D
D
D
IIISSSCCCUUUSSSSSSIIIOOONNNEEEL’aterosclerosi, la patologia delle grandi arterie responsabile attraverso gli eventi coronarici e cerebro-vascolari del 50% delle morti, è una condizione di infiammazione cronica che può generare eventi clinici acuti per l’intervenire del processo trombotico a livello di una placca instabile (507). Si tratta di un processo in continuo divenire caratterizzato da accumulo di lipidi e di componenti fibrose a livello della parete vascolare (87). Le lesioni aterosclerotiche originano preferenzialmente in punti critici dell’albero arterioso, quali le biforcazioni, le diramazioni e le pareti convesse di tratti arteriosi ad andamento curvo. In queste sedi, lo shear stress, cioè l’insieme delle forze emodinamiche applicate in senso tangenziale alla parete del vaso per effetto del flusso ematico, è basso o intermittente e verosimilmente tale da consentire il trasferimento passivo di componenti ematiche nella parete vascolare (508). In queste sedi si formano le lesioni precoci, le strie lipidiche, preludio alla formazione della placca intramurale, cioè precursori di lesioni più avanzate caratterizzate dall’accumulo di lipidi extracellulari provenienti dalla apoptosi delle cellule schiumose e dalla proliferazione delle cellule muscolari lisce.
Le strie lipidiche sono aree di ispessimento intimale sostenute dall’accumulo sottoendoteliale di macrofagi carichi di lipidi (cellule schiumose, foam cells) presenti già nella prima decade di vita a livello dell’aorta, a livello delle coronarie nella seconda decade, nella terza e quarta decade compaiono nelle arterie cerebrali. L’inizio del processo aterosclerotico coincide quindi con gli eventi patogenetici che regolano il formarsi della stria lipidica. Il primo evento che è possibile osservare nella parete arteriosa in ratti deficienti in Apolipoproteina E (ApoE) o in recettore per le lipoproteine a bassa densità (LDL) sottoposti a dieta ricca di colesterolo è l’accumulo di aggregati di particelle di lipoproteine a livello dell’intima (509, 510). Entro giorni o settimane, si osservano l’adesione dei monociti alla superficie dell’endotelio, successivamente, la loro migrazione attraverso il monostrato di cellule endoteliali, il loro accumulo nell’intima, la proliferazione, la differenziazione in macrofagi e, infine, la cattura delle lipoproteine con trasformazione in cellule schiumose. Con il tempo, l’apoptosi delle cellule schiumose costituisce il core necrotico della lesione ateromatosa a cui contribuiscono la migrazione dalla media, la proliferazione e l’accumulo delle cellule muscolari lisce (SMCs) (87).
S
SShhheeeaaarrrssstttrrreeessssss,,,ssstttrrreeessssssooossssssiiidddaaatttiiivvvooo,,,pppeeerrrmmmeeeaaabbbiiillliiitttàààeeennndddooottteeellliiiaaallleeeeeellleeeLLLDDDLLL
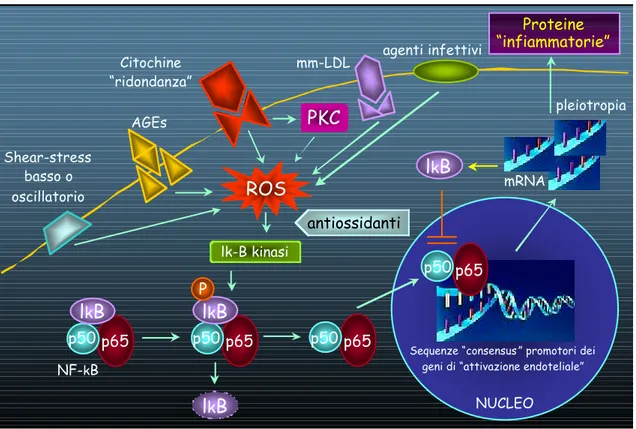
La localizzazione delle lesioni aterosclerotiche in tratti arteriosi a geometria particolare, in cui il flusso è più “complesso”, suggerisce un ruolo importante delle forze emodimaniche locali nell’aterogenesi. Le forze emodinamiche regolano la morfologia delle cellule endoteliali. Le cellule endoteliali che rivestono tratti “rettilinei” dell’albero arterioso, dove il flusso è laminare ed uniforme, presentano forma ellissoide e allineamento nella direzione del flusso. Le cellule delle biforcazioni, dei tratti di diramazione e delle “curvature”, dove il flusso è turbolento, hanno aspetto poligonale e nessun particolare orientamento. In queste zone, sedi preferenziali di lesione, la permeabilità dell’endotelio alle macromolecole (ed alle LDL) è aumentata (511). Evento primario del processo aterogenetico è l’accumulo di LDL a livello della matrice subendoteliale; il trasporto e la ritenzione delle LDL sono maggiori nelle sedi a flusso “disturbato” e quanto più elevata è la concentrazione delle LDL circolanti. Sensori primari di flusso (recettori) sono accoppiati ad eventi nucleari attraverso elementi responsivi allo shear stress (SSREs, shear stress response elements) presenti nelle regioni promoters di geni sensibili agli stimoli biomeccanici (512). Lo shear stress ridotto o oscillatorio o pulsatile induce un aumento dello stress ossidativo intracellulare a cui consegue l’attivazione di uno o pochi fattori di trascrizione tra cui nuclear factor-kB (NF-kB) che legandosi agli SSREs inducono la trascrizione “concertata” di numerosi geni capaci di attivare l’endotelio (paradigma “bio-meccanico” di attivazione endoteliale). NF-kB è un fattore di trascrizione ubiquitario la cui forma attivata, un eterodimero, è costituita dalle subunità p65 e p50 (figura 56). In cellule non attivate, NF-kB si localizza nel citoplasma dove il legame con IkBa e IkBb ne impedisce l’ingresso nel nucleo. Quando la cellula è stimolata, specifiche chinasi fosforilano IkB causandone la rapida degradazione; l’eterodimero NF-kB è libero di entrare nel nucleo per legarsi a sequenze specifiche nelle regioni promoter dei geni bersaglio. Poiché il gene per IkBα
presenta una sequenza capace di riconoscere kB nella propria regione promoter, NF-kB induce la sintesi di IkBa che, entrato nel nucleo, lega l’NF-kB attivato, lo riconduce nel citoplasma, interrompendo l’attivazione dell’espressione genica (513).
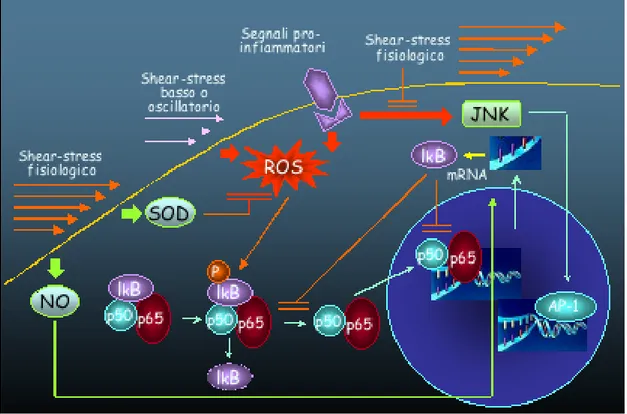
Lo shear stress laminare è in grado di indurre selettivamente geni “ateroprotettivi” quali la NO (nitric oxide) sintetasi costitutiva con aumentata produzione di NO. È inoltre capace di induzione della espressione di IkBa, inibizione della attivazione citoplasmatica di NF-kB e accelerazione della sua disattivazione a livello del nucleo (508). Inoltre, lo shear stress fisiologico stimola l’espressione di enzimi ad attività antiossidante con riduzione dello stress ossidativo (figura 57).
Figura 56 - Il sistema di trascrizione NF-kB comprende un eterodimero p50/p65 normalmente sequestrato nel citoplasma ad opera dell’inibitore IkB. Sotto l’influenza dello shear stress, delle LDL minimamente ossidate, di molte citochine, dei prodotti avanzati della glicazione (AGEs), della PKC (proteina chinasi C) e di altri stimoli capaci di indurre attivazione endoteliale, la generazione intracellulare di specie reattive dell’ossigeno (ROS) porta alla degradazione enzimatica di IkB. I complessi p50/p65, liberi dall’inibitore, migrano nel nucleo, dove si legano a sequenze di riconoscimento nella regione promoter di geni (molecole di adesione, citochine, enzimi) alla cui trascrizione consegue l’attivazione endoteliale.
L’attività endoteliale
Due aspetti dell’attivazione endoteliale hanno particolare rilevanza nel determinismo del processo aterosclerotico: l’aumentata permeabilità endoteliale e l’aumentata adesività per i leucociti (514). Numerose evidenze dimostrano che lo stress ossidativo aumenta la permeabilità endoteliale. Le specie reattive dell’ossigeno (ROS) causano la formazione di gap nel monostrato delle cellule endoteliali, modificazioni nella morfologia cellulare, riorganizzazione della rete dei filamenti di actina; a queste alterazioni conseguono la compromissione delle giunzioni (adherens e tight junctions) e della adesività intercellulare quali determinanti primari dell’aumentata
p65 ROS Citochine “ridondanza” mm-LDL Shear-stress basso o oscillatorio AGEs PKC lk-B kinasi p50 lkB NF-kB p65 p50 lkB p65 p50 lkB p65 p50 NUCLEO
Sequenze “consensus” promotori dei geni di “attivazione endoteliale”
P mRNA Proteine “infiammatorie” lkB antiossidanti pleiotropia agenti infettivi
Figura 57 - Meccanismi pro-infiammatori ed anti-infiammatori dello shear stress. Lo shear stress turbolento (ridotto, oscillatorio, pulsatile), così come segnali proinfiammatori, aumentano lo stress ossidativo intracellulare a cui consegue attivazione di NF-kB (nuclear factor-kB). Lo shear stress laminare invece stimola la produzione di NO che, attraverso l’induzione della espressione di IkBa determina inibizione della attivazione di NF-kB e accelera la disattivazione dell’eterodimero p50/p65 a livello del nucleo. Lo shear stress fisiologico, inoltre, stimola l’espressione della SOD (superoxide dismutase) con conseguente riduzione dello stress ossidativo intracellulare e blocco della JNK (c-Jun N-terminal kinase) pathway attivata in risposta a stimoli pro-infiammatori (508).
