1. Le cellule staminali
Le cellule staminali sono cellule indifferenziate capaci sia di auto-rinnovarsi (self-renewal), che di differenziare verso diverse filiere. Rappresentano il presupposto per l’esistenza degli organismi multicellulari in quanto sono responsabili sia della capacità di un embrione di generare diversi tipi cellulari, sia del rinnovamento dei tessuti adulti lungo il corso della vita dell’individuo (Fuchs and Segre, 2000). Esistono molteplici tipi di cellule staminali classificate in base alla loro potenzialità. Al vertice di questa gerarchia vengono poste le cellule staminali totipotenti, presenti nelle fasi iniziali della vita embrionale e in grado di generare un organismo intero, compresi gli annessi extra-embrionali. Nel corso delle divisioni embrionali, le cellule staminali restringono gradualmente la loro potenzialità diventando capaci di generare solo i tre foglietti embrionali (endoderma, ectoderma e mesoderma): queste cellule sono chiamate cellule staminali embrionali e sono pluripotenti. Le cellule staminali sono presenti anche nell’organismo adulto dove ne sono state trovate due tipologie:
multipotenti, capaci di produrre più tipi cellulari appartenenti a un dato tessuto, e unipotenti,
capaci di produrre un solo tipo cellulare (Brignier and Gewirtz, 2010).
Fig.1.1 Diversa potenzialità delle cellule staminali.
Sebbene alcuni studiosi sostengano l’esistenza di un modello gerarchico, il cui vertice è rappresentato dalle cellule staminali totipotenti e la base dalle cellule differenziate
terminalmente, all’interno del quale la perdita di potenzialità è progressiva e irreversibile, recenti studi hanno dimostrato che questa non è l’unica ipotesi possibile. Infatti, sono state generate cellule pluripotenti manipolando cellule somatiche, tramite espressione forzata di alcuni fattori di trascrizione o tramite trasferimento del nucleo della cellula somatica in oociti enucleati. In questa maniera si ha la reversione di modificazioni epigenetiche e l’attivazione di fattori di trascrizione chiave coinvolti nella staminalità.
1.1 Le cellule staminali embrionali
Uno zigote è, per definizione, una cellula staminale dalle potenzialità illimitate, in grado di generare sia le componenti embrionali sia quelle extra-embrionali. Questa condizione di totipotenza è mantenuta fino allo stadio a 8 blastomeri. Dopo la compattazione, infatti, si realizza il primo evento differenziativo che porta alla formazione di due popolazioni distinte: il trofoblasto, più esterno e costituito da cellule dall’aspetto epiteliale dalle quali deriverà la componente embrionale della placenta; e le cellule della massa interna (Inner Cell Mass,
ICM), che mantengono una maggiore potenzialità. Il secondo evento differenziativo si
realizza a carico dell’ ICM e porterà alla formazione dell’ipoblasto, dal quale deriverà il sacco vitellino, e dell’epiblasto, precursore delle cellule appartenenti ai tre lineages embrionali (endoderma, mesoderma ed ectoderma) (Gilbert, 3a ed.). A questo punto, l’embrione pre-impianto è composto da tre diverse popolazioni cellulari: trofoectoderma, ipoblasto e epiblasto. Da ognuna di queste popolazioni possono essere derivate cellule staminali dalla diversa potenzialità, rispettivamente si avranno: Trophoblast Stem cells (TS) (Tanaka et al., 1998), Extraembryonic Endoderm cells (XEN) (Kunath et al., 2005), ed Embryonic Stem cells (ES) (Martin, 1981; Evans and Kaufman, 1981). TS, XEN e ES sono capaci di self-renewal in vitro quando al mezzo di coltura vengono aggiunti fattori specifici. TS e XEN, sia in vitro che in vivo, generano rispettivamente cellule del trofoectoderma e dell’endoderma primitivo (Ralston and Rossant 2005). Le ES, invece, possono differenziare verso i tre foglietti embrionali sia in vitro, formando i corpi embrioidi, che in vivo, generando teratomi quando iniettati in un topo immunosoppresso. Inoltre, quando introdotte in una blastocisti ospite, contribuiscono alla formazione della linea germinale (Beddington and Robertson, 1989). Queste caratteristiche hanno reso le ES interessanti sia per i possibili impieghi terapeutici, sia soprattutto per lo studio delle diverse fasi dello sviluppo e dei meccanismi molecolari che le regolano sia per la produzione di topi transgenici.
Fig. 1.2 Cellule staminali embrionali coltivate in vitro.
