INDICE
RIASSUNTO
1ABSTRACT
3INTRODUZIONE
5 1.1 LE NEUROTOSSINE BOTULINICHE 51.1.1 Struttura delle BoNT 6
1.1.2 Funzione delle BoNT 8
1.1.2.1 Legame alla membrana delle cellule target 8
1.1.2.2 Internalizzazione dei neuroni 11
1.1.2.3 Traslocazione attraverso la membrana vescicolare 12
1.1.2.4 Attività endopeptidasica Zinco-dipendente 13
1.1.3 Target delle neurotossine botuliniche 17
1.1.3.1 Sintaxina 17
1.1.3.2 SNAP-25 17
1.1.3.3 VAMP/sinaptobrevina 20
1.1.4 La giunzione neuromuscolare 20
1.1.5 Il Sistema Nervoso Centrale 25
1.1.5.1 Azione delle BoNT nel Sistema Nervoso Centrale in vitro 25
1.1.5.2 Azione delle BoNT nel Sistema Nervoso Centrale in vivo 27
1.1.5.3 Durata d’azione delle neurotossine 28
1.1.6 Le neurotossine botuliniche come agenti terapeutici 29
1.1.6.1 Effetto antiepilettico di BoNT/E 31
1.2 L’ISCHEMIA CEREBALE 32
1.2.1 Forme e modelli di ischemia cerebrale 33
1.2.1.1 Ischemia cerebrale globale 33
1.2.1.2 Ischemia cerebrale focale 34
1.2.2 Patogenesi dell’ischemia cerebrale 35
1.2.2.1 Modificazioni molecolari e cellulari 35
1.2.3.1 Neuroprotezione da agenti Ca -antagonisti
e farmaci chelanti lo ione Ca2+ 39
1.2.3.2 Neuroprotezione da agenti glutammato-antagonisti 39
1.2.3.3 Potenziamento della neurotrasmissione GABA-ergica 40
1.2.3.4 Neuroprotezione da antiossidanti 40
1.3 OBIETTIVI 41
MATERIALI E METODI
43
2.1 TRATTAMENTO DEGLI ANIMALI 43
2.1.1 Iniezione di BoNT/A e BoNT/E 43
2.1.2 Ischemia cerebrale focale 43
2.2 ELETTROFISIOLOGIA 44
2.2.1 Registrazione dei potenziali d’azione 44
2.2.2 Analisi EEG 45
2.3 IMMUNOBLOTTING 46
2.4 ISTOLOGIA 47
2.4.1 Fissazione dei tessuti 47
2.4.2 Rivelazione di SNAP-25 tagliata 48
2.4.3 Valutazione della degenerazione neuronale a livello della lesione ischemica 49
2.4.3.1 Doppia marcatura con anticorpi anti-NeuN e anti-GFAP 49
2.4.3.2 Colorazione di Nissl 50
2.4.3.3 Calcolo dell’estensione della lesione ischemica in CA1 50
2.5 ANALISI STATISTICA 50
RISULTATI
52
3.1 CARATTERIZZAZIONE DELL’AZIONE DI BoNT/A E BoNT/E NELL’IPPOCAMPO 52
3.1.1 Effetto di BoNT/E sull’attività spontanea ippocampale 52
3.1.2 Durata dell’effetto di BoNT/E 53
3.1.2.1 Analisi della durata dell’azione proteolitica di BoNT/E su SNAP-25 55
3.1.3 Durata dell’effetto di BoNT/A 57
3.1.3.1 Analisi della durata dell’azione proteolitica di BoNT/A su SNAP-25 59
3.1.4 Localizzazione immnoistochimica della forma tagliata di SNAP-25 in seguito all’iniezione di BoNT/A e di BoNT/E 61
3.1.5 Il meccanismo d’azione di BoNT/E è dipendente dall’attività elettrica? 64
3.2 AZIONE NEUROPROTETTIVA DI BoNT/E IN UN MODELLO DI ISCHEMIA CEREBRALE FOCALE 67
3.2.1 Caratterizzazione della lesione ischemica provocata dall’iniezione di ET-1 67
3.2.2 Valutazione delle proprietà neuroprotettive di BoNT/E 69
DISCUSSIONE
70
4.1 EFFETTI DI BoNT/A E BoNT/E NELL’IPPOCAMPO 70
4.1.1 Silenziamento dell’attività elettrica 70
4.1.2 Diversa durata d’azione delle due neurotossine 71
4.1.3 Trasporto di BoNT/A o del suo prodotto di taglio nell’ippocampo non iniettato 73
4.1.4 Effetto paradossale del trattamento con KA sull’azione proteolitica di BoNT/E 74
4.1.4.1 Ruolo di SNAP-25 nell’eccitablità neuronale 76
4.2 EFFETTO NEUROPROTETTIVO DI BoNT/E IN UN MODELLO DI ISCHEMIA CEREBRALE FOCALE 77
4.2.1 Modello di ischemi cerebrale focale utilizzato 77
4.2.2 Finestra terapeutica di BoNT/E 78
4.2.3 L’effetto neuroprotettivo di BoNT/E è a lungo termine? 78
Riassunto
Le neurotossine botuliniche (BoNT) sono endopeptidasi prodotte dai batteri del genere
Clostridium che esercitano un'azione proteolitica altamente selettiva su molecole del
complesso SNARE, proteine di membrana implicate nel processo di rilascio del neurotrasmettitore alla terminazione presinaptica. Il taglio proteolitico rende inattive le SNARE e determina il blocco della trasmissione sinaptica.
L’azione delle tossine è stata ampiamente caratterizzata a livello del Sistema Nervoso Periferico.
In particolare, a livello di placca motrice bloccano il rilascio di acetilcolina causando paralisi neuromuscolare. Meno si conosce sull’azione delle BoNT nel Sistema Nervoso Centrale.
Nel mio lavoro di tesi abbiamo valutato gli effetti in vivo delle neurotossine nel Sistema Nervoso Centrale. In particolare abbiamo studiato l’azione dei serotipi A ed E (BoNT/A e BoNT/E) a livello dell’ippocampo dei roditori.
BoNT/A e BoNT/E esercitano la loro attività proteolitica sulla stessa proteina del complesso SNARE, la proteina associata ai sinaptosomi dal peso di 25 KDa (SNAP-25), localizzata preferenzialmente nei terminali glutammatergici. Studi in vitro mostrano infatti che sono in grado di inibire reversibilmente il rilascio di glutammato.
Abbiamo effettuato un’iniezione unilaterale e stereotassica della BoNT/E o della BoNT/A nell’ippocampo dorsale di topo adulto e ne abbiamo valutato gli effetti a livello elettrofisiologico e biochimico.
Le registrazioni in vivo di potenziali d’azione dall’ippocampo mostrano che entrambi i serotipi determinano un silenziamento dell’attività di scarica spontanea dei neuroni piramidali. Tuttavia la durata di quest’effetto inibitorio persiste per periodi diversi: il silenziamento indotto da BoNT/A dura per almeno 60 giorni mentre quello determinato da BoNT/E, meno di 14 giorni. Questi dati elettrofisiologici sono confermati da analisi di espressione di SNAP-25 tagliata tramite Western blot. Esperimenti di immunoistochimica mostrano una localizzazione similare delle due forme di SNAP-25 tagliata da BoNT/A o da BoNT/E. Tuttavia a partire dal terzo giorno dall’iniezione si rivela la marcatura per SNAP-25 tagliata da BoNT/A anche nell’ippocampo controlaterale, soprattutto a livello della CA3. Questo dato viene supportato dagli
esperimenti di Western blot che confermano la diffusione dell’azione proteolitica di BoNT/A anche nell’emisfero non iniettato.
È noto che nel Sistema Nervoso Periferico, l’azione delle BoNT è attività-dipendente: è stato osservato che la paralisi indotta dalla somministrazione di BoNT/A avviene più efficacemente se il terminale nervoso viene stimolato ad alta frequenza. Abbiamo quindi voluto verificare se l’azione di BoNT/E fosse attività-dipendente nel Sistema Nervoso Centrale. Per far questo abbiamo valutato l’attività proteolitica di BoNT/E in una situazione di aumentata attività elettrica cerebrale, causata dalla somministrazione intraippocampale di KA, un agonista del glutammato, nel ratto adulto. I risultati preliminari ottenuti dimostrano una diminuizione paradossale dell’attività proteolitica di BoNT/E su SNAP-25 negli animali trattati con KA rispetto ai controlli.
Abbiamo infine esaminato il possibile effetto neuroprotettivo della BoNT/E in un modello di ischemia cerebrale focale nel ratto.
L’ischemia cerebrale è una delle patologie neurologiche più diffuse e si verifica quando viene interrotto il flusso sanguigno in una porzione dell’encefalo. L’ischemia causa la morte dei neuroni ipossici con formazione di un’area di necrosi (core ischemico) e di una penombra perilisionale in cui la degenerazione avviene, almeno in parte, con meccanismi attivi (apoptosi). La nostra ipotesi è che la degenerazione neuronale sia innescata dall’eccessivo rilascio di glutammato dai neuroni danneggiati.
Il modello di ischemia focale da noi utilizzato, si basa sulla microiniezione di un potente vasocostrittore, l’Endotelina-1 (ET-1), nella regione CA1 dell’ippocampo di ratti adulti. Gli animali hanno ricevuto dapprima ET-1 e circa 20 minuti dopo BoNT/E o la soluzione di controllo e sono stati perfusi con paraformaldeide dopo 24 ore o dopo un mese dall’iniezione.
L’analisi stereologica su sezioni istologiche trattate mediante colorazione di Nissl, ha rivelato una netta riduzione nell’ estensione del danno negli animali trattati con BoNT/E rispetto ai controlli.
