Capitolo 3 – Risultati
3.1 – Ragioni del lavoro di tesi
In passato, al fine di ottenere oligomeri di-idrossiterminati, diversi autori hanno studiato la polimerizzazione e la copolimerizzazione di eteri ciclici in presenza di dioli. In questi vari lavori è sempre stato assunto che la polimerizzazione procede con il tipico meccanismo di propagazione di catena, grazie al quale si nota una diminuzione della frazione di oligomeri ciclici. Tali autori hanno quindi constato tale fatto senza però darne una spiegazione razionale.
Più recentemente, in un lavoro di Kuzaev e Olkahova, si legge che l’epicloridrina (ECH) è stata polimerizzata in presenza di un 10%mol di glicoletilenico(EG) e catalizzata con BF3·THF assumendo che tutto il EG si consuma nel primo stadio di reazione. Analisi condotte sul campione ottenuto tramite GPC hanno mostrato che la quantità di oligomeri ciclici presenti è praticamente assente e che praticamente tutte le catene hanno propagato in modo lineare.
In un lavoro di Biedron, Kubisa e Penczek infine, è spiegato che il meccanismo di polimerizzazione degli eteri ciclici in presenza di dioli procede attraverso il meccanismo del monomero attivato (AMM). Questo meccanismo è favorito da una lenta aggiunta di monomero alla soluzione di reazione; infatti aggiungendo istantaneamente il monomero il meccanismo predominante è quello con terminazione di catena (ACE) di cui abbiamo già discusso nel Capitolo 1. Grazie al meccanismo AMM, sostengono ancora gli autori, si hanno prodotti a peso molecolare controllato con una bassa polidispersione e in cui sono praticamente assenti oligomeri ciclici. Nel caso che la polimerizzazione proceda realmente con il meccanismo AMM, l’agente di quench usato per terminare la reazione non dovrebbe influenzare il prodotto finale.
Proprio su quest’ultimi aspetti si è incentrato il presente lavoro di tesi al fine di produrre leganti energetici per propellenti solidi. In particolar modo la nostra attenzione si è focalizzata sulla sintesi e caratterizzazione della pECH (1 e 2) e del copolimero ECH/BBrMO(50:50).
3.1.1 – Leganti energetici:
Come già accennato nel paragrafo 3.1 esistono due strade percorribili per la produzione di polimeri energetici: la polimerizzazione di monomeri energetici o la modifica di polimeri precostituiti.
Il grande vantaggio della seconda via è che l’introduzione di gruppi energetici direttamente sul polimero consente di evitare la manipolazione di un monomero altamente instabile ovvero di limitare i rischi nello stadio più pericoloso della preparazione. Ad esempio, il GAP (figura 3.1.1), derivato azidico della poli-epicloroidrina (pECH), è preparato tramite questa via perché, oltre ad essere quella più sicura, è anche l’unica percorribile; il monomero azidato infatti è molto instabile e quindi difficile da produrre.
O C l O N3 A zid atio n P o lim erizatio n H O O H C l n H O O H N3 n
Figura 3.1.1 – Due vie per la sintesi del GAP.
L’utilizzo della seconda soluzione, però, non è facilmente praticabile per tutti i polimeri energetici destinati all’impiego nei propellenti solidi. In alcuni casi, infatti, il cloro non si rivela gruppo uscente sufficientemente buono da garantirne una sostituzione completa con la funzionalità azidica.
Al fine di approfondire le potenzialità di sviluppo di questa strategia sintetica, si sono sintetizzati copolimeri random ECH/BBrMO (50:50).
Il monomero BBrMO ha il vantaggio di contenere ben due gruppi azidabili per unità monomerica (con BAMO si intende il corrispondente monomero azidato, figura 3.2), ma l’inconveniente di dar luogo ad un polimero con struttura altamente cristallina, il che ne limita la possibilità di impiego come legante polimerico. La presenza dell’ECH come comonomero (seppur meno efficace del BBrMO per la percentuale ponderale di gruppi azidici potenzialmente ottenibile) è necessaria per introdurre delle irregolarità strutturali e dar luogo alla formazione di un polimero (azidato o meno) con caratteristiche amorfe.
3.2 – pECH(1)
3.2.1 – Caratterizzazione e sintesi
La procedura di sintesi di questo polimero è stata illustrata nel paragrafo 2.2.1 a cui si rimanda. Dopo questa fase di reazione abbiamo ottenuto 4 campioni: a) pECH(1) gocciolato e sottoposto a quench con acqua (sigla pECH(1) G Q W); b) pECH(1) gocciolato e sottoposto a quench con metanolo (sigla pECH(1) G Q M); c) pECH(1) non gocciolato e sottoposto a quench con acqua (sigla pECH(1) NG Q W); d) pECH(1) non gocciolato e sottoposto a quench con metanolo (sigla pECH(1) NG Q M); che sono stati caratterizzati tramite le varie analisi descritte nel paragrafo 2.5. Vediamo campione per campione i risultati ottenuti.
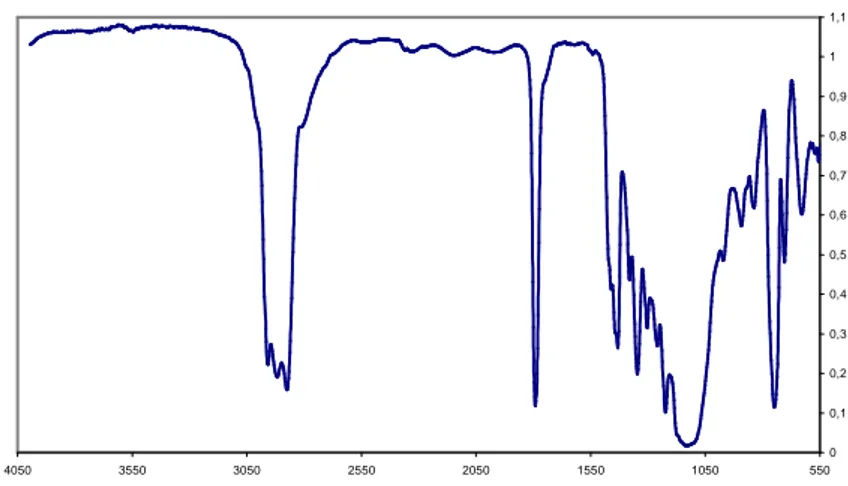
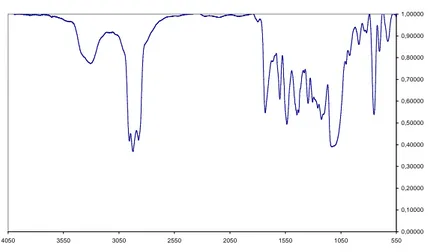
Come prima cosa abbiamo analizzato i campioni tramite spettrografia ad infrarosso (IR) ottenendo il seguente spettro(uguale per tutti e 4 i campioni):
pECH(1) gocciolato quench acqua
0,000 0,200 0,400 0,600 0,800 1,000 550 1050 1550 2050 2550 3050 3550 4050
Come si vede dallo spettro IR riportato in figura 3.2.1, tra 3400 e 3700 cm-1 si ha un picco il quale ci conferma la presenza di gruppi OH all’interno del polimero; infatti questi valori di frequenza sono tipici dell’assorbimento dei suddetti gruppi.
Sempre dallo spettro precedente possiamo dire che il materiale ottenuto a seguito della reazione di polimerizzazione è effettivamente un polimero; infatti confrontando questo spettro con quello del monomero di ECH (Figura 3.2.2) si vede che la banda a 990 cm-1 relativa al monomero, nello spettro del polimero compare a 1100 cm-1. Inoltre la banda che compare a 435 cm-1 ci mostra che il legame C-Cl subisce uno stretching.
Figura 3.2.2 – Spettro IR della ECH.
Questo fatti ci confermano quindi che la reazione di polimerizzazione ha effettivamente avuto luogo, però non è in grado di fornirci risultati quantitativi riguardo la composizione del materiale polimerico, poiché dallo spettro non è esclusa la presenza di oligomeri ciclici.
Al fine di caratterizzare anche quantitativamente il nostro campione, abbiamo funzionalizzato i gruppi OH terminali (Figura 3.2.3) come illustrato nel paragrafo 2.3 e lo abbiamo analizzato tramite spettrografia 1H-NMR in CDCl3 (Figura 3.2.4). La funzionalizzazione del campione risulta necessaria poiché, nello spettro 1H-NMR del polimero tal quale, il segnale relativo al protone del gruppo ossidrilico è di difficile individuazione, in quanto di debole intensità e sovrapposto all'intenso assorbimento protonico nella regione compresa tra 3,4 e 3,7 ppm. A seguito della reazione di
acetilazione, il segnale del gruppo CF3COO-, corrispondente a quello del protone ossidrilico nello spettro del tal quale, va a collocarsi in una regione spettrale ben definita (5,3 ppm) e sgombra da altri assorbimenti, triplicando anche la propria intensità.
pECH(1) gocciolato quench acqua funzionalizzato
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.2.3 – Spettro IR del pECH(1) G Q W.
Come si può vedere dal grafico precedente, il nuovo picco che si ha a 1800 cm-1 è quello dovuto alla funzionalizzazione di cui sopra, dato confermato anche dalla scomparsa del picco degli OH.
Figura 3.2.4 – Spettro 1H-NMR del pECH(1) G Q W.
Come si può vedere in figura 3.3.4 il quintupletto che cade a 5,3 ppm rappresenta la risposta che danno i gruppi OH funzionalizzati da anidride trifluoroacetica; il picco a
1,6 ppm rappresenta la risposta che ci dà il butandiolo in catena e il picco grande a 3,5 ppm rappresenta il contributo che da il polimero.
Per gli altri 3 campioni di pECH(1) gli spettri IR del campione tal quale e del campione funzionalizzato con anidride trifluoroacetica hanno mostrato risultati identici a quelli appena visti per il pECH(1) G Q W; per quanto riguarda gli spettri 1H-NMR invece i valori integrati delle aree dei picchi di interesse sono stati i seguenti:
Area picco a 5,3 Area picco a 3,5 Area picco a 1,2
pECH(1) G Q W 1 131,07 0,74
pECH(1) G Q M 1 159,88 0,5
pECH(1) NG Q W 1 204,81 1,05
pECH(1) NG Q M 1,46 165,94 1,63
Tabella 3.2.1 – Aree dei picchi di interesse ottenuti dagli spettri 1H-NMR.
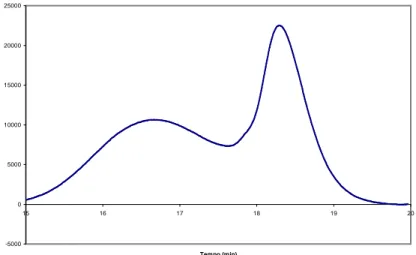
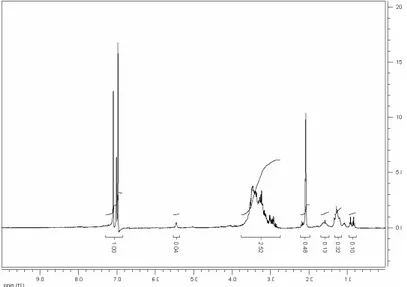
A questo punto, abbiamo determinato il peso molecolare medio numerale (Mn ) tramite cromatografia a permeazione di gel (GPC) ottenendo il seguente spettro:
-10000 0 10000 20000 30000 40000 50000 0 5 10 15 20 25 30 Tempo (min) In te n s it à
Figura 3.2.5 – Risposta GPC del pECH(1) G Q W.
Del grafico precedente riportiamo di seguito i picchi di nostro interesse, corrispondenti ai valori che vanno da 14,5 a 19,5 min circa:
0 2000 4000 6000 8000 10000 12000 14000 16000 18000 20000 10 100 1000 10000 MW In te n s it à
Figura 3.2.6 – Risposta GPC picchi di interesse del pECH(1) G Q W.
Il picco più grande rappresenta la risposta del polimero; quello più piccolo invece è il contributo degli oligomeri ciclici (in questi campioni, dimeri privi di gruppi OH) che, come si può vedere, sono in numero limitato. Infatti, da un confronto tra il campione precedente (gocciolato al fine di favorire il meccanismo AMM) e un campione non gocciolato in cui predomina il meccanismo ACE (ad esempio pECH(1) NG Q M), si vede che il gocciolamento effettivamente ci permette di diminuire il numero di oligomeri ciclici che si vengono a formare durante la fase di polimerizzazione.
0 2000 4000 6000 8000 10000 12000 14000 16000 10 100 1000 10000 MW In te n s it à
Figura 3.2.7 – Risposta GPC picchi di interesse del pECH(1) NG Q M.
Come si può vedere dalle figure precedenti, il primo picco (quello relativo agli oligomeri ciclici) di figura 3.2.6 è molto più basso del relativi picco di figura 3.2.7 e quindi questo ci conferma che i ciclici presenti nel campione gocciolato sono di meno rispetto a quelli che si riscontrano nel campione non gocciolato.
Dai grafici delle GPC riportati precedentemente, si è ottenuto che il valore del Mn e della polidispersione (DP) dei 4 campioni ottenuti sono:
n
M catene lineari Mn oligomeri ciclicic DP
pECH(1) G Q W 1611 141 1,42
pECH(1) G Q M 1974 143 1,34
pECH(1) NG Q W 944 181 1,30
pECH(1) NG Q M 1359 170 1,31
Tabella 3.2.2 – Risultati ottenuti dagli spettri GPC.
Per conferma del valore del peso molecolare ottenuto con la GPC, abbiamo analizzato il campione di pECH(1) NG Q W tramite VPO. Il valore risultante è stato Mn = 950 confermando così il valore ottenuto dalla GPC e confermando il fatto che i valori ottenuti da queste analisi sono affidabili.
3.2.2 – Determinazione dei gruppi OH
Alla luce di quanto detto, e usando lo schema di calcolo riportato in seguito, siamo passati a determinare il numero di gruppi OH.
La loro quantificazione è necessaria in termini applicativi, dal momento che le funzionalità ossidriliche saranno indispensabili per procedere alla reticolazione. Inoltre, la presenza di due idrossili terminali per macromolecola rappresenterebbe un'ulteriore prova che la polimerizzazione ha seguito il meccanismo AMM.
Quindi, conoscendo Mn e la composizione media percentuale del copolimero, è possibile calcolare il numero medio di ossidrili per macromolecola.
La composizione media del polimero in esame è ricavabile come segue:
) 5 ( 4 2 . 1 5 . 3 2 . 1 3 . 5 3 . 5 A A A A A XOH − + +
= = Frazione dei gruppi OH per catena
) 5 ( 4 4 2 . 1 5 . 3 2 . 1 3 . 5 2 . 1 A A A A A YBDO − + +
) 5 ( 4 5 2 . 1 5 . 3 2 . 1 3 . 5 2 . 1 5 . 3 A A A A A A ZECH − + −
= = Frazione di ECH per catena
dove xOH, yBCMO, zECH, rappresentano rispettivamente le frazioni medie di composizione del terminale ossidrilico, dell'unità di BDO in catena e di quella di ECH; le aree A5.3 (relativa a 5,3 ppm dei gruppi OH), A1.2 (relativa a 1,2 ppm del butandiolo), A3.5 (relativa a 3,5 ppm del monomero ECH), sono ottenute per integrazione dei picchi dello spettro 1H-NMR dei campioni.
Il numero di gruppi ossidrilici per macromolecola è ricavabile risolvendo il seguente sistema di equazioni: BDO BDO OH OH ECH L n n PM n PM n PM M = + + OH OH ECH L n X Z n = OH OH BDO BDO n X Y n =
dove nECH, nBDO e nOH sono rispettivamente il numero medio delle due unità monomeriche di ECH e BDO e degli ossidrili terminali presenti in una macromolecola di peso molecolare Mn .
Svolti i calcoli sopra riportati abbiamo determinato quindi che il numero di gruppi OH presenti in media per catena relativi ai quattro campioni sono:
Gruppi OH medi per catena
pECH(1) G Q W 0,77
pECH(1) G Q M 0,76
pECH(1) NG Q W 0,32
pECH(1) NG Q M 0,77
Tabella 3.2.3 – Numero di gruppi OH medi per catena.
In questo modo, è stato calcolato che in ciascun polimero sintetizzato sono presenti, mediamente, tra 0,7 e 0,8 gruppi ossidrilici per macromolecola (eccetto che per il pECH(1) NG Q W). Questo valore si colloca coerentemente nell’intervallo compreso
tra 0,5 e 1,5, riportato in letteratura per la poli-ECH a basso peso molecolare polimerizzata per mezzo del sistema iniziante TFBE/BDO. Infatti, questo sistema, seppur sia evidente che abbia permesso di ottenere delle rese in polimero piuttosto elevate e, nonostante consenta di esercitare un buon controllo sul peso molecolare del prodotto, volutamente basso, e sulla relativa dispersione, non esclude completamente la formazione di oligomeri ciclici non contenenti ossidrili terminali, consentendo la propagazione di alcune catene macromolecolari secondo il “classico” meccanismo di apertura di anello.
I dati bassi di gruppi OH medi per catena sopra riportati quindi stanno a significare che per molte catene il meccanismo che ha predominato è il meccanismo ACE il quale ci da catene in cui è presente un solo gruppo OH terminale, catene lineari prive di gruppi OH o oligomeri ciclici.
Alla luce di quanto detto avremmo dovuto notare delle differenze nei campioni a seconda degli agenti di quench utilizzati (acqua o metanolo) visto che il meccanismo ACE è influenzato dalla terminazione; in realtà questo non lo abbiamo riscontrato probabilmente perché molte delle catene che hanno propagato con tale meccanismo hanno dato oligomeri ciclici.
3.3 – Sintesi e caratterizzazione del pECH(2)
3.3.1 – Caratterizzazione e sintesi
Visti i risultati ottenuti con il pECH(1), al fine di controllare meglio la fase di gocciolamento, e quindi il meccanismo di reazione, e al fine di avere una maggiore quantità di materiale polimerico col quale lavorare, abbiamo deciso di condurre una nuova reazione di polimerizzazione, con quantità maggiori rispetto a quelle usate per la sintesi del pECH(1), mantenendo però costanti le concentrazioni dei reagenti usati. La procedura di sintesi di questo polimero è stata illustrata nel paragrafo 2.2.2 a cui si rimanda. Dopo questa fase di reazione abbiamo ottenuto 4 campioni: a) pECH(2) gocciolato e sottoposto a quench (in soluzione) con acqua (sigla pECH(2) G Q (in soluz) W); b) pECH(2) gocciolato e sottoposto a quench (in soluzione) con metanolo (sigla pECH(2) G Q (in soluz) M); c) pECH(2) gocciolato e sottoposto a quench (dopo aver svaporato) con acqua (sigla pECH(2) G Q (dopo svap) W); d) pECH(2) gocciolato
e sottoposto a quench (dopo aver svaporato) con metanolo (sigla pECH(2) G Q (dopo svap) M); che sono stati caratterizzati tramite le varie analisi descritte nel paragrafo 2.5. Per quanti riguarda la fase di caratterizzazione la metodologia usata è stata la stessa che abbiamo illustrato nel paragrafo 3.1.1 e cioè abbiamo analizzato i nostri 4 campioni tramite IR, 1H-NMR e GPC.
Gli spettri IR dei campioni tal quali, come si può vedere in figura 3.3.1, hanno dato risultati sostanzialmente simili a quelli ottenuti per il pECH(1):
pECH(2) gocciolato quench dopo svaporazione metanolo
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1 1,1 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.3.1 – Spettro IR del pECH(2) G Q (dopo svap) M tal quale.
Come ormai è noto dal pECH(1), la sola analisi ad infrarosso ci da notizie puramente qualitative; quindi, al fine di caratterizzare anche quantitativamente il nostro campione, abbiamo funzionalizzato i gruppi OH terminali (Figura 3.3.2) come illustrato nel paragrafo 2.3 e lo abbiamo analizzato tramite spettrografia 1H-NMR in toluene-d8.
pECH(2) gocciolato quench dopo svaporazione metanolo funzionalizzato
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1 1,1 550 1050 1550 2050 2550 3050 3550 4050
Le analisi 1H-NMR, però, hanno dato problemi riguardo alla fasatura dello spettro ottenuto dando risultati poco attendibili in quanto troppo soggetti alla sensibilità dell’analista; quindi per il calcolo dei gruppi OH, ci siamo concentrati principalmente sull’analisi dei dati ottenuti dalla GPC (per i calcoli relativi si rimanda al paragrafo 3.3.2 qui di seguito riportato).
Abbiamo cercato, quindi, di determinare il peso molecolare medio numerale (Mn ) dal seguente spettro: 0 5000 10000 15000 20000 25000 0 5 10 15 20 25 30 Tempo (min) In te n sit à
Figura 3.3.3 – Risposta GPC della pECH(2).
Per tutti e 4 i campioni analizzati, il risultato ottenuto è stato molto simile a quello riportato in figura 3.3.3. Di tale grafico riportiamo di seguito i picchi di nostro interesse, corrispondenti ai valori che vanno da 15 a 20 min circa:
-5000 0 5000 10000 15000 20000 25000 15 16 17 18 19 20 Tempo (min) In te n s it à
Il picco più basso, in questo caso, rappresenta la risposta che da il polimero, mentre quello più alto è il contributo dovuto agli oligomeri ciclici.
A prima occhiata, confrontando il grafico precedente con quello a pagina 53 (figura 3.2.7), si notano subito delle differenze che in linea teorica non ci saremmo assolutamente aspettati. Come abbiamo già affermato in precedenza, infatti, uno degli scopi per cui abbiamo condotto la seguente prova è quello di controllare in modo migliore la formazione dei ciclici in fase di gocciolamento (tanto da spettarci una totale scomparsa di questi); in realtà, come si vede in figura 3.3.4, questo non è accaduto e il picco relativo ai ciclici è assai superiore a quello che avevamo ottenuto nella prova a piccole quantità (pECH(1)) gocciolata.
Questo è accaduto perché, una volta levato il bagno di ghiaccio a 0°C, abbiamo notato un forte sviluppo di calore (non notato nella prova con le piccole quantità) probabilmente dovuto ad un peggiore smaltimento del calore di reazione dovuto al volume maggiore della soluzione di reazione. Quindi possiamo concludere che la reazione di polimerizzazione ha avuto effettivamente inizio a 25°C e in modo istantaneo non risentendo assolutamente della operazione di gocciolamento e quindi procedendo principalmente tramite il meccanismo ACE (il quale ricordiamo che favorisce la formazione dei ciclici).
Detto questo, dai grafici delle GPC riportati precedentemente, si è ottenuto che il valore del Mn e della polidispersione (DP) dei 4 campioni ottenuti sono:
n M catene lineari n M oligomeri ciclicic DP
pECH(2) G Q (in soluz) W 848 173 1,4
pECH(2) G Q (in soluz) M 824 172 1,4
pECH(2) G Q (dopo svap) W 832 173 1,4
pECH(2) G Q (dopo svap) M 832 172 1,35
Tabella 3.3.1 – Risultati ottenuti dagli spettri GPC.
Per conferma del valore del peso molecolare ottenuto con la GPC, abbiamo analizzato il campione di pECH(2) G Q (dopo svap) M tramite VPO. Il valore risultante è stato Mn
= 850 confermando così il valore ottenuto dalla GPC e confermando il fatto che i valori ottenuti da questo tipo di analisi sono affidabili.
3.3.2 – Determinazione dei gruppi OH
Come già anticipato nel paragrafo precedente, il metodo di calcolo dei gruppi OH medi per catena adottato per questo tipo di polimero è diverso rispetto a quello usato per il pECH(1).
Come prima cosa, a seguito della deconvoluzione dello spettro GPC, ci siamo ricavati la percentuale di oligomeri ciclici; quindi, dall’analisi delle aree dei picchi dell’ 1H-NMR, ci siamo ricavati la percentuale di segnale dovuto alle sole catene lineari.
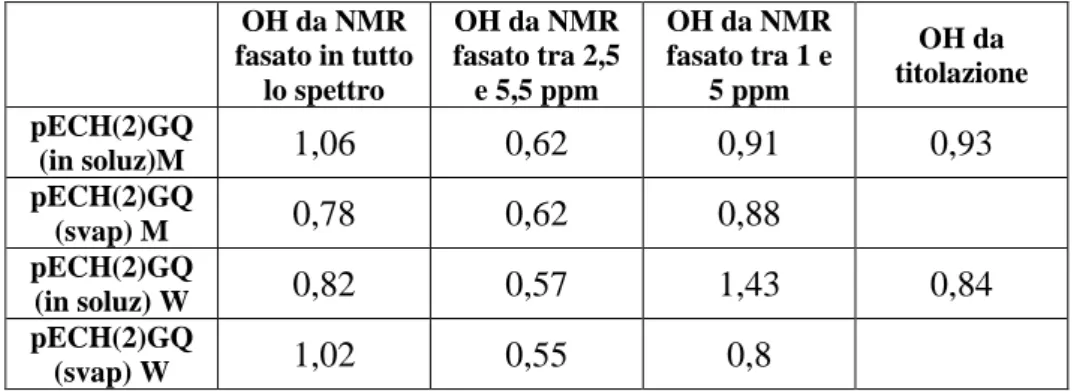
Pertanto, tramite un foglio di calcolo informatico, ci siamo determinati il numero medio di gruppi OH per catena (tabella 3.3.2):
OH da NMR fasato in tutto lo spettro OH da NMR fasato tra 2,5 e 5,5 ppm OH da NMR fasato tra 1 e 5 ppm OH da titolazione pECH(2)GQ (in soluz)M 1,06 0,62 0,91 0,93 pECH(2)GQ (svap) M 0,78 0,62 0,88 pECH(2)GQ (in soluz) W 0,82 0,57 1,43 0,84 pECH(2)GQ (svap) W 1,02 0,55 0,8
Tabella 3.3.2 – Numero di gruppi OH medi per catena.
Come si nota dai valori riportati nella tabella precedente, il numero medio di gruppi OH per catena è ancora inferiore ai due che avremmo dovuto ottenere se la polimerizzazione fosse avvenuta col solo meccanismo di propagazione AMM.
Come già precedentemente detto, la tecnica sperimentale adottata, lascia qualche perplessità a causa dell’utilizzo dello spettro 1H-NMR; infatti, i dati ottenuti col suddetto metodo, dipendono inevitabilmente dalla sensibilità dell’operatore il quale deve fasare il sistema e scegliere gli estremi di integrazione per i picchi d’interesse. Questi due aspetti ci fanno quindi capire bene che, fino a quando i dati sono ricavati da picchi molto intensi, come ad esempio quello a 3,5 negli spettri da noi ottenuti, i risultati sono attendibili perché l’errore commesso è di poche parti percentuali; diventa però inammissibile quando il picco studiato è poco intenso, come ad esempio il picco a 1,6 visto nei nostri spettri.
Per ovviare a questi inconvenienti, si è pensato di determinare il numero dei gruppi OH terminali tramite titolazione. Posto che questa tecnica richiede grosse quantità di materiale, riportiamo di seguito la procedura usata per realizzarla:
Preparazione della soluzione:
Si mettono 7,5 g (o 7 ml) di anidride acetica, con una precisione di ± 0,1 mg, in un recipiente graduato e si diluiscono con 50 ml di dicloroetano. La soluzione fresca deve essere preparata il giorno stesso in cui viene condotta l’analisi. La soluzione così ottenuta deve essere mantenuta lontana da fonti di luce, quindi il recipiente usato per la preparazione (che è di vetro) deve essere avvolto completamente con un foglio di alluminio. A questo punto si esegue un test di titolazione in bianco per la corretta valutazione del peso di anidride acetica. A tale scopo si usa un indicatore composto da uno 0,04% in acqua di un sale di sodio e bromotimolo. La determinazione della normalità di NaOH è fatta tramite la titolazione della soluzione acida.
Procedura per la determinazione analitica:
1) Circa 5-5,3 g del polimero analizzato vengono pesati in un contenitore di vetro da 300 ml;
2) 50 ml di 1,2-dicloroetano anidro vengono aggiunti al polimero e miscelati per 30 minuti con un agitatore magnetico fino a completa solubilizzazione del polimero. Come noto, la pECH e meno solubile del corrispondente polimero azidato (GAP), quindi ci dobbiamo assicurare che questo sia completamente disciolto nel solvente. A questo punto si avvolge completamente il contenitore con un foglio di alluminio;
3) Si aggiungono 3 ml di N-metil imidazolo;
4) Si aggiungono 5 ml di anidride acetica (in eccesso);
5) Il contenitore, a questo punto, deve essere chiuso e bloccato poiché durante la fase di riscaldamento la pressione all’interno del contenitore aumenta e se non è ben chiuso il campione prende aria e rischia di compromettere l’esito della prova;
6) Il campione viene riscaldato a 45 °C per 3 ore in un bagno con olio siliconico. Dopo questa fase il contenitore deve essere agitato dolcemente due volte per rendere la soluzione omogenea.
7) A questo punto il contenitore deve essere raffreddato per 5 minuti (o comunque fintato non sia totalmente raffreddato) in un bagno di ghiaccio.
8) Si aggiungono quindi 25 ml di acqua distillata al fine di annullare l’eccesso di anidride acetica;
9) Il contenitore viene quindi nuovamente chiuso e riscaldato per 10 minuti a 45 °C agitandolo occasionalmente;
10)Il contenitore quindi è nuovamente raffreddato con la stessa metodologia illustrata al punto 7;
11)A questo punto si aggiungono 50 ml di cloroformio e 50 ml di metanolo e si agita nuovamente per ottenere una soluzione completamente omogenea; 12)Si aggiunge l’indicatore alla soluzione e si titola (il CH3COOH ottenuto dalla
reazione) con una soluzione 0,1 molare di NaOH fino a quando la soluzione non cambia colore.
Al fine di chiarire meglio questa tecnica, si riportano di seguito le reazioni che avvengono: HO OH + (CH 3CO)2O CH3 C O O O + CH3COOH + (CH3CO)2O (CH3CO)2O + H2O 2CH3COOH 1) 2) Eccesso
3) Titolo CH3COOH ottenuti con una soluzione standard di NaOH 0.1 M
OH O C
O CH3
Figura 3.3.5 – Schema di reazione per la titolazione.
Alcune prove di titolazione sono al momento in atto sul pECH(2) (risultati in tabella 3.3.2) e su un nuovo campione pECH(3), sintetizzato tramite la stessa procedura vista per il pECH(2), ma senza bagno di ghiaccio e con un gocciolamento più lento. Dalle analisi di titolazione condotte su questo nuovo materiale polimerico, ipotizzando un peso molecolare simile a quello ottenuto per il pECH(2) (e cioè ~900), abbiamo ricavato che i gruppi OH terminali per macromolecola sono 2; infatti, le moli di OH ricavate da tale analisi sono 0,012 (contro le 0,005 del pECH(2) che danno un numero di gruppi OH
macromolecola. In futuro si dovranno fare prove di GPC su tale materiale al fine di confermare l’ipotesi fatta che comunque, alla luce delle vari analisi condotte sui vari materiali, sembra lecita in quanto per ottenere lo stesso numero di gruppi OH ottenuti per il pECH(2) dovremmo avere un PM di 400 (e ciò vorrebbe dire che la polimerizzazione, in pratica, non è avvenuta).
3.4 – Metodi di reticolazione studiati
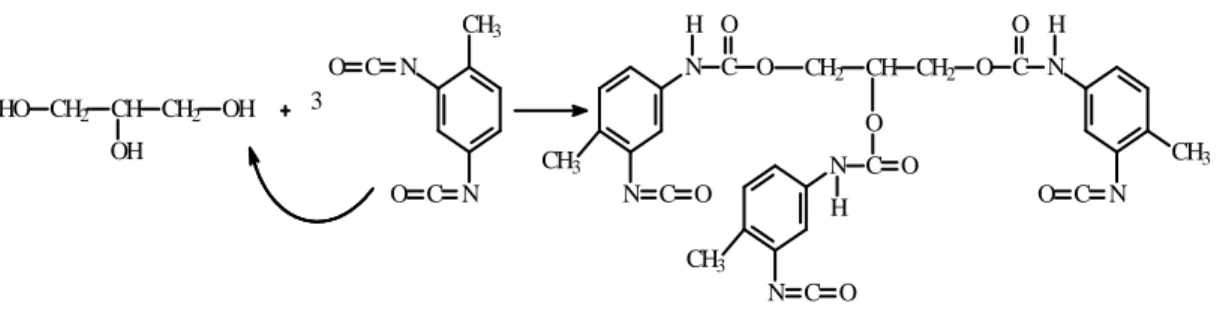
Come già accennato nel paragrafo 2.4 le vie usate per la fase di reticolazione sono state due: la reticolazione di tipo A e quella di tipo B. Riportiamo qui di seguito le due reazioni: Reazione di tipo A: HO CH2 CH CH2 OH OH 3 CH3 N N C O C O N CH2 CH CH2 N C O C H O O CH3 N N C O C O H O CH3 N N C O C O H O CH3
Figura 3.4.1 – Produzione dell’agente reticolante.
Per comodità di rappresentazione indicheremo l’agente reticolante e il polimero come segue: N N N C O C O C O [ ]n HO OH
N N N C O C O C O [ ]n HO OH N N N C O C O H O H O C O O [ ]n HO [ ]n OH ]n [ OH
Figura 3.4.3 – Reazione di reticolazione.
Reazione di tipo B: [ ]n HO OH CH3 N N C O C O 2 [ ]n N N C O H O C O CH3 N N C O H O C O CH3
Figura 3.4.4 – Reazione di TDI+pECH.
Per comodità indicheremo il prodotto ottenuto con la seguente formula:
[ ]n N C O
N C O
Figura 3.4.5 – Prodotto in forma schematica.
[ ]n N C O N C O HO CH2 CH CH2 OH OH O CH2 CH CH2 O O [ ]n N C O N C O H [ ]n N C O N C O H ]n [ N C O N C O H O CH2 CH CH2 O O A A
( )
= AFigura 3.4.6 – Reazione di reticolazione.
Queste due vie sono entrambe percorribili, ed entrambe permettono l’ottenimento di materiale reticolato. Delle due strade però ci siamo concentrati maggiormente sulla seconda poiché il meccanismo di reazione è più facilmente controllabile. Si riportano di
3.4.1 – Reticolazione tipo A
Questo tipo di procedura di reticolazione è stata illustrata nel paragrafo 2.4.1 a cui si rimanda. Vediamo i risultato ottenuti per i vari campioni:
3.4.1.1 – pECH(1) NG Q W
Dopo questa fase di reazione abbiamo ottenuto 6 campioni che a seguito della prova di solubilità quantitativa hanno mostrato i seguenti risultati:
Peso totale (mg)
Peso parte non sciolta (mg)
Peso parte sciolta (mg)
THF 49,89 25,57 24,32
Acetone 50,80 30,00 20,80
DCM 49,71 26,41 23,30
Tabella 3.4.1 – Prova di solubilità quantitativa.
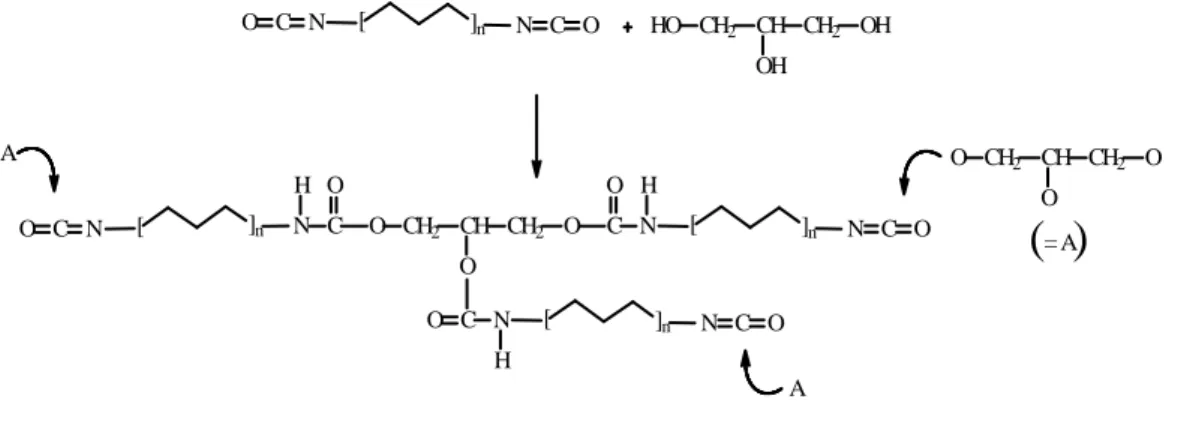
A questo punto abbiamo cercato di caratterizzare i vari campioni. Per quanto riguarda la parte non sciolta, abbiamo usato il microscopio ottico e scaldando i vari campioni da temperatura ambiente a 350°C si è notato che tutti e tre i campioni subiscono ignizione alla temperatura di 250°C. Questo fatto e il fatto che i campioni non si sciolgo più in niente, ci confermano che si hanno dei prodotti reticolati. Inoltre, al fine di caratterizzare a livello qualitativo i tre campioni, sono stati fatti degli spettri IR:
pECH(1)NGQW reticolato tipo A parte non sciolta in THF
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
pECH(1)NGQW reticalato tipo A parte non sciolta in acetone
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050 Figura 3.4.7 – Spettro IR della
parte non sciolta in THF.
Figura 3.4.8 – Spettro IR della parte non sciolta in acetone.
pECH(1)NGQW reticolato tipo A parte non sciolta in diclorometano
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Negli spettri precedenti si può notare che il picco tra 3400 e 3700 cm-1 è aumentato rispetto allo spettro del polimero non reticolato; questo è dovuto al fatto che i gruppi NH (dovuti alla reazione con l’agente reticolante) assorbono alla stessa frequenza dei gruppi OH, sovrapponendosi. Al fine di capire meglio quanto appena affermato, in figura 3.4.10, si riporta lo spettro dell’agente reticolante TDI+Glicerolo:
TDI_Glicerolo recuperato svaporando
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.4.10 – Spettro IR dell’agente reticolante TDI+Glicerolo.
Come si vede dallo spettro IR tra 3400 e 3700 si ha un picco dovuto appunto alla presenza di gruppi NH dell’agente reticolate ottenuto dalla reazione tra TDI e Glicerolo. Quindi, a seguito delle analisi appena descritte, vediamo che la quantità di campione reticolato per ogni prova è: 51% per la prova di solubilità con tetraidrofurano; 59% per la prova di solubilità con acetone; 53% per la prova di solubilità con diclorometano. Per quanto riguarda invece la caratterizzazione della parte che si è sciolta (in cui sono presenti anche gli oligomeri ciclici) abbiamo fatto delle analisi IR che hanno dato il seguente risultato:
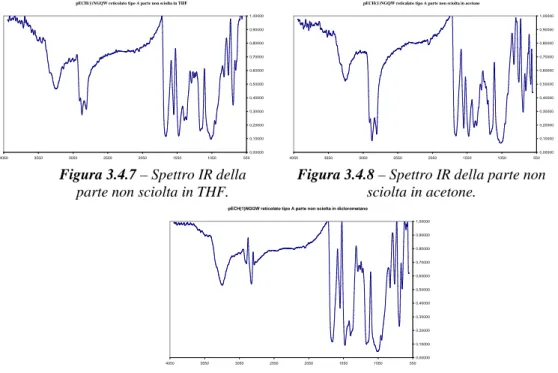
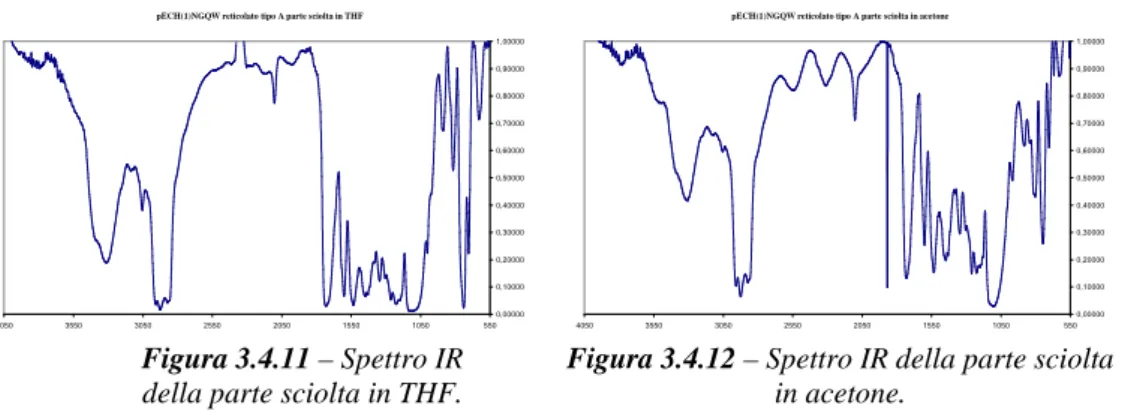
pECH(1)NGQW reticolato tipo A parte sciolta in THF
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
pECH(1)NGQW reticolato tipo A parte sciolta in acetone
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050 Figura 3.4.11 – Spettro IR
della parte sciolta in THF.
Figura 3.4.12 – Spettro IR della parte sciolta in acetone.
pECH(1)NGQW reticolato tipo A parte sciolta in diclorometano 0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.4.13 – Spettro IR della parte sciolta in DCM.
Anche in questi spettri, come nei precedenti, le bande degli OH sono più grandi a causa della presenza dell’agente reticolante (gruppi NH).
La presenza dell’agente reticolante nello spettro è dovuta probabilmente a reazioni di allungamento di catena che sono intercorse tra catene aventi un solo gruppo OH terminale (ottenuti quindi tramite meccanismo ACE).
3.4.1.2 – pECH(2) G Q (dopo svap) W
Dopo la fase di reticolazione abbiamo ottenuto 6 campioni che a seguito della prova di solubilità quantitativa hanno mostrato i seguenti risultati:
Peso totale (mg)
Peso parte non sciolta (mg)
Peso parte sciolta (mg)
THF 50,0 32,5 17,5
Acetone 49,7 28,7 21,0
DCM 49,8 29,3 20,5
Tabella 3.4.2 – Prova di solubilità quantitativa.
La quantità che non si è sciolta è anche la quantità di campione reticolato che abbiamo ottenuto, pertanto vediamo che la percentuale di reticolato per ogni prova è:
65,0% per la prova di solubilità con tetraidrofurano; 58,8% per la prova di solubilità con acetone;
3.4.1.3 – pECH(2) G Q (dopo svap) M
Dopo la fase di reticolazione abbiamo ottenuto 6 campioni che a seguito della prova di solubilità quantitativa hanno mostrato i seguenti risultati:
Peso totale (mg)
Peso parte non sciolta (mg)
Peso parte sciolta (mg)
THF 50,2 39,2 11,0
Acetone 50,3 31,2 19,1
DCM 49,7 31,8 17,9
Tabella 3.4.3 – Prova di solubilità quantitativa.