Per mantenere la pluripotenza in vitro, le ES sono coltivate in presenza di un feeder layer, costituito da fibroblasti murini mitoticamente inattivati (MEFs) (Evans and Kaufman, 1981). Il frazionamento del mezzo condizionato ha permesso di individuare la citochina chiave prodotta dal feeder layer: il Leukemia Inhibitor Factor (LIF). LIF può sostituire totalmente il feeder layer, appartiene alla famiglia delle Interleuchine 6, attiva la via JAK/STAT, coinvolgendo in particolare STAT3 (Niwa et al., 1998). In un mezzo serum-free, però, LIF da solo non è in grado di prevenire il differenziamento. Si rivela necessaria, infatti, la segnalazione delle Bone Morphogenetic Proteins (BMPs) e di Wnt (Nakashima, Colamarino and Gage, 2004). BMP4 porta, tramite la via di SMAD, all’attivazione degli Inhibitor of Differentiation genes (Id) che inibiscono il differenziamento in senso neurale (Ying et al., 2003). La via canonica di Wnt, invece, porta alla stabilizzazione della β-catenina e alla trascrizione finale di geni coinvolti nella proliferazione cellulare (vedi 3.2) (Kleber and Sommer, 2004). Come queste vie integrino le loro segnalazioni ancora non è chiaro, ma il loro effetto finale è l’attivazione di fattori di trascrizione come Oct4, Nanog, Sox2 e FoxD3. Questi fattori chiave, una volta attivati, oltre ad agire sui propri targets, formano un network di regolazione che stimola o limita i livelli di espressione degli altri componenti (Pan and Thomson, 2007). Mantenere un livello adeguato di espressione è, infatti, fondamentale in
quanto alcuni fattori chiave, come Oct4, agiscono in maniera dose-dipendente (Niwa et al., 2001). La pluripotenza in vivo è mantenuta solo per un breve periodo, in quanto, in seguito all’impianto nella parete uterina, ha inizio la gastrulazione che porta alla formazione dei foglietti embrionali. Le ES sono state ottenute anche da blastocisti umane derivate da fecondazione in vitro (Thomson et al., 1998). Esistono però delle differenze fra le mES e le hES, sia per quanto riguarda la morfologia e gli antigeni di superficie, sia per le segnalazioni estrinseche richieste per il mantenimento della pluripotenza in vitro: la segnalazione di LIF nelle cellule murine, infatti, viene sostituita da quella di bFGF e TGFβ/Activin/Nodal nelle ES umane (Wei et al., 2005; Hoof et al., 2006; Yu and Thomson, 2008). Nonostante l’ampia potenzialità e il conseguente possibile impiego in campo clinico, l’uso delle cellule staminali embrionali è improbabile sia in quanto pone non pochi problemi etici, sia perché, il trapianto in posizione ectopica causa la formazione di teratomi. Alcuni di questi ostacoli potrebbero essere superati grazie alle cellule pluripotenti indotte (iPS), ottenute dalla riprogrammazione di fibroblasti tramite introduzione di specifici fattori usando vettori virali: Oct4, Sox2, c-Myc e Klf4 (Takahashi and Yamanaka, 2006, Takahashi et al., 2007) oppure Oct4, Sox2, Nanog e Lin28 (Yu et al., 2007). Le iPS sono simili alle ES in morfologia, espressione genica, stato epigenetico e differenziazione in vitro. Inoltre, se usate per l’ingegneria tissutale, potrebbero rappresentare un espediente per il superamento di problemi legati al rigetto, in quanto sarebbero le cellule somatiche del paziente stesso a venire riprogrammate. Siamo, però, ancora molto lontani dall’ottenere un protocollo applicabile alla clinica, in quanto le iPS possono rivelarsi tumorigeniche, probabilmente come conseguenza dell’uso di vettori virali e dell’oncogene c-myc. Nuove strategie sono state recentemente testate al fine di ottimizzare la loro produzione: i vettori virali sono stati sostituiti con plasmidi e il numero di geni da introdurre è stato ridotto (Nakagawa et al., 2008; Okita et al., 2008). Questi passi in avanti non sono comunque ancora sufficienti per garantire un soddisfacente protocollo d’impiego clinico, in quanto non sono ancora chiari i targets sui quali agiscono i fattori introdotti e i meccanismi alla base delle riprogrammazione, limitando quindi la capacità di prevedere eventuali effetti collaterali.