Da questi risultati si può concludere che BoNT/E previene la morte neuronale in un modello di ischemia cerebrale focale.
Abstract
Botulinum neurotoxins (BoNTs) are endopeptidases produced by bacteria of the genus
Clostridium that exert a highly selective proteolytic action on a group of protein
involved in the the exocytotic process, the SNARE proteins. The specific cleavage inactivates SNAREs and causes the block of exocytosis. The action of BoNTs has been widely characterized at the level of the Peripheral Nervous System (PNS), where they inhibit achetilcoline release from cholinergic terminals at the neuromuscular junction. Much less is known on BoNT action in the Central Nervous System (CNS).
In this thesis we have characterized the effects of BoNTs in the CNS in vivo. In particular we have studied the action of serotype A and E (BoNT/A and BoNT/E) in the rodent hippocampus.
BoNT/A and BoNT/E cleave the same protein of SNARE complex, the Synaptosomal Associated Protein of 25 KDa (SNAP-25) that is preferentially localized in glutamatergic terminals. Indeed, in vitro studies show that they are able to inhibit glutamate release.
We have performed a unilateral and stereotaxic injection of either BoNT/E or BoNT/A into the dorsal hippocampus of adult mice and we have characterized their effects at the electrophysiological and biochemical level.
The in vivo recordings of action potentials in the hippocampus show that both serotypes are able to completely silence spontaneous hippocampal activity. However, this strong inhibition of hippocampal spontaneous discharges persists for different times: the BoNT/A silencing effect persists for at least 60 days, while BoNT/E effect persists for about 14 days. This electrophysiological data are in agreement with the expression of cleaved SNAP-25 as determined by Western blot analysis. Immunostaining for cleaved SNAP-25 show a similar localization of the two SNAP-25 forms cleaved by BoNT/A or by BoNT/E. Remarkably, BoNT/A cleaved SNAP-25, but not BoNT/E cleaved SNAP-25, is detectable in the contralateral hippocampus, especially in the CA3 region, starting from 3 days after neurotoxin injection. This spread of BoNT/A proteolytic activity to the contralateral hemisphere is demostrated by both immunostaining and Western blot experiments.
It is known that BoNT action is activity-dependent in the PNS: paralysis occurs more rapidly if the nerve is stimulated at high frequency. So we have investigated whether BoNT/E action is activity-dependent in the CNS.
To address this issue, we have evaluated BoNT/E proteolytic activity when synaptic activity is elevated. In order to increase synaptic activity we have injected the glutamate agonist Kainic Acid (KA) into the rat hippocampus. The preliminary results show a paradoxical reduction of the proteolytic activity in animals treated with KA as compared with controls.
Finally, we have examined the possible neuroprotective effect of BoNT/E in a model of rat focal cerebral ischemia.
Ischaemic stroke is one of the more diffuse neuropathological condition and results from a transient or permanent reduction in cerebral blood flow in a portion of the brain. Ischaemia causes neuronal death by necrosis in the ischaemic core. Between this lethally damaged core and the normal brain lies the penumbra, an area in which neuronal death resembles apoptosis. We probe the hypothesis that neuronal degeneration is caused by excessive glutamate release from ischaemic neurons.
In our model we have injected Endothelin-1 (ET-1, a powerful vasoconstrictor) into the CA1 region of the rat hippocampus to induce the ischaemic stroke.
All the animals received hippocampal injection of ET-1 and after 20 minutes they were injected with BoNT/E or with control solution and they were perfused the day after. Stereological analysis of tissue loss on Nissl-stained histological sections reveals a significant reduction of cerebral damage in the animal treated with BoNT/E. We conclude that BoNT /E prevents neuronal death in a model of focal cerebral ischaemia.
Introduzione
1.1 Le neurotossine botuliniche
"Les poisons peuvent etre employes soit comme moyens pour la destruction
de la vie, soit comme agents pour le traitment du malade"
(I veleni possono essere usati sia come mezzo per distruggere la vita sia come agenti per il trattamento delle malattie).
Claude Bernard (1875)
Molte migliaia di specie viventi producono tossine per modificare la fisiologia di altre specie in modo da aumentare le proprie possibilità di sopravvivenza.
Considerando che il sistema nervoso svolge un ruolo essenziale nella fisiologia animale, non sorprende che le tossine più conosciute siano quelle selettive per molecole del tessuto nervoso.
Tutte le neurotossine esistenti sono molto specifiche per il proprio bersaglio molecolare, sono infatti il risultato di una lunga co-evoluzione tra le specie che le producono e le specie target. Per questo, lo studio del loro meccanismo d’azione può rivelare alcuni aspetti fondamentali della fisiologia nervosa.
Le neurotossine botuliniche (BoNT) prodotte dal batterio Clostridium Botulinum bloccano la trasmissione sinaptica interferendo direttamente e selettivamente sul rilascio del neurotrasmettitore (Schiavo et al., 2000).
Sono infatti metalloproteasi che agiscono selettivamente su un gruppo di proteine di membrana implicate nel processo di rilascio del neurotrasmettitore alla terminazione presinaptica.
Queste tossine sono la causa di tutti i sintomi di una malattia neuroparalitica, nota come botulismo. Nel botulismo si ha una paralisi flaccida perchè le tossine inbiscono il rilascio di acetilcolina a livello della giunzione neuromuscolare.
Sono stati identificati sette sierotipi diversi di BoNT, denominati con le lettere da A a G. I sierotipi A, B ed E (rarissimamente F) sono coinvolti nel botulismo umano mentre i
sierotipi C e D sono associati quasi esclusivamente al botulismo animale. Le differenti neurotossine sono in genere prodotte da differenti ceppi di Cl. botulinum, ma recentemente sono stati identificati ceppi che contengono geni codificanti per due diverse tossine ed anche un ceppo che produce una tossina chimerica C-D.
La scoperta del blocco della giunzione neuromuscolare da parte delle BoNT e della loro elevata specificità d’azione, ha promosso un uso crescente delle tossine come farmaci nel trattamento di patologie caratterizzate da iperattività della giunzione neuromuscolare.
1.1.1 Struttura delle BoNT
Per capire il meccanismo d’azione delle BoNT è necessario conoscere le loro principali caratteristiche strutturali.
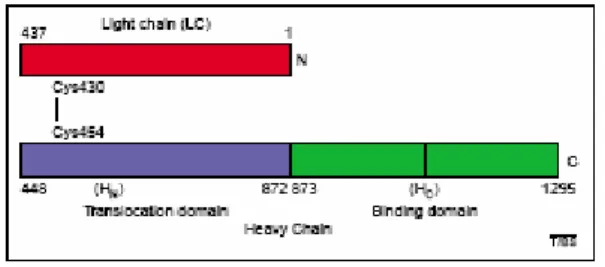
Le tossine botuliniche vengono sintetizzate nel citosol del batterio e rilasciate nel mezzo di coltura in seguito a lisi cellulare come singola catena polipeptidica di 150 kDa. La forma inattiva della proteina viene poi processata da diverse proteinasi batteriche o tissutali (Turton et al., 2002) che tagliano la tossina in un singolo punto trasformandola nella forma bicatenaria biologicamente attiva. Essa è composta di una catena pesante (H, 100 kDa) che consiste di due domini, ciascuno di 50 kDa e di una catena leggera (L, 50 kDa). Le due catene sono tenute assieme da un singolo ponte disolfuro intercatena, da un segmento della catena H che gira attorno alla catena L e da forze non covalenti (Fig. 1 a). L’ integrità del ponte disolfuro è essenziale per la neurotossicità (Schiavo et al., 1990).
Recentemente è stata determinata la struttura terziaria tridimensionale della BoNT/A (Fig. 1 b) (Lacy et al., 1998).
Si possono riconoscere tre distinti domini funzionali ciascuno di 50 kDa.
-un dominio contenente l’attività endopeptidasica Zinco-dipendente nella catena L; -un dominio di traslocazione nella metà N-terminale della catena H;
-il dominio di legame nella metà C-terminale della catena H;
Questi tre domini funzionali sono strutturalmente distinti e orientati in modo che tra di loro non ci sia contatto.
•
Fig. 1 a: Struttura di una neurotossina botulinica (BoNT/A). La catena pesante e la catena leggera sono
legati da un singolo legame disulfidico Cys430-Cys454. La catena leggera mostrata in rosso, funziona come Zinco-endopeptidasi. La catena pesante comprende due domini funzionali di 50 kDa: HN, mostrato in blu, è il dominio di traslocazione; HC, mostrato in verde, è invece il dominio di legame (immagine tratta da Turton et al., 2002).
Fig. 1 b: Struttura terziaria di BoNT/A. I tre domini funzionali sono distinti strutturalmente e orientati in
modo che tra loro non ci siano contatti. Il HC di legame ha una struttura a 3 foglietti β ripiegati; il dominio HN di traslocazione è composto da due lunghe eliche e da un loop che occlude il sito attivo del dominio catalitico; il dominio catalitico alloggia l’atomo di Zinco e possiede il motivo conservato HExxH caratteristico delle endopeptidasi Zinco-dipendenti (immagine tratta da Turton et al., 2002).