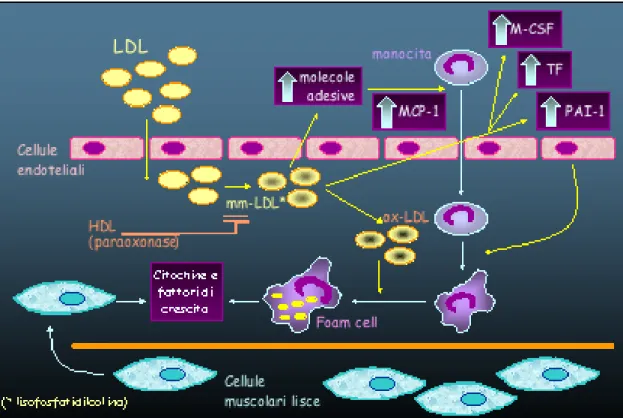
Le LDL diffondono passivamente attraverso le giunzioni intercellulari e la loro ritenzione subendoteliale sembra coinvolgere una interazione tra la Apolipoproteina B (ApoB) ed i proteoglicani della matrice. Oltre le LDL, altre lipoproteine contenenti ApoB quali la lipoproteina (a), capace di effetti sulla fibrinolisi e sulla proliferazione delle SMCs, ed i remnants possono accumularsi nell’intima. Le LDL native non sono captate dai macrofagi con avidità sufficiente a generare le foam cells. Tuttavia, le LDL intrappolate subiscono modificazioni (ossidazione, lipolisi, proteolisi, aggregazione) capaci di favorire la captazione da parte dei monociti, di attivare/alimentare il processo infiammatorio e di condurre alla formazione delle cellule schiumose. La modificazione ossidativa delle LDL che si verifica nell’intima arteriosa, in microambienti protetti dagli antiossidanti circolanti, sembra essere il momento critico nella attivazione del processo infiammatorio.
Questa modificazione dà origine inizialmente a LDL “minimamente ossidate” che hanno capacità pro-infiammatoria, ma non sono sufficientemente alterate dall'essere riconosciute dai recettori scavenger dei macrofagi. La lipo-ossigenasi è verosimilmente una importante fonte di ROS per la ossidazione delle LDL; l’enzima produce molecole quali l’acido idroperossi-eicosatetranoico (HPETE) che viene trasferito attraverso la membrana cellulare trascinando le LDL extracellulari (515). L’ossidazione delle LDL è inibita dalle lipoproteine ad alta densità (HDL) che trasportano una proteina anti-ossidante, la paraoxonasi (PON1), capace di degradare fosfolipidi ossidati biologicamente attivi (428).
L’adesione dei monociti e dei linfociti, ma non dei polimorfonucleati, ad un endotelio morfologicamente indenne, caratterizza le fasi iniziali dell’aterosclerosi. Un evento trigger per l’attivazione del processo infiammatorio è l’accumulo di LDL minimamente ossidate che stimolano le sovrastanti cellule endoteliali a produrre molteplici famiglie di proteine che, ognuna con una distinta funzione, forniscono “segnali di traffico” per i leucociti. L’attività biologica delle LDL minimamente ossidate è esercitata principalmente dalla componente fosfolipidica, ma le LDL ossidate possono anche inibire la produzione di NO. Oltre alle LDL ossidate, numerosi altri fattori possono modulare lo stress ossidativo e il processo infiammatorio comprese le forze emodinamiche, l’omocisteina, gli steroidi sessuali, il glucosio e i prodotti avanzati della glicazione (AGEs, advanced glycation endproducts). Le LDL devono essere ampiamente modificate (altamente ossidate) perché possano essere captate dai macrofagi con l’avidità necessaria alla formazione delle foam cells (figura 58) (516, 517).
L’uptake delle LDL modificate è mediato da recettori scavenger (SR-A, CD36, CD68) la cui espressione è regolata dal PPAR-γ (peroxisome proliferator-activated receptor-γ), un fattore di trascrizione che riconosce gli acidi grassi ossidati tra i suoi ligandi, e da citochine quali il TNF-α
e l’interferone-γ (IFN-γ). I macrofagi producono attivamente ApoE e questo può promuovere l’efflusso di colesterolo verso le HDL, inibendo la trasformazione dei macrofagi in cellule schiumose.
La stria lipidica progredisce attraverso l’aumento della deposizione intra- ed extra-cellulare di lipidi (colesterolo ed esteri), l’infiltrazione di SMCs migrate dalla media o proliferate in situ e la deposizione di matrice prodotta dalle SMCs. L’iperespressione delle molecole di adesione ed il reclutamento leucocitario continuano ad alimentare la lesione ateromatosa; citochine e fattori di crescita secreti dai macrofagi e dalle cellule T sostengono la migrazione e la proliferazione delle SMCs e la produzione della matrice extracellulare.
L’alterazione delle proprietà antitrombotiche dell’endotelio (alterata capacità di produrre sostanze antipiastriniche quali prostaciclina e NO, ridotta produzione di trombomodulina e di attivatori del plasminogeno) può consentire la deposizione piastrinica a cui consegue l’aumentata produzione di un fattore di crescita di derivazione piastrinica, il platelet-derived growth factor (PDGF), un potente chemoattrattante per le SMCs.
Gli effetti aterogenici dell’ipertensione, dell’ipercolesterolemia e di altri fattori di rischio cardiovascolare sembrano almeno in parte mediati dall’attivazione del sistema renina-angiotensina.
Figura 58 - Le modificazioni ossidative delle LDL a livello dell’intima arteriosa sono alla base della aterogenicità di queste lipoproteine. Le LDL minimamente modificate (mmLDL) e le β-lipoproteine a densità molto bassa (β-VLDL) contribuiscono alla adesività monocitaria e alla produzione di MCP-1, M-CSF, fattore tissutale (TF) e inibitore dell’attivatore del plasminogeno (PAI-1). Le LDL altamente ossidate, captate dai macrofagi, inducono la formazione delle cellule schiumose e successivamente, l’accumulo di lipidi extracellulari ed il reclutamento delle cellule muscolari lisce.
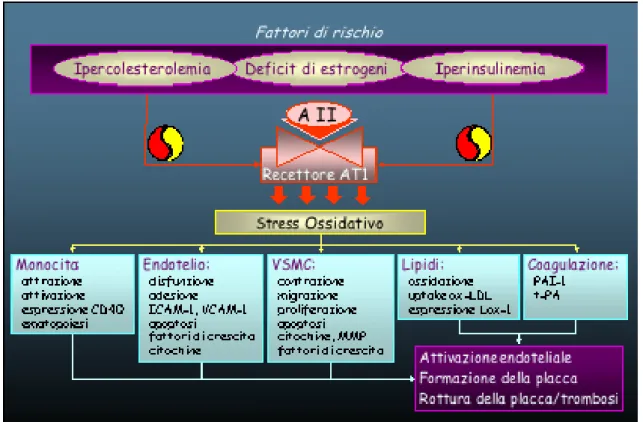
L’angiotensina II stimola direttamente la proliferazione delle cellule muscolari lisce (iperespressione di PDGF) e la deposizione della matrice. Fattori di rischio quali l’ipercolesterolemia, la carenza di estrogeni, l’iperinsulinemia possono indurre iperespressione del recettore AT1 per l’angiotensina II; all’iperespressione del recettore AT1 conseguono aumentata produzione di ROS, proliferazione delle SMCs e vasocostrizione. L’iperespressione del recettore AT1 insieme alla upregulation dell’ACE vascolare contribuiscono ad aumentare la pressione arteriosa, ad indurre attivazione e disfunzione endoteliale (adesione dei monociti, vasocostrizione, pro-coagulazione) e a favorire la progressione del processo aterosclerotico (figura 59) (518, 519).
Figura 59 - L’attivazione del recettore AT1 per l’angiotensina II aumenta la produzione di ROS. Lo stress ossidativo è coinvolto nella attrazione/attivazione dei monociti, nella attivazione delle cellule endoteliali (disfunzione, adesione, apoptosi), nella migrazione, proliferazione, sintesi di matrice ed apoptosi delle SMCs. L’ossidazione delle LDL e l’uptake delle LDL ossidate sono aumenati, così come l’espressione del PAI-1.
La funzione endoteliale nel diabete mellito
Il diabete mellito è caratterizzato da disfunzione endoteliale la cui espressione preminente è una ridotta attività biologica di monossido d’azoto (NO). Una ridotta sintesi di NO, già peraltro in presenza di insulino-resistenza, non solo si associa a ridotta vasodilatazione, ma anche ad aumentata aggregazione piastrinica e ad un’aumentata adesività leucocitaria alla parete arteriosa. L’NO è un radicale libero prodotto da un aminoacido essenziale, L-arginina, che viene convertito a L-citrullina con produzione di NO. La reazione è catalizzata dalla sintasi dell’ossido nitrico, che prevede tre differenti isoforme: la sintasi neuronale (nNOS o NOS1) espressa prevalentemente nei neuroni, la sintasi inducibile (iNOS o NOS2), la cui espressione è indotta solamente da alcuni specifici stimoli, e la sintasi endoteliale (eNOS o NOS3) espressa nell’endotelio, nei cardiomiociti e nelle piastrine. L’evento fisiologico che porta a un aumento dell’attività di NOS3, e quindi della sintesi di NO, è lo shear stress, ovvero quella forza che viene prodotta dal flusso ematico per unità di superficie della parete vascolare.