1.2 Le cellule staminali adulte
Dopo la formazione di un organismo completo, in molti tessuti e organi si realizza un turnover cellulare noto come omeostasi, un processo che permette il rimpiazzo di cellule
morte o comunque danneggiate con cellule di nuova formazione. Questo fenomeno è sempre stato sotto i nostri occhi, basti pensare alle ferite che si rimarginano o ai peli che ricrescono, ma solo recentemente, si è potuto affermare che l’omeostasi è mantenuta grazie alla presenza di cellule staminali adulte. Negli ultimi decenni, infatti, è stato possibile riscontrare la presenza di cellule staminali adulte praticamente in tutti i tessuti, anche in quelli che apparentemente non subiscono ricambio cellulare, come il tessuto nervoso e quello cardiaco. Le cellule staminali adulte sono generalmente delle cellule quiescenti che, rispondendo a segnali ambientali, possono dividersi in maniera simmetrica, ripopolando il pool di cellule staminali, oppure in maniera asimmetrica, generando una cellula staminale e un progenitore di transito. È compito dei progenitori dividersi ripetutamente per un breve arco temporale, in modo da espandere il pool cellulare che potrà differenziare poi verso un lineage definito. Questo è un fine meccanismo di prevenzione dell’ accumulo di mutazioni al DNA nelle cellule staminali, in quanto queste le trasmetterebbero alla progenie (Fuchs and Segre, 2000). Le cellule staminali adulte suscitano molto interesse dal punto di vista terapeutico in quanto non possiedono gli svantaggi delle ES, ma hanno la potenzialità di rigenerare tutti i tipi cellulari presenti in specifici tessuti. Potrebbero, inoltre, essere usate per la produzione ex vivo di determinate popolazioni cellulari utili per il trattamento di malattie neurodegenerative o di infarto del miocardio. Ad oggi, vengono ampiamente usate in campo medico le cellule staminali ematopoietiche (HSCs), usate attraverso trapianti di midollo osseo sia nelle terapie di patologie del sistema ematopoietico che per il recupero di pazienti immunodepressi in seguito a trattamento chemioterapico. Il loro uso è stato possibile in quanto sono le cellule staminali meglio caratterizzate: infatti la loro nicchia è ben definita e vengono selezionate positivamente per i markers di superficie c-Kit, Sca-1, CD43 e CD45; e negativamente per i markers di lineage (lin). Nonostante le HSCs siano le cellule staminali adulte per prime isolate, problema non da poco risulta l’impossibilità di coltivarle in vitro. Questa incapacità rende difficile comprendere a pieno la biologia delle HSCs e quindi limita il loro impiego in campo clinico.
Negli ultimi anni si è fatta largo l’idea dell’esistenza di cellule staminali anche all’interno di tumori. Queste cellule vengono chiamate cancer stem cells e condividono le stesse caratteristiche delle cellule staminali adulte: possono autorinnovarsi, sono quiescenti e danno vita a progenitori che poi differenziano terminalmente. Questo pool di cellule è molto ristretto e pare sfuggire all’azione dei chemioterapici, dal momento che questi agiscono sulle cellule
proliferanti. Le cancer stem cells sopravvivono così ai trattamenti e sembrano quindi essere le responsabili di eventuali recidive (Reya et al., 2001; Clarke et al., 2006; Costa et al., 2007; Lee and Herlyn, 2007).
1.3 Plasticità delle cellule staminali adulte
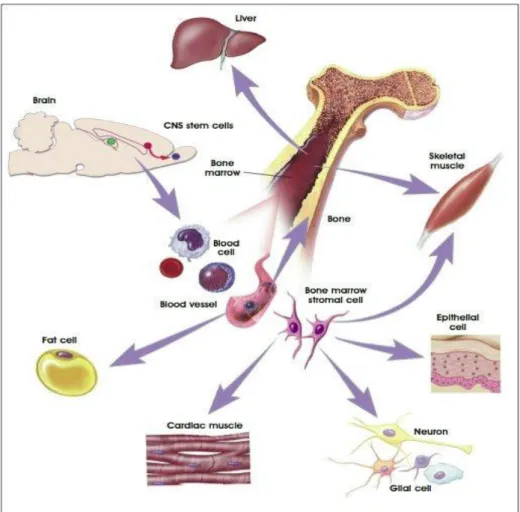
Fin dalla loro scoperta, si è ritenuto che le cellule staminali adulte potessero contribuire solo alla formazione del tessuto nel quale risiedono, senza poter valicare i confini fra i vari tessuti, né tantomeno partecipare al mantenimento di sistemi diversi. Studi dell’ultimo decennio hanno però posto in discussione questa visione ipotizzando che le cellule staminali adulte, in determinate condizioni ambientali, possano generare anche cellule atipiche Questi dati pongono le fondamenta per la nascita del concetto di plasticità delle cellule staminali adulte: il loro destino non è rigido e pre-stabilito, ma, piuttosto, flessibile e dinamico. Prime evidenze sulla plasticità sono state ottenute da esperimenti condotti sul sistema ematopoietico. Trapianti di midollo osseo totale o di cellule ematopoietiche arricchite da midollo osseo in modelli murini letalmente irradiati e genotipicamente o fenotipicamente distinguibili mostrano il coinvolgimento di queste cellule nella formazione di cellule non-ematopoietiche come pelle, polmone, intestino, rene, fegato, pancreas, muscolo, endotelio, miocardio e neuroni. Se realmente presente, la plasticità sarebbe di enorme importanza in quanto garantirebbe una condizione vicina alla pluripotenza per le cellule staminali adulte, permettendo il superamento dei problemi etici e politici legati all’uