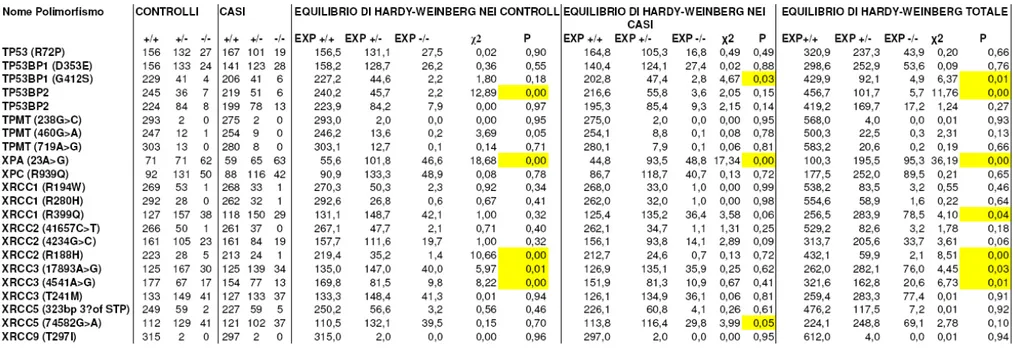
Come si nota dalle tabelle 2, 3, 4 5, i polimorfismi nei geni XRCC1, XRCC2, XRCC3, XRCC5, XRCC9/FANG, FANCD2, ERCC1, XPD/ERCC2, XPF/ERCC4, XPG/ERCC5, XPA, XPC, LIG3, LIG4, POLB, APEX/APE1, PCNA, OGG1, MGMT/AGT, GADD45A, MLH1, MSH2, MSH3, MYH, PMS2, RAD9, RAD 23, RAD51, RAD52, RAD54B, NBS1, RECQL, BARD1, BRCA1, BRCA2, PARP/ADPRT, TPMT, TP53, CDKN1A/P21, CDKN2A/P16, CDKN1B, CDKN2B, CCND1, CDK7, CHEK2, MDM2, CASP-3, CASP-8, CASP-9, CASP-10, RB1 e ATR non hanno mostrato associazioni statisticamente significative (tabelle 6 a, 6 b, 6 c, 7 a, 7 b, 7 c).
Risultati
Risultati
Risultati
Risultati
Risultati
Risultati
Risultati
Discussione
4. DISCUSSIONE
Il cancro è una malattia multi-fattoriale, la sua insorgenza non può essere messa in relazione solo all’esposizione alle sostanze cancerogene, ma dobbiamo considerare anche altre componenti che possono influenzare il rischio di sviluppare il tumore. Infatti nonostante sia un dato di fatto che l’esposizione a fattori ambientali come il fumo di tabacco e la dieta può significativamente determinare lo sviluppo del cancro, la variabilità genetica e quindi l’interazione gene-ambiente può spiegare la suscettibità individuale alla malattia. Il nostro organismo è dotato di un complesso sistema di enzimi atto a biotrasformare molti composti endogeni presenti nelle cascate metaboliche che stanno alla base dei processi naturali quali la crescita, lo sviluppo, la differenziazione, l’omeostasi, l’apoptosi, etc. Ma questi enzimi svolgono anche l’importantissimo ruolo di detossificazione di molti composti esogeni, in genere facilitandone l’escrezione, anche se durante questi processi si producono degli intermedi molto reattivi che hanno una notevole capacità tossica e cancerogena.
Poiché gli enzimi del metabolismo di queste sostanze esogene, chiamate collettivamente xenobiotici, presentano più di una variante polimorfica, ne deriva che il corretto funzionamento di tutto il sistema enzimatico dipende strettamente dai fattori genetici che determinano il grado di attività di ciascun enzima polimorfico. Perciò è chiaro che a parità di esposizione, gli individui che hanno un’alterata capacità detossificante corrono un rischio più alto di sviluppare un tumore rispetto agli individui che presentano una capacità “normale”. Ad esempio, una diversa capacità di metabolizzare i composti contenuti nel fumo di tabacco può spiegare perché, all’interno di un gruppo di fumatori elevato, solo una frazione (1/10 circa) si ammala di cancro al polmone (Kiyohara et al., 2002).
Nel nostro studio abbiamo considerato i polimorfismi di geni della fase I e II e ci siamo chiesti se l’elevato grado di polimorfismo di questi geni influenzi la suscettibilità dei tumori polmonari, per i quali il principale fattore eziopatologico è il fumo di tabacco.
Per quanto riguarda i geni di fase I del metabolismo, dall’analisi dei dati è emersa un’associazione significativa con: CYP1B1, CYP1A2 e CYP2A6.
Discussione
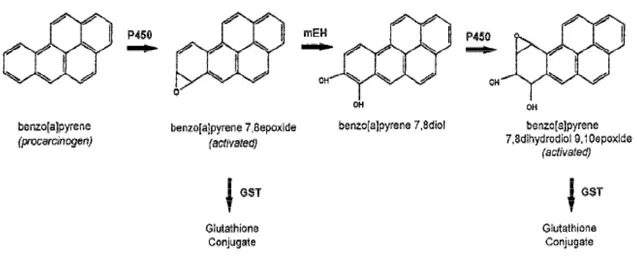
CYP1B1, CYP2A6 e CYP1A2 fanno parte della grande famiglia dei citocromi P450, enzimi che sono implicati in svariate reazioni cellulari volte alla funzionalizzazione di numerose molecole endogene ed esogene. Questi enzimi hanno la capacità di idrossilare i substrati mediante una reazione di mono-ossigenazione. Per farè ciò necessitano di ossigeno e del coenzima (fattore riducente) NADPH, necessario per donare i protoni. La reazione catalizzata è riportata nella figura 22.
Fig. 22. Rappresentazione ciclica della reazione catalizzata da un citocromo p450. Il substrato (RH) da idrossilare è specificamente diverso da citocromo a citocromo.
Si è visto che gli omozigoti per l’allele variante C4326G del gene CYP1B1 mostrano un rischio più alto rispetto alle altre categorie. Questo gene presenta inducibilità da parte degli idrocarburi aromatici planari (Sutter et al., 1994; Otto et al., 1991; 1992). Nel topo sono stati evidenziati due tipi di regolazione, in base al tessuto: in tessuti dove si ha la sintesi di ormoni steroidei la regolazione di questo enzima è sopratutto ormonale, mentre in altri tessuti come nel caso del polmone, l’induzione è maggiormente legata al recettore per gli idrocarburi policiclici aromatici (IPA) (Bhattacharyya et al., 1995).
Discussione
CYP1B1 è coinvolto nel metabolismo degli xenobiotici (Otto et al., 1991; Shimada et al., 1996), ma in particolare degli ormoni steroidei (Walker et al., 1995; Spink, et al., 1990; Hayes et al., 1996): infatti la formazione del 4-idrossi-estradiolo è specificamente catalizzata dal CYP1B1 (Zheng et al., 2000). Inoltre, CYP1B1 appare indotto dall’estradiolo medesimo (Tsuchiya, 2004).
La variazione C4326G, che nel nostro studio è risultata associata ad un aumento di rischio di cancro polmonare, causa la sostituzione di una leucina con una valina al codone 432 dell’esone 3. La stessa variazione è stata in precedenza associata ad incrementati rischi di cancro della prostata (Tang et al., 2000). Preparati microsomali di polmone umano sembrano indicare che la variante Val432 costituisca l’allele ad incrementata attivita’ (Tang et al., 2000), così possiamo supporre che l’incremento della formazione del 4-idrossi-estradiolo, e/o in altrernativa, del metabolismo di alcuni IPA, porti ad aumenti importanti nella formazione di intermedi cancerogeni e quindi, in definitiva, che il genotipo Val432 costituisca un fattore di rischio per alcune forme di cancro, incluso quello polmonare.
Anche per l’enzima CYP1A2, in base ai nostri risultati, è stata evidenziata un’associazione con il rischio di tumore al polmone: i portatori dell’allele A-164C presentano un incremento di rischio di cancro. In questo caso l’allele variante è associato ad una aumentata inducibilità. È assai probabile invece che l’associazione del polimorfismo T1545C dello stesso gene come fattore di rischio, essendo una variazione silente (Asn516Asn), sia dovuta al linkage disequilibrium con l’altro polimorfismo. La proteina codificata da questo gene è localizzata nel reticolo endoplasmatico. Non si conosce in modo preciso il suo specifico ruolo sul metabolismo dei substrati endogeni. Il citocromo CYP1A2 come CYP1A1 e CYP1B1 è indotto dagli IPA attraverso i recettori Ah (Bartsch et al., 2000), e dalle ammine aromatiche (Pavanello e Clonfero, 2000). CYP1A2 ha un ruolo importante nel metabolismo delle amine aromatiche e degli IPA e un’accelerazione del loro metabolismo può portare ad un accumulo di composti cancerogeni (Landi, 1999).
Altro citocromo che nel nostro studio è stato associato al cancro al polmone è il CYP2A6. L’allele di suscettibilità determina diminuzione dell’attività dell’enzima.
Il citocromo CYP2A6 è stato isolato per la prima volta nel 1990, quando si scoprì la sua attività di 7-idrossilasi della cumarina (Miles et al., 1990; Yamano et al., 1990; Yun
Discussione
et al., 1991). Questo enzima è espresso prevalentemente nel fegato (Koskela et al., 1999) ed è responsabile del metabolismo di molti farmaci droghe e sostanze chimiche ambientali (Oscarson, 2001). È uno degli enzimi maggiormente coinvolti nel metabolismo delle nitrosamine: NNK [4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone], NNAL [4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanol], NDEA [N-nitrosodiethylamine], e NNN [N9-nitrosonornicotine] (Oscarson et al., 1998; Pelkonen et al., 2000). Inoltre studi in vivo hanno dimostrato che il CYP2A6 è il più importante citocromo responsabile della C-ossidazione della nicotina (Cashman et al., 1992; Nakajina et al., 1996; Messina et al., 1997), ma è anche coinvolto nella successiva ossidazione della cotinina a numerosi prodotti differenti (Nakajina et al., 1996; Murphy et al., 1999).
Proprio per il suo specifico ruolo nell’eliminazione della nicotina è stato proposto che questo enzima possa influenzare l’abitudine al fumo. Inoltre se si riuscirà a dimostrare ciò, è già stato suggerito di utilizzare l’inibizione di questo citocromo come terapia per curare la dipedenza dalla nicotina contenuta nel tabacco (Sellers et al., 2000). Secondo quanto detto fino ad ora, l’assenza o la diminuzione dell’attività dell’enzima CYP2A6 causate da specifici polimorfismi, dovrebbero, teoricamente, modulare il rischio di cancro al polmone in due modi. Il primo, se l’ipotesi di Pianezza e dei suoi collaboratori (1998) è corretta, è che gli individui che sono deficienti dell’enzima siano appagati da un numero ristretto di sigarette. Il secondo è che, siccome molti precancerogeni trovati nel fumo di tabacco come NNK, NDEA (nitrosodietilamina) e NNN sono metabolicamente attivati da CYP2A6 (Smith et al., 1992; Camus et al., 1993), una riduzione dell’attività dell’enzima potrebbe essere protettivo perchè si genererebbero meno metaboliti attivati e meno addotti del DNA. I nostri risultati sono in accordo con questa ultima ipotesi, in analogia ad altri studi precedenti. Infatti la variante enzimatica che abbiamo studiato presenta una diminuzione di espressione del 58% a causa della sostituzione di una guanina al posto di una timina in posizione -48 della regione TATA box (Pitarque et al., 2001; Tricker, 2002). La presenza di questo polimorfismo pare correlata con una diminuzione di rischio di cancro al polmone.
A seguire le reazioni di fase I, gli enzimi di fase II come le glutatione transferasi (GSTs), sono responsabili della detossificazione di forme attivate di procancerogeni
Discussione
come gli epossidi dei IPA. Le GSTs sono costitutivamente presenti in molti tessuti, con differenti isoenzimi che catalizzano reazioni di coniugazione con glutatione, perossidazioni, e isomerizzazioni (Pavanello, Clonfero, 2000). I geni GST formano una superfamiglia di 13 geni che viene divisa in cinque distinte classi chiamate alfa (GSTA), sigma (GSTS), mu (GSTM), pi (GSTP) e theta (GSTT) (Kiyohara et al., 2002). Alcuni componenti delle famiglie GSTM, GSTT, GSTP, sono polimorfici. e varianti a ridotta attività enzimatica sono state associata con l’istotipo SCC dato che l’eziologia di tale tipo di tumore polmonare è maggiormente legato all’esposizione agli idrocarburi aromatici (Risch et al., 2001). Il nostro studio ha evidenziato la presenza di un’associazione tra variante allelica Thr111Ser del gene GSTA2 e l’aumento di rischio di cancro al polmone. Non è nota l’attività di questa variante allelica, però in base al ruolo biologico di questo enzima, che lo vede coinvolto nella detossificazione di metaboliti attivati abbiamo ipotizzato che sia legato ad una diminuzione di attività, visto che la sua presenza rappresenterebbe un fattore di rischio. In letteratura sono apparentemente assenti studi di associazione volti a indagare il ruolo del GSTA2 nella suscettibilità al cancro al polmone.
Altro enzima che è stato in letteratura poco studiato ma che noi abbiamo trovato associato è GSTM3. La variante allelica Val224Ile presenta una maggiore attività (Tetlow, 2004) rispetto al selvatico e nel nostro studio è risultata essere un fattore di protezione per il cancro al polmone in individui di giovane età. I dati da noi ottenuti hanno una plausibilità biologica, infatti avere un attività potenziata di questo enzima aumenterebbe anche la capacità del tessuto polmonare di detossificare l’organismo dai metaboliti attivati e genotossici, scongiurando o comunque rendendo minore il rischi della formazione di addotti del DNA. Alcuni studi suggeriscono infatti che la proteina corrispondente del gene GSTM3 abbia, come GSTM1, un ruolo importante nel primo passaggio del metabolismo degli IPA (Nakajima et al., 1995)
Secondo i nostri risultati esiste anche una associazione tra cancro al polmone e la variante His139Arg (o A24448G) del gene EPHX1, il cui prodotto (la epossido idrolasi microsomale) è un enzima protettivo coinvolto nella generale difesa ossidativa contro molte sostanze ambientali epossidate (Oesch et al., 1973). Questo enzima però è anche coinvolto nell’attivazione metabolica di cancerogeni del tabacco (Kiyohara et al., 2002). Questa seconda azione si esplica nell’abilità di idrossilare metaboliti epossidici degli
Discussione
irocarburi aromatici a cancerogeni ancora più tossici come IPA diolo-epossido (Smith et al., 1997) (Gelboin, 1980; Miyata et al., 1999). L’epossido idrolasi microsomiale è altamente espressa in molti tessuti inclusi i polmoni dove catalizza l’idrolisi di vari intermedi reattivi epossidici in composti meno tossici e più solubili in soluzioni acquose che vengono successivamente escreti dal corpo (Hassett et al., 1994; Smith et al., 1997). Infatti catalizza l’addizione di una molecola di acqua a un epossido per formare un diidrodiolo (Pavanello et al., 2000). La reazione catalizzata è riportata in figura 23.
Recentemente un importante studio americano ha dimostrato che, a seconda della durata dell’esposizione al fumo di sigaretta, il fenotipo lento di questo enzima può generare un diverso grado di rischio. Infatti mentre nei non fumatori e fumatori passivi la presenza del polimorfismo che conferisce minore attività catalitica è legato ad un aumento di suscettibilità al cancro, negli accaniti fumatori con una esposizione prolungata diventa un fattore di protezione (Zhou et al., 2001).
Fig. 23. Reazioni catalizzate dai citocromi P450 e dall’Epossido Idrolasi (mEH).
Probabilmente l’accumulo dei prodotti del fumo di sigaretta in condizioni di prolungata esposizione, fa sì che venga accentuata l’importanza di questo enzima come metabolizzatore di fase I.
Discussione
Il nostro polimorfismo His139Arg determina invece un aumento di attività dell’enzima ed è risultato essere un fattore di protezione, infatti la sua presenza sembra associata con una diminuzione del rischio di cancro al polmone. Questi risultati si discostano da alcuni studi che hanno indicato il polimorfismo mEH4 come sfavorevole nei forti fumatori (Pavanello et al., 2000; Wu et al., 2001; Persson et al., 1999). In realtà ancora una volta dobbiamo ricordare che data l’età giovane dei nostri casi non si può parlare di una prolungata e massiccia esposizione alle IPA e che quindi in queste condizioni probabilmente prevale ancora l’importanza dell’enzima come detossificatore e non come attivatore.
Infine la variante allelica Ile349Val o SspI dell’alcol deidrogenasi 3 (ADH3/ADH1C) che presenta attività enzimatica minore è risultata associata ad una diminuzione del rischio di tumore polmonare. Questo enzima è implicato nel metabolismo dell’alcol e catalizza l’ossidazione dell’etanolo ad acetaldeide (Freudenheim et al., 2003). L’acetaldeide è un composto capace di interagire col DNA con il quale forma degli addotti (Yang et al., 2002).
Esistono sei geni ADH chiamati ADH1, ADH2, ADH3, ADH4, ADH5, ADH6. La classe I costituita dai geni ADH1, ADH2, ADH3, è quella maggiormente coinvolta nel metabolismo dell’alcol (Freudenheim et al., 2003) (Fig. 24).
NADPH + H+ NADP+ Etanolo H H C OH CH3 Acetaldeide H C O CH3
ADH3
Discussione
Tra gli isoenzimi ADH, solo ADH3 è geneticamente polimorfico nei caucasici (Shen et al., 1997). Studi precedenti hanno mostrato che il genotipo di ADH3 in condizioni di esposizione ad alcol è in grado di modulare il rischio di varie malattie croniche (Freudenheim et al., 1999; Day et al., 1991; Fan et al., 1997), cancro al seno (Freudenheim et al., 1999), cancro alla bocca e alla faringe (Harty et al., 1997; Coutelle et al., 1997). Per ritenere valida la nostra associazione dobbiamo supporre che insieme al consumo di tabacco ci sia anche un consumo di alcol e che l’alcol anche se in minor grado rispetto al fumo possa essere considerato un fattore di rischio di cancro polmonare (Potter et al., 1984; Levi, 1999). In letteratura sono presenti studi che supportano questa ipotesi. Infatti alcuni ricercatori hanno dimostrato che la variante allelica ADH3*1 è associata ad una elevata attività ed espressione enzimatica nel polmone e porta ad una alta quantità di addotti aldeide-DNA (Yang et al., 2002). Ciò ha fatto ipotizzare un possibile ruolo di questo enzima nel modulare il rischio di cancro al polmone in uno studio del 2003 condotto dal gruppo di Freudenheim. Tuttavia i risultati non sono stati significativi e quindi la possibile associazione tra varianti alleliche di ADH3 e il rischio di cancro al polmone ha ancora bisogno di ulteriori indagini (Fig. 24). In letteratura si possono rinvenire innumerevoli studi di associazione tra il cancro al polmone e i polimorfismi riguardanti geni implicati nella riparazione del DNA e nel controllo del ciclo cellulare. Alcuni di questi polimorfismi sono sospettati di alterare la capacità individuale di riparare il DNA, tuttavia le loro conseguenze biologiche e funzionali rimangono incerte.
Il gene XRCC1 (x- ray repair cross-complementing group1) codifica per una proteina multidominio che interagisce con geni implicati nel sistema di riparazione del DNA tramite l’eliminazione della base danneggiata (BER). Cellule in cui XRCC1 è mutato sono sensibili ad un gran numero di agenti tossici e hanno numerose instabilità genetiche come traslocazioni cromosomiche o delezioni (Ratnasinghe et al., 2001). In questo gene sono stati identificati alcuni polimorfismi e tre di questi sono stati esaminati in studi di associazione con il cancro al polmone: Arg194Trp, Arg399Gln, Arg280His (Ratnasinghe et al., 2001). E’ stato dimostrato che la variante allelica 194Trp è protettiva nei confronti della patologia tra coloro che abusano di sostanze alcoliche (David-Beabes and London, 2001). Per quanto riguarda il polimorfismo Arg399Gln
Discussione
alcuni studi suggeriscono associazione tra questa variante e aumento del rischio di cancro (Ratnasinghe et al., 2001; David-Babes e London, 2001). Zhou et al. suggeriscono, attraverso studi epidemiologi su Caucasici (180 casi), Afro-Americani (154 casi) e Coreani (192 casi), che tale polimorfismo sia associato ad un alto rischio solo tra coloro che fumano poco e ad un basso rischio tra quelli che fumano tanto. Inoltre il genotipo Gln/Gln rivela un marcato aumento di cancro al polmone in individui giovani piuttosto che negli anziani (Park et al., 2002 ). Il polimorfismo Arg280His tra i bevitori di alcool risulta associato al carcinoma (Rotnasinghe et al.,2001) anche se altri studi non indicano nessun tipo di associazione (Vogel et al., 2004). Analizzando i nostri risultati non abbiamo trovato associazione tra questi polimorfismi e il cancro al polmone anche se è opportuno evidenziare il fatto che per lo SNP Arg194Trp il genotipo eterozigote risulta protettivo nei confronti della malattia (OR: 0.62 e 95%CI compreso tra 0.39 e 1.00), ma il valore del trend test non raggiunge formalmente la significativita’ statistica (p=0.06). Per il polimorfismo Arg280His il basso numero di campioni omozigoti per l’allele variante non permette di stabilire con fiducia il rischio per questa categoria.
Un altro gene implicato nella riparazione del DNA è XRCC3 (x-ray repair cross-complementing group 3). Uno studio su 463 casi di tumore al polmone (204 adenocarcinomi e 212 carcinomi a cellule squamose) e 460 controlli (individui sani), ha evidenziato nel sottogruppo degli adenocarcinomi una correlazione tra il polimorfismo XRCC3 241Met e aumento del rischio del carcinoma (Popanda et al., 2004). Nel nostro studio non abbiamo trovato nessuna associazione tra la variazione allelica di questo gene e il tumore, presumibilmente perché abbiamo effettuato l’analisi statistica sulla totalita’ dei casi senza dividere i nostri campioni nei diversi istotipi. Ci riserviamo di verificare questo aspetto nelle analisi che verranno fatte in futuro.
Il gene XPD (xeroderma pigmentosum complementary group D) chiamato anche ERCC2 (excision repair cross-complementary group 2) ha 23 esoni ed è localizzato nel cromosoma 19, nella regione 13.2-q13.3. Esso codifica per una elicasi, una subunità di TFIIH essenziale per la trascrizione e per la riparazione per escissione di nucleotidi (NER). Mutazioni di XPD impediscono che la proteina tradotta interagisca con p44, un’altra subunità di TFIIH e diminuiscono la capacità di riparare il DNA (Taylor et al., 1997). Mutazioni in diversi siti di questo gene producono differenti fenotipi clinici: lo
Discussione
Xeroderma Pigmentoso (XP), una malattia genetica recessiva caratterizzata da ipersensibilità alla luce solare, in particolare alle radiazioni ultraviolette; la Tricotia Distrofica (TTD), causata da un difetto in proteine che portano zolfo e che provoca fragilità dei capelli e ritardo psicomotorio; la Sindrome di Cockayne, che colpisce il sistema nervoso provocando demielinizzazione delle fibre nervose (Eveno et al., 1995). Sono stati identificati alcuni polimorfismi riguardanti il gene ERCC2 tra i quali Asp312Asn e Lys751Gln (Spitz et al., 2001 ).
Il polimorfismo Lys751Gln, dovuto ad una sostituzione di una adenina con una citosina al codone 751, è stato associato con un alto livello di aberrazioni cromatidiche e di addotti nel DNA in persone che non fumano (Xing et al.,2003). Sono stati condotti studi in popolazioni etniche diverse per evidenziare l’associazione dei diversi polimorfismi di XPD e il cancro al polmone (Spitz et al., 2001). Tra essi ricordiamo uno studio effettuato in Svezia su 185 pazienti affetti da tale malattia (97 fumatori e 88 non fumatori) e 162 controlli (83 fumatori e 79 non fumatori): è stato visto che la presenza di una sola o di entrambe le varianti alleliche è associata con aumento del rischio di tumore solo tra i fumatori, in particolare al di sotto dei 70 anni (Hou et al., 2002).
L’analisi di nove studi su 3725 casi di cancro al polmone e di 4152 controli dimostra che individui con un genotipo 751CC hanno il 21% di probabilità in più di contrarre la patologia se paragonati con individui 751AA e che individui con il polimorfismo Asp312Asn, dovuto ad una sostituzione di una guanina con una adenina al codone 312, hanno una probabilità del 27% (Hu et al., 2004).
Liang et al. hanno riportato che il genotipo XPD 751CC è associato con aumento del rischio di carcinoma con cellule squamose ma non con adenocarcinoma.
Il diverso background genetico in popolazioni diverse spiega le variazioni del rischio di tumore associato ai polimorfismi di ERCC2: la frequenza dell’allele 312A varia da 0.28 a 0.40 nei Caucasici (Zhou et al., 2002) ed è solo di 0.067 tra gli Asiatici (Liang et al., 2003), mentre la frequenza dell’allele 751C varia da 0.35 a 0.41 nei Caucasici , è di 0.26 tra gli Afro-Americani, ma è molto meno comune negli Asiatici (David-Beabes et al., 2001).
I nostri risultati non indicano associazione tra i due polimorfismi e la malattia e questo potrebbe essere dovuto al fatto che nel nostro studio non abbiamo tenuto conto né della divisione dei campioni in istotipi diversi, come precedentemente è stato detto,
Discussione
né della proporzioni di individi che fumano in modo da identificare fattori genetici di rischio che fossero comuni a tutti i campioni.
La prima difesa cellulare per minimizzare l’accumulo di 8-idrossiguanina nel genoma prodotta da radicali liberi include antiossidanti e enzimi come la glutatione perossidasi; dopodichè l’8-idrossiguanina DNA glicosilasi 1 (OGG1) codifica una proteina che elimina la base danneggiata nel doppio filamento di DNA (Vogel et al., 2003). Il polimorfismo Ser326Cys del gene OGG1 è stato associato con il rischio di tumore al polmone (Goode et al, 2002). Tuttavia uno studio caso-controllo di 265 persone affette dal tumore (il 20% costituito da carcinoma a piccole cellule, il 33% da adenocarcinoma, il 22% da carcinoma a cellule squamose, il 6% da carcinoma a cellule grandi e il rimanente 19% da altri tipi istologici) e 272 controlli con età ed esposizione al fumo simili a quelle dei casi, selezionati da una coorte di 54020 Danesi, non ha confermato l’associazione tra il polimorfismo e il cancro al polmone (Vogel et al., 2003), similmente a quanto avviene nel nostro studio. Riguardo al gene che codifica per la proteina soppressore tumorale p53 nell’uomo sono noti tredici polimorfismi: cinque in sequenze esoniche e otto in sequenze introniche (Murata et al., 1996). Tra i primi cinque, tre di questi non causano sostituzione aminoacidica (codone 21, 36 e 213), mentre al codone 47 abbiamo una sostituzione di una prolina con una serina e al codone 72 (esone 4) una sostituzione di una arginina con una prolina (Harris et al., 1986). Tuttavia il polimorfismo al codone 47 è raro, al contrario di quello al codone 72 che è invece frequente (Weston et al.,1992).
Sebbene la relazione tra il polimorfismo Arg72Pro e il cancro al polmone sia stata molto studiata i risultati sono inconsistenti: tre studi (Weston et al.,1992; Jin et al.,1995; Biros et al.,2001) non trovano nessuna associazione con il tumore, tuttavia Fan et al. hanno trovato che i genotipi Pro/Pro e Pro/Arg, paragonati con il genotipo omozigote Arg/Arg, sono associati con un aumento di adenocarcinoma nei Caucasici. Noi riteniamo che il polimorfismo Arg72Pro non sia associato ad aumento del rischio di cancro al polmone, ma forse ad una sua diminuizione nei soli individui eterozigoti anche se i valori di OR e 95%CI risultano essere statisticamente non significativi (OR: 0.71 e 95%CI: 0.51 e 1.00)
Studi simili sono stati fatti su due polimorfismi trovati negli introni 3 e 6 di TP53 e anche in questo caso i risultati sono contrastanti: Birgander et al. (1995) non hanno
Discussione
trovato associazione tra i due polimorfismi e il rischio di tumore; Biros et al. (2001) riportano un’ alta frequenza del polimorfismo nell’introne 6 sia in pazienti affetti che in soggetti di controllo. In uno studio su 635 persone affette dal tumore e 635 soggetti sani si è evidenziato che la frequenza dei genotipi e aplotipi di TP53 dipende fortemente dalla diversa etnicità: la frequenza dell’allele variante è alta tra gli Afro-Americani (29.1%) e più bassa tra i Messicani (12.2 %). Tutti e tre i polimorfismi di TP53 sono associati con un aumento del rischio di tumore direttamente proporzionale al numero di copie della variante stessa, in tutti i gruppi etnici (De Jong et al., 2002).
In molti tumori l’mRNA di MDM2, gene che codifica per un importante regolatore negativo di p53, è sovraespresso. Variazioni di sequenze nel promotore di questo gene risultano in una alterata espressione della proteina MDM2 e quindi dell’attività di p53 (Michael and Oren, 2003). Sono stati trovati due polimorfismi di singoli nucleotidi: uno di questi dovuto ad una sostituzione di una timina con una guanina al codone 309, è stato trovato con alta frequenza nella popolazione. Un altro polimorfismo, dovuto ad una sostituzione di una timina con una adenina al codone 344, è raro (è stato trovato solo allo stato eterozigote nell’8% della popolazione) (Bond et al., 2004). La presenza dello SNP 309T>G nel promotore di MDM2 aumenta l’affinità dell’attivatore trascrizionale Sp1 e ciò determina un alto livello della proteina codificata da MDM2 e quindi una attenuazione della risposta di p53 al danno al DNA, in concordanza dell’osservazione precedentemente fatta che MDM2 è un regolatore negativo di p53 (Bond et al.,2004).
L’inibizione del pathway di p53 da parte del polimorfismo 309T>G di MDM2, potenzialmente, può andare ad agire sul numero di mutazioni richieste per lo sviluppo del tumore stesso, sulla frequenza di mutazioni per mitosi e sulla frequenza di proliferazione delle cellule affette (Jin and Levine, 2001; Lain and Lane, 2003). Nonostante la plausibilità di questo meccanismo quale fattore di rischio questo polimorfismo non è stato trovato associato con la neoplasia da noi studiata negli individui sotto i 50 anni.
Altri geni polimorfici, soppressori tumorali, che sono frequentemente mutati nei tumori sono p16 e p21, tuttavia il loro potenziale ruolo nello sviluppo del cancro al polmone non è stato ancora chiarito.
Discussione
Dall’analisi dei risultati da noi ottenuti è emersa una associazione significativa con un polimorfismo C>G nell’introne 48 del gene ATM: tale polimorfismo risulta protettivo nei confronti del cancro al polmone.
Il prodotto di questo gene è una fosfoproteina di 370 kDa che risiede principalmente nel nucleo di cellule proliferanti (Lavin, 1998). ATM contiene 3056 aminoacidi con una regione carbossi terminale che ha una grande affinità con il dominio catalitico del fosfatidil-inositolo-3-chinasi. La proteina codificata da ATM ha un ruolo importante nella regolazione dei segnali che innescano la riparazione della rottura del doppio filamento di DNA (Lavin, 1998). Inoltre tale proteina è implicata nella regolazione dei “checkpoint” del ciclo cellulare andando ad agire su p53 e sulle chinasi Chk1 e Chk2: ATM blocca il ciclo cellulare in modo che i danni al DNA possano essere riparati (Hoekstra, 1997). Allo stesso tempo ATM media l’attivazione di fattori di trascrizione per geni implicati nella risposta allo stress cellulare (Schafman et al., 1995).