L’enzima che sintetizza NO, richiede la fisiologica presenza di alcuni cofattori, quali la tetraidrobiopterina: in corso di diabete il loro deficit “disaccoppia” la eNOS che non produce più NO quanto radicali nitrosilati, prodotti altamente tossici sia per le funzioni cellulari sia per le fisiologiche funzioni del vaso arterioso. NO, aumentando la vasodilatazione, favorisce l’utilizzazione di substrati ossidabili, quali il glucosio, da parte dei tessuti insulino-sensibili: pertanto l’NO funge da “ponte” tra fisiologia vascolare e metabolismo intermedio.
Disfunzione endoteliale e diabete
L’iperglicemia è in grado di causare disfunzione endoteliale, la maggior parte delle osservazioni suggerisce che il danno da iperglicemia sull’endotelio sia secondario a stress ossidativo (520). Sono stati evidenziati dei composti che inattivano o alterano il normale rapporto precursore/prodotto: uno di questi è la dimetil arginina asimmetrica (ADMA): è stato recentemente riscontrato che nel diabete di tipo 2 l’ADMA aumenta significativamente (521). Alcuni fattori sono in grado di ridurre l’espressione di NOS: tra questi l’iperglicemia stessa, l’ipossia, elevate concentrazioni di TNF alfa, elevate concentrazioni di lipoproteine a bassa densità (LDL) ossidate, tutte condizioni queste che caratterizzano il milieu metabolico del diabetico di tipo 2.
I prodotti di glicazione tardiva sono in grado di interferire con la capacità di NO di diffondere e quindi di svolgere la propria azione vasodilatatrice a livello delle cellule muscolari lisce. Un ruolo particolarmente importante è rivestito dalla disponibilità di cofattori, in particolare della tetraidrobiopterina (BH4): in corso di iperglicemia è stata osservata una riduzione di questo cofattore. Una diretta conseguenza dell’alterata funzione di NOS è non solo la formazione di radicali liberi ma anche di perossinitriti (-OONO), composti estremamente tossici per la cellula che rappresentano la diretta conseguenza della “distruzione” dell’NO. Infatti, in presenza di ione superossido in eccesso e di ridotte capacità antiossidanti l’NO viene convertito a perossinitrito, un potente ossidante il quale produce radicali idrossilici e NO2. In condizioni normali la formazione di perossinitriti è estremamente bassa mentre in condizioni quali il diabete vi è un significativo aumento di questi radicali. Al di là della gran messe di lavori sperimentali che confermano un danno glucosio-mediato a livello endoteliale, nel diabetico di tipo 2 l’osservazione di una franca alterazione endoteliale non è univoca. Alcuni autori non hanno osservato una riduzione dell’aumento di flusso mediato dall’acetilcolina in diabetici privi di altri fattori di rischio coronarici (522). Qualora invece si prendano in considerazione diabetici di tipo 2 in cui coesistano uno o più fattori di rischio vi è un netto deficit della funzione endoteliale. Nei diabetici di tipo 2, una ridotta funzione endoteliale è stata di volta in volta associata, non tanto alla malattia diabetica quanto alla dislipidemia, all’ipertensione, all’obesità addominale. L’insulina è in grado di stimolare la sintesi di NO da parte delle cellule endoteliali. L’insulina, mediante l’interazione con il suo recettore, è in grado di stimolare a livello delle cellule endoteliali, sia la sintesi di NO che l’utilizzazione di glucosio. L’NO, a sua volta, non solo è in grado di vasodilatare ma anche di aumentare, nelle cellule muscolari, il trasporto di glucosio. Appare pertanto evidente come i messaggi metabolico ed emodinamico dell’insulina siano tra loro strettamente correlati: pertanto difetti nella transduzione del messaggio metabolico possono predire difetti nella transduzione del messaggio emodinamico (523). Studi nell’uomo suggeriscono un ruolo primario della disfunzione endoteliale nella genesi della resistenza insulinica.
Il legame tra insulino-resistenza e disfunzione endoteliale sembra essere più consistente quando l’insulino-resistenza cosegrega con un qualsiasi elemento della classica sindrome metabolica: una ridotta produzione/attività di NO è stata riscontrata in pazienti con ipertensione arteriosa, con ipertrigliceridemia, bassi livelli di colesterolo HDL, obesità addominale, elevati livelli di acidi grassi liberi e citochine infiammatorie.
Recentemente è stato dimostrato che una moderata iperinsulinemia causa disfunzione endoteliale mediante stress ossidativi (524). Si può pertanto affermare che l’insulino-resistenza non solo causa disfunzione endoteliale mediante l’azione negativa dei molteplici fattori di rischio che cosegregano con questa patologia, ma anche mediante un’azione negativa diretta dell’ormone sul letto endoteliale.
L’endotelio gioca un ruolo importante nella regolazione del flusso sanguigno ai tessuti insulino-sensibili ed è stata evidenziata una correlazione diretta fra la vasodilatazione endotelio-mediata e la sensibilità insulinica. Infatti, l’insulina agisce come vasodilatatore e, negli stati di insulino-resistenza che si associano ad obesità, ipertensione e diabete mellito tipo 2, un’alterata vasodilatazione è stata descritta in presenza di un ridotto numero di capillari a livello del muscolo scheletrico. Vi sono evidenze in letteratura che un’attivazione della NO sintetasi endoteliale (eNOS) aumenta il flusso ematico a livello muscolare e che l’effetto è mediato attraverso un’attivazione di IP3 chinasi e AKT chinasi. L’aumentata perfusione muscolare mediata dall’ossido nitrico fa aumentare l’arrivo di glucosio alle cellule muscolari.
La dislipidemia è un’altra alterazione tipica della sindrome da insulino-resistenza e rappresenta uno dei più importanti fattori di rischio per la malattia cardiovascolare. E’ stato evidenziato come una disfunzione endoteliale a livello dei capillari possa giocare un ruolo importante nel modulare le modificazioni della concentrazione delle lipoproteine, attraverso un’alterata attività della lipoprotein lipasi endoteliale, un enzima legato ai glicosaminoglicani che idrolizza i trigliceridi ed ha come sito di azione la superficie dell’endotelio capillare.
Una disfunzione della lipoprotein lipasi endoteliale comporta aumentati livelli di trigliceridi, ridotta concentrazione di HDL e aterosclerosi prematura. È stato proposto che diversi insulti a livello dell’endotelio, quali prodotti del fumo di sigaretta, l’esposizione ai radicali liberi, alle lipoproteine ossidate e incrementi di shear stress, possono causare perdita dei glicosaminoglicani e un’alterazione dell’attività della lipoprotein lipasi endoteliale. Questo può rappresentare un interessante meccanismo per spiegare l’ipertrigliceridemia caratteristica del paziente diabetico di tipo 2 e con sindrome da insulino-resistenza.
Endotelio e complicanze microvascolari (microalbuminuria)
Sia nel diabete tipo 1 che nel tipo 2, l’aumento della secrezione urinaria di albumina è fattore predittivo di malattie cardiovascolari indipendentemente dalla presenza dei convenzionali fattori di rischio, della durata e del grado di controllo del diabete e prima della comparsa di nefropatia conclamata. Se la microalbuminuria deve essere considerata una componente della sindrome plurimetabolica e quindi essere come tale predittivo di complicanze cardiovascolari è ancora incerto dato che, in studi recenti (Horn Study), essa sembra correlare più strettamente con l’aumento della pressione arteriosa e con la ridotta tolleranza al glucosio (diabete tipo 2) che con l’insulino-resistenza (525).
I meccanismi patogenetici che legano microalbuminuria e aterogenesi non sono chiari. L’associazione tra microalbuminuria, ipertensione ed alterazione aterogene di lipidi e lipoproteine spiega solo in parte l’elevato rischio cardiovascolare di questi pazienti. Indubbiamente, la comparsa della microalbuminuria si associa ad un aumento dei valori della pressione arteriosa sistolica e diastolica (158), ma le modificazioni nel profilo lipidico che emergono chiaramente nei soggetti con macroalbuminuria (aumento dei trigliceridi, del colesterolo totale e LDL, del rapporto tra colesterolo totale e HDL e, nelle donne, la riduzione del colesterolo HDL, come registrato - nel diabete tipo 1 – dallo studio EURODIAB) sono tutt’altro che evidenti nei pazienti con microalbuminuria. In questi ultimi, lo studio EURODIAB evidenzia soltanto un aumento dei trigliceridi spiegato, in parte, dalla presenza di fattori confondenti (188). La “Steno hypothesis” postula che la microalbuminuria sia specchio di disfunzione vascolare generalizzata (e di successiva aterosclerosi) (526) riconducibile ad alterazioni strutturali della parete quali riduzione del contenuto e della solfatazione degli eparansolfati della matrice extracellulare. Aumentate concentrazioni sieriche di acido sialico, predittive di patologia aterosclerotica (527), potrebbero essere espressione di una aumentata incorporazione nelle glicoproteine della matrice extracellulare con conseguenti alterazioni di numerosi fattori emoreologici, della permeabilità transvascolare e dell’accumulo di lipidi nella parete arteriosa (528). Aumentate concentrazioni sieriche di acido sialico sono state descritte in diabetici tipo 1 con microalbuminuria (529, 530) e con nefropatia conclamata (530).
Se accettiamo che la microalbuminuria faccia parte o tenda ad associarsi a insulino-resistenza, allora altri fattori oltre l’ipertrigliceridemia e i bassi livelli delle HDL, quali elevate concentrazioni di LDL piccole e dense e aumentata lipemia post-prandiale, possono contribuire all’eccesso di rischio cardiovascolare.
La microalbuminuria può porsi quale marker di disfunzione delle cellule endoteliali. La microalbuminuria potrebbe rappresentare l’espressione e l’amplificazione a livello renale di alterazioni più generalizzate della permeabilità endoteliale e dell’emodinamica del microcircolo. La disfunzione endoteliale è il candidato più probabile a spiegare perché un indice di disfunzione glomerulare (l’aumento dell’escrezione urinaria dell’albumina) si comporta come un marker di aterosclerosi associandosi ad un eccesso di complicanze extrarenali, soprattutto cardiovascolari (531-532). La capacità di lavoro aerobico è ridotta nei diabetici tipo 1 con microalbuminuria, l’indice di massa ventricolare sinistra e la prevalenza di ipertrofia ventricolare sinistra, in associazione ad aumenti nel rapporto notte/giorno della pressione sistolica per aumento della pressione sistolica notturna, sono maggiori nei diabetici tipo 2 con microalbuminuria. Aumentate concentrazioni plasmatiche di fibrinogeno e di fattore di von Willebrand e ridotta attività fibrinolitica, presenti nei diabetici di tipo 1 con microalbuminuria e nefropatia diabetica iniziale, suggeriscono l’esistenza di un danno endoteliale generalizzato (531,533). Lo studio IRAS (Insulin Resistance Atherosclerosis Study) dimostra una associazione tra microalbuminuria e livelli plasmatici del fibrinogeno (r=0,14; p=0,0001). Tale correlazione risulta indipendente dall’associazione che pur esiste tra microalbuminuria e ipertensione arteriosa, circonferenza alla vita, glicemia a digiuno (534). I microalbuminurici diabetici e non diabetici presentano aumentate concentrazioni plasmatiche di proteina C-reattiva (CRP), ma l’associazione tra CRP e microalbuminuria (r=0,17; p<0,0001) non permane dopo correzione per pressione arteriosa, circonferenza alla vita, glicemia e fibrinogenemia (534). Altri studi hanno documentato l’associazione tra microalbuminuria sICAM-1 (soluble intercellular adhesion molecule-1) e sVCAM-1 (soluble vascular cell adhesion molecule-1) (535), ACE (angiotensin converting enzyme), endotelina e PAI-1 (394). Particolarmente interessante in questo senso è il riscontro in diabetici tipo 1 microalbuminurici di aumentati valori di transcapillary escape rate di albumina (TERalb) (536) - espressione di una aumentata permeabilità del microcircolo - e di alterata vasodilatazione endotelio-dipendente - espressione di alterazione dei meccanismi NO-mediati (531,537,538).
Studi più recenti, in pazienti con diabete tipo 1 confermano la presenza di alterazioni nella funzione endoteliale e dei marker di danno endoteliale nei soggetti con microalbuminuria ed estendono queste osservazioni anche ai diabetici con normale escrezione urinaria di albumina; così i diabetici tipo 1 microalbuminurici (con maggior intensità), ma anche i diabetici tipo 1 normoalbuminurici (possibilmente quelli destinati a progredire verso la microalbuminuria) presentano ridotta vasodilatazione flusso-mediata (539) e ridotta risposta vasodilatatoria all’acetilcolina (540).
Dogra e coll. (541), dimostrano che diabetici tipo 1 microalbuminurici in condizioni di “near-normoglycemia” presentano ridotta vasodilatazione flusso mediata (ma anche ridotta vasodilatazione endotelio-indipendente) rispetto ai normoalbuminurici. Questi ultimi, a loro volta, presentano ridotta vasodilatazione endotelio-dipendente rispetto ai controlli. La presenza di disfunzione endoteliale nei diabetici microalbuminurici suggerisce che essa può precedere la comparsa della microalbuminuria e porsi come marker precoce di danno cardiovascolare. Aumentati livelli di PAI-1, aumentata attività del TFPI (tissue factor pathway inhibitor), ridotta distensibilità arteriosa (542), aumentata permeabilità del microcircolo (543) e aumentata sintesi, secondaria alla aumentata inattivazione da parte delle specie reattive dell’ossigeno, di ossido nitrico (NO) (544,545) sono stati descritti nei soggetti con diabete tipo 1 e microalbuminuria. L’aumentata biosintesi ed attività di NO potrebbe concorrere a generare iperfiltrazione glomerulare e microalbuminuria persistente. Aumentate concentrazioni plasmatiche di VEGF (vascular endothelial growth factor) sono state descritte, in un follow-up di 8 anni, in diabetici tipo 1 normoalbuminurici destinati a sviluppare la microalbuminuria e la nefropatia incipiente. Analogamente, nel diabete tipo 2, la microalbuminuria è associata ad aumentate concentrazioni di fattore di von Willebrand, trombomodulina, t-PA, PAI-1 e collagene tipo IV (531), ad aumentati livelli sierici di fattore VII, fibronectina, fibrinogeno e alfa-2-microglobulina (546) e, sebbene non in tutti gli studi (188), con aumentati valori di TERalb (547). Più recentemente sono state descritte riduzioni nel release della lipoprotein lipasi endoteliale (a cui consegue un ridotto catabolismo delle lipoproteine ricche in trigliceridi) (548), alterazioni nella risposta vascolare endotelio-dipendente delle arterie retiniche ed intrarenali (549), riduzioni nella risposta vasodilatatoria endotelio-dipendente indotta da acetilcolina (somministrata per iontoforesi) a livello del microcircolo cutaneo, aumenti nelle concentrazioni plasmatiche di sICAM e sVCAM non solo in soggetti con microalbuminuria, ma anche in diabetici tipo 2 con normale escrezione urinaria di albumina (550).
In studi prospettici, lo svilupparsi della microalbuminuria è accompagnato da un aumento del fattore di von Willebrand (valore di riferimento: 50-150%) da 121 a 203% nel diabete tipo 1 (533) e da 116 a 219% nel diabete tipo 2, mentre nessuna associazione è stata riscontrata con la retinopatia (551). Inoltre, un suggestivo studio prospettico, dimostra che nel diabete tipo 1 l’aumento dei livelli plasmatici di vWF precede la comparsa della microalbuminuria di circa 3 anni (552). Un marker di disfunzione endoteliale più sensibile dell’aumento del vWF potrebbe essere la compromissione della vasodilatazione endotelio-dipendente riportata in diabetici tipo 1 anche in assenza di complicanze (553,554).
Alcune osservazioni sono tuttavia in conflitto con questa ipotesi. Il diabete tipo 1 non complicato è caratterizzato da vasodilatazione piuttosto che vasocostrizione a livello del microcircolo (555) e del macrocircolo (556); inoltre studi in cui la stratificazione in accordo con la presenza o alla assenza di complicanze è stata eseguita più accuratamente suggeriscono che la vasodilatazione endotelio-dipendente e quella non endotelio-dipendente dei vasi di resistenza e di conduttanza non sono né compromesse né accentuate in assenza di complicanze (556-558). La vasodilatazione endotelio-dipendente non è stata estesamente studiata nel diabete tipo 2, soprattutto in relazione alla presenza e assenza di microalbuminuria. La funzione endoteliale potrebbe essere compromessa (559-560), anche in soggetti con normale escrezione urinaria di albumina (561), o rimanere inalterata. D’altra parte, sebbene non sia noto quanto specificamente siano di origine endoteliale, sia i livelli plasmatici di endotelina che quelli del PAI-1 sono aumentati in diabetici tipo 2 non complicati.
In conclusione, la disfunzione endoteliale (562) che caratterizza il diabete tipo 1 e il tipo 2 in presenza di micro (e macroalbuminuria) è generalizzata e coinvolge numerosi aspetti delle funzioni dell’endotelio che vanno dall’alterazione della permeabilità (563) che caratterizza le fasi precoci del processo aterosclerotico, all’attivazione dei processi di coagulazione responsabile dell’intervenire di eventi critici (564). Lo stretto legame tra microalbuminuria e disfunzione endoteliale, sebbene non universalmente confermato, fornisce una spiegazione suggestiva dell’eccesso di rischio cardiovascolare che caratterizza i microalbuminurici non diabetici (565) così come i diabetici microalbuminurici (566) ancor prima dei pazienti con nefropatia conclamata (159). È tuttavia a questo punto necessario ricordare, come discusso sopra, che alterazioni della funzione endoteliale sono state osservate, sebbene non sempre, anche in soggetti diabetici in assenza di albuminuria (538) e addirittura in soggetti normoglicemici a rischio di futuro sviluppo di diabete di tipo 2 quali sono le donne con storia positiva per diabete gestazionale (567), suggerendo che la disfunzione endoteliale può precedere la comparsa del diabete di tipo 2 (568). È a questo punto utile ricordare che una interessante ipotesi pone la disfunzione endoteliale nel ruolo di fattore causale non solo del processo aterosclerotico, ma anche della insulino-resistenza (569). La disfunzione endoteliale “centrale”, a livello dei grossi vasi gioca un ruolo fondamentale nella patogenesi dell’aterosclerosi. La disfunzione endoteliale “periferica” a livello arteriolare e capillare, potrebbe giocare un ruolo chiave nella patogenesi sia dell’insulino-resistenza che di numerose delle componenti che caratterizzano la sindrome plurimetabolica.