La quantità che non si è sciolta è anche la quantità di campione reticolato che abbiamo ottenuto, pertanto vediamo che la percentuale di reticolato per ogni prova è:
78,1% per la prova di solubilità con tetraidrofurano; 62,0% per la prova di solubilità con acetone;
63,9% per la prova di solubilità con diclorometano.
3.4.14 – pECH(2) G Q (in soluz) M
Dopo la fase di reticolazione abbiamo ottenuto 6 campioni che a seguito della prova di solubilità quantitativa hanno mostrato i seguenti risultati:
Peso totale (mg)
Peso parte non sciolta (mg)
Peso parte sciolta (mg)
THF 50,2 35,1 15,1
Acetone 50,2 33,0 17,2
DCM 50,3 29,9 20,4
Tabella 3.4.4 – Prova di solubilità quantitativa.
La quantità che non si è sciolta è anche la quantità di campione reticolato che abbiamo ottenuto, pertanto vediamo che la percentuale di reticolato per ogni prova è:
65,7% per la prova di solubilità con tetraidrofurano; 59,4% per la prova di solubilità con acetone;
3.4.1.5 – pECH(2) G Q (in soluz) W
Dopo la fase di reticolazione abbiamo ottenuto 6 campioni che a seguito della prova di solubilità quantitativa hanno mostrato i seguenti risultati:
Peso totale (mg)
Peso parte non sciolta (mg)
Peso parte sciolta (mg)
THF 50,1 29,3 20,8
Acetone 50,1 27,1 23,0
DCM 49,7 24,9 24,8
Tabella 3.4.5 – Prova di solubilità quantitativa.
La quantità che non si è sciolta è anche la quantità di campione reticolato che abbiamo ottenuto, pertanto vediamo che la percentuale di reticolato per ogni prova è:
58,5% per la prova di solubilità con tetraidrofurano; 54,1% per la prova di solubilità con acetone;
50,1% per la prova di solubilità con diclorometano.
Come si può notare dai dati precedenti, il materiale reticolato ottenuto va dal 50 al 65% in media per ogni campione. Questo è un risultato che abbiamo giudicato essere basso, quindi, al fine di aumentarlo, abbiamo cercato di indagare meglio sulla seconda via di reazione di reticolazione (giudicata essere migliore).
3.4.2 – Reticolazione tipo B
Questo tipo di procedura di reticolazione è stata illustrata nel paragrafo 2.4.2 a cui si rimanda.
La prova è stata condotta solamente sul pECH(2) poiché, grazie ai dati ottenuti dalla reticolazione di tipo A, abbiamo visto che il numero di catene diOH terminate per questo tipo di campioni sono maggiori rispetto a quello riscontrato nel pECH(1).
Vediamo di seguito nel dettaglio i risultati ottenuti.
3.4.2.1 – pECH(2) G Q (dopo svap) M
Dopo la reazione di reticolazione e il lavaggio con etanolo, abbiamo ottenuto due campioni: una fase solida ed una liquida.
La fase solida, in quanto insolubile in ogni solvente, non è stata caratterizzata a livello qualitativo, ciò che abbiamo recuperato dalla fase liquida (dopo aver evaporato il
solvente) invece è stato caratterizzato tramite analisi IR. I risultati ottenuti sono riportati qui di seguito:
pECH(2) gocciolato quench (dopo svaporazione) con metanolo parte non scioltadella reticolazione di tipo B
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.4.14 – Spettro IR della fase non reticolata.
Confrontando questo spettro con quello relativo al polimero tal quale si nota che questi sono praticamente identici salvo per il fatto che la banda a 3500 (quella degli OH) risulta più accentuata. Questa maggiore intensità della banda è dovuta alla sovrapposizione del segnale dei gruppi NH inseriti in catena dalla reazione di reticolazione.
Al fine di caratterizzare anche quantitativamente le due fasi, abbiamo condotto su queste un’analisi parallela tra i dati ottenuti dalla GPC del campione tal quale (a seguito di deconvoluzione) e l’analisi quantitativa condotta sul campione in esame.
Dalla GPC abbiamo ottenuto che la percentuale in peso delle catene lineari è 86,7% (il 13,3% sono catene cicliche); dall’analisi quantitativa condotta sui campioni invece i risultati ottenuti sono stati i seguenti:
Polimero NCO terminato (g) Reticolato (g) Non reticolato (g) %peso delle catene lineari 2,007 1,439 0,568 71,7
Tabella 3.4.6 – Analisi quantitativa.
Il fatto che la % in peso dopo reticolazione è minore della % peso ottenuta dalla GPC conferma che non tutte le catene sono diOH terminate. Quindi parte delle catene lineari le andiamo a perdere nella fase liquida.
Per avere conferma del fatto che non tutte le catene sono diOH terminate, sulla parte non reticolata sono state condotte delle analisi 1H-NMR in toluene-d8 (Figura 3.4.15) ottenendo i seguenti risultati:
Figura 3.4.15 – Spettro 1H-NMR della fase non reticolata.
Dalle analisi precedenti vediamo che lo spettro 1H-NMR della parte che non ha reticolato è sostanzialmente diverso da quello del polimero tal quale; infatti il picco a 5,3 ppm relativo ai gruppi OH è scomparso quasi del tutto segno che i gruppi OH hanno reagito in modo quantitativo con l’agente reticolante. Quelli ancora presenti sono dovuti ai gruppi OH del glicerolo che non hanno reagito.
Il picco a 3,5 (relativo agli assorbimenti del polimero) è molto meno intenso di quello relativo al polimero tal quale. Questo perché è causato dalla presenza di oligomeri ciclici e di catene che hanno subito allungamento di catena che come già detto sono in numero minore rispetto a quelle diOH terminate. Questa maggiore intensità la si nota poiché la parte di spettro tra 1,8 e 0 (relativa ad impurezze) risulta più intensa.
3.4.2.2 – pECH(2) G Q (dopo svap) W
Dopo la reazione di reticolazione e il lavaggio con etanolo, abbiamo ottenuto due campioni: una fase solida ed una liquida.
La fase solida, in quanto insolubile in ogni solvente, non è stata caratterizzata a livello qualitativo, ciò che abbiamo recuperato dalla fase liquida (dopo aver evaporato il solvente) invece è stato caratterizzato tramite analisi IR. I risultati ottenuti sono riportati qui di seguito:
pECH(2) gocciolato quench dopo svaporazione con acqua parte non sciollta della reticolazione tipo B 0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.4.16 – Spettro IR della fase liquida.
Confrontando questo spettro con quello relativo al polimero tal quale si nota che questi sono praticamente identici salvo per il fatto che la banda a 3500 (quella degli OH) risulta più accentuata. Questa maggiore intensità della banda è dovuta alla sovrapposizione del segnale dei gruppi NH inseriti in catena dalla reazione di reticolazione.
Al fine di caratterizzare anche quantitativamente le due fasi, abbiamo condotto su queste un’analisi parallela tra i dati ottenuti dalla GPC del campione tal quale (a seguito di deconvoluzione) e l’analisi quantitativa condotta sul campione in esame.
Dalla GPC abbiamo ottenuto che la percentuale in peso delle catene lineari è 86,3% (il 13,7% sono catene cicliche); dall’analisi quantitativa condotta sui campioni invece i risultati ottenuti sono stati i seguenti:
Polimero NCO terminato (g) Reticolato (g) Non reticolato (g) %peso delle catene lineari 1,375 0,931 0,444 67,7
Tabella 3.4.7 – Analisi quantitativa.
Il fatto che la % in peso dopo reticolazione è minore della % peso ottenuta dalla GPC conferma che non tutte le catene sono diOH terminate. Quindi parte delle catene lineari le andiamo a perdere nella fase liquida.
Per avere conferma del fatto che non tutte le catene sono diOH terminate, sulla parte non reticolata sono state condotte delle analisi 1H-NMR in toluene-d8 (Figura 3.4.17) ottenendo i seguenti risultati:
Figura 3.4.17 – Spettro 1H-NMR della fase non reticolata.
Dalle analisi precedenti vediamo che lo spettro 1H-NMR della parte che non ha reticolato è sostanzialmente diverso da quello del polimero tal quale; infatti il picco a 5,3 ppm relativo ai gruppi OH è scomparso del tutto segno che i gruppi OH hanno reagito in modo quantitativo con l’agente reticolante.
Il picco a 3,5 (relativo agli assorbimenti del polimero) è molto meno intenso di quello relativo al polimero tal quale. Questo perché è causato dalla presenza di oligomeri ciclici e di catene che hanno subito allungamento di catena che come già detto sono in numero minore rispetto a quelle diOH terminate. Questa maggiore intensità la si nota poiché la parte di spettro tra 1,8 e 0 (relativa ad impurezze) risulta più intensa.
3.4.2.3 – pECH(2) G Q (in soluz) M
Dopo la reazione di reticolazione e il lavaggio con etanolo, abbiamo ottenuto due campioni: una fase solida ed una liquida.
La fase solida, in quanto insolubile in ogni solvente, non è stata caratterizzata a livello qualitativo, ciò che abbiamo recuperato dalla fase liquida (dopo aver evaporato il solvente) invece è stato caratterizzato tramite analisi IR. I risultati ottenuti sono riportati qui di seguito:
pECH(2) gocciolato quench in soluzione con metanolo parte sciolta della reticolazione di tipo B 0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.4.18 – Spettro IR della fase liquida.
Confrontando questo spettro con quello relativo al polimero tal quale si nota che questi sono praticamente identici salvo per il fatto che la banda a 3500 (quella degli OH) risulta più accentuata. Questa maggiore intensità della banda è dovuta alla sovrapposizione del segnale dei gruppi NH inseriti in catena dalla reazione di reticolazione.
Al fine di caratterizzare anche quantitativamente le due fasi, abbiamo condotto su queste un’analisi parallela tra i dati ottenuti dalla GPC del campione tal quale (a seguito di deconvoluzione) e l’analisi quantitativa condotta sul campione in esame.
Dalla GPC abbiamo ottenuto che la percentuale in peso delle catene lineari è 85,4% (il 14,6% sono catene cicliche); dall’analisi quantitativa condotta sui campioni invece i risultati ottenuti sono stati i seguenti:
Polimero NCO terminato (g) Reticolato (g) Non reticolato (g) %peso delle catene lineari 1,299 0,85 0,499 65,4
Tabella 3.4.8 – Analisi quantitativa.
Il fatto che la % in peso dopo reticolazione è minore della % peso ottenuta dalla GPC conferma che non tutte le catene sono diOH terminate. Quindi parte delle catene lineari le andiamo a perdere nella fase liquida.
Per avere conferma del fatto che non tutte le catene sono diOH terminate, sulla parte non reticolata sono state condotte delle analisi 1H-NMR in toluene-d8 (Figura 3.4.19) ottenendo i seguenti risultati:
Figura 3.4.19 – Spettro 1H-NMR della fase non reticolata.
Dalle analisi precedenti vediamo che lo spettro 1H-NMR della parte che non ha reticolato è sostanzialmente diverso da quello del polimero tal quale; infatti il picco a 5,3 ppm relativo ai gruppi OH è scomparso quasi del tutto segno che i gruppi OH hanno reagito in modo quantitativo con l’agente reticolante. Quelli ancora presenti sono dovuti ai gruppi OH del glicerolo che non hanno reagito.
Il picco a 3,5 (relativo agli assorbimenti del polimero) è molto meno intenso di quello relativo al polimero tal quale. Questo perché è causato dalla presenza di oligomeri ciclici e di catene che hanno subito allungamento di catena che come già detto sono in numero minore rispetto a quelle diOH terminate. Questa maggiore intensità la si nota poiché la parte di spettro tra 1,8 e 0 (relativa ad impurezze) risulta più intensa.
3.4.2.4 – pECH(2) G Q (in soluz) W
Dopo la reazione di reticolazione e il lavaggio con etanolo, abbiamo ottenuto due campioni: una fase solida ed una liquida.
La fase solida, in quanto insolubile in ogni solvente, non è stata caratterizzata a livello qualitativo, ciò che abbiamo recuperato dalla fase liquida (dopo aver evaporato il solvente) invece è stato caratterizzato tramite analisi IR. I risultati ottenuti sono riportati qui di seguito:
pECH(2) gocciolato quench in soluzione con acqua parte scolate della eticolazione di tipo B 0,50000 0,55000 0,60000 0,65000 0,70000 0,75000 0,80000 0,85000 0,90000 0,95000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.4.20 – Spettro IR della fase liquida.
Confrontando questo spettro con quello relativo al polimero tal quale si nota che questi sono praticamente identici salvo per il fatto che la banda a 3500 (quella degli OH) risulta molto più accentuata. Questa maggiore intensità della banda è dovuta alla sovrapposizione del segnale dei gruppi NH inseriti in catena dalla reazione di reticolazione.
Al fine di caratterizzare anche quantitativamente le due fasi, abbiamo condotto su queste un’analisi sui dati ottenuti dalla GPC del campione tal quale (a seguito di deconvoluzione) e abbiamo ottenuto che la percentuale in peso delle catene lineari è 84,9% (il 15,1% sono catene cicliche); l’analisi quantitativa purtroppo non è stato possibile condurla poiché si sono avute perdite notevoli del campione esaminato.
Per indagare sul fatto che non tutte le catene sono diOH terminate (come fatto per i campioni precedenti), sulla parte non reticolata sono state condotte delle analisi 1 H-NMR in toluene-d8 (Figura 3.4.21) ottenendo i seguenti risultati:
Dalle analisi precedenti vediamo che lo spettro 1H-NMR della parte che non ha reticolato è sostanzialmente diverso da quello del polimero tal quale; infatti il picco a 5,3 ppm relativo ai gruppi OH è scomparso quasi del tutto segno che i gruppi OH hanno reagito in modo quantitativo con l’agente reticolante. Quelli ancora presenti sono dovuti ai gruppi OH del glicerolo che non hanno reagito.
Il picco a 3,5 (relativo agli assorbimenti del polimero) è molto meno intenso di quello relativo al polimero tal quale. Questo perché è causato dalla presenza di oligomeri ciclici e di catene che hanno subito allungamento di catena che come già detto sono in numero minore rispetto a quelle diOH terminate. Questa maggiore intensità la si nota poiché la parte di spettro tra 1,8 e 0 (relativa ad impurezze) risulta più intensa.
3.4.2.5 – Discussione
Dalle prove di solubilità, quindi, abbiamo ottenuto che la percentuale di reticolato ottenuto in media è pari ad un 70% del materiale di partenza (30 % materiale non reticolato), mentre dall’analisi derivante dalla deconvoluzione (fatta con Origin 6) dei picchi ottenuti dalla GPC (Figura 3.4.22) si ha che le catene lineari sono l’85% e quelle cicliche il 15%.
Figura 3.4.22 – Analisi ottenuta dalla deconvoluzione.
Alla luce di questo abbiamo quindi un’ulteriore conferma del fatto che anche tra le catene lineari ve ne sono alcune in cui è presente un solo gruppo OH o addirittura nessuno; infatti se tutte le catene fossero state diOH terminate, la percentuale di materiale reticolato dovrebbe essere stata pari alla percentuale delle catene lineari e nella parte non sciolta avremmo dovuto trovare solo catene cicliche.
Alla luce dei dati ottenuti dalle reazioni di reticolazione si ha ulteriore conferma del fatto che entrambi i meccanismi di polimerizzazione (AMM e ACE) hanno avuto luogo contemporaneamente, fatto che ci è confermato dalla presenza di oligomeri ciclici e di catene lineari che non reticolano.
3.5 – Sintesi e caratterizzazione del Cpo ECH/BBrMO (50:50)
La procedura di sintesi di questo copolimero è stata illustrata nel paragrafo 2.2.3 a cui si rimanda. Dopo questa fase di reazione abbiamo ottenuto 4 campioni: a) copolimero ECH/BBrMO(5) gocciolato e sottoposto a quench con acqua (sigla ECH/BBrMO(5) G Q W); b) copolimero ECH/BBrMO(5) gocciolato e sottoposto a quench con metanolo (sigla ECH/BBrMO(5) G Q M); c) copolimero ECH/BBrMO(5) non gocciolato e sottoposto a quench con acqua (sigla ECH/BBrMO(5) NG Q W); d) copolimero ECH/BBrMO(5) non gocciolato e sottoposto a quench con metanolo (sigla ECH/BBrMO(5) NG Q M); che sono stati caratterizzati tramite alcune delle varie analisi descritte nel paragrafo 2.5 (analogamente a quanto visto nel paragrafo 3.1 riguardo alla poliepicloridrina). Vediamo campione per campione i risultati ottenuti. Come prima cosa abbiamo analizzato i campioni tramite spettrografia ad infrarosso (IR) ottenendo il seguente spettro:
Cpo ECH/BBrMO(5) gocciolato quench metanolo
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.5.1 – Spettro IR del Cpo ECH/BBrMO(5) G Q M.