1.1.2 Funzione delle BoNT
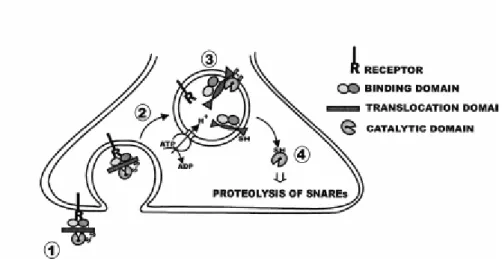
Il meccanismo di intossicazione neuronale delle BoNT è strettamente correlato con l’organizzazione strutturale in tre domini distinti e consiste di quattro stadi (Fig. 2):
1. legame alla membrana presinaptica delle cellule neuronali, mediato dal dominio C-terminale della catena H (HC);
2. entrata nelle cellule all’interno di vescicole;
3. traslocazione attraverso la membrana vescicolare mediata dal dominio N-terminale della catena H (HN);
4. blocco della neuroesocitosi, dovuto all’attività enzimatica della catena L.
Fig. 2: Meccanismo di intossicazione neuronale delle BoNT : 1)legame a un recettore della membrana
presinaptica mediato dal dominio HC; 2)entrata nelle cellule all’interno di vescicole; 3)traslocazione attraverso la membrana vescicolare mediata dal dominio HN; 4)blocco della neuroesocitosi, dovuto all’attività enzimatica della catena L (immagine tratta da Schiavo et al., 2000).
1.1.2.1 Legame alla membrana di cellule target
Dal sito di produzione o di assorbimento le BoNT diffondono nei fluidi del corpo e raggiungono la membrana presinaptica dei neuroni colinergici.
Sebbene risulti che la metà carbossi-terminale della catena H abbia un ruolo preponderante nel legame neurospecifico (Shone et al., 1985), pare che altre regioni delle BoNT siano implicate nel legame. Infatti un’immunizzazione con il frammento HC
mostra solo una protezione parziale dall’intossicazione con BoNT intatta (Poulain et al., 1991).
Un gran numero di studi ha stabilito che l’estrema specificità delle BoNT per le cellule nervose è in parte dovuta alla loro capacità di riconoscere specifici polisialogangliosidi, largamente distribuiti nel tessuto nervoso (Kitamura et al., 1980; Montecucco et al., 1988). I gangliosidi sono glicosfingolipidi sialiati implicati nello sviluppo, nella funzione e nel mantenimento del sistema nervoso e sono abbondantemente presenti nella membrana presinaptica (Ledeen et al., 1978; Simons and Ikonen, 1997; Vyas et al., 2002.). Dal momento che questi gangliosidi non sono le uniche molecole presenti nella membrana del terminale nervoso, potrebbero non essere i soli responsabili della neurospecificità del legame presinaptico delle BoNT. È stato infatti dimostrato che sono coinvolte anche alcune proteine delle vescicole sinaptiche.
Per questo motivo è stato proposto un modello a ‘doppio recettore’ secondo il quale le BoNT legano fortemente e specificatamente la membrana presinaptica perché sono in grado di interagire contemporaneamente con entrambi i tipi di recettori, gangliosidici e proteici (Schiavo e Montecucco, 2004). In particolare, i polisialogangliosidi determinerebbero l’accumulo iniziale delle BoNT sul piano della membrana plasmatica ed in seguito, le neurotossine riconoscerebbero i propri recettori proteici. Tale modello è sostenuto dal fatto che nel dominio HC sono presenti un sottodominio di legame per gli zuccheri e un sottodominio di legame per le proteine (Umland et al., 1997; Lacy et al., 1998).
L’identità del recettore proteico è stata riconosciuta soltanto per alcuni serotipi. BoNT/B e BoNT/G interagiscono con il dominio luminale della proteina vescicolare Sinaptotagmina Ι e ΙΙ (Syt-Ι, Syt-ΙΙ). BoNT/B ha un’affinità maggiore per Syt-ΙΙ mentre BoNT/G interagisce preferenzialmente con Syt-Ι (Nishiki et al., 1994; Dong et al., 2003).
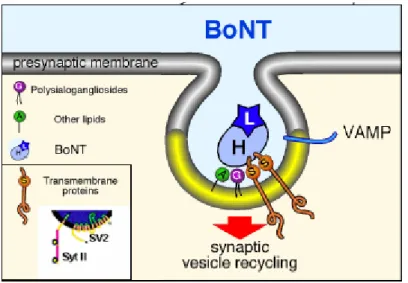
Molto recentemente è stato dimostrato che si verifica un’interazione molto specifica tra BoNT/A e il dominio luminale di SV2 (Synaptic Vesicle protein 2; Dong et al., 2006). SV2 è altamente glicosilato e possiede dodici putativi domini transmembrana (Fig. 3). La fuzione di SV2 nel tessuto nervoso non è ancora stata definita ma è noto che nelle cellule endocrine esso è implicato nel meccanismo di recupero della membrana vescicolare (Iezzi et al., 2005).
Fig. 3: Rappresentazione schematica del recettore SV2. Si pensa che SV2 possieda 12 regioni transmembrana, estremità N-terminale e C-terminale esposte al citosol e un grosso dominio intravescicolare, critico per il legame di BoNT/A (immagine tratta da Dong et al., 2006).
Nei vertebrati esistono tre geni SV2 distinti che codificano per proteine altamente omologhe, chiamate SV2A, SV2B ed SV2C (Bajjalieh et al., 1992 e 1993).
SV2A è presente in quasi tutti i neuroni indipendentemente da tipo di neurotrasmettitore, mentre SV2B non è presente nei neuroni GABAergici del cervelletto, del globus pallidus e dei nuclei reticolari del talamo (Bajjalieh et al., 1994; Janz and Sudhof, 1999). SV2C è stato identificato solamente in alcuni neuroni del cervello anteriore e non è rilevabile nell’ippocampo tranne che in una sottopopolazione di neuroni inibitori (Verderio et al., dati non pubblicati).
BoNT/A è in grado di interagire con tutti e tre i tipi di recettore ma mostra un’affinità maggiore con il loop luminale L4 di SV2C (Dong et al., 2006). Nonostante questo, SV2A ed SV2B sono comunque responsabili del legame di BoNT/A a neuroni ippocampali in coltura e nella giunzione neuromuscolare (Dong et al., 2006). In contrasto a questo risultato Mahrhold et al. (2006) non riscontrano un significativo contributo di SV2A ed SV2B all’intossicazione da parte di BoNT/A dei terminali dei motoneuroni.
Lo studio dei vari recettori per le neurotossine rappresenta un importante campo di indagine, ancora aperto, in quanto l’identificazione dei recettori delle diverse BoNT rappresenterebbe non soltanto un progresso per le neuroscienze, ma anche un notevole contributo per il miglioramento dei protocolli terapeutici basati sulle BoNT.
1.1.2.2 Internalizzazione nei neuroni
Tutti i dati disponibili riguardo il meccanismo di internalizzazione nel terminale presinaptico indicano che le BoNT non entrano nella cellula direttamente attraverso la membrana plasmatica, ma vengono endocitate all’interno di compartmenti cellulari acidi. Studi con il microscopio elettronico hanno mostrato che, dopo il legame alla membrana, le BoNT entrano nel lume di strutture vescicolari attraverso un processo temperatura ed energia dipendente (Critchley et al., 1985; Black and Dolly, 1986; Matteoli et al., 1996).
Esperimenti effettuati su neuroni del midollo spinale di topo (Lalli et al., 1999) hanno stabilito che il dominio HC di BoNT/A, /B, e /E, è sufficiente per il processo di internalizzazione.
È noto che a livello della giunzione neuromuscolare, l’attività delle neurotossine botuliniche è influenzata dalla stimolazione nervosa (Simpson et al., 1980) in quanto la paralisi neuromuscolare indotta dalla somministrazione delle BoNT avviene molto più rapidamente se il terminale nervoso viene stimolato ad alta frequenza. L’aumento dell’internalizzazione delle BoNT conseguente all’aumento dell’attività sinaptica suggerisce che le strutture vescicolari responsabili dell’entrata delle neurotossine potrebbero essere le vescicole sinaptiche SSV (small synaptic vesicle) implicate nel rilascio del neurotrasmettitore.
A livello del Sistema Nervoso Centrale, esperimenti svolti su neuroni ippocampali in coltura, mostrano che le BoNT entrano nei neuroni centrali attraverso un processo endocitotico non mediato dalle SSV (Verderio et al., 1999). Verderio e collaboratori hanno intossicato neuroni ippocampali e astrociti in coltura con BoNT/B e con BoNT/F e ne hanno esaminato il meccanismo di entrata e l’attività proteolitica. Se i neuroni venivano incubati con le due tossine dopo depolarizzazione, non si rivelava nessuna differenza nel taglio del target. Questo risultato ha portato gli autori alla conclusione che l’internalizzazione delle BoNT non è un meccanismo attività-dipendente.
Al contrario, Dong e collaboratori, (2006) utilizzando come modello gli stessi neuroni ippocampali in coltura, mostrano che BoNT/A viene internalizzata in maniera attività-dipendente insieme ad un anticorpo che riconosce il dominio luminale della proteina vescicolare sinaptotagmina. Questi risultati e la scoperta che alcuni serotipi legano la porzione luminale di proteine vescicolari indicano che le neurotossine botuliniche
utilizzano le vescicole sinaptiche come “cavallo di Troia” per entrare nel terminale nervoso (Fig. 4).
Come discuterò in seguito, dati recenti mostrano promettenti effetti benefici delle BoNT sul Sistema Nervoso Centrale. Per questo motivo, il meccanismo e la cinetica di entrata delle tossine nei neuroni del Sistema Nervoso Centrale richiede una approfondita indagine.
Fig. 4:. Rappresentazione schematica del meccanismo di entrata delle BoNT nella terminazione
presinaptica. Le BoNT si legano alla membrana presinaptica attraverso l’interazione con gangliosidi e con il dominio luminare di proteine vescicolari. In seguito al processo di riciclaggio delle SSV, le BoNT raggiungono il citosol (Immagine tratta da Verderio et al., 2006).