Sindrome da insulino-resistenza e molti aspetti dell’aterogenesi possono essere visti come conseguenze diverse della disfunzione endoteliale a livello di differenti letti vascolari. In base a questa ipotesi, microalbuminuria e cardiopatia ischemica possono essere interpretate come risultati, spesso coesistenti, di un determinante comune capace di indurre danno vascolare.
Presupposti della ricerca
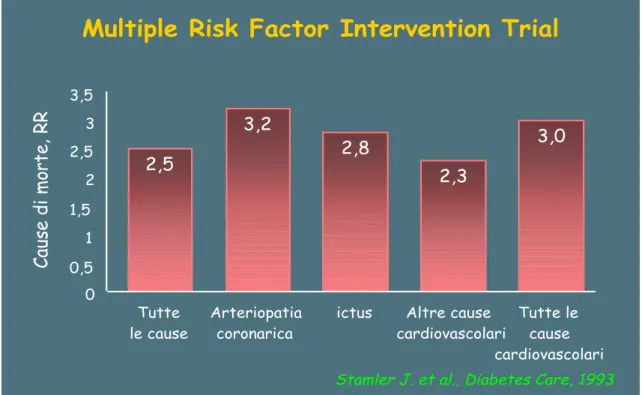
L'aterosclerosi rappresenta la principale causa di morbilità e mortalità nella popolazione generale e ancor più nel paziente con diabete mellito. Nel diabete, le lesioni aterosclerotiche appaiono più precocemente, sono più gravi e più estese. La mortalità dovuta alla malattia coronarica ed alla patologia cerebrovascolare è almeno doppia nel diabete rispetto alla popolazione generale. Il rischio relativo di malattia cardiovascolare dei diabetici va da due (maschi) a quattro volte (femmine) il rischio della popolazione non-diabetica. Tra i dati più convincenti sono quelli dello studio MRFIT (Multiple Risk Factor Intervention Trial) in cui, dalla popolazione generale dei soggetti reclutati costituita da 347978 maschi di età compresa tra 35 e 57 anni, è stata estratta una coorte di 5163 diabetici (27).
I risultati ottenuti, in termini di mortalità corretti per età, colesterolemia, pressione arteriosa e fumo di sigaretta, sono riportati in figura 60.
Figura 60 - Studio MRFIT. Cause di morte e rischio relativo nel diabete mellito tipo 2 (27).
Multiple Risk Factor Intervention Trial
Stamler J. et al., Diabetes Care, 1993
2,5 3,2 2,8 2,3 3,0 0 0,5 1 1,5 2 2,5 3 3,5 Tutte le cause Arteriopatia coronarica
ictus Altre cause cardiovascolari Tutte le cause cardiovascolari Cause di morte, RR
I rapporti tra malattia cardiovascolare e diabete sono sottesi da una serie di fenomeni. Nella popolazione diabetica, rispetto alla popolazione generale, è descritta una maggiore prevalenza dei "tradizionali" fattori di rischio aterogeno (maggior prevalenza di ipertensione, di dislipidemia, etc.). Nei diabetici assume particolare significato e rilevanza la presenza di fattori di rischio "modificati" dalla condizione diabete (ossidazione delle lipoproteine LDL, glicazione di alcune apolipoproteine, glicazione di componenti strutturali dell'endotelio, etc.). Sono infine presenti fattori di rischio "specifici" del diabete (iperglicemia, iperinsulinemia) (5).
Tutti questi aspetti sono particolarmente evidenti nei diabetici con nefropatia. I pazienti con nefropatia diabetica in fase conclamata presentano un'esasperazione del rischio cardiovascolare e una prevalenza particolarmente elevata dei fattori di rischio sia "tradizionali" che "modificati" o "specifici".
Inoltre, già i pazienti con microalbuminuria persistente (nefropatia incipiente) sono caratterizzati da un maggior rischio di malattia cardiovascolare e riconoscono un cluster di fattori di rischio per aterosclerosi ben prima della comparsa della nefropatia conclamata.
Un’ipotesi suggestiva collega l'aumentato rischio aterogeno dei diabetici ad un "terreno comune" che potrebbe spiegare, su base dismetabolica e/o su base genetica, la comparsa sia delle malattie cardiovascolari che della microangiopatia e, in particolare, della nefropatia diabetica. Un primo terreno comune potrebbe essere rappresentato dal danno endoteliale, un fenomeno (o un meccanismo patogenetico) che potrebbe precedere la comparsa della nefropatia e della microalbuminuria (552), ma anche giustificare l’aumentato rischio di malattia cardiovascolare (570). Al danno endoteliale possono essere ricondotte in un complesso meccanismo di causa-effetto una serie di alterazioni aterogene (ipertensione arteriosa, ipertrigliceridemia, riduzione del colesterolo HDL, insulino-resistenza e iperinsulinemia, migrazione ed adesione dei monociti, modificazione dell'architettura e della funzione delle cellule muscolari lisce) che possono esprimersi sia nel danno aterosclerotico che nell’alterazione del microcircolo. Al potenziale aterogeno della disfunzione endoteliale, si sono via via aggiunti l'ipercoagulabilità e la ridotta fibrinolisi (aumento dei livelli di PAI-1), l'aumento della permeabilità vascolare, il riscontro di LDL piccole e dense e la stessa microalbuminuria (571).
Non sorprende quindi che gli studi dedicati ad indagare la base genetica delle complicanze del diabete si siano rivolti non soltanto allo studio dei processi che regolano il metabolismo glucidico, ma anche e soprattutto a valutare i meccanismi che, a livello genetico, modulano l'espressione, l'attività e la funzione di proteine coinvolte nei meccanismi di danno endoteliale (ipertensione, dislipidemia, ipercoagulabilità, etc.) che strettamente coincidono sia con i fattori di rischio cardiovascolare che con i determinanti del danno microangiopatico (figura 61).
Di fatto, i marcatori genetici del rischio macroangiopatico sono stati pressochè sistematicamente candidati a spiegare il rischio di sviluppare le complicanze microangiopatiche del diabete e, in particolare, a spiegare la suscettibilità alla nefropatia diabetica (572). Un secondo terreno comune potrebbe essere rappresentato dall’insulino-resistenza (573). L’iperinsulinemia e l’insulino-resistenza emergono sia come un fattore di rischio indipendente che come un denominatore comune del cluster di fattori di rischio cardiovascolare che caratterizza il paziente con diabete mellito (574), ma recentemente sono state confermate anche come fattori di rischio indipendenti sia nell’incidenza della retinopatia (575) che della microalbuminuria persino nel diabete tipo 1.
In particolare, la dislipidemia associata al diabete e all’insulino resistenza (dislipidemia aterogena) è caratterizzata da una costellazione di anomalie lipoproteiche rappresentate da: 1. aumentati livelli di lipoproteine a densità molto bassa (VLDL); 2. presenza di particelle LDL piccole e dense; 3. bassi livelli di colesterolo HDL (576).
Queste tre anomalie sono metabolicamente interconnesse. Elementi chiave di queste interconnessioni sono le HDL, l’enzima LCAT (Lecitin-Colesterolo-Acil-Transferasi) e la Proteina di Trasferimento degli Esteri del Colesterolo (CETP). Queste componenti hanno un ruolo fondamentale nel modellare la composizione del core e della superficie delle LDL durante la loro produzione. Elevati livelli di acidi grassi si producono in condizioni di insulino resistenza per l’incapacità dell’insulina di sopprimere il rilascio di acidi grassi liberi (NEFA) da parte del tessuto adiposo, soprattutto quello distribuito centralmente, il più metabolicamente attivo.
D’altra parte, la lipasi ormono-sensibile, presente negli epatociti è infatti attivata dalla insulino-resistenza o dalla carenza di insulina. L’insulino-insulino-resistenza e l’iperinsulinemia compensatoria sono associate ad un’aumentata produzione epatica di VLDL e ad un incremento dei trigliceridi plasmatici. In fase post prandiale, quando una notevole quota dei trigliceridi è inglobata nei chilomicroni, l’azione fisiologica dell’insulina dovrebbe essere quella di sopprimere la produzione epatica di VLDL.
Nelle condizioni di insulino-resistenza (o di carenza insulinica), la normale soppressione della sintesi epatica delle VLDL è compromessa. La competizione tra chilomicroni e VLDL endogene per le comuni vie di rimozione, che in queste condizioni tendono ad essere sature, comporta un allungamento del tempo di residenza in circolo delle lipoproteine ricche in trigliceridi. La lipoprotein-lipasi, l’enzima che regola la rimozione dei trigliceridi è attivata dall’insulina, ma la sua attivazione è ridotta negli stati di insulino-resistenza. L’aumento della permanenza in circolo delle lipoproteine ricche in trigliceridi porta ad un eccessivo scambio di trigliceridi ed esteri del colesterolo tra queste lipoproteine da una parte e LDL e HDL dall’altra. La trasformazione è operata dalla CETP (Proteina di Trasferimento degli Esteri del Colesterolo) che sposta gli esteri del colesterolo (CE) dalle HDL alle lipoproteine ricche in trigliceridi con trasferimento in direzione opposta dei trigliceridi. Le HDL TG-rich/CE poor, che hanno un rapporto TG/CE più alto delle HDL di origine, sono capaci di convertire le LDL in LDL TG-rich/CE poor mediante trasferimento CETP-mediato di trigliceridi alle LDL e di CE dalle LDL. La LCAT (Lecitin-Colesterolo-Acil-Transferasi), localizzata sulle HDL produce CE a partire da colesterolo libero e fosfolipidi presenti sulle stesse HDL e incrementa la generazione di HDL deplete di lipidi di superficie. Tali HDL possono rimuovere fosfolipidi e colesterolo libero dalle LDL. I trigliceridi presenti sulle LDL così modificate sono idrolizzati dalla triglicerido-lipasi epatica con generazione di LDL piccole e dense (455). La maggior aterogenicità delle LDL piccole e dense sarebbe legata in parte alla maggior rapidità con cui esse filtrano nella parete arteriosa rispetto alle LDL di dimensioni normali, ma soprattutto alla più spiccata tendenza ad essere ossidate e ritenute nella matrice extracellulare (577).
La matrice extracellulare dell’intima della parete vascolare e il mesangio del glomerulo renale sono le sedi in cui si realizzano i processi patogenetici fondamentali che portano all’aterosclerosi ed alla glomerulosclerosi: gli eventi molecolari e cellulari che intervengono in queste sedi presentano somiglianze molto marcate.
Non sorprende quindi che le alterazioni lipidiche della dislipidemia aterogena (la così detta triade lipidica) strettamente associata alla condizione di insulino-resistenza ed iperinsulinemia e soprattutto la generazione di LDL-ossidate emergono come momenti patogenetici comuni delle complicanze aterosclerotiche e delle complicanze microvascolari del diabete mellito e, in particolare, della nefropatia diabetica. D'altra parte, lo stress ossidativo gioca un ruolo importante, sebbene non pienamente compreso, nello sviluppo delle complicanze vascolari del diabete mellito e soprattutto del diabete tipo 2. Nella maggior parte degli studi l’attenzione è stata focalizzata sulla ossidazione dei lipidi e soprattutto sulla ossidazione delle lipoproteine a bassa densità (LDL). Indagini più recenti hanno rivelato che non solo la componente lipidica, ma anche la molecola lipoproteica delle LDL diventa modificata in senso ossidativo, contribuendo alla formazione di aggregati insolubili a livello della matrice extracellulare (496).
Polimorfismo ε2, ε3, ε4 dell'ApoE e complicanze del diabete
L’Apolipoproteina E (ApoE) è una glicoproteina di 299 aminoacidi che svolge un ruolo centrale nel metabolismo lipidico. Essa si lega con elevata affinità al recettore delle LDL consentendo l’endocitosi delle particelle lipoproteiche ad essi associate. ApoE è polimorfica e presenta tre isoforme comuni (E2, E3, E4) codificate da tre alleli (ε2, ε3, ε4) localizzati nell’esone 4 del gene codificante. Come precedentemente descritto, le tre isoforme differiscono per una singola sostituzione aminoacidica (in due distinte posizioni) che modifica la struttura e la funzione della proteina (578). I portatori di ApoE4 presentano più elevati livelli plasmatici di colesterolo totale e di colesterolo LDL rispetto ai portatori dell’apoE2 e agli omozigoti per ApoE3. D’altra parte, il legame dell’ApoE2 ai recettori per le lipoproteine è difettoso rispetto al legame di apoE3 e ApoE4 e determina una ridotta clearance delle lipoproteine ricche in trigliceridi che quindi rimangono più a lungo in circolo. ApoE4 è un fattore di rischio di moderata intensità per cardiopatia ischemica e influenza gli stadi precoci del processo aterosclerotico a livello carotideo (579). Analoghi risultati sono stati osservati nella coorte di diabetici tipo 2 da noi studiata. A parità di età, durata del diabete, BMI ed emoglobina glicata, i portatori dell'allele ε4 (il 10.8% dell'intera coorte) presentavano più elevati livelli di colesterolo LDL e di colesterolo non-LDL
I portatori di ε4, inoltre, presentavano più bassi livelli di colesterolo HDL sia rispetto ai portatori di ε2 che agli omozigoti per ε3. I portatori di ε2 presentavano invece il profilo lipidico più favorevole: più bassi livelli di colesterolo LDL e di colesterolo totale e più elevati livelli di colesterolo HDL. È quindi evidente come il rapporto tra colesterolo LDL e colesterolo HDL (LDL/HDL) è massimo nei portatori di ε4 (3.17±0.85; p=0.0006 vs ε3/ε3 e p=0.0003 vs portatori di ε2), intermedio negli omozigoti per ε3 (2.92±1.07) e più bassa nei portatori di ε2 (2.42±0.81). In accordo con questi risultati è il comportamento delle apolipoproteine ApoB e ApoA1. ApoA1 è più alta e ApoB più bassa nei portatori dell'allele ε2. A differenza di quanto rilevato in letteratura, nessuna differenza è stata osservata nei livelli dei trigliceridi a digiuno. Il modesto aumento dei trigliceridi rilevato nei portatori di ε2, in cui è ipotizzabile una ridotta clearence delle lipoproteine ricche in trigliceridi non è risultato, nel nostro studio, né statisticamente, né clinicamente significativo.
L’espressione del gene della Apolipoproteina E, la sintesi della proteina, la secrezione del colesterolo cellulare e la ricaptazione del colesterolo mediata dalla ApoE legata alla superficie cellulare sono tutti eventi genotipo-dipendenti. Tali eventi hanno sede nei macrofagi, elementi cellulari fondamentali del processo aterosclerotico. L’espressione del gene e la secrezione di ApoE sono ε4ε4>ε3ε3>ε2ε2, la secrezione del colesterolo è ε2ε2>ε3ε3 >ε4ε4, mentre la
ricaptazione del colesterolo è ε4ε4>ε3ε3>ε2ε2.
I portatori dell’allele ε2 ed ε3 sono protetti dall’eccessivo accumulo di colesterolo nei macrofagi per un efficace meccanismo di efflusso, mentre i portatori di ε4 sono esposti ad un’aumentata formazione di cellule schiumose nella parete arteriosa. Con questa pur semplificata ipotesi del ruolo della ApoE nel metabolismo lipidico stanno in accordo gli studi clinici ed autoptici che dimostrano che l’allele ε4 è correlato ad un aumentato rischio di aterosclerosi coronarica, carotidea e aortica. Dalla meta-analisi di Wilson e coll. relativa a 9 studi clinici e 5 studi autoptici sulla associazione tra ApoE e rischio di infarto miocardico, coronaropatia e morte coronarica, risulta che l’odds ratio (95% CI) per cardiopatia ischemica, rispetto a ε3, è per ε2 0.96 (0.81−1.13) e per ε4 1.44 (1.27-1.62). ε4 è quindi associato, in uomini e donne, con un aumentato rischio di coronaropatia (580). Nel Framingham Offspring Study l’odd ratio per coronaropatia associato all’allele ε4, dopo aggiustamento per età, fumo, obesità, ipertensione, diabete e colesterolo LDL e HDL, risulta pari a 1.44 (1.00-2.06). Il 15% della prevalenza della cardiopatia ischemica poteva essere attribuito all’allele ε4 (435).
Il significato di ε4 quale fattore di rischio cardiovascolare indipendente dall’assetto lipidico, suggerisce che l’effetto di ε4 non è solo mediato dalle concentrazioni dei lipidi plasmatici associate, ma anche da altri meccanismi. Le diverse isoforme di ApoE sono dotate di differente potere antiossidante che risulta essere maggiore per ε2 rispetto a ε3 e ε4. L’allele ε4 potrebbe quindi associarsi anche a più elevate concentrazioni di LDL ossidate sia in circolo sia a livello della parete vascolare.
Il ruolo che il processo ossidativo gioca nel determinismo delle complicanze del diabete, e la potenziale influenza negativa dell’assetto lipidico aterogeno sia nelle fasi iniziali che nella progressione della nefropatia diabetica (581) rappresentano il razionale di numerosi studi che hanno indagato il ruolo del polimorfismo ε2, ε3, ε4 della ApoE quale possibile fattore di rischio per le complicanze del diabete. Alcuni di questi studi suggeriscono che l’allele ε2 è associato ad un aumentato rischio di nefropatia diabetica.
Nel nostro studio, l’allele ε2 del gene dell’ApoE, associato a più bassi livelli di colesterolo LDL e non-HDL, a più elevate concentrazioni del colesterolo HDL, ma non a più elevati livelli di trigliceridi (come atteso in base alla ipotesi di una ridotta clearance delle lipoproteine ricche in trigliceridi), non sembra essere un fattore di rischio per la nefropatia nel diabete tipo 2. Infatti, la frequenza dell’allele ε2 non è risultata maggiore nei soggetti con nefropatia conclamata o con microalbuminuria rispetto ai soggetti senza danno glomerulare. Il risultato non si modifica anche dopo aver escluso dal sottogruppo dei normoalbuminurici i soggetti con più breve durata del diabete (<15 anni) che, come tali, sono potenzialmente a rischio pieno di sviluppare la nefropatia diabetica. Del resto, questo “accorgimento” è forse meno utile nei soggetti con diabete tipo 2 rispetto a quelli con diabete tipo 1. Infatti, la microalbuminuria ed anche la proteinuria clinica sono relativamente frequenti anche alla diagnosi del diabete tipo 2 e nei soggetti con breve durata nota del diabete.
Inoltre la relazione che esiste tra durata della malattia ed incidenza e prevalenza della nefropatia nel diabete tipo 2, non riflette l’andamento caratteristico osservato nel diabete tipo 1. Nel diabete tipo 1, ma non nel diabete tipo 2, infatti l’incidenza della nefropatia è trascurabile nei primi 5-10 anni della diagnosi, aumenta rapidamente e raggiunge il picco nel decennio successivo per declinare poi quando la durata del diabete è superiore a 20-25 anni. Anche l’analisi di regressione logistica non assegna un ruolo di rilievo al polimorfismo dell’ApoE nella nefropatia diabetica, mentre viene confermato il significato di fattori di rischio quali il sesso maschile, la pressione arteriosa sistolica, l’emoglobina glicata, la durata del diabete e, seppur marginalmente, del BMI (124).