Come si vede dallo spettro IR riportato in figura 3.5.1, tra 3400 e 3700 cm-1 si ha un picco il quale ci conferma la presenza di gruppi OH all’interno del polimero; infatti
Sempre dallo spettro precedente possiamo dire che il materiale ottenuto a seguito della reazione di polimerizzazione è effettivamente un polimero; infatti confrontando questo spettro con quello del monomero di ECH (Figura 3.5.2) e con quello del BBrMO (Figura 3.5.3) si vede che la banda a 990 cm-1 relativa al monomero ECH, nello spettro del polimero compare a 1100 cm-1; inoltre le bande che compaiono a 435 cm-1 (relative al legame C-Cl) e quella a 605 cm-1 (relativa al legame C-Br) ci mostrano che tali legami subiscono uno stretching.
Figura 3.5.2 – Spettro IR della ECH.
Figura 3.5.3 – Spettro IR del monomero BBrMO.
Questo fatti ci confermano quindi che la reazione di polimerizzazione ha effettivamente avuto luogo, però non è in grado di fornirci risultati quantitativi riguardo la
composizione del materiale polimerico, poiché dallo spettro non è esclusa la presenza di oligomeri ciclici.
Al fine di caratterizzare anche quantitativamente il nostro campione, abbiamo funzionalizzato i gruppi OH terminali (Figura 3.5.4) come illustrato nel paragrafo 2.3 (e per le ragioni già spiegate nel paragrafo 3.1) e lo abbiamo analizzato tramite spettrografia 1H-NMR in toluene-d8 (Figura 3.5.5).
Cpo ECH/BBrMO(5) gocciolato quench metanolo funzionalizzato
0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.5.4 – Spettro IR del Cpo ECH/BBrMO(5) G Q M.
Come si può vedere dal grafico precedente, il nuovo picco che si ha a 1800 cm-1 è quello dovuto alla funzionalizzazione di cui sopra, dato confermato anche dalla scomparsa del picco degli OH.
Figura 3.5.5 – Spettro 1H-NMR del Cpo ECH/BBrMO(5) G Q M.
Come si può vedere in figura 3.3.4 il quintupletto che cade a 5,3 ppm rappresenta la risposta che danno i gruppi OH funzionalizzati da anidride trifluoroacetica; il picco a
1,6 ppm rappresenta la risposta che ci dà il butandiolo in catena e il picco grande a 3,5 ppm rappresenta il contributo che da il polimero.
Per gli altri 3 campioni di Cpo gli spettri IR del campione tal quale e del campione funzionalizzato con anidride trifluoroacetica e gli spettri 1H-NMR hanno mostrato risultati identici a quelli appena visti per il Cpo ECH/BBrMO G Q M.
Al fine di determinare la composizione del copolimero, lo spettro 1H-NMR, non c’è utile poiché sotto al picco a 3-4 ppm cascano tutti i protoni e proprio per questo motivo non si riesce a determinare la composizione monomerica delle catene. Dalla letteratura si è visto che per determinare suddetta composizione possiamo ricorrere alla analisi fatta attraverso 13C-NMR (Figura 3.5.6):
Figura 3.5.6 – Spettro 13C-NMR del Cpo ECH/BBrMO(5) G Q M.
Da questo spettro si evince che partendo da un rapporto monomerico 50:50, riusciamo ad ottenere un materiale polimerico composto dal 70% di BBrMO e dal 30% di ECH il quale può essere completamente azidato. Pertanto atteniamo un copolimero energetico composto dal 70% di BAMO e dal 30% di GAP.
Il composto suddetto ha una % di gruppi N3 pari al 48% (ricordiamo che il GAP ne ha 42% e il pBAMO 50%) e quindi è molto energetico; inoltre le proprietà meccaniche non sono assolutamente compromesse poiché si ottiene un materiale ancora amorfo. Pertanto, seguendo questa strada, si possono ottenere leganti che mostrato un buon compromesso (il migliore tra quelli studiati), tra proprietà meccaniche e % di N3 in catena.
Continuando con la determinazione del materiale polimerico in esame, abbiamo riscontrato notevoli difficoltà nel determinare il peso molecolare per mezzo della GPC, poiché il materiale ottenuto si è dimostrato poco solubile nel solvente di analisi (cloroformio). Questo accade perché le unità di BBrMO rendono le catene più stereoregolari con l’ottenimento di un materiale più cristallino e pertanto meno solubile. Per ovviare a questo fatto in futuro la determinazioni dei gruppi OH su questo tipo di copolimeri verrà fatta per titolazione dopo che i campioni sono stati azzidati; infatti la solubilità del polimero azidato è molto maggiore di quella del corrispondente polimero alogenato.
Una caratterizzazione più approfondita di questi copolimeri non è stata fatta poiché, come si può vedere dalla trattazione fino a qui fatta, ci siamo concentrati maggiormente sullo studio della pECH.
3.6 – Stabilità di polimero pECH(1) e copolimero
Al fine di studiare la sensibilità dei polimeri studiati, nei laboratori dell’AVIO di Colleferro sono state condotte due tipi di prove:
test di impatto: consiste nel far cadere un peso di 5 o 1 Kg da diverse altezze su un campione di alcuni milligrammi e riporta il minimo valore (espresso in Joule e calcolato come prodotto dei Kg peso per altezza di caduta) necessario per innescare l’esplosione.
test di frizione: consiste nel sottoporre a sfregamento il campione mediante applicazione di una forza di taglio che viene gradualmente incrementata e riporta il valore minimo (espresso in Kg) necessario per osservare l’intervento di fenomeni degradativi.
Queste prove sono state fatte su polimero e copolimero energetici ottenuti tramite reazione di azidazione con NaN3.
Come prima cosa abbiamo analizzato il campione di pECH(1) tramite spettrografia ad infrarosso (IR) ottenendo il seguente spettro:
pECH(1) azidato 0,00000 0,10000 0,20000 0,30000 0,40000 0,50000 0,60000 0,70000 0,80000 0,90000 1,00000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.6.1 – Spettro IR del campione azidato in esame.
Come si vede dallo spettro IR riportato in figura 3.6.1 tra 2000 e 2100 si ha un picco il quale ci conferma la presenza di gruppi azidici all’interno del polimero; infatti questi valori di frequenza sono tipici dell’assorbimento dei suddetti gruppi.
I valori ottenuti dalle prove suddette sono riportati nelle seguenti tabelle:
Carico applicato
(Kg)
Crepitio Innesco Decomposizione Nessuna
reazione 36 X 36 X 36 X 36 X 19.2 X 19.2 X 19.2 X 19.2 X
Tabella 3.6.1 – Test di frizione su pECH(1) azidato.
Peso Massa (Kg) Altezza caduta (mm)
Esplosione Disgregazione Nessuna
reazione 5 1000 X 5 1500 X 5 2000 X 5 500 X 5 750 X 5 750 X 5 850 X 5 800 X 5 800 X 5 800 X
In seguito abbiamo analizzato il campione di Cpo ECH/BBrMO(5) tramite spettrografia ad infrarosso (IR) ottenendo il seguente spettro:
Cpo ECH/BBrMO azidato
-0,10000 0,10000 0,30000 0,50000 0,70000 0,90000 1,10000 550 1050 1550 2050 2550 3050 3550 4050
Figura 3.6.2 – Spettro IR del campione azidato in esame.
Come si vede dallo spettro IR riportato in figura 3.6.2 tra 2000 e 2100 si ha un picco il quale ci conferma la presenza di gruppi azidici all’interno del polimero; infatti questi valori di frequenza sono tipici dell’assorbimento dei suddetti gruppi.
I valori ottenuti dalle prove suddette sono riportati nelle seguenti tabelle:
Carico applicato
(Kg)
Crepitio Innesco Decomposizione Nessuna
reazione 36 X 36 X 36 X 19.2 X 19.2 X
Tabella 3.6.3 – Test di frizione su Cpo ECH/BBrMO(5) azidato.
Peso Massa (Kg) Altezza caduta (mm)
Esplosione Disgregazione Nessuna
reazione 5 1000 X 5 1500 X 5 2000 X 5 2000 X 5 2000 X
Quindi, confrontando i valori riportati nelle tabelle precedenti con quelli di tabella 3.6.5, vediamo che il monomero azidato è effettivamente più instabile del corrispondente polimero e quindi azzidare polimeri precostituiti risulta senza dubbio la via più sicura per la reazione di azzidazione (nella tabella seguente i valori della ECH azzidata non sono riportati poiché ricordiamo che la produzione del GAP, il corrispettivo polimero energetico della ECH, si fa solo passando dal polimero, per le ragioni già dette nel paragrafo 3.1.1). Test d’impatto (J) Test di frizione (kg) ECH - - BAMO 0.49 4