1.1.2.3 Traslocazione attraverso la membrana vescicolare.
Il dominio con attività endopeptidasica (catena L) deve raggiungere il citosol perché è qui che esplica la sua attività catalitica.
Per far ciò la catena L deve attraversare la barriera idrofobica della membrana vescicolare e l’ambiente acido all’interno della vescicola favorisce la traslocazione transmembrana. A pH acido le BoNT subiscono un cambiamento conformazionale che le fa passare da una struttura “neutra”, solubile in acqua, a una struttura “acida”, caratterizzata dall’esposizione superficiale di regioni idrofobiche. Queste zone di idrofobicità favoriscono l’inserzione della tossina all’interno del bilayer lipidico
(Bouquet e Duflot, 1982; Cabiaux et al., 1985; Roa e Boquet, 1985; Montecucco et al., 1986; Menestrina et al., 1989; Schiavo et al., 1991). In seguito all’inserzione nella membrana, le BoNT formano canali ionici selettivi per alcuni cationi e per molecole più piccole di 700 Da (Bouqet e Duflot, 1982; Hoch et al., 1985; Blaustein et al., 1987; Shone et al., 1987; Gambale e Montal, 1988; Menestrina et al., 1989; Rauch et al., 1990; Schmid et al., 1993; Fisher e Montal; 2006).
Molte evidenze indicano che questi canali sono formati dall’oligomerizzazione del dominio HN (Donovan et al., 1986; Menestrina et al.,1989; Shmid et al., 1993; Shone et al., 1987).
Molto probabilmente la formazione di questi canali è correlata al meccanismo di traslocazione della catena L verso il citosol cellulare. Come questo processo avvenga, non è ancora chiaro ma si suppone che la catena L non si ripieghi a bassi pH e che permei attraverso il poro transmembrana formato dalle catene H. Inoltre, secondo questa ipotesi (“tunnel” model), il ponte disolfuro viene ridotto cosicchè la catena viene rilasciata nel citosol cellulare dove il pH neutro le fa assumere la conformazione necessaria per l’attività catalitica (Bouqet e Duflot, 1982).
Un secondo modello (avanzato da Beise et al., 1994) prevede che il canale ionico formato dalle neurotossine alteri il gradiente elettrochimico creato dalla pompa protonica ATP-ase di tipo vacuolare.
Questi cambiamenti di permeabilità causerebbero la lisi osmotica della vescicola e la catena L si ritroverebbe così nel citosol.
1.1.2.4 Attività endopeptidasica Zinco-dipendente
L’attività catalitica delle tossine fu scoperta in seguito al sequenziamento del gene corrispondente (Mintone et al., 1995). In seguito ad un’analisi comparativa delle sequenze amminoacidiche delle neurotossine botuliniche, è stato notato che nelle catene L era assolutamente conservata la breve sequenza Istidina-Glutammico-Xaa-Xaa-Istidina tipica delle Zinco-endopeptidasi, dove essa media il legame dello Zinco alla proteina (Montecucco, 1986).
Le endopeptidasi Zinco-dipendenti costituiscono una grande famiglia di proteasi di enorme interesse dato che enzimi di questo tipo controllano la pressione arteriosa, sono
coinvolte nella risposta infiammatoria, nei processi di riparo tissutale, nei processi d'invasione e metastasi dei tumori maligni e nella morfogenesi durante l’embriogenesi. Proprio a causa dei loro importanti e molteplici ruoli, di queste proteasi si conosce molto: dalla struttura tridimensionale di alcune di esse, al meccanismo di scissione del legame peptidico, in cui gioca un ruolo fondamentale lo Zinco. Seguendo questa osservazione iniziale, Montecucco e collaboratori hanno misurato il contenuto in metalli pesanti di preparazioni altamente purificate di neurotossine botuliniche ed hanno trovato che tutte contengono un atomo di Zinco, per molecola di tossina, legato alla catena L attraverso le due istidine del segmento centrale conservato. E’ stata quindi avanzata l’ipotesi che, se le neurotossine botuliniche sono delle metallo-proteasi che bloccano il rilascio del neurotrasmettitore contenuto nelle vescicole sinaptiche, allora è probabile che esse taglino una proteina della membrana delle vescicole stesse oppure una proteina della faccia citosolica della membrana presinaptica coinvolta nel legame e/o nella fusione della vescicola con la membrana. Quest’ipotesi si è rivelata fruttuosa perché ha permesso di identificare il bersaglio dell’attività proteolitica di queste neurotossine. Le BoNT effettuano un taglio proteolitico altamente specifico sulle proteine di un complesso detto SNARE (soluble NSF (Nethylmaleimide-sensitive fusion) attachment
protein receptor).
L’interazione tra proteine integrali della membrana vescicolare (v-SNARE) e recettori proteici presenti nella membrana target (t-SNARE) porta la membrana della vescicola, contenente il neurotrasmettitore, in stretto contatto con la membrana plasmatica, in una situazione predisponente alla fusione (Rizo e Sudhof, 1998) delle membrane. La fusione viene poi indotta nell’istante in cui gli ioni Ca2+ entrano nel citosol attraverso i canali al Ca2+ voltaggio-dipendenti
.
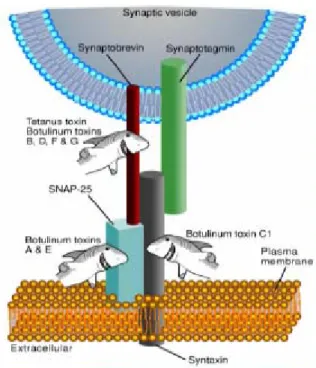
Nel tessuto cerebrale sono state identificate due t-SNARE: la sintaxina, proteina integrale di membrana e la SNAP-25 (proteina associata ai sinaptosomi dal peso di 25 kDa), proteina di membrana periferica. La proteina integrale di membrana vescicolare VAMP (o sinaptobrevina) è stata invece classificata come v-SNARE. La sintaxina, SNAP-25 e VAMP formano un complesso ternario estremamente stabile (Fig. 5). Per un efficiente meccanismo di eso ed endocitosi delle viscicole sinaptiche il complesso deve essere disassemblato. Il legame di una proteina citoplasmatica solubile, la NSF permette la scomposizione del trimero.
Fig. 5: Complesso SNARE formato da SNAP-25, VAMP e sintaxina
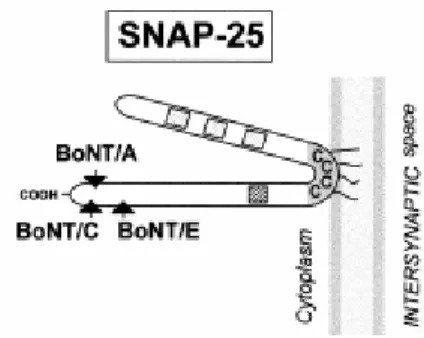
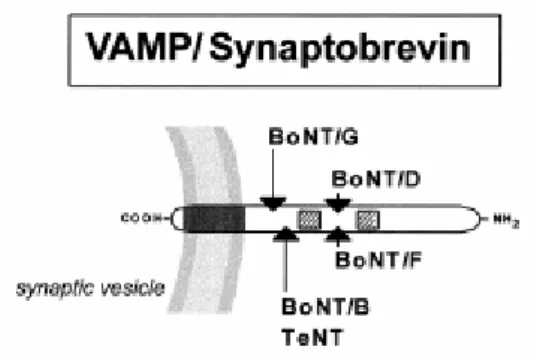
I vari sierotipi delle neurotossine botuliniche tagliano diverse proteine SNARE o clivano la stessa proteina in punti diversi. I sierotipi B, D, F e G tagliano VAMP in differenti siti, i sierotipi A ed E tagliano la SNAP-25 ed il sierotipo C taglia sia la SNAP-25 che la sintaxina (Schiavo et al., 1992 ; Schiavo et al., 1993; Schiavo et al., 1994).
La VAMP/sinaptobrevina viene tagliata in un solo punto, diverso per ciascuna delle quattro neurotossine botuliniche VAMP specifiche: in ogni caso viene rimossa una gran parte della porzione citosolica della proteina e questo previene la formazione del complesso trimerico. Lo stesso avviene nel caso della sintaxina. Diverso è il caso di SNAP-25, da cui la neurotossina botulinica A rimuove un piccolo segmento carbossi-terminale di nove amminoacidi. Quindi, l'eliminazione di meno del cinque per cento della massa totale di SNAP-25 è sufficiente a bloccarne la funzione, indicando che questa porzione della molecola gioca un ruolo fondamentale nel funzionamento dell'apparato di neuroesocitosi. Questi risultati contribuiscono a spiegare anche la tremenda potenza di queste tossine. Infatti, se la loro neurospecificità contribuisce in
modo rilevante a determinarne la potenza, si deve anche considerare che una sola molecola di catena L può tagliare, una dopo l'altra, tutte le molecole bersaglio, VAMP/sinaptobrevina o SNAP-25 o sintaxina, presenti nelterminale nervoso.
VAMP, SNAP-25 e sintaxina ricombinanti vengono tagliati nello stesso modo delle corrispondenti proteine cellulari, indicando che non sono necessari fattori endogeni aggiuntivi per l’attività proteolitica delle BoNT.
A livello di struttura primaria e secondaria le neurotossine sono molto simili ed è noto che nè i variabili siti di taglio né le regioni fiancheggianti rappresentano la specificità per le tre proteine SNARE.
Queste considerazioni suggeriscono che le SNARE potrebbero avere un comune elemento strutturale che funzioni da motivo di riconoscimento. Il confronto tra sequenze amminoacidiche di proteine SNARE neuroesocitosi-specifiche di specie diverse ha rivelato la presenza di un motivo di nove residui, caratterizzato dalla presenza di tre residui carbossilati alternati a residui idrofobici e idrofilici, chiamato poi “motivo SNARE”(Rossetto et al., 1994).
Molte evidenze sperimentali hanno poi dimostrato l’importanza di questo motivo per la specificità delle BoNT verso le tre proteine SNARE (Foran et al., 1994; Shone et al., 1993).
Per la selettività e per la resistenza di legame della tossina è comunque necessaria un’ulteriore interazione con regioni specifiche di ciascuna SNARE.
La regione delle BoNT implicata nel riconoscimento del substrato non è ancora stata identificata. È stato proposto che la sequenza conservata di 100 residui ammino-terminale sia in qualche modo coinvolta. Infatti la rimozione di più di otto di questi residui porta alla completa perdita di attività.
Sono comunque necessarie ulteriori investigazioni per stabilire il ruolo di questo dominio.
1.1.3 Target delle neurotossine botuliniche.
1.1.3.1 Sintaxina
La sintaxina di tipo ΙΙ è una proteina di membrana dal peso di 35 kDa localizzata nel plasmalemma neuronale (Fig. 6). Ha una porzione ammino-terminale esposta verso il citosol, un dominio transmembrana e alcuni residui extracellulari (Bennet et al., 1992). Sulla sintaxina, bersaglio di BoNT/C, ci sono due copie del motivo SNARE, X1 e X2. Le sintaxine appartengono ad una grande famiglia di proteine con più di 20 isoforme nei mammiferi e con omologhi nel livito e nelle piante (Bennet et al., 1993). Nel tessuto nervoso ne esiste un elevato polimorfismo.
Nelle zone attive del terminale sinaptico la sintaxina è associata con vari tipi di canali per il Ca2+ (Bezprozvanny et al.,1995) edinteragisce con le proteine Sec-1/ Munc-18 (proteine SM), proteine implicate negli eventi intracellulari di fusione delle membrane. Pare che tale interazione sia responsabile dei meccanismi di attacco e di fusione vescicolare mediati dal complesso SNARE (Südhof et al., 1995).
Durante il potenziamento a lungo termine alcune isoforme subiscono un complesso pattern di splicing alternativo. Quest’osservazione suggerisce un probabile coinvolgimento della sintaxina nei meccanismi di plasticità sinaptica (Hicks et al., 1997).
Fig. 6: Struttura schematica della sintaxina. La sintaxina è inserita nella membrana plasmatica ed è
per la maggior parte esposta nel citosol. La freccia indica il sito di taglio di BoNT/C mentre i due piccoli quadrati grigi rappresentano i motivi X1 e X2.
1.1.3.2 SNAP-25
SNAP-25 è una proteina di membrana periferica presente nel plasmalemma neuronale. Data l’assenza di un dominio transmembrana, si pensa che la sua localizzazione sia
mediata dalla palmitolazione di alcuni residui di Cys presenti nel mezzo della catena polipeptidica (Hess et al., 1992). SNAP-25, che è conservata dal lievito all’uomo, è in grado di autoassemblarsi in un dimero legato da un ponte disolfuro sia in vivo che in
vitro (Sadoul et al., 1997; Fig. 7).
SNAP-25 ha un importante ruolo nella formazione del complesso SNARE in quanto in sua assenza la sinaptotagmina si lega molto debolmente alla sintaxina e tale associazione è notevolmente potenziata da SNAP-25 (Hayashi et al., 1994).
Questa evidenza può spiegarsi col fatto che SNAP-25 contiene molti siti di fosforilazione per la Protein Kinasi C (PKC), da molti anni riconosciuta come modulatore positivo del processo esocitotico.
Sembra che l’effetto di PKC sul rilascio del neurotrasmettitore sia mediato dalla fosforilazione di SNAP-25 sulla Ser187. Pare che tale fosforilazione sia in grado di modificare l’interazione tra le proteine SNARE. In particolare, SNAP-25 subisce un cambiamento conformazionale e si dissocia dalla sintaxina più velocemente. (Shimazaki et al., 1996). L’ accelerata cinetica di dissociazione del complesso potrebbe favorire l’efficienza della neuroesocitosi. Sono tuttavia necessari ulteriori lavori per definire con esattezza le conseguenze biochimiche della fosforilazione di Ser 187 sulla funzione di SNAP-25.
Esperimenti svolti su colture ippocampali organotipiche mostrano che la fosforilazione di SNAP-25 da parte della PKC è attività-dipendente (Genoud et al., 1999). Gli autori hanno utilizzato la bicucullina, un agonista del neurotrasmettitore inibitorio GABA per incrementare un’attività spontanea di tipo epilettico. In queste condizioni si verifica un’attivazione della PKC che determina modificazioni sul rilascio dei neurotrasmettitori eccitatori accompagnate da un aumento di SNAP-25 fosforilata.
Il fatto che la fosforilazione di SNAP-25 potrebbe aumentare in condizioni patofisiologiche nell’ippocampo, viene confermato dai risultati non ancora pubblicati di Matteoli e collaboratori. Da questi dati risulta che si verifica in vivo un aumento di SNAP-25 fosforilata sul residuo Ser187 nell’ippocampo di topi trattati con una somministrazione sistemica di acido Kainico (KA). Il KA è un agonista del glutammato che determina un aumento dell’attività sinaptica e viene utilizzato per indurre sperimentalmente le crisi epilettiche. L’aumentata presenza di SNAP-25 fosforilata in
animali trattati con il KA porta gli autori alla conclusione che la fosforilazione della Ser187 è un evento attività-dipendente.
SNAP-25 forma un complesso con il sensore per il Ca2+ sinaptotagmina e questa interazione sembra essere importante per la fase Ca2+-dipendente del rilascio del neurotrasmettitore (Banerjee et al., 1996). Infatti, i recenti risultati ottenuti da Verderio e collaboratori (2004) mostrano che SNAP-25 è cruciale per la dinamica intracellulare del Ca2+ e suggeriscono che l’interazione SNAP-25/sinaptotagmina potrebbe essere coinvolta in una regolazione negativa di tale dinamica. Dal momento che la sinaptotagmina interagisce con i canali per il Ca2+ (Sheng et al., 1997; Charvin et al., 1997), il meccanismo di regolazione potrebbe avvenire attraverso una modulazione dell’attività dei canali oppure attraverso il diretto legame del complesso allo ione Ca2+(Verderio et al., 2004).
SNAP-25 è quindi una proteina multifunzionale che partecipa al processo endocitotico sia a livello di meccanica (si lega alle altre proteine del complesso SNARE) sia a livello di regolazione.
Oltre a queste proprietà, SNAP-25 è richiesta per la crescita assonale durante lo sviluppo neuronale ed è coinvolta nella plasticità dei terminali nervosi nel sistema nervoso maturo (Osen-Sand et al., 1993).
Probabilmente entrambe le isoforme di SNAP-25, indicate con le lettere A e B, sono coinvolte nei meccanismi di plasticità sinaptica. Risulta infatti la loro sintesi è sovraregolata durante il potenziamento a lungo termine in neuroni ippocampali (Roberts et al., 1998).
Su SNAP-25 sono presenti 4 copie del motivo SNARE S1, S2, S3 ed S4, per il riconoscimento da parte di BoNT/A e di BoNT/E.
Fig. 7: Struttura schematica della SNAP-25. La freccie indicano i siti di taglio di BoNT/A e
BoNT/E mentre i quattro piccoli quadrati irappresentano i motivi S1, S2, S3 e S4 .
1.1.3.3 VAMP/sinaptobrevina
VAMP è una proteina di 13 kDa localizzata nelle vescicole sinaptiche. È ancorata alla membrana vescicolare attraverso un dominio transmembrana e dal lato extravescicolare espone un dominio ben conservato che è sito di fosforilazione da parte della Ca2+/calmodulina chinasi di tipo ΙΙ (Fig. 8).
Come precedentemente affermato, VAMP è il bersaglio molecolare di BoNT/B, /D, /F, /G, e possiede due copie del motivo SNARE,V1 e V2.
Sulla membrana vescicolare VAMP è associata con la sinaptofisina, proteina di membrana, e con la v-ATPase (Calakos et al., 1994). La sua funzione è fondamentale per il meccanismo di rilascio del neurotrasmettitore.
Fig. 8: Struttura schematica di VAMP. VAMP ha una piccola coda C-terminale all’interno del lume
vescicolare, un dominio transmembrana e un segmento citosolico di 60 residui, la cui terminazione N-terminale è ricca di residui di prolina. La freccie indicano i siti di taglio di BoNT/D, /B, /G e /F mentre i quattro piccoli quadrati irappresentano i motivi V1 e V2 (Fig 6, 7, 8: immagini tratte da Schiavo et al., 2000).
1.1.4 La giunzione neuromuscolare
L’azione delle tossine è stata ampiamente caratterizzata a livello del Sistema Nervoso Periferico.
In particolare, a livello di placca motrice bloccano il rilascio di acetilcolina causando una profonda, seppur transitoria, paralisi neuromuscolare (Fig. 9).
Il primo studio elettrofisiologico sugli effetti delle BoNT sulla giunzione neuromuscolare è stato condotto da Burgen (Burgen et al., 1949) su preparazioni di emidiaframma di ratto.