È interessante inoltre osservare come la coesistenza di retinopatia, analogamente a quanto osservato nel diabete tipo 1 (575) rappresenta un importante fattore di rischio indipendente per la nefropatia diabetica. A conferma del ruolo della retinopatia, è stato recentemente dimostrato che la velocità di progressione del danno renale in diabetici tipo 2 con proteinuria è maggiore nei soggetti con retinopatia rispetto a quelli senza segni di danno retinico (186). I nostri risultati arricchiscono una letteratura relativamente ampia che ha valutato il ruolo di questo polimorfismo sia nel diabete tipo 1, che nel diabete tipo 2. Alcuni studi dedicati al diabete tipo 1. Secondo Onum e coll., la frequenza degli alleli e la distribuzione dei genotipi era simile nei normo-, nei micro- e nei macroalbuminurici in un piccolo studio che ha reclutato 146 diabetici tipo 1 con durata di diabete compresa tra 15 e 21 anni.
Il genotipo dell’ApoE è risultato non associato con le fasi iniziali o quelle avanzate della nefropatia diabetica anche nel GENEDIAB study (582) e in più recenti studi realizzati in Danimarca (439) e in Russia (583).
Almeno altrettanti studi suggeriscono invece che l’allele ε2 del gene dell’ApoE può essere considerato uno dei molteplici fattori di rischio per la nefropatia diabetica (353,442,443a).
Nello studio di Werle e coll., l’allele ε2 è associato a più bassi valori della clearance della creatinina e a più elevati livelli dell’escrezione urinaria di albumina e di immunoglobuline IgG (443a).
Nello studio di Chowdhury e coll., l’odds ratio per nefropatia associato alla presenza dell’allele ε2 era 3.91 (95% CI 3.12-4.84) rispetto ai soggetti con diabete tipo 1, lunga durata di malattia e normoalbuminuria; infatti, tra i pazienti con nefropatia, il 23.4% possedeva almeno un allele ε2 rispetto al 13.7% dei soggetti con diabete tipo 1 di recente diagnosi ed il 6.6% dei non-nefropatici con lunga durata di malattia (442). È tuttavia necessario notare che, benché il risultato sia significativo, solo poco più del 20% dei diabetici con nefropatia è portatore dell’allele ε2. Ciò dimostra che questo locus dell’ApoE non è l’unico né il più rilevante fattore genetico che contribuisce alle nefropatia diabetica.
Nello studio di Araki e coll., la frequenza dell’allele ε2 era significativamente più alta nei soggetti con nefropatia rispetto ai normoalbuminurici con lunga durata del diabete (9% vs 3%) e il rischio di nefropatia era 3.1 volte più alto (95% CI 1.6-5.9) nei portatori di ε2 rispetto ai non portatori. Nello studio familiare, i genitori eterozigoti per l’allele ε2, trasmettono preferibilmente ε2 ai figli con nefropatia diabetica (64% vs 36%) (353).
Altri autori hanno valutato gli effetti dell’allele ε2 sul rischio di nefropatia nel diabete tipo 2. Eto e coll. hanno descritto in diabetici tipo 2 di origine giapponese portatori dell’allele ε2 un odds ratio per nefropatia pari a 3.0 (95% CI=1.2-7.7); la frequenza di ε2 era sensibilmente più elevata nei nefropatici (7.2%) o nei soggetti con insufficienza renale (9.7%) che nei diabetici senza nefropatia (2.6%) o nella popolazione generale (3.7%) (348). Kimura e coll, ancora in una coorte di diabetici tipo 2 di origine giapponese, suggeriscono, in un follow-up di circa 10 anni, che i soggetti con funzione renale stabile, presentano ε4 più frequentemente dei soggetti con declino della funzione renale, suggerendo un ruolo protettivo per l’allele ε4 (349).
Nessuna associazione tra polimorfismo ApoE e markers di nefropatia (clearance della creatinina ed albuminuria) è stata rilevata in una piccola coorte di soggetti con diabete tipo 2 di origine tedesca (443a). Non mancano osservazioni che tendono in direzione opposta. Nello studio di Yauhkonen e coll., il polimorfismo dell’ApoE non si associa con le complicanze microvascolari del diabete, ma, tra i controlli, i portatori dell’allele ε4 presentano una più elevata frequenza di microalbuminuria. Infine, nello studio di Boize e coll., condotto in 134 diabetici tipo 2 di origine caucasica, la prevalenza di valori di AER>20 mg/24 ore, era più bassa nei portatori dell’allele ε2 (36%) rispetto ai non portatori dell’allele ε2 (69%) (350).
In qualche modo, in accordo con questi ultimi studi è l’osservazione che emerge dai nostri dati di una tendenza ad una minor durata del diabete nei soggetti con nefropatia conclamata portatori dell’allele ε4, quasi a suggerire un possibile ruolo di ε4 nel favorire o accelerare la comparsa della macroalbuminuria.
Il nostro studio non ha rilevato alcuna associazione tra polimorfismo ApoE e retinopatia diabetica. Nessuna associazione emergeva anche dalla regressione logistica che permetteva invece di confermare il ruolo indipendente di fattori di rischio di retinopatia quali, durata del diabete, età anagrafica, pressione arteriosa sistolica ed emoglobina glicata. La scarsa letteratura disponibile è concordante con la nostra osservazione. Nessuna associazione tra gene ApoE e retinopatia è stata rilevata nel diabete tipo 1 (439,583), mentre nel diabete tipo 2, in una piccola coorte studiata da Santos e coll., l’allele ε4 sembra associarsi al rischio di edema maculare (584).
A differenza di quanto osservato da Ukkola e coll., nessuna associazione emerge dal nostro studio tra polimorfismo dell’ApoE e arteriopatia coronarica o ipertensione arteriosa in soggetti con diabete tipo 2 (585). In quest’ultimo studio, in accordo con dati relativi alla popolazione non diabetica (580), l’allele ε2 tende a proteggere dalla patologia cardiovascolare i soggetti con diabete tipo 2.
Inoltre, l’arteriopatia coronarica è risultata più frequente tra i diabetici tipo 1 nefropatici portatori dell’allele ε4 (36%) rispetto ai soggetti con il genotipo ε3/ε3 (12%) (439). Infine in soggetti con diabete tipo 2, a differenza di quanto osservato nel gruppo di controllo, il genotipo ε4 non ha mostrato alcuna associazione con l’intervenire di eventi macrovascolari o con lo spessore dell’intima-media a livello carotideo.
Le ragioni per le discrepanze tra studi caso-controllo, soprattutto quelli più numerosi dedicati alla nefropatia diabetica non sono facilmente individuabili. Numerose sono le possibilità: differenti criteri diagnostici per la nefropatia; grado variabile di linkage disequilibrium in diverse popolazioni se l’allele ε2 non è un diretto fattore di rischio, ma solo un marker di suscettibilità; interazione con altri fattori genetici ed ambientali possibilmente eterogenea in diverse popolazioni e, infine, la stratificazione della popolazione. Tra queste possibilità, l’effetto della stratificazione della popolazione può essere escluso dallo studio delle famiglie tramite il transmission disequilibrium test. In effetti, nello studio di Araki S e coll., la trasmissione dell’allele ε2 dai genitori eterozigoti ai soggetti con nefropatia diabetica è risultata significativamente più frequente della trasmissione attesa in base alla casualità (50%) e significativamente più frequente della trasmissione di tale allele da genitori eterozigoti ai figli senza nefropatia diabetica. Tale osservazione riguarda però i pazienti con diabete tipo 1 (535). Nel diabete tipo 1, le osservazioni che attribuiscono un ruolo all’allele ε2 nel modulare la suscettibilità alla nefropatia diabetica sono in gran parte concordanti e a conclusioni analoghe giunge anche da un ampio studio caso-controllo realizzato nel nostro laboratorio (586). Difficile determinare quali stadi della nefropatia sono eventualmente influenzati dalle isoforme dell’ApoE. Poiché, negli studi positivi, il rischio per proteinuria e per ESRD è simile nei portatori di ApoE2, è ipotizzabile che questa isoforma influenzi il rischio nelle fasi più precoci della nefropatia (insorgenza e/o progressione della microalbuminuria). L’ipotesi di un ruolo delle isoforme dell’ApoE non soltanto nel processo aterosclerotico, ma anche nella patogenesi della microangiopatia diabetica, al di là della discordanza degli studi di epidemiologia genetica, è tuttavia plausibile, anche se i meccanismi che possono modulare lo sviluppo delle complicanze e, in particolare, della nefropatia diabetica, non sono chiari.
Almeno due possibilità devono essere considerate. La prima è correlata alle anomalie lipidiche associate alle isoforme dell’ApoE. A confronto dei soggetti con ApoE3, i portatori di ApoE2 presentano più bassi livelli di colesterolo, ma più elevate concentrazioni di trigliceridi dovute alla persistente presenza in circolo di lipoproteine ricche in trigliceridi.
Più elevate concentrazioni di particelle ricche in trigliceridi sono state descritte in diabetici tipo 1 con microalbuminuria (587) e l’ipertrigliceridemia rappresenta un fattore di rischio indipendente nell’incidenza della microalbuminuria (575). Così, la dislipidemia correlata al genotipo ApoE potrebbe promuovere lo sviluppo della nefropatia. Nel nostro studio, tuttavia né ipertrigliceridemia, né nefropatia sono più frequenti nei portatori dell'allele ε2.