Mutazioni in ATM sono la causa dell’Ataxia Telangiectasia (AT), una malattia autosomica recessiva caratterizzata da atassia cerebellare, telangiectasia (dilatazione dei vasi sanguigni nella congiuntiva e nel bulbo oculare), immunodeficienza, ritardo di crescita e immaturità sessuale (Easton et al., 1994). Approssimativamente il 70% di pazienti con atassia telangiectasia (AT) possiede una proteina tronca e instabile, mentre il rimanente 30% contiene inserzioni/delezioni o mutazioni (Gilad et al., 1996).
Sono stati descritti ottantotto SNPs in questo gene: ventisei si trovano in regioni codificanti e sessantadue in regioni non codificanti. Dei ventisei polimorfismi localizzati negli esoni, otto sono silenti e diciotto provocano cambiamenti aminoacidici (Thorstenson et al., 2001). Alcuni di queste variazioni alleliche sono associate con aumento del rischio di cancro al seno (Thorstenson et al., 2003); questi polimorfismi potrebbero avere un effetto sull’interazione di ATM con altre proteine alterando la normale funzione di questa molecola (Scott et al., 2002).
Polimorfismi in ATM sono stati descritti aumentare il rischio di cancro alla prostata (Hall et al., 1998): uno studio caso-controllo ha dimostrato una relazione tra il polimorfismo 3161C>G e questo tipo di tumore (Angèle et al., 2004).
Studi epidemiologi in famiglie con AT hanno suggerito che individui eterozigoti sono particolarmente predisposti al cancro al seno (Olsen et al., 2001). Non è stato trovato invece un incremento significativo per il rischio di cancro al polmone, all’ovaio,
Discussione
al pancreas, allo stomaco e al colon-retto in individui che hanno mutazioni di ATM (Geoffroy-Perez et al., 2001). Sulla base di queste considerazioni l’associazione da noi descritta va presa con estrema cautela e andrà necessariamente verificata per escludere che si tratti di un risultato “falso positivo”.
Abbiamo trovato anche una associazione tra aumento del rischio del cancro al polmone e due polimorfismi del gene LIG1: uno di questi si trova nella regione 5’UTR ed è dovuto alla sostituzione di una citosina con una timina, mentre l’altro è localizzato nell’introne 9 dove una adenina è stata sostituita con una guanina.
La DNA ligasi1 è un enzima nucleare di 102 kDa che è implicato nel sistema di riparazione per escissione di nucleotidi (Nocentini, 1999) e nel “long-patch” del sistema di riaparazione per escissione di basi (Levin et al, 2000). Inoltre il prodotto di questo gene è stato identificato come un componente del complesso di replicazione del DNA (Levin et al, 2000).
Non sono stati effettuati studi precedenti sulle variazioni alleliche da noi prese in considerazione e il cancro al polmone: l’unico studio che è stato fatto riguarda la sostituzione di una adenina con una citosina all’esone 6 che risulta essere molto comune nella popolazione generale. Tuttavia non è stata trovata nessuna associazione tra questo SNP e la neoplasia (Shen et al., 2002).
Poichè la DNA ligasi 1 è implicata in svariati meccanismi connessi con la riparazione del DNA, è presumibile che variazioni alleliche in questo gene abbiano un ruolo importante per la suscettibilità al cancro: potremmo supporre che i polimorfismi da noi studiati siano associati ad un aumento del rischio di tumore al polmone almeno in pazienti al di sotto dei 50 anni. E’ probabile che la variante intronica IVS9-21 entro lo stesso gene, risulti associata alla malattia in maniera statisticamente “borderline” (tabella 1), poiche’ i due polimorfismi di LIG1 risultano avere un elevato livello di Linkage Disequilibrium. L’altro polimorfismo appare associato in misura leggermente maggiore e potrebbe avere un ruolo funzionale.
Il polimorfismo 655 A→G nel gene MLH1, che provoca un cambiamento di una Isoleucina con una Valina al codone 219, è risultato protettivo nei confronti del cancro al polmone. La proteina codificata da MLH1 si lega a PMS1 e forma un complesso eterodimerico omologo a MutL, che interagisce con MSH2-MSH6 e MSH2-MSH3 (Prolla et al., 1994) come in E.Coli. Poiché, nel sistema di riparazione eucariotico,
Discussione
mancano proteine omologhe a MutH, il ruolo del complesso MLH1-PMS1 è anche quello di analizzare il DNA alla ricerca di tagli e di degradare il filamento inciso a partire dal sito di taglio, estendendo la propria azione oltre il sito del disappaiamento (Habraken et al., 1997).
Mutazione ereditarie in MLH1 sono la causa maggiore del carcinoma del colon ereditario senza poliposi (HNPCC) (Peltomaki et al, 1997). Guerrette et al. hanno esaminato l’effetto di una mutazione nonsenso in MLH1 nella regione implicata nell’interazione con PMS1 confermando che essa riduce la formazione del complesso (Guerrette et al., 1998).
Sono stati studiati molti polimorfismi in questo gene tra cui anche quello analizzato da noi: questo SNP è stato identificato in popolazioni Europee e Nord-Americane, con una frequenza del 34% in Svedesi con HNPCC (Curia et al., 1999). Inoltre in una popolazione Coreana il polimorfismo Ile219Val è stato trovato prevalentemente in individui che non hanno predisposizione al cancro, suggerendo che in questa popolazione sia una comune variante polimorfica (Kim et al., 2004). In molti tumori tra cui al polmone, al colon e all’endometrio sono state evidenziate mutazioni somatiche e ipermetilazioni del promotore (Kane et al., 1997).
Anche se i dati su alterazioni di geni del mismatch repair nel cancro al polmone sono scarsi, attraverso uno studio caso controllo è stato dimostrato che il 58.6% di pazienti con carcinoma a cellule non piccole ha una ridotta espressione del livello della proteina MLH1. (Xinarianos et al., 2000).
Alla luce di queste considerazioni è probabile che il polimorfismo Ile219Val sia associato al cancro al polmone solo in pazienti al di sotto dei 50 anni e che in tali soggetti sia protettivo nei confronti della malattia.
Nel nostro studio è stato trovato associato anche un polimorfismo in un altro gene del MMR: lo SNP 540 (C→T) nell’esone 3 del gene MSH6 è risultato essere associato con aumento del rischio di cancro al polmone.
Come abbiamo già descritto MSH6 è una proteina del complesso MutSα (MSH2-MSH6) del “mismatch repair” che ha un ruolo importante nella riparazione di basi male appaiate o di piccole inserzioni o delezione (Genshel et al, 1998): una mutazione di MSH6 causa un aumento della frequenza di mutazione somatica con incrementi delle sostituzioni di basi e in alcuni casi anche di inserzione o delezione di una singola base
Discussione
(Marsischky et al, 1996). Mutazioni ereditarie in MSH6 sono state trovate in una piccola porzioni di casi di carcinoma del colon ereditario senza poliposi (HNPCC) e nella maggior parte di casi familiari di cancro colon-rettale (Berends et al, 2002).
In questo gene sono state trovate quindici variazioni alleliche di cui sei localizzate in esoni e le rimanenti in introni. Due di queste alterazioni provocano un cambiamento della sequenza della proteina codificata da questo gene: Gly39Glu e Leu395Val; mentre le altre non provocano cambiamenti aminoacidici (Parc et al, 2000).
Non ci sono studi precedenti riguardanti i polimorfismi di MSH6 e il cancro al polmone, tuttavia poiché gli SNPs nei geni del “mismatch repair” sono teoricamente un potenziale fattore di rischio per il cancro colon rettale (Houlston et al, 2001) sono stati fatti studi caso controllo ed è stato dimostrata associazione con aumento del rischio per questo tipo di tumore. Poiché non sono stati fatti studi funzionali del polimorfismo da noi preso in esame l’associazione da noi descritta va presa con estrema cautela e si può solo supporre che questa variazione allelica possa inibire la formazione del complesso MutSα alterando il normale meccanismo di riparazione, come avviene nel cancro colon-rettale (Guerrette et al, 1998).