Le conseguenze della somministrazione delle BoNT, osservate in questo lavoro, possono essere così elencate:
-forte e persistente blocco dell’EPP (potenziale di placca) dovuto all’interruzione della trasmissione sinaptica nei terminali intossicati;
-riduzione della frequenza ma non dell’ampiezza dell’mEPP (potenziale di placca in miniatura);
- la sintesi, la ricaptazione e l’immagazzinamento del neurotrasmettitore, non vengono danneggiati;
-si mantiene un’attività sinaptica spontanea residua ma non sufficiente per far si che l’EPP raggiunga l’appropriato livello di potenziale di membrana per l’instaurarsi del potenziale d’azione sulla fibra muscolare.
L’iniezione di BoNT nel muscolo striato di mammifero causa severi cambiamenti istologici (Borodic et al., 1994). Il primo segnale patologico è l’accumulo delle vescicole sinaptiche nel lato citosolico della membrana plasmatica (Neale et al., 1989, 1999). Come discusso precedentemente, questa è la conseguenza immediata del taglio proteolitico: le vescicole non sono più in grado di fondersi alla membrana plasmatica per il rilascio del neurotrasmettitore.
Il contatto anatomico tra il nervo e il muscolo viene mantenuto e non si verifica perdita di assoni motori.
Nel muscolo le BoNT determinano alterazioni simili a quelle documentate per i fenomeni di denervazione come un’atrofia della fibra muscolare. Le acetilcolina esterasi e i recettori per l’acetilcolina diffondono dalla giunzione ad altre zone della membrana muscolare.
In risposta alla paralisi indotta da BoNT i terminali dei motoneuroni sono in grado di produrre nuovi processi che formano nuove sinapsi immature (sprouting assonale). Analisi immunoistochimiche sulla giunzione neuromuscolare dopo un mese dal trattamento con BoNT/A hanno rlevato un intenso sprouting dal terminale nervoso paralizzato. Questi nuovi processi possiedono molte proteine chiave per l’esocitosi
come SNAP-25, VAMP, sintaxina, sinaptotagmina, sinaptofisina e canali per il Na+, per il Ca2+ e per il K+ (Angaut-Petit et al., 1990).
Durante il primo mese dopo l’iniezione di BoNT/A si verifica un cambiamento dei canali per il Ca2+ voltaggio-dipendenti accoppiati al rilascio del neurotrasmettitore. Nei terminali nervosi non trattati di topo adulto sono normalmente presenti i canali per il Ca2+ di tipo P/Q. Dopo l’iniezione di BoNT/A oltre a questi canali si ritrovano anche quelli di tipo L e N, funzionalmente associati al rilascio del neurotrasmettitore.
Tutte queste scoperte indicano che i nuovi processi originatisi dal terminale nervoso paralizzato possiedono il macchinario molecolare per il rilascio di acetilcolina. Pare quindi che lo sprouting abbia il ruolo di rimpiazzare i terminali parentali durante il recupero della trasmissione neuromuscolare.
Il recente sviluppo di coloranti fluorescenti che permettono la quantificazione dell’attività sinaptica in vivo in terminali nervosi è stato fondamentale per dimostrare il ruolo dello sprouting per il recupero funzionale dalla paralisi (de Paiva et al., 1999). Nei terminali dei motoneuroni trattati con il colorante fluorescente FM1-43, dopo simolazione, si verifica un accumulo del marker nelle vescicole sinaptiche, cosa che non si verifica nelle preparazioni trattate con BoNT/A.
Ventotto giorni dopo l’iniezione di BoNT/A soltanto i nuovi processi formatesi dai terminali che subiscono l’azione della tossina mostrano un accumulo attività-dipendente del colorante all’interno delle vescicole sinaptiche.
Nelle successive due settimane continuano a formarsi nuovi processi e solo dopo quarantadue giorni comincia a diminuire la marcatura per FM1-43. Contemporaneamente nei terminali originari si ha un aumento della marcatura che cresce sempre di più durante le successive quattro settimane.
Tre mesi dopo la somministrazione di BoNT/A, la placca motrice originaria riacquista la tipica morfologia e il pattern di marcatura attività-dipendente per FM1-43 ritorna identico a quello visualizzato prima del trattamento con la tossina.
Questa tecnica ha permesso di dimostrare che durante la paralisi indotta da BoNT/A i nuovi processi sono in grado di dar luogo ad appropriati processi di eso-endocitosi in
vivo, probabilmente associati al rilascio di acetilcolina. Non è tuttavia ancora chiaro se
questo rilascio dia luogo a potenziali di placca postsinaptici e possa quindi contribuire ad un recupero della funzionalità della giunzione neuromuscolare.
In seguito allo sprouting assonale e alla riformazione di una giunzione neuromuscolare funzionale, il muscolo torna alla forma originaria. Successivamente la placca motrice riacquista tutte le sue funzioni e le acetilcolinesterasi e i recettori per l’acetilcolina tornano ad essere concentrati esclusivamente a livello della giunzione neuromuscolare (Borodic et al., 1994; De Paiva et al., 1999).
Gli aspetti molecolari di questo fenomeno complesso non sono ancora conosciuti. In generale, la lunghezza del periodo di tempo necessario per il recupero della funzionalità dopo la paralisi indotta da BoNT, dipende soprattutto dal tipo di BoNT e dal tipo di terminale nervoso.
Il fatto che l’atrofia muscolare indotta dalle BoNT in modelli animali e nell’uomo sia reversibile, anche dopo ripetute iniezioni di neurotossina, risulta importante per le applicazioni terapeutiche delle BoNT.
Fig. 9: Azione delle tossine nella giunzione neuromuscolare. L’assone del motoneurone innerva una zona
specializzata della membrana muscolare che è detta placca motrice. In assenza di BoNT l’acetilcolina (Ach) liberata dalle vescicole sinaptiche si lega al proprio recettore presente in particolari strutture della membrana postsinaptica, dette pieghe giunzionali. In seguito a questo legame la membrana della placca motrice si depolarizza rapidamente creando il cosiddetto potenziale di placca. Tale potenziale sinaptico è sufficientemente ampio per attivare i canali per il sodio presenti nelle pieghe giunzionali, dando origine al potenziale d’azione che si propaga per tutta la fibra neuromuscolare determinandone la contrazione. In presenza di BoNT, tutto questo non può verificarsi per mancanza del rilascio del neurotrasmettitore Ach. Si ha quindi paralisi della fibra muscolare. In quest’immaggine viene anche mostrato il passaggio della tossina prodotta dal batterio dallo stomaco all’intestino. BoNT può così raggiungere la circolazione sistemica per transcitosi dal lato apicale a quello basale delle cellule epiteliali intestinali. Per diffusione poi raggiunge il terminale presinaptico del motoneurone, viene internalizzata per endocitosi e, come descritto precedentemente, blocca il rilascio di Ach agendo sulle proteine SNARE.
1.1.5 Il Sistema Nervoso Centrale
1.1.5.1 Azione delle BoNT nel Sistema Nervoso Centrale in vitro
Sebbene nel botulismo le BoNT non raggiungano il Sistema Nervoso Centrale (SNC) in quantità significative, ci sono evidenze che le neurotossine sono in grado di intossicare i neuroni del SNC in vitro.
La prima dimostrazione che le BoNT possono inibire il rilascio del neurotrasmettitore nelle sinapsi centrali venne effettuata da Bigalke e collaboratori. Essi dimostrarono che BoNT/A inibiva il rilascio di acetilcolina da sinaptosomi preparati dal cervello di ratto adulto (Bigalke et al., 1981a).
Gli stessi autori descrissero anche l’effetto inibitorio di BoNT/A sul rilascio di noradrenalina da sinaptosomi dello striato e di glicina da sinaptosomi del modollo spinale (Bigalke et al., 1981b).
Anche altri serotipi sono in grado di interferire sul rilascio del neurotrasmettitore dai neuroni centrali: BoNT/B inibisce il rilascio di glutammato e noradrenalina (Ashton e Dolly, 1991; McMahon et al., 1992; Takei et al., 1998), BoNT/C blocca il rilascio dei neurotrasmettitori glicina (inibitorio) e glutammato (eccitatorio) (Williamson et al., 1999) e BoNT/F riduce la secrezione Ca2+-dipendente ed evocata da depolarizzazione di acetilcolina, noradrenalina e dopamina (Fassio et al., 1999).
Esperimenti eseguiti su sinaptosomi cerebrocorticali di ratto hanno dimostrato che BoNT/A e BoNT/E sono capaci di inibire il rilascio Ca2+-dipendente K+ evocato di vari neurotrasmettitori, tra i quali, glutammato, acetilcolina, noradrenalina e dopamina (Ashton and Dolly, 1988; Foran et al., 1996). Il rilascio di GABA da sinaptosomi cerebrali intossicati con BoNT/E diminuisce del 40% , quello di glutammato, invece, si riduce del 90%. Questa grande differenza si può spiegare considerando la localizzazione preferenziale del target di BoNT/A e BoNT/E, SNAP-25, nei terminali gutammatergici (Verderio et al., 2004). Esperimenti di immunoistochimica su sezioni istologiche di ippocampo di ratto adulto hanno dimostrato che SNAP-25 è largamente presente nei terminali gutammatergici dello strato oriens e radiatum, mentre non si ritrova nei terminali inibitori dei neuroni di tipo ΙΙΙ che sinaptano sulle cellule piramidali della CA1.
Dal momento che i terminali inibitori esprimono livelli, seppur bassi, di recettori per BoNT/A (Verderio et al., dati non pubblicati), la relativa resistenza dei terminali GABAergici a BoNT/A e BoNT/E potrebbe risultare da due diversi scenari:
-nell’esocitosi del neurotrasmettitore GABA potrebbe essere coinvolta una isoforma di SNAP-25 BoNT/A e BoNT/E resistente;
- nelle sinapsi inibitorie ci sono piccole quantità di SNAP-25 totalmente impegnate nel complesso SNARE e non disponibili all’azione delle tossine (Hayashi et al., 1994; Montecucco et al., 2005).