La seconda ipotesi fa riferimento a funzioni diverse dell’ApoE. L’ApoE tende a localizzarsi a livello della matrice extracellulare della fibra nervosa danneggiata, suggerendo un possibile ruolo nei processi di riparazione tissutale. Nel rene, studi in immunofluorescenza dimostrano una massiva colorazione per ApoE a livello delle aree mesangiali di soggetti con patologie renali, compresa la nefropatia diabetica. Inoltre, l’ApoE2 è associata con la glomerulopatia da lipoproteine, una rara malattia renale recentemente individuata che, tra l’altro tende a recidivare nel rene trapiantato (588). Questo suggerisce che l’anomalia primaria della glomerulopatia da lipoproteine non è intrinseca al rene, ma dipende da fattori sistemici, compreso il fenotipo apoE2. È così possibile ipotizzare che le proteine apoE2 tendono ad accumularsi nell’area mesangiale del glomerulo del paziente diabetico modificando le proprietà della matrice extracellulare e la funzione cellulare. Numerosi studi hanno dimostrato la relazione tra genotipo ApoE e risposta della colesterolemia al trattamento con inibitori della HMG-CoA reduttasi. In questi studi, i portatori dell’allele ε2 e in alcuni studi anche i soggetti con genotipo ε3/ε3 sembrano rispondere più favorevolmente alla terapia con statine dei soggetti portatori dell’allele ε4. Il trattamento con statine (488) sembra rallentare la progressione del danno glomerulare. È possibile che anche questo effetto possa essere modulato delle isoforme dell’ApoE.
La paraoxonasi: polimorfismi ed attività
Le basse concentrazioni del colesterolo HDL rappresentano uno dei fattori di rischio cardiovascolare maggiori identificati dagli studi epidemiologici.
Per ciascun livello di colesterolo LDL, esiste una correlazione inversa tra concentrazione delle HDL e rischio per malattia coronarica e patologia cerebrovascolare.
L'effetto protettivo delle HDL è complesso. La maggior parte delle ricerche in questo settore ha valorizzato il ruolo delle HDL nel trasporto dei lipidi ed in particolare nel trasporto inverso del colesterolo e nell'efflusso del colesterolo dalla parete arteriosa. Tuttavia, più recentemente è emersa un'altra area di interesse. Questa ha esplorato il ruolo delle HDL quale fattore di protezione delle LDL e delle membrane cellulari nel danno indotto dalla perossidazione delle componenti lipidiche. In questo modo, le HDL proteggono la parete vascolare e si oppongono ai processi che mediano l'iniziare ed il progredire della lesione ateromatosa. Dati questi meccanismi, non è possibile escludere, anzi è logico ipotizzare un ruolo importante per le HDL anche nella induzione del danno microvascolare del diabete mellito. Studi recenti, infatti, tendono a mettere in evidenza, sia nel diabete tipo 1 che nel diabete tipo 2, il ruolo dell'insulino-resistenza e delle componenti della sindrome metabolica, e tra queste le basse concentrazioni del colesterolo HDL, nel determinismo delle complicanze renali e retiniche del diabete. Attività enzimatiche e tra queste le paraoxonasi associate alle particelle delle HDL svolgono un ruolo critico nel processo di protezione del danno macrovascolare e microvascolare mediato dai meccanismi antiossidanti. La paraoxonasi 1 (PON1, la più studiata e conosciuta) la paraoxonasi 2 e la paraoxonasi 3 (PON2 e PON3 molecole meno studiate e meno note) sono espressione di una famiglia di geni, ampiamente diffusi e strettamente conservati tra i mammiferi, localizzati, nell'uomo, nel braccio lungo del cromosoma 7 nelle posizioni comprese tra q21.3 e q22.1. Nel siero, la paraoxonasi 1 è localizzata esclusivamente sulle HDL. PON1 rappresenta il più efficiente tra i vari meccanismi localizzati sulle HDL (ApoA1, LCAT) capaci di prevenire e contrastare l'ossidazione delle LDL. La PON1 localizzata sulle HDL agisce mobilizzando i lipoperossidi ed impedendone l'accumulo sulle LDL. Il gene PON1 riconosce almeno cinque polimorfismi nella regione promoter e due polimorfismi nella regione codificante per la proteina. Di questi ultimi, uno è in posizione 55 (metionina/leucina; M/L), l'altro in posizione 192 (arginina/glutamina; R/Q). L'allele R del polimorfismo arginina/glutamina sembra codificare una proteina più efficiente nel proteggere le LDL dall'ossidazione.
L'analisi dei due polimorfismi è stata eseguita su pazienti con diabete tipo 2 e su una popolazione di soggetti sani.
Mentre la distribuzione genotipica del polimorfismo PON1-Gln192Arg è risultata in equilibrio di Hardy-Weinberg sia nei controlli che nei soggetti con diabete tipo 2, la distribuzione genotipica del polimorfismo PON1-Leu55Met è risultata in equilibrio di Hardy-Weinberg nei controlli ma non nei soggetti con diabete tipo 2. Questo ultimo risultato ci permette di ipotizzare che nella popolazione con diabete di tipo 2 almeno una delle assunzioni del principio di Hardy-Weinberg non sia rispettata.
Le assunzioni del principio di Hardy-Weinberg sono le seguenti: 1. L'organismo studiato è diploide
2. La riproduzione è sessuale 3. L'accoppiamento è casuale
4. La dimensione della popolazione è sufficientemente grande 5. La migrazione è trascurabile
6. La mutazione è trascurabile 7. La selezione è trascurabile
8. Le generazioni non si sovrappongono 9. Il locus studiato non è legato al sesso.
Soddisfatte queste condizioni, le frequenze alleliche entro una popolazione rimarranno costanti per un periodo di tempo indefinito. Per la legge di Hardy-Weinberg, cinque sono i fattori principali che governano i mutamenti evolutivi a carico di una certa popolazione: mutazione, migrazione, ridotte dimensioni del campione, accoppiamento non casuale e selezione naturale. Difficile è capire il motivo della deviazione dall'equilibrio Hardy-Weinberg osservata nella distribuzione genotipica del polimorfismo Leu55Met nei pazienti diabetici da noi studiati. Ovviamente, il mancato rispetto di alcune condizioni quali quelle riportate al punto 1 (organismo diploide), 2 (riproduzione sessuale), 3 (accoppiamento casuale - data l'estensione della popolazione studiata) e 9 (locus non legato al sesso) è facilmente escludibile. L'ipotesi più probabile rimane quella di una insufficiente dimensione della popolazione studiata o di una selezione del campione reclutato.
Indagini future, non ultima l'estensione dello screening per i polimorfismi di PON1 all'intera popolazione di diabetici tipo 2 reclutata, potranno forse permetterci di capire l'origine di questo fenomeno.
A tutt'oggi infatti l'analisi dei polimorfismi di PON1 (Leu55Met e Gln192Arg) è stata eseguita in circa la metà dei soggetti con diabete tipo 2 inclusi nello studio.
Sebbene una prima analisi non abbia permesso di mettere in evidenza differenze nella distribuzione di PON1 L/M tra normoalbuminurici, microalbuminurici e macroalbuminurici, anche quando tra i normoalbuminurici sono stati esclusi i soggetti con più breve durata del diabete (<15 anni), differenze marginalmente significative (p=0.07) o sia pur debolmente significative (p=0.04) sono state osservate quando si sono confrontati direttamente i microalbuminurici con i normoalbuminurici e, rispettivamente, i macroalbuminurici con i normoalbuminurici. In pratica nei microalbuminurici, e soprattutto nei macroalbuminurici, è stata registrata una maggiore frequenza dell'allele L e del genotipo omozigote L/L. Ovviamente significativa la differenza che emerge confrontando i normoalbuminurici con l'insieme dei diabetici tipo 2 con aumentati valori dell'escrezione urinaria dell'albumina (micro- + macroalbuminurici). L'analisi di regressione multipla assume particolare significato in relazione alle differenze in alcuni parametri clinici rilevata in funzione del genotipo M/L. Infatti i soggetti con omozigosi L/L presentavano una durata del diabete tendenzialmente maggiore in associazione a valori significativamente più bassi sia del BMI che dell'HbA1c senza differenza alcuna per quanto riguarda età e livelli della pressione arteriosa. In regressione logistica, pressione sistolica, coesistenza di retinopatia (soprattutto se proliferante), ma anche il genotipo L/L sono risultati associati in maniera indipendente con la presenza di aumentati valori dell'albuminuria. Nessuna associazione è stata invece osservata tra polimorfismo PON1 Leu55Met e retinopatia diabetica anche dopo aver confrontato i non retinopatici con l'insieme dei soggetti che presentavano una qualunque forma di danno retinico. In regressione logistica solo la durata del diabete e la coesistenza di nefropatia, soprattutto se conclamata, ma non il polimorfismo L55M emergevano come correlati indipendenti della retinopatia. Analogamente, nella nostra coorte, il polimorfismo Leu55Met di PON1 non risulta associato con la presenza di ipertensione, o con la patologia coronarica. Una analisi simile è stata eseguita per descrivere l'eventuale associazione tra complicanze micro- e macrovascolari del diabete tipo 2 e polimorfismo Gln192Arg di PON1. I tre genotipi, pressoché ugualmente rappresentati Q/Q e Q/R, meno frequente il genotipo R/R, non differivano per nessuno dei parametri clinici esaminati. Nessuna differenza nella distribuzione dei genotipi è stata rilevata in funzione della presenza e/o dello stadio sia della nefropatia diabetica che della retinopatia diabetica.