2. MATERIALI E METODI
2.1 Siti di campionamento e trattamento dei campioni
Gli individui di Aristeus antennatus analizzati in questo lavoro provengono da tre località del bacino occidentale del Mar Mediterraneo: Calella de Palafrugell, Genova e Palermo (Fig. 2.1, Tab. 2.1). Il termine “campione” da qui in poi verrà utilizzato per indicare gli individui di una determinata località. Gli animali ottenuti da fonti commerciali sono stati conservati in barattoli con etanolo al 95%. In laboratorio, per ogni individuo è stata asportata, con il bisturi, una piccola sezione di tessuto muscolare addominale, (nei casi di Calella de Palafrugell e Genova), o di tessuto muscolare contenuto nei pleopodi (Palermo). I campioni di tessuto muscolare venivano poi trasferiti con pinzette in provette siglate da 10 ml contenenti etanolo assoluto. I bisturi e le pinzette venivano accuratamente pulite con alcool prima di ogni prelievo, al fine di non contaminare i tessuti prelevati. I campioni di tessuto così fissati sono stati conservati in etanolo assoluto a –20 °C.
Fig. 2.1. Siti di campionamento di A. antennatus nel Mediterraneo Occidentale
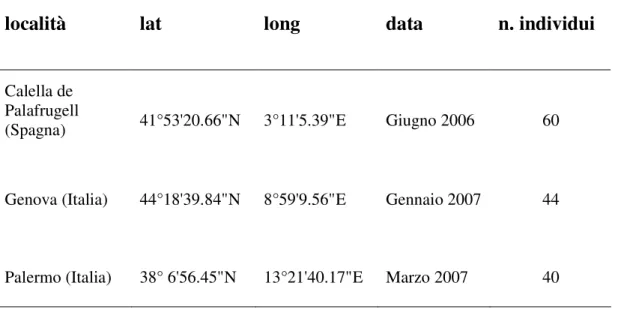
Tab. 2.1. Località di campionamento di A. antennatus, coordinate geografiche del sito di campionamento, data di campionamento e numero di individui raccolti.
località
lat
long
data
n. individui
Calella de Palafrugell
(Spagna) 41°53'20.66"N 3°11'5.39"E Giugno 2006 60
Genova (Italia) 44°18'39.84"N 8°59'9.56"E Gennaio 2007 44
2.2. Le analisi genetiche
Nel processo di amplificazione mediante reazione a catena della polimerasi (PCR) possono essere riconoscibili tre fasi: 1) la pre-PCR, in cui vengono effettuati la preparazione del campione, l’estrazione del DNA e l’inizio della reazione di amplificazione; 2) la PCR stessa, con amplificazione del DNA templato; e 3) la post-PCR, con la messa in evidenza dei prodotti di amplificazione. Nella pre-PCR assumono notevole importanza l'omogeneità dei campionamenti, fattore pregiudizievole la riproducibilità dei dati, le condizioni del DNA estratto, dipendenti dall'efficienza dei metodi di estrazione (resa e purezza) e la definizione precisa della modalità di stima della concentrazione di DNA genomico destinato alla PCR. Dai campioni trattati si procede quindi all'estrazione del DNA.
2.2.1. Estrazione del DNA
L’estrazione di acidi nucleici è il primo passo nelle applicazioni di biologia molecolare. L’estrazione di acidi nucleici da materiale biologico richiede la lisi cellulare,
l’inattivazione delle nucleasi cellulari e la separazione dell’acido nucleico dai residui cellulari.
Esistono vari metodi, la cui scelta si effettua in base a:
- tipo di acido nucleico (ssDNA, dsDNA, RNA totale, mRNA, etc.); - organismo studiato (tessuti animali o vegetali, eucarioti, procarioti, virus); - materiale di partenza (organo intero, tessuto, coltura cellulare, sangue, etc.); - risultato desiderato (quantità, purezza, tempo richiesto);
- applicazione post-estrazione (PCR, cloning, marcatura, restrizione enzimatica, southern blotting, RT-PCR, sintesi di cDNA, etc.).
In letteratura esistono diversi protocolli di estrazione del DNA che si possono dividere in tre categorie fondamentali: 1) estrazione chimica, 2) estrazione mediante ultracentrifugazione e purificazione, 3) estrazione tramite colonne e kit di separazione. L’estrazione chimica è la tecnica normalmente più utilizzata, l’ultracentrifugazione è utilizzata per ottenere DNA altamente purificato, mentre i kit vengono prodotti ed ottimizzati per specifici tessuti animali o vegetali, permettendo normalmente di ottenere, in maniera piuttosto veloce, un prodotto puro, ma a basso peso molecolare (Procaccini & Maltagliati, 2003).
2.2.2. Estrazione chimica – protocollo “fenolo/cloroformio”
Tutti i protocolli di estrazione del DNA iniziano dalla lisi chimica ed enzimatica delle cellule in cui esso è contenuto. La dissoluzione della membrana cellulare solitamente avviene per mezzo di detergenti quali SDS TRITON X: nel caso di tessuti animali le cellule sono immerse in una matrice proteica (collagene) e quindi occorrono trattamenti con una enzima, la proteinasi K che la idrolizzino. Tutte queste attività non hanno nessuna influenza sul DNA. A pH fisiologico gli acidi nucleici assumono una carica fortemente negativa e formano complessi stabili con diverse forme di cationi, oligoammine e proteine basiche. Occorre quindi, una volta solubilizzate le membrane cellulari, rompere queste forti interazioni molecolari.
Il protocollo “fenolo/cloroformio” (Sambrook et al.,1989) è un metodo classico di estrazione del DNA genomico che utilizza fenolo acquoso, in modo che le proteine denaturate rimangano in soluzione nella fase fenolica o nell'interfaccia fenolo-acqua, mentre gli acidi nucleici rimangono nella fase acquosa. Solitamente si ripete due volte
l'estrazione e la si fa seguire da un "lavaggio" con cloroformio allo scopo di eliminare dalla fase acquosa eventuali tracce di fenolo. Questa sostanza è infatti particolarmente attiva come denaturante delle proteine e la sua presenza nella miscela di reazione potrebbe fortemente inibire l'attività polimerasica. L'ultima fase consiste nel far precipitare il DNA a freddo con alcool etilico, ciò porta alla riduzione dell'attività dell'acqua e consente, soprattutto in presenza di sodio acetato, l'eliminazione dei contaminanti a basso peso molecolare (Fig. 2.2).
Fig.2.2 Gli step di estrazione con fenolo-cloroformio (A), la successiva precipitazione con etanolo assoluto (B), il lavaggio con etanolo (C) del pellet ottenuto dopo centrifugazione, l’essiccamento all’aria (D) e la risospensione in acqua.
Il protocollo da noi utilizzato è quello riportato in Vinas et al. (2004) opportunamente modificato. Una piccola porzione di tessuto era posta in una provetta contenente 600 µl di TENS (0,5 M EDTA, 0,2 M Tris-Nacl pH 8, 2% SDS) e 40µl di Proteinasi K. La miscela, posta su un agitatore meccanico, veniva messa a incubare in stufa a 37 °C finché il tessuto non veniva digerito completamente. Al lisato venivano
aggiunti 650 µl di fenolo sotto cappa sterile e il tutto centrifugato per 4 min a 13.000 rpm. Al sovranatante prelevato venivano aggiunti 325 µl di fenolo e 325 µl di cloroformio e messo nuovamente a centrifugare per 4 min a 13.000 rpm Si aspirava poi il sovranatante, al quale venivano aggiunti 2 volumi di etanolo assoluto (900 µl) e così messo a centrifugare per 15 min. A questo seguivano 2 lavaggi con etanolo al 70% fino ad ottenere il “pellet “, asciugato in stufa a 60 °C e reidratato con 75 µl di acqua. Allo scopo di verificare l’avvenuta estrazione, una piccola aliquota dei campioni di DNA estratti era sottoposta a elettroforesi su gel di agarosio (1%), contenente bromuro di etidio, in un tampone di migrazione TAE1X. Il DNA era visualizzato mediante l’utilizzo di un transilluminatore a luce ultravioletta, fotografato con una fotocamera digitale e archiviato in un PC.
A
B C
Fig. 2.3 (A) trans-illuminatore a luce ultravioletta collegato al pc; (B-C) Esempi di visualizzazione al transilluminatore di campioni di Genova e Palermo.
2.3. Sintesi di oligonucleotidi
Un primer è un filamento di acido nucleicoche serve come punto di innesco per la replicazione del DNA. I primer sono necessari perché molte DNA-polimerasi (enzimi che catalizzano la replicazione del DNA) non possono iniziare la sintesi di un nuovo filamento "da zero", ma possono solo aggiungere nucleotidi ad un filamento pre-esistente. Esistono primer “forward” e primer “reverse”, a seconda che siano complementari al filamento 3'-5' o a quello 5'-3'. Nella costruzione dei primer è necessario prestare attenzione al fatto che sia il primer forward sia quello reverse devono avere la stessa temperatura di “melting”, cioè quella temperatura sufficiente a
tenere separate il 50 % delle molecole, e di “annealing”, temperatura alla quale i primers si
attaccano al filamento di DNA, (o temperature molto vicine, al massimo 1 o 2 °C di differenza), quindi i due primer devono avere all'incirca lo stesso contenuto in AT (o in CG). Gli oligonucleotidi usati come primer nelle reazioni di amplificazione per il gene 16SrRNA nel presente lavoro sono stati disegnati da Palumbi et al (1991) e modificati in accordo alle sequenze del peneide Penaeus disponibili in GENBANK.
16SarL pan –T 5’- TGC CTG TTT ATC AAA AAC AT – 3’
16SbrH pan 5’ – CCG GTC TGA ACT CAA ATC ATG T – 3’
Per trovare i primer idonei alla subunità I del citocromo c ossidasi (COI) sono stati effettuati diversi tentativi, in quanto, nè quelli disegnati da Palumbi et al. (1991), nè quelli modificati secondo Penaeus monodon, fornivano risultati evidenti. Primer più idonei sono stati ridisegnati in laboratorio utilizzando il programma PRIMERS3 (Rozen & Skaletsky, 2000).
COIL 5’ GGT GAC CCA GTC CTT TAC CA -3’
COIHmeu 5’ GTC TGG ATA ATC AGA ATA CCG AC – 3’
Gli oligonucleotidi usati come primer interni nelle reazioni di sequenziamento diretto dei prodotti amplificati sono gli stessi della PCR ad una diversa concentrazione.
2.4. Reazione di amplificazione e sequenziamento
La tecnica della reazione a catena della polimerasi o PCR (Polymerase Chain Reaction) è stata introdotta da Kary Mullis alla metà degli anni ’80 e ha rivoluzionato la genetica molecolare permettendo l’amplificazione di una regione specifica di DNA. La PCR ricostruisce in vitro uno specifico passaggio della riproduzione cellulare: la sintesi di un segmento di DNA "completo" (a doppia elica) a partire da un filamento a singola elica. Il filamento mancante viene ricostruito a partire da una serie di nucleotidi (i "mattoni" elementari che costituiscono gli acidi nucleici) che vengono disposti nella corretta sequenza, complementare a quella del DNA interessato.
Composizione tipica di una reazione di PCR:
• una quantità, anche minima di DNA templato;
• miscela dei quattro nucleotidi precursori (dNTPs: 2’ desossinucleosidi 5’
trifosfato)
• opportuni primer complementari al segmento da amplificare;
• altri elementi di supporto (ad es. ioni Mg++), necessari per costituire l'ambiente
adatto alla reazione;
• DNA polimerasi
Nel presente lavoro la miscela di reazione di un volume di 50µl conteneva 2 µl di DNA templato, 5 µl di GeneAmp 10x PCR Buffer II (Applied Biosystem), 2 µl di MgCl2 (50 mM), 4 µl di dNTP (10 mM), 2 µl per ogni primer (10 µM), 0,25µl di
Amplitaq Dna polimerasi (Applied Biosystems). Questa soluzione va incontro a tre fasi: viene portata a una temperatura compresa tra 94 e 99 °C. Ci si trova, di conseguenza, in una situazione in cui la doppia elica del DNA viene completamente scissa ed i due filamenti di cui essa è composta sono liberi (fase di denaturazione). Successivamente la
temperatura viene abbassata fino a 30-55 °C circa, al fine di permettere l’attacco dei
primer alle regioni loro complementari dei filamenti di DNA templato denaturati (fase
di annealing). Infine la temperatura viene alzata fino a 65-72 °C al fine di massimizzare l'azione della DNA polimerasi che determina l’allungamento dei primer legati, utilizzando come stampo il filamento singolo di DNA (fase di prolungamento). Il ciclo descritto viene ripetuto generalmente per circa 20-30 volte. In genere non si superano i 50 cicli in quanto ad un certo punto la quota di DNA ottenuto raggiunge un plateau. Ciò avviene, ad esempio, per carenza degli oligonucleotidi usati come inneschi o per diminuzione dei dNTP.
Fig.2.4. Fasi della PCR. Fonte: internet
La reazione della PCR sono state condotte nel termociclatore programmabile PTC – 100; MJ Research, Inc. che esegue automaticamente i cambi di temperatura necessari per la PCR seguendo il programma LLUC–II; questo prevede una fase iniziale di denaturazione di 1 min a 94 °C, seguito da 35 cicli comprendenti una fase di
denaturazione di 1 min a 94 °C, una fase di annealing di 1 min a 52 °C e una fase di estensione di 2 min a 72 °C con una estensione finale di 5 min a 72 °C.
Fig 2.5. Termociclatore programmabile PTC – 100; MJ Research, Inc.
I frammenti amplificati sono stati verificati attraverso corsa elettroforetica con un opportuno “ladder” di riferimento, cioè un campione di frammenti di DNA di peso molecolare noto. Vengono inoltre allestiti un controllo positivo, che consiste in un campione in cui la sequenza bersaglio è contenuta e un controllo negativo, cioè un campione in cui la sequenza bersaglio manca. Essi servono per evidenziare eventuali contaminazioni che potrebbero riferirsi sia all'estrazione del materiale genomico, sia a una delle fasi di preparazione della PCR.
A) B)
Fig. 2.6 A) corsa elettroforetica del DNA caricato su gel di agarosio , B) Stampa di una visualizzazione delle bande di DNA per mezzo di trans-illuminatore a luce ultravioletta.
Una volta verificata l’avvenuta amplificazione del frammento di DNA specifico si procede con la purificazione per eliminare eventuali residui di primer e desossinucleotidi attraverso l’uso di GFX PCR DNA and gel Band Purification Kit (Amersham Biosciences).Per quanto riguarda la reazione di sequenziamento, si procede come un normale processo di PCR salvo alcune differenze. I due filamenti di DNA (3’-5’ e (3’-5’-3’) vengono divisi e amplificati separatamente. Oltre alle normali basi vengono aggiunte ddA-Fc (didesossiadenina +fluorocromo), ddG-Fc, ddC-Fc, ddT-Fc, che sono le quattro basi private di un ulteriore ossigeno dalla molecola di ribosio e combinate con sostanze, dette fluorocromi, capaci, se sollecitate da un raggio laser, di emettere fasci luminosi di colori differenti. Queste particolari basi interrompono l’opera della DNA polimerasi in quanto mancando di due ossigeni impediscono la formazione di un ponte ossigeno con un’altra base. In tal modo bloccano la costruzione del filamento, che viene così a terminare con un fluorocromo. La proporzione tra la quantità di d-basi e dd-basi, basata su leggi della probabilità, è studiata in modo tale che vengano a formarsi filamenti di numero crescente di basi, da una a tutte quelle comprese nel frammento, senza salti. La reazione di sequenziamento dei prodotti amplificati sono state eseguite utilizzando un dye terminator (dRhodamine, Applied Biosystems) seguendo le istruzioni della ditta fornitrice. I primer utilizzati per il sequenziamento sono gli stessi usati per la PCR. In ultimo i prodotti del sequenziamento sono stati caricati in un sequenziatore automatico multicapillare ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
2 .5. Trattamento statistico dei dati
2.5.1. Allineamento sequenze
Le sequenze nucleotidiche ottenute appaiono come un grafico a quattro colori (secondo convenzioni internazionali all’adenina corrisponde il colore verde, alla guanina il blu, alla timina il rosso, alla citosina il giallo) contenente una successione di picchi che rappresenta la successione di basi del filamento sequenziato. Inoltre particolari lettere indicano la copresenza in una stessa banda di basi diverse (compresenza che può verificarsi, per esempio, nel caso che la regione di DNA nucleare sequenziata provenga da un individuo eterozigote, o in caso di errori o presenza di impurità). Le sequenze sono state allineate usando il programma SeqScape versione 2.5 (Applied Biosystems) usando come sequenza di riferimento quella del peneide Penaeus disponibile in GENBANK. Gli allineamenti sono stati visualizzati con il programma BioEdit (Hall, 1999). La conversione delle sequenze nucleotidiche della COI in amminoacidiche è stata eseguita con il programma MEGA versione 3.1 (Kumar
et al. 2004). È stato utilizzato il programma MODELTEST 3.7 (Posada e Crandall,
1998) 1) per verificare il modello di sostituzione nucleotidica che soddisfacesse meglio i nostri dati e 2) per applicare un test di omogeneità alle due regioni analizzate in modo da valutare se le due sequenze potessero essere unite e trattate come un’unica regione mitocondriale.
2.5.2. Stime di diversità genetica all’interno dei campioni
La diversità genetica presente all’interno di ciascuna località è stata stimata calcolando la diversità aplotipica (h) e la diversità nucleotidica (π). Il primo parametro è analogo all’eterozigosità attesa, ed è definita come la probabilità che due aplotipi scelti a caso siano differenti nel campione ed è ottenuta dalla seguente formula:
dove n è il numero di copie del gene del campione, K è il numero di aplotipi e pi è la
frequenza dell’i-esimo aplotipo nel campione (Nei, 1987)
Se c’è completa omogeneità e tutti gli individui hanno lo stesso aplotipo, il valore di h è nullo, mentre se tutti gli individui hanno aplotipi differenti, h assume il suo massimo valore, cioè 1. La diversità nucleotidica (π) è, invece, la probabilità che due nucleotidi omologhi scelti a caso siano differenti. È equivalente alla diversità aplotipica a livello nucleotidico:
dove dijè una stima del numero di mutazioni che sono avvenute dalla divergenza degli
aplotipi i e j, k è il numero di aplotipi, pi è la frequenza dell’i-esimo aplotipo e L è il
numero di loci (Tajima, 1983; 1993).
2.5.3 Stime di diversità genetica tra i campioni
Per stimare l’eterogeneità genetica totale tra i campioni è stato usato l’indice di fissazione FST di Weir e Hill (2002). I valori di FST possono andare da 0, completa
omogeneità, a 1, totale eterogeneità. È stata inoltre applicata l’analisi della varianza molecolare (AMOVA) (Excoffier et al., 1992), che distribuisce la diversità genetica nei suoi diversi livelli gerarchici. Nel presente lavoro sono state considerate le componenti di varianza presenti all’interno e tra i campioni utilizzando il programma ARLEQUIN ver.3.1 (Excoffier et al., 2005).
Le distanze genetiche di Tamura e Nei (1993) all’interno e tra i campioni sono state calcolate mediante il programma MEGA vers. 3.1 (Kumar et al. 2004).
Le relazioni esistenti tra i vari aplotipi sono state analizzate costruendo “network” con l’algoritmo del “median-joining” (Bandelt et al., 1999; 2000). Per questa analisi è stato utilizzato il programma NETWORK ver. 4.1.1.2 (disponibile gratuitamente al sito internet http://www.fluxus-technology.com). Si è preferito usare i network invece degli alberi e dei dendrogrammi, in quanto i processi microevolutivi intraspecifici e i rapporti filogeografici tra gli individui e le popolazioni non necessariamente seguono le dicotomie caratteristiche degli alberi e dei dendrogrammi (Clement et al., 2000; Posada & Crandall, 2001, Makarenkov et al., 2006). I network quindi forniscono una rappresentazione migliore delle relazioni di tipo “reticolato” che caratterizzano i rapporti filogenetici e filogeografici presenti tra popolazioni naturali con specifiche (Bandelt et al., 1999; Clement et al., 2000; Posada & Crandall, 2001; Makarenkov et al., 2006). Essi rappresentano una topologia tale per cui non è individuabile un punto principale di divergenza, ma illustrano una distribuzione spaziale di dati molecolari intraspecifici.
Il modello dell’“isolamento da distanza”, cioè la dipendenza tra divergenza genetica e distanza geografica per ciascuna coppia di campioni, è stata vagliata con un’analisi di regressione dei valori di FST sui valori di distanza geografica. Le distanze
geografiche tra le località sono state misurate in miglia nautiche utilizzando GOOGLE EARTH (disponibile al sito web http://earth.google.it/). Per valutare la significatività della regressione è stato applicato il test non parametrico di Mantel (1967) per il confronto di due matrici. Questo test permette di determinare l’eventuale dipendenza delle due matrici. Il valore del parametro di associazione Z (coefficiente di Mantel) è calcolato a partire dai dati reali e poi comparato alla serie di pseudovalori ottenuti per permutazione di righe (o colonne) di una delle due matrici. L’ipotesi nulla è l’indipendenza tra le due matrici. Se non c’è relazione tra le due matrici, il valore di Z
ottenuto sui dati reali non si discosterà significativamente dalla distribuzione degli pseudo-Z ottenuti con le permutazioni. Nel caso contrario, sarà rigettata l’ipotesi nulla e quindi le matrici risulteranno significativamente correlate. I valori di probabilità nel presente lavoro sono stati ottenuti mediante 10000 permutazioni. Il test di Mantel e i grafici di regressione sono stati ottenuti mediante il programma IBD Web Service ver. 2.1 (Jensen et al., 2005).
Per le stime di flusso genico il programma DnaSP (Rozas et al., 2003) ci fornisce diversi parametri, tra cui γST (Nei, 1982), NST (Lynch & Crease, 1990), FST
(Hudson et al., 1992) legati al valore di Nm dalla seguente relazione:γST (oppure NST,
oppure FST) = 1 / (1 + 2Nm). Il parametro Nm rappresenta il numero di migranti efficaci
per ogni generazione; se Nm è minore di uno, come regola empirica, le popolazioni sono considerate isolate, se compreso tra 1 e 4 rappresenta una situazione intermedia, per valori maggiori di 4 si rientra nel caso di panmissia.