A livello elettrofisiologico, Bigalke e collaboratori furono i primi a dimostrare che BoNT/A blocca sia il potenziale postsinaptico eccitatorio sia quello inibitorio in neuroni del midollo spinale di topo in coltura (Bigalke et al., 1985).
Questi dati concordano con i successivi risultati ottenuti da Neale et al., (1999): non si verificano correnti sinaptiche né spontanee né K+ evocate in neuroni del midollo spinale trattati con BoNT/A.
In fettine di ippocacampo di ratto è stato dimostrato che BoNT/A, /C ed /E prevengono l’instaurarsi del potenziale postsinaptico eccitatorio (EPSP), sia spontaneo che evocato (Capogna et al., 1997) e registrazioni eseguite su neuroni ippocampali mostrano che BoNT/A è in grado di ridurre drammaticamente la frequenza della corrente postsinaptica eccitatoria (EPSC) (Sutton et al., 2004).
Infine, esperimenti eseguiti su colture primarie di neuroni mostrano che BoNT/A è in grado di inibire anche la corrente postsinaptica inibitoria (IPSC), sia spontanea che evocata (Trudeau et al., 1998).
Da tutti questi risultati si può concludere che i diversi serotipi della BoNT sono in grado di interferire sulla trasmissione sinaptica (sia eccitatoria che inibitoria) in neuroni centrali in vitro.
Sono stati valutati anche gli effetti delle BoNT sul differenziamento e sulla sopravvivenza dei neuroni centrali.
In neuroni ippocampali in coltura BoNT/A inibisce la crescita assonale e dendritica, blocca il meccanismo di riciclaggio vescicolare e previene la formazione delle sinapsi, mentre BoNT/B non ha nessun effetto sulla crescita assonale e sulla sinaptogenesi.
(Osen-Sand et al., 1996; Grosse et al., 1999). È stato anche documentato che BoNT/C, al contrario di BoNT/A, induce una rapida morte neuronale (Osen-Sand et al., 1996). Quest’effetto neurocitotossico di BoNT/C ma non di BoNT/A indica che la sintaxina, lo specifico substrato di BoNT/C, ha un ruolo fondamentale nella sopravvivenza cellulare.
1.1.5.2 Azione delle BoNT nel Sistema Nervoso Centrale in vivo.
Fino ad ora pochi studi hanno descritto gli effetti delle BoNT in vivo (Bozzi et al., 2006). I primi risultati sugli effetti di una somministrazione ippocampale delle BoNT derivano da esperimenti di microdialisi in vivo su ratti adulti.
Da uno studio recente risulta che la pre-somministrazione di BoNT/A, /B e /C riduce il rilascio basale e K+ evocato di serotonina ma pare che BoNT/B e /C siano più potenti di BoNT/A (Okada et al., 2001). Un successivo lavoro di Murakami et al. (2001) mostra che la pre-somministrazione di BoNT/C e /B nell’ippocampo riduce anche il rilascio di dopamina, risultato confermato da un lavoro del 2002 di Bergquist et al. in cui si dimostra l’effetto acuto di BoNT/C e di BoNT/A sul rilascio di dopamina, dopo iniezione in vivo delle neurotossine nello striato di ratti adulti.
Topi trattati con un’iniezione intracerebroventricolare di BoNT/A e /B riportano molti segni di disfunzioni colinergiche (Luvisetto et al., 2003) e a dosi sub-letali, le tossine inducono danni comportamentali come una riduzione della capacità di distinguere oggetti nuovi all’interno di un ambiente familiare (Luvisetto et al., 2004).
Un recente lavoro del gruppo con cui ho svolto il mio lavoro di tesi, (Costantin et al., 2005) ha analizzato gli effetti di BoNT/E dopo iniezione unilaterale nell’ippocampo di ratto. Esperimenti di Western Blot e di immunoistochimica per SNAP-25 tagliata mostrano che la proteina è rapidamente ed efficientemente proteolizzata in vivo dalla neurotossina.
Le conseguenze funzionali del taglio di SNAP-25 da parte di BoNT/E sono:
-drammatica riduzione del rilascio Ca2+-dipendente/K+-evocato del glutammato osservato su sinaptosomi ippocampali in superfusione;
-silenziamento dell’attività di scarica spontanea dei neuroni ippocampali dimostrato con registrazioni elettrofisiologiche di potenziali d’azione nelle regioni CA1 e in CA3 dell’ippocampo (Costantin et al., 2005).
Attraverso il Morris water maze, test comportamentale ippocampo-dipendente, è stata dimostrata la reversibilità degli effetti di BoNT/E. Durante il periodo d’azione di BoNT/E (1-7 giorni) gli animali trattati mostrano profondi deficit di apprendimento spaziale. Tuttavia dopo circa cinque settimane, quando l’azione della tossina è ormai completamente teminata, gli stessi animali esibiscono un apprendimanto normale nel Morris water maze (Costantin et al., 2005). Si può quindi concludere che l’azione della neurotossina è reversibile e che non causa deficit permanenti nel comportamento ippocampo-dipendente.
Sulla base di queste scoperte si può affermare che BoNT/E rappresenta un nuovo strumento per silenziare transitoriamente l’attività neurale del SNC. Come discuterò in seguito, quest’affermazione suggerisce un possibile utilizzo terapeutico della neurotossina per quelle condizioni patologiche caratterizzate da iperattività cerebrale, come l’epilessia.
1.1.5.3 Durata d’azione delle neurotossine.
La durata dell’effetto neuroparalitico nelle sinapsi centrali è profondamante diversa a seconda del serotipo di BoNT.
Esperimenti svolti su neuroni cerebellari in coltura mostrano che l’intossicazione da parte di BoNT/E e /F termina prima rispetto a quella da parte di BoNT/A e /C (Foran et al., 2003). Questi risultati concordano con quelli ottenuti precedentemente da Keller et al., (1999) in cui si dimostra la diversa durata d’azione di BoNT/E e BoNT/A. Neuroni del midollo spinale vengono intossicati con le due tossine e viene monitorata la presenza di SNAP-25 tagliata col passare di giorni. Nel caso di BoNT/E, SNAP-25 tagliata scompare verso il diciottesimo giorno dal trattamento con la tossina. Nel caso di BoNT/A, invece, i livelli di SNAP-25 tagliata rimangono inalterati per più di ottanta giorni.
Perché queste due neurotossine mostrano questa grande differenza nella durata d’azione se possiedono lo stesso bersaglio molecolare SNAP-25? Esistono due possibili risposte:
a. Per i diversi siti di taglio di BoNT/A e BoNT/E su SNAP-25. b. Per la diversa stabilità delle due neurotossine nel citosol neuronale.
a) BoNT/E taglia un frammento di 26 amminoacidi dall’estremità carbossi-terminale di SNAP-25, mentre BoNT/A rimuove solamente nove residui (Tab.1).
Si potrebbe quindi ipotizzare che SNAP-25 tagliata da BoNT/A, pur non essendo funzionale per la neuroesocitosi, non sia così alterata da essere rimossa rapidamente dai terminali presinaptici. Al contrario, quella processata da BoNT/E, molto più danneggiata, verrebbe rimossa rapidamente e subito rimpiazzata da molecole di SNAP-25 di nuova sintesi. Per ciò nel caso di BoNT/E si ottiene un recupero più rapido della funzionalità neuronale (Eleopra et al., 1998).
Tab 1(Keller et al.,1999):
b) Sembra che, nel citosol neuronale, BoNT/A sia significativamente più stabile rispetto a BoNT/E. Risulta infatti che la maggior durata di azione di BoNT/A sia dovuta alla sua continua attività proteolitica su SNAP-25 di nuova sintesi (Keller et al., 1999; Foran et al., 2003). È stato inoltre suggerito che questa maggiore stabilità sia dovuta ad una localizzazione di BoNT/A sulla membrana presinaptica, al contrario di BoNT/E che rimane nel citosol (Fernandez-Salas et al., 2004).
1.1.6 Le neurotossine botuliniche come agenti terapeutici
L’evidenza che l’inibizione dell’impulso nervoso è seguita da un recupero funzionale della giunzione neuromuscolare, ha fornito le basi scientifiche per la rapida crescita dell’utilizzo delle BoNT nella terapia di quelle malattie umane caratterizzate da
iperfunzionalità dei terminali colinergici. In un lavoro pioneristico, pubblicato nel 1973, Alan Scott e collaboratori dimostrarono che si potevano selettivamente indebolire i muscoli oculari di scimmie inoculando localmente quantità minutissime di tossina botulinica di tipo A. Vista l’efficacia di questo trattamento, tale pratica si è diffusa dapprima molto lentamente, forse a causa di un timore nei confronti di un "farmaco" tanto potente, ma poi in modo esponenziale. L’uso terapeutico della neurotossina botulinica tipo A è stato progressivamente esteso a distonie di muscoli più grandi e, più recentemente, ad alcuni tipi di strabismo (Jankovic et al., 1994). La tossina è stata anche usata con successo nel trattamento di rughe del viso, dovute a contrattura di alcuni muscoli facciali. L’uso terapeutico della tossina botulinica sierotipo A non dà luogo a controindicazioni particolari e gli unici effetti collaterali noti sono dovuti ad una ridotta diffusione della tossina a terminali "sani" attorno alla zona da trattare con conseguente leggera paralisi. Il vero problema irrisolto in questo tipo di intervento terapeutico è costituito dal fatto che la paralisi indotta dalla tossina è reversibile in tempi che variano a seconda del muscolo trattato, della dose, del paziente, e di altri fattori non noti per cui dopo 2-6 mesi dal trattamento di terminali scheletrici il paziente deve essere trattato nuovamente. Un altro problema è che si può verificare una immunizzazione con produzione da parte del paziente di anticorpi anti BoNT/A ad azione neutralizzante. Inoltre, alcuni rari soggetti sono resistenti alla neurotossina botulinica di tipo A dall’inizio.