Tuttavia è anche possibile che questa variazione allelica provochi una diminuita espressione di MutS. In effetti è stato dimostrato attraverso studi di clonaggio e sequenziamento che la regione del promotore di questo gene possiede sette “GC boxes”, localizzate tra la posizione -454 e -159, potenzialmente capaci di legare i fattori di trascrizione Sp1 e Sp3, c-JUN, E2F e E4TF1 (Courey et al., 1998) e che le varianti polimorfiche in questa regione alterino l’attività del promotore stesso (Gazzoli and Kolodner, 2003). Studi simili dimostrano anche che questo gene contenga molte sequenze GC localizzate centinaia di paia di basi a valle dei siti di inizio della trascrizione e che tali sequenze vengano riconosciute dalla proteina Sp1 che attiva la RNA polimerasi II per iniziare la trascrizione (Dynan et al., 1985). Si potrebbe supporre che la variazione allelica (C→T) nell’esone 3 alteri uno di questi siti riconosciuti da Sp1 con conseguente riduzione della trascrizione del gene da noi preso in esame, riduzione della formazione del complesso MutS e quindi dell’efficienza del “Mismatch repair”.
Discussione
Nel nostro studio è risultato essere associato alla neoplasia un polimorfismo nel gene NOD2/CARD15 : individui omozigoti per l’allele variante (G→A, nel promotore) hanno una minore predisposizione al cancro al polmone.
NOD2 (Nucleotide oligomerization bindin domain 2) codifica una proteina costituita da un dominio N-terminale implicato nel reclutamento di caspasi e nell’attivazione del fattore di trascrizione NFkB, il quale attiva Bcl2, inibitore dell’apoptosi (Ogura et al., 2001). Inoltre questa proteina possiede un dominio di legame ai nucleotidi e un dominio LRR (Leucine-rich repeat) che ha la funzione di riconoscere componenti batteriche (Inohara et al., 2001).
La più comune mutazione in questa regione, 3020insC, risultante in una sostituzione di una Leucina con una Prolina al codone 1007, favorisce l’introduzione di un codone di stop all’esone 11 generando una proteina tronca (Ogura et al., 2001). Questa variante è associata con un aumento dell’attività di NFkB (Ogura et al., 2001), con una maggiore predisposizione al cancro colon-rettale (Kurzawski et al., 2004) e al Morbo di Crohn, un infiammazione dell’apparato digerente che colpisce in particolare le porzioni dell’ ileo e del colon (Hampe et al., 2001). E’ stato dimostrato che mutazioni in NOD2 sono associate anche al rischio di cancro al seno (Huzarski et al., 2005).
Sono stati identificati in letteratura otto polimorfismi nella regione del promotore tra cui lo SNP di nostro interesse (-926G>A): alcune di queste variazioni alleliche sono associate con una maggiore suscettibilità alla tubercolosi polmonare (Stockton. et al., 2004).
Per quanto riguarda la neoplasia presa da noi in considerazione in letteratura non risultano studi precedenti: potremo perciò supporre che almeno in pazienti giovani tale polimorfismo sia associato con il cancro al polmone, anche se dovranno essere eseguiti ulteriori studi per confermare le nostre ipotesi.
Un altro polimorfismo risultato positivo nel nostro studio è Asp298Ser nel gene XRCC4: individui con genotipo eterozigote hanno una maggiore predisposizione al cancro al polmone. Il polimorfismo Asp298Ser insieme ad altri polimorfismi di questo gene è stato analizzato anche in studi caso-controllo sul cancro al seno risultando associato ad un aumento del rischio anche per questo tipo di tumore (Fu et al., 2003). Il prodotto del gene XRCC4 è responsabile della formazione di un complesso con la ligasi IV implicato nel completamento del processo di riparazione NHEJ (Hoeijmakers,
Discussione
2001). Studi precedenti dimostrano che alterazioni genetiche nelle proteine implicate nel NHEJ possono alterare la capacità di riparare rotture del doppio filamento di DNA e quindi predisporre l’individuo ad un alto rischio di cancro, tuttavia non sono necessariamente la causa del tumore (Ruttan et al, 2002).
Si può ipotizzare che il polimorfismo G245A in XRCC4 risulta associato ad un aumento del rischio di cancro poiché il sistema di riparazione di cui fa parte questo gene è importante nel mantenere la stabilità genetica: variazioni alleliche in XRCC4 potrebbero portare ad una diminuita attività della riparazione del DNA e quindi ad instabilità genomica. L’accumulo di DNA non riparato potrebbe portare a mutazioni genetiche e in seguito alla formazione di tumori. Questo processo potrebbe essere velocizzato a causa di esposizione a sostanze cancerogene o di inibizione dell’apoptosi dovuta ad una mutazione di p53.
Infine attraverso l’analisi dei nostri risultati abbiamo trovato una associazione tra il polimorfismo Ala222Val, dovuto ad una sostituzione di una citosina con una timina nell’esone 4 (C677T), nel gene MTHFR (metilene-tetraidrofolato reduttasi) e il cancro al polmone: la variazione allelica è associata con un aumento del rischio di cancro.
Numerosi studi epidemiologi hanno dimostrato che il consumo di frutta e verdura è associato con una diminuizione del rischio di cancro (Steinmetz et al., 1996). Tra tutti i nutrienti studiati, il folato esibisce il più consistente effetto protettivo contro il cancro al polmone (Voorips et al., 2000).
MTHFR è uno degli enzimi implicati nel metabolismo dei folati: questa proteina ha la funzione di ridurre il 5,10-metilene-tetraidrofolato a 5- metil-tetraidrofolato, un substrato per la rimetilazione di omocisteina a metionina, implicata nella produzione di S-adenosylmetionina, il donatore universale di metili richiesti per la metilazione del DNA (Kim et al., 2000). La metilazione del DNA è uno dei meccanismi molecolari con i quali è regolata l’espressione del gene (Jacob et al., 1998): l’ipometilazione è associata con attivazione di oncogeni e l’ipermetilazione del promotore è associata con perdita di funzione di geni soppressori tumorali (Kim et al., 1997).
MTHFR è anche implicato nella produzione di dTMP e nella sintesi delle purine e inoltre ha un ruolo importante nell’accumulo di nucleotidi essenziali per la sintesi del DNA e per la riparazione (Choi et al., 1998). (Figura 25).
Discussione
Figura 25. Ruolo di MTHFR.
E’ stato dimostrato che individui omozigoti per il polimorfismo C677T hanno una ipometilazione del DNA genomico nei loro leucociti periferici, una caratteristica di molti tipi di tumore (Stern et al., 2000). Studi precedenti dimostrano una diminuita attività del gene dovuta a tale SNP con un conseguente aumento del rischio di cancro al polmone (Siemianowicz et al., 2003). Tuttavia uno studio effettuato su una popolazione Cinese suggerisce che individui con un genotipo omozigote per l’allele variante abbiano una diminuizione del rischio per questo tipo di neoplasia (Jeng et al., 2003).
Uno studio condotto a San Francisco ha dimostrato che il polimorfismo 677T è associato anche ad aumento del rischio di linfoma follicolare (Skibola et al., 2004). Un possibile meccanismo è che la variazione allelica produca una riduzione dell’attività del gene e quindi un elevato livello di omocisteina che provoca ipometilazione globale del DNA che può portare ad instabilità cromosomica, fattore che contribuisce ad aumento del rischio di linfoma (Stamatopoulos et al., 2000).
Mettendo in evidenza il fatto che popolazioni dell’ Europa dell’Est hanno una dieta povera in folati e alta incidenza di tumori colorettali, potremmo concludere che la
Discussione
carenza di questo nutriente insieme ad una ridotta attività dell’enzima dovuta allo SNP da noi considerato potrebbero aumentare il rischio di cancro al polmone.
Indubbiamente sarà necessario condurre una analisi più ampia, che permetta di confermare le osservazioni riportate. Nel futuro provvederemo all’analisi delle interazioni gene-gene nel tentativo di identificare quei profili genetici che potrebbero conferire, grazie alla combinazione di varianti genetiche differenti, un aumento del rischio per questo tipo di tumore.
Bibliografia
5. Bibliografia
Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15: 2177-2196.
Adair GM, Rolig RL, Moore Faver D, Zabelshansky M, Wilson JH and Nairn RS. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000, 19: 5552-5561
Agundez JA. Cytochrome P450 gene polymorphism and cancer. Curr Drug Metab. 2000, 5: 211-24.
Allen DJ, Makhov A, Grilley M, Taylor J, Thresher R, Modrich P, Griffith JD. MutS mediates heteroduplex loop formation by a traslocation mechanism. EMBO J. 1997, 16: 4467-4476. Anderson CW and Lees-Miller SP. The nuclear serine/threonine protein kinase DNA-PK. Crit. Rev. Eukaryot. Gene Expr.1992, 2: 283-314.
Angèle S, Falconer A, Edwards SM, Dòrk T, Bremer M, Moullan N, Chapot B, Muir K, Houlston R, Norman AR, Bullock S, Hope Q, Meitz J, Dearnaley D, Dowe A, Southgate C, Ardern-Jones A, Easton DF, Eeles RA, Hall J. ATM polymorphisms as risk factors for prostate cancer development. British Journal of Cancer 2004, 91: 783-787.
Armitage AK, Dollery CT, George CF, Houseman TH, Lewis PJ, Turner DM. Absorption and metabolism of nicotine from cigarettes. Br Med J. 1975, 8: 4313-6.
Aspinwall R, Rothwell DG, Roldan Arjona T, Anselmino C, Ward CJ, Cheadle JP, Sampson JR, Lindahl T, Harris PC, Hickson ID. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl. Acad. Sci. U.S.A.1997, 94: 109-114. Au KG, Welsh K, Modrich P. Initiation of methyl-directed mismatch repair. J. Biol.Chem.1992, 267: 12142-12148.
Barnes CJ, Wahl AF, Shen B, Park MS, Bambara RA. Mechanism of tracking and cleavage of adduct-damaged DNA substrates by the mammalian 5’- to 3’- exonuclease/endonuclease RAD2 homologue 1 or flap endonuclease 1. J. Biol. Chem.1996, 271: 29624-29631.
Bartsch H, Nair U, Risch A, Rojas M, Wikman H, Kroum A. Genetic polymorphism of CYP genes, alone or in combination, as a risk modifier of tobacco-related cancers. Cancer Epidemiol Biomarkers Prev. 2000,1: 3-28.
Benowitz NL, Jacob P 3rd. Nicotine and cotinine elimination pharmacokinetics in smokers and nonsmokers. Clin Pharmacol Ther. 1993, 53: 316-23.
Benowitz NL, Jacob P 3rd. Metabolism of nicotine to cotinine studied by a dual stable isotope method. Clin Pharmacol Ther. 1994, 5: 483-93.
Benowitz NL, Jacob P 3rd. Individual differences in nicotine kinetics and metabolism in humans. NIDA Res Monogr. 1997, 173: 48-64.
Bibliografia
Berends MJ, Wu Y, Sijmons RH, Mensink RG, van der Sluis T, Hordijk-Hos JM, de Vries EG, Hollema H, Karrenbeld A, Buys CH, van der Zee AG; Hofstra RM, Kleibeuker JH. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am. J. Hum. Genet. 2002, 70: 26-37.
Bhattacharyya KK, Brake PB, Eltom SE, Otto SA, Jefcoate CR. Identification of a rat adrenal cytochrome P450 active in polycyclic hydrocarbon metabolism as rat CYP1B1. Demonstration of a unique tissue-specific pattern of hormonal and aryl hydrocarbon receptor-linked regulation. J Biol Chem. 1995, 270: 11595-602.
Bindoli A, Cavallini L. I radicali liberi aspetti biochimici e medici. Piccin Editore Padova 1980. Bidoli P, Robustelli della Cuna G, Pastorino U. Neoplasie dei polmoni. Medicina oncologica, 2003.
Bill CA, Yu Y, Miselis NR, Little JB and Nickoloff JA. A role for p53 in DNA end rejoining by human cell extracts. Mutat. Res.1997, 385: 21-29.
Birgander R, Sjalander A, Rannug A, Alexandrie AK, Sundberg MI, Seidegard J, et al. P53 polymorphisms and haplotypes in lung cancer. Carcinogenesis 1995,16: 2233-6.
Biros E, Kalina I, Kohut A, Stubna J, Salagovic J. Germline polymorphisms of the tumor suppressor gene p53 and lung cancer. Lung Cancer 2001, 31: 157-62.
Bjoras M, Luna L, Johnsen B, Hoff H, Haug T, Rognes T, Seeberg E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997, 16: 6314-6322.
Blander G, Kipnis J, Leal JF, Yu CE, Schellenberg GD. Oren M. Physical and functional interaction between p53 and the Werner's syndrome protein. J. Biol. Chem.1999, 274: 29463-29469.
Blot WJ, Fraumeni JF, Jr. Cancers of the lung and pleura. In: Schottenfeld D, Fraumeni JF, Jr., eds. Cancer epidemiology and prevention. 2nd ed. Oxford: Oxford University Press, 1996; pp. 637-665.
Boffetta P, Kogevinas M, Simonato L, Wilbourn J, Saracci R. Current Perspectives on Occupational Cancer Risks. Int J Occup Environ Health. 1995 , 4: 315-325.
Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang S, Strong LC, Lozano G, Levine AJ. A single nucleotide polymorphisms in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119: 591-602.
Bootsma D, Kraemer KH, Cleaver J, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Vogelstein B, Kinzler KW. (Eds.), The Genetic Basis of Human Cancer. McGraw-Hill, New York: 1998, 245-274. Bradsher J, Auriol J, Proietti de Santis L, Iben S, Vonesch JL, Grummt I, Egly JM. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10: 819-829.
Bibliografia
Butkiewicz D, Rusin M, Enewold L, et al. Genetic polymorphisms in DNA repair genes and risk of lung cancer. Carcinogenesis 2001, 22: 593-7.
Butt, AJ, Harvey NL, Parasivam G, Kumar S. Dimerization and autoprocessing of the Nedd2 (caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J. Biol. Chem. 1998, 273: 6763-6768.
Camus AM, Genestre O, Honkakoski P., Bereziat JC, Henderson CJ, Wolf CR. High variability of nitrosamine metabolism among individuals: role of cytochrome P450 2A6 and 2E1 in the dealkylation of N-nitroso dimethylamine and N-nitrosodiethylamine in mice and humans. Mol Carcinogenesis 1993; 7: 268-75.
Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K, Frosina G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem. 1997, 272: 23970-23975.
Cashman JR, Park SB, Yang ZC, Wrighton SA, Jacob P 3rd, Benowitz NL. Metabolism of nicotine by human liver microsomes: stereoselective formation of trans-nicotine N'-oxide. Chem Res Toxicol. 1992, 5: 639-46.
Carmella SG, Kagan SS, Kagan M, Foiles PG, Palladino G, Quart AM, Quart E, Hecht SS. Mass spectrometric analysis of tobacco-specific nitrosamine hemoglobin adducts in snuff dippers, smokers, and nonsmokers. Cancer Res. 1990, 17: 5438-45.
Carmella SG, Akerkar S, Hecht SS. Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers' urine. Cancer Res. 1993, 53: 721-4. Carmella SG, Borukhova A, Desai D, Hecht SS. Evidence for endogenous formation of tobacco-specific nitrosamines in rats treated with tobacco alkaloids and sodium nitrite. Carcinogenesis. 1997, 3: 587-92.
Carmella SG, Le Ka KA, Upadhyaya P, Hecht SS. Analysis of N- and O-glucuronides of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human urine. Chem Res Toxicol. 2002, 4: 545-50.
Carmines EL. Evaluation of the potential effects of ingredients added to cigarettes. Part 1: cigarette design, testing approach, and review of results. Food Chem Toxicol. 2002, 1: 77-91. Cecconi F, Gruss P. Apaf1 in developmental apoptosis and cancer: how manyway s to die? Cell. Mol. Life Sci. 2001, 58: 1688-1697.
Chan DW, Lees-Miller SP. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem. 1996, 15: 8936-41.
Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002, 21: 2827-2832.
Choi SW, Kim YI, Weitzel JN, Mason JB. Folate depletion impairs DNA excision repair in the colon of the rat. Gut 1998, 43: 93-99.
Bibliografia
Choi JH, Anh KS, Kim J, Hong YS. Enhanced induction of Bax gene expression in H460 and H1299 cells with the combined treatment of cisplatin and adenovirus mediated wt-p53 gene transfer. Exp Mol Med 2000, 32: 23-8.
Chu G. Double strand break repair. J. Biol. Chem.1997, 272: 24097-24100.
Cohen BS, Harley NH, Tso TC. Clearance of polonium-210-enriched cigarette smoke from the rat trachea and lung. Toxicol Appl Pharmacol. 1985, 2: 314-22.
Conklin BS, Zhao W, Zhong DS, Chen C. Nicotine and cotinine up-regulate vascular endothelial growth factor expression in endothelial cells. Am J Pathol. 2002, 2:413-8.
Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000, 1: 80-84.
Courey AJ, Tjian R. Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 1998, 55: 887-898.
Coutelle C, Ward PJ, Fleury B, Quattrocchi P, Chambrin H, Iron A, Couzigou P, Cassaigne A. Laryngeal and oropharyngeal cancer, and alcohol dehydrogenase 3 and glutathione S-transferase M1 polymorphisms. Hum Genet. 1997, 3: 319-25.
Cox LS. Who binds wins: competiton for PCNA rings out cell-cycle changes. Trends Cell Biol. 1997, 7: 493-498.
Cryns V, Yuan J. Proteases to die for. Genes Dev.1998, 12: 1551-1570.
Curia MC, Palmirotta R, Aceto G et al. Unbalanced germ-line expression of hMLH1 and hMSH2 alleles in hereditary nonpolyposis colorectal cancer. Cancer Res 1999, 59: 3570-5. Datta A, Bagchi S, Nag A, Shiyanov P, Adami GR, Yoon T, Raychaudhuri P. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat. Res.2001, 486: 89-97.
David-Beabes GL, London SJ. Genetic polymorphism of XRCC1 and lung cancer risk among African-Americans and Caucasians. Lung cancer 2001 34: 333-339.
David-Beabes GL, Lunn RM, London SJ. No Association between the XPD (Lys751G1n) polymorphism or the XRCC3 (Thr241Met) polymorphism and lung cancer risk. Cancer Epidemiol. Biomarkers Prev. 2001,10: 911-912.
De Flora S, Bennicelli C, D'Agostini F, Izzotti A, Camoirano A. Cytosolic activation of aromatic and heterocyclic amines. Inhibition by dicoumarol and enhancement in viral hepatitis B. Environ Health Perspect. 1994, 6:69-74.
Day CP, Bashir R, James OF, Bassendine MF, Crabb DW, Thomasson HR, Li TK, Edenberg HJ. Investigation of the role of polymorphisms at the alcohol and aldehyde dehydrogenase loci in genetic predisposition to alcohol-related end-organ damage. Hepatology. 1991, 5: 798-801. De Jong MM, Nolte IM, te Meerman GJ, van der Graaf WT, de Vries EG, Sijmons RH, Hofstra RM, Kleibeuker JH. Low-penetrance genes and their involvement in colorectal cancer
Bibliografia
DeMarini DM. Genotoxicity of tobacco smoke and tobacco smoke condensate: a review. Mutat Res. 2004, 2-3: 447-74.
Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994, 63: 915-948.
Denison MS, Whitlock JP. Jr Xenobiotic-inducible transcription of cytochrome P450 genes. J Biol Chem. 1995, 31:18175-8.
Devlin TM. Biochimica con aspetti clinici, terza edizione; 1997 Wiley-Liss, Inc.
Dianov GL, Jensen BR, Kenny MK, Bohr VA. Replication protein A stimulates proliferating cell nuclear antigen-dependent repair of abasic sites in DNA by human cell extracts. Biochemistry 1999, 38: 11021-11025.
Dianova II, Bohr VA, Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry 2001, 40: 12639-12644.
Divine KK, Gilliland FD, Crowell RE, et al. The XRCC1 399 glutamine allele is a risk factor for adenocarcinoma of the lung. Mutat Res. 2001, 461: 273-8.
Doll R, Hill AB. Smoking and carcinoma of the lung. Preliminary report. Br Med J 1950; 2:739.
Doll R. The first reports on smoking and lung cancer. Clio Med 1998; 46:130-142.
Drummod JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 1995, 268: 1909-1912.
Dynan WS, Tjian R. Control of eukaryotic messenger RNA synthesis by sequence-specific DNA-binding proteins. Nature 1985, 316: 774-778.
Easton DF. Cancer risks in AT heterozygotes. Int. J. Radiat. Biol. 1994, 66: 177-182.
Eckert KA, Kunkel TA. High fidelity DNA synthesis by the Thermus aquaticus DNA polymerase. Nucleic Acid Res. 1990, 18: 3739-3744.
Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res.1995, 23: 2715-2723.
Elliott B, Jasin M. Double-strand breaks and translocations in cancer. Cell. Mol. Life Sci.2002, 59: 373-385.
Emmendoerffer A, Nakamura M, Rothe G, Spekermann K, Lohmann-Matthers ML, Roesler J. Evaluation of flow cytometric methods for diagnosis of chronic granulomatous disease variants under routine laboratory conditions. Cytometry 1994, 15: 147-155.
Emmendoerffer A, Hecht M, Boeker T, Mueller M, Heinrich U. Role of inflammation in chemical-induced lung cancer. Toxicol Lett. 2000,112-113:185-91.
Bibliografia
Eveno E, Bourre F, Quilliet X, et al. Different removal of ultraviolet photoproducts in genetically related xeroterma pigmentosum and trichothiodystropy doseases. Cancer Res.1995, 55: 4325-4332.
Fabricius P, Lange P. Diet and lung cancer. Monaldi Arch Chest Dis. 2003, 3: 207-11.
Fan YC, Edenberg HJ, et al. Polymorphism of ADH and ALDH genes among four ethnic groups in China and effects upon the risk for alcoholism. Alcohol Clin Exp Res 1997, 21:1272/ 7.
Fan R, Wu MT, Miller D, Wain JC, Kelsey KT, Wiencke JK, et al. The p53 codon 72 polymorphism and lung cancer risk. Cancer Epidemiologic Biomarkers Prev.2000, 9: 1037-42. Fischer S, Spiegelhalder B, Eisenbarth J, Preussmann R. Investigations on the origin of tobacco-specific nitrosamines in mainstream smoke of cigarettes. Carcinogenesis. 1990, 5: 723-30.
Forgacs E, Zochbauer-Muller S, Olah E, Minna JD Molecular Genetic Abnormalities in the Pathogenesis of Human Lung Cancer. Pathology Oncology Res. 2001, Vol 7. No 1.
Freudenheim JL, Ambrosone CB, Moysich KB, Vena JE, Graham S, Marshall JR, Muti P, Laughlin R, Nemoto T, Harty LC, Crits GA, Chan AW, Shields PG. Alcohol dehydrogenase 3 genotype modification of the association of alcohol consumption with breast cancer risk. Cancer Causes Control. 1999, 5: 369-77.
Freudenheim Jo L, Ram M, Nie J, Muti P, Trevisan M, Goldman R. Lung cancer in humans is not associated with lifetime total alcohol consumption or with genetic variation in alcohol dehydrogenase 3 (ADH3). Nutritional epidemiology 2003, 3619-3624.
Friend SH, Tapscott SJ. Sibling rivalry, arrested development and chromosomal mayhem. Nat. Genet.1998, 19: 9-10.
Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996, 271: 9573-9578.
Fu YP, Yu JC, Cheng TC, Lou MA, Hsu GC, Wu CY, Chen ST, Wu HS, Wu PE, Shen CY. Breast cancer risk associated with genotypic polymorphism of the nonhomologous end-joining genes: a multigenic study on cancer susceptibility. Cancer Res. 2003, 10: 2440-6.
Fujieda M, Yamazaki H, Saito T, Kiyotani K, Gyamfi MA, Sakurai M, Dosaka-Akita H, Sawamura Y, Yokota J, Kunitoh H, Kamataki T. Evaluation of CYP2A6 genetic polymorphisms as determinants of smoking behavior and tobacco-related lung cancer risk in male Japanese smokers. Carcinogenesis. 2004 12: 2451-8.
Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem. 1995, 49: 29270-8.
Gazzoli I, Kolodner RD. Regulation of the Human MSH6 gene by the Sp1 transcription factor and alteration of promoter activity and expression by polymorphisms. Molecolar and cellular biology 2003, 7992-8007.