2.5.4. Aspetti di demografia storica
La comparazione di sequenze nucleotidiche all’interno delle specie fornisce un valido approccio per determinare aspetti rilevanti della loro storia evolutiva (Cann, 2001). Variazioni della dimensione di una popolazione lasciano nel DNA tracce che possono essere individuate nelle sequenze nucleotidiche. Ciò ha consentito lo sviluppo di test statistici per valutare l’espansione demografica di una popolazione. Per valutare la possibilità di eventi recenti di espansione demografica in A. antennatus è stata eseguita un’analisi della distribuzione “mismatch” che si basa sul modello dell’espansione improvvisa di Rogers & Harpending (1992). Questo modello, applicabile a sequenze di DNA non ricombinante, risultando quindi particolarmente adatto nel trattamento di sequenza di DNA mitocondriale, prevede una popolazione di
femmine, con dimensione iniziale N0, in equilibrio tra mutazione e deriva genetica; si
suppone poi che la popolazione cresca (o si restringa) fino alla dimensione finale N1
dopo t generazioni. Solo la dimensione della popolazione femminile è importante ai fini del modello, perché i maschi non trasmettono il genoma mitocondriale. Nello specifico,
N0 e N1 non si riferiscono al numero assoluto di femmine nella popolazione ma al loro
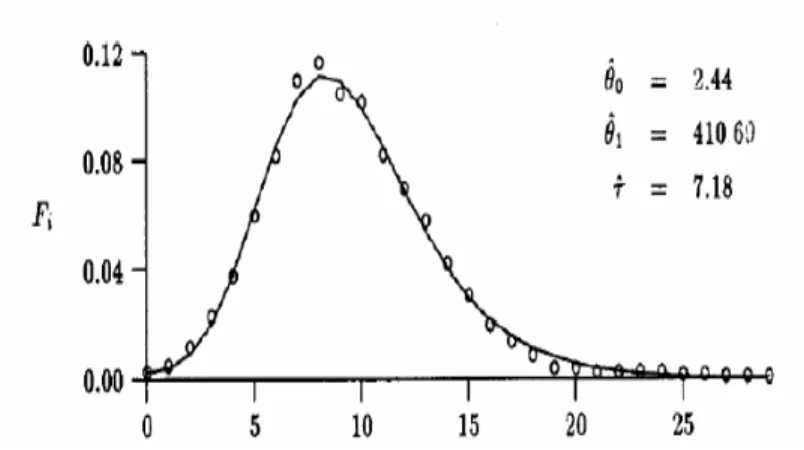
“numero efficace”, definito come il reciproco della probabilità che due individui scelti a caso abbiano la stessa madre (Rogers, 1995). Il modello dell’espansione improvvisa mantiene una buona affidabilità, non solo nel caso dell’espansione improvvisa, ma anche in quello di una crescita continua della popolazione (Rogers & Harpending, 1992). Il programma DnaSP (Rozas et al., 2003) mostra in forma di tabella e di grafico la distribuzione delle differenze nucleotidiche per ciascuna coppia di sequenze (distribuzione mismatch) e i valori attesi per popolazioni in espansione o regressione demografica. I valori attesi per una popolazione in espansione si dispongono graficamente a formare una curva dalla forma approssimabile ad un’onda (Rogers & Harpending, 1992). Popolazioni stabili dovrebbero, in teoria, originare una disposizione rettilinea ma generalmente presentano una disposizione disordinata, una spezzata, scarsamente attinente alla disposizione attesa. Nel caso di popolazioni in espansione, la curva delle osservazioni segue più o meno fedelmente la distribuzione attesa o comunque presenta onde evidenti (Fig. 2.3) (Rogers & Harpending, 1992).
Fig. 2.7. Esempio di curva relativa ad una popolazione in espansione: la linea continua indica la distribuzione attesa, i cerchietti indicano la distribuzione osservata. Da Rogers & Harpending, 1992.
Per valutare la significatività statistica delle espansioni demografiche, è stato applicato il test R2 (Ramos-Onsins & Rozas, 2002), utilizzando il programma DnaSP (Rozas et al.,
2003). Questo metodo si basa sulla frequenza delle mutazioni segreganti all’interno delle sequenze, tipiche dei fenomeni di espansione demografica. La statistica R2 rileva
l’eccesso di mutazione sui rami esterni di una linea genealogica e quindi quel eccesso di mutazioni recenti e proprie di una singola sequenza (“mutazioni private”) che deriva dalla crescita di una popolazione. Recentemente è stato dimostrato che i due test più potenti per individuare l’espansione demografica delle popolazioni sono il test FS di Fu
(1997) e quello R2 di Ramos-Onsins & Rozas (2002). Il comportamento di quest’ultimo
risulta però superiore all’altro nel caso di campioni a bassa numerosità (Ramos-Onsins & Rozas, 2002). Esiste un altro test considerato meno potente, quello D di Tajima (1989a).