Per superare questi problemi, sono stati testati altri serotipi di BoNT. BoNT/B, BoNT/F e BoNT/E sono risultati molto efficienti nell’indurre un effetto paralizzante (Eleopra et al., 1998). Tuttavia il loro effetto è più breve rispetto a quello di BoNT/A e quindi queste neurotossine non sono da considerarsi come valide alternative, mentre con BoNT/C si sono ottenuti risultati incoraggianti (Eleopra.et al., 1997).
Ultimamente è stato esteso l’uso delle BoNT anche a disturbi che non hanno basi neuromuscolari come l’iperidrosi ascellare (Heckmann et al., 2001), i dolori miofasciali (Porta et al., 2000) e la cefalea emicrania e muscolo-tensiva (Silberstein et al., 2000).
1.1.6.1 Effetto antiepilettico di BoNT/E.
L’epilessia dei lobi temporali è la forma più frequente di epilessia umana ed è caratterizzata da crisi ricorrenti, perdita neuronale nell’ippocampo e ridotte funzionalità di memoria.
Dal momento che l’eccessiva attività sinaptica è la causa primaria delle crisi epilettiche, potrebbe rivelarsi terapeutico l’utilizzo di farmaci in grado di colpire il macchinario di rilascio del neurotrasmettitore.
I risultati ottenuti da Costantin et al. (2005), sugli effetti di BoNT/E nell’ippocampo di ratto hanno suggerito un possibile effetto anticonvulsivo di BoNT/E.
BoNT/E è stata iniettata nell’ippocampo e dopo uno o due giorni le crisi sono state indotte attraverso una somministrazione intraippocampale di Kainato (KA), un agonista del glutammato.
Attraverso analisi elettroencefalografiche è stato dimostrato che la somministrazione profilattica di BoNT/E nell’ippocampo riduce fortemente il numero e la durata delle crisi focali indotte dal KA. Questi risultati sono stati ottenuti su un modello di crisi focali acute e l’epilessia è una malattia cronica caratterizzata da crisi spontanee ricorrenti. Per validare l’utillizzo terapeutico di BoNT/E come nuovo strumento per interferire con il macchinario alla base dell’epilessia, è necessario determinare l’effettiva durata d’azione della tossina in vivo.
L’eccessiva attività sinaptica risulta critica anche per un’altra neuropatologia umana: l’ischemia cerebrale.
L’ipotesi che la degenerazione neuronale, caratteristica dell’ischemia, sia innescata dallo sproporzionato rilascio di glutammato dai neuroni danneggiati, suggerisce la possibilità di utilizzare BoNT/E come agente neuroprotettivo.
1.2
L’ischemia cerebrale
L’ischemia rappresenta uno dei più seri problemi di salute pubblica ed è una delle maggiori cause di morte e di invalidità nel mondo. È stata definita come “rapido sviluppo di segnali clinici con disturbi delle funzioni cerebrali, caratterizzati da sintomi che durano ventiquattro ore o fino alla morte, con apparentemente nessun’altra causa se non quella di origine vascolare” (Organizzazione Mondiale Della Santà, 1988). Infatti, per ischemia si intende un blocco più o meno protratto del flusso sanguigno. Il cervello, essendo privo di riserve di ossigeno e/o riserve di glucosio, risulta essere particolarmente sensibile e vulnerabile al danno ischemico. I neuroni privati di ossigeno e delle altre sostanze nutrienti, anche per pochi minuti, esauriscono rapidamente le riserve energetiche e iniziano a morire.
Il mancato apporto di sangue può essere l’esito di eventi distinti, poiché sono svariati i motivi per cui il sangue può non riuscire a raggiungere il cervello. Tra i più importanti è l’ischemia che si verifica quando la circolazione viene interrotta a causa di un’ostruzione del vaso, in questo caso un’arteria, che irrora il tessuto cerebrale. L’ischemia rende conto dell’85% degli ictus, una minoranza di casi è invece attribuibile a emorragia, e per quanto meno comune è più frequentemente fatale.
L’ischemia cerebrale è la terza causa di morte nei Paesi occidentali con una incidenza di 250-400/100,000 ed una mortalità che supera il 30%. Negli Stati Uniti è stato stimato in oltre 3 milioni il numero di pazienti sopravvissuti ad un attacco ischemico cerebrale e l'ischemia cerebrale è la prima causa di handicap mentali e fisici. È inoltre responsabile di circa la metà dei ricoveri ospedalieri per patologie neurologiche acute.
Per molti anni si è ricercata una valida terapia per l’ischemia cerebrale, ma le molte sperimentazioni cliniche non hanno avuto successo. Rimane ancora da scoprire un’adeguata, effettiva e sicura terapia neuroprotettiva.
Clinicamente l’ischemia è uno stato notevolmente variabile per quanto riguarda la localizzazione, la severità, i sintomi, l’eziologia, le dimensioni dell’area infartuata e i fattori di complicanza. I modelli sperimentali di ischemia di solito non sono in grado di
riprodurre tutti i tipi di situazioni possibili ma sono comunque necessari per la ricerca di strategie terapeutiche.
1.2.1 Forme e modelli di ischemia cerebrale
L’obiettivo dei modelli di ischemia cerebrale è quello di ridurre il rifornimento di ossigeno e di glucosio al cervello. Questo processo determina un danno cerebrale attraverso una varietà di meccanismi molecolari e cellulari.
Comunemente i modelli di ischemia cerebrale vengono distinti in modelli globali e in modelli focali.
L’ischemia globale avviene quando il flusso sanguigno cerebrale è ridotto in tutto il cervello o nella maggior parte di esso, quella focale invece determina una diminuzione del flusso sanguigno in una regione cerebrale specifica.
1.2.1.1 Ischemia cerebrale globale
I modelli di ischemia cerebrale globale simulano ciò che avviene nel cervello, o in una grande porzione di esso, dopo un arresto cardiaco o dopo una severa ipotensione. Tipicamente il flusso di sangue al cervello viene interrotto per un periodo di tempo transitorio e vengono analizzati gli effetti di morte alcuni giorni dopo.
In questo tipo di danno ischemico, soltanto alcune selettive popolazioni neuronali, tra cui le cellule piramidali dell’ippocampo, i neuroni neocorticali degli strati 3, 5, 6, i neuroni dello striato e le cellule cerebellari del Purkinje, risultano danneggiate (Brierley, 1979; Pulsinelli et al., 1982).
Il danno ischemico si sviluppa lentamente dopo un periodo di latenza ed è quindi conosciuto come “fenomeno di maturazione” (Ito et al., 1975) o “morte neuronale ritardata” (Kirino, 1982; Pulsinelli et al., 1982).
Tra questi modelli ci sono quelli in cui l’ischemia globale viene prodotta occludendo le arterie che portano il sangue al cervello come l’arteria carotide comune (CCA) e le arterie vertebrali. Il modello CCAO (occlusione bilaterale delle arterie carotidi comuni) del porcellino d’India (Meriones unguiculatus) è probabilmente il più utilizzato perché implica una chirurgia molto semplice. In questo animale non esiste comunicazione tra le
arterie carotidi e vertebrali e quindi l’ischemia può essere provocata nel cervello anteriore occludendo soltanto la CCA.
Nel ratto, i modelli di ischemia globale includono il 4-VO (occlusione di quatto vasi) e il 2-VO (occlusione di due vasi). Anche in questi modelli l’occlusione è temporanea e seguita da riperfusione.
Il primo può essere riprodotto in ratti in libero movimento ma richiede una procedura a due stadi. Inizialmente vengono posizionati dei clamp atraumatici intorno a ciascuna arteria carotide e vengono resi accessibili sul collo dell’animale. Le arterie vertebrali vengono invece elettrocauterizzate. Il giorno seguente vengono occluse le arterie carotidi mentre il ratto è in libero movimento.
Nel modello 2-VO, invece, viene effettuata una occlusione bilaterale delle arterie carotidi comuni ed una ipotensione sistemica provocando una emorragia con agenti farmacologici (Smith et al., 1984b). Rispetto al modello 4-VO, questo ha il vantaggio di richiedere una preparazione chirurgica più semplice e di avere una riperfusione più immediata.
Un grosso problema di tutti questi modelli è la loro elevata variabilità negli esiti sperimentali, principalmente dovuta alle varianti presenti nella circolazione cerebrale.
1.2.1.2 Ischemia cerebrale focale
L’ischemia cerebrale focale risulta dalla completa occlusione di un’arteria cerebrale. L’occlusione embolica dell’arteria prossimale mediana (MCA) clinicamente è una delle forma più comuni di ischemia (1986; Karpiak et al., 1989). La maggior parte dei modelli di ischemia focale simulano quest’evento.
Nel ratto, i modelli di l’occlusione transiente o permanente della MCA sia distale che prossimale (MCAO) sono largamente utilizzati per studi di neuroprotezione farmacologica e per la caratterizzazione dei meccanismi implicati nel danno ischemico. La MCAO può essere indotta con varie tecniche come, ad esempio, la cauterizzazione o l’elettrocoagulazione della MCA (Tamura et al., 1981; Tyson et al., 1984).
Esistono anche alcune varianti di questo modello come quella in cui la MCA viene occlusa insieme all’arteria carotide comune ipsilaterale (Chen et al., 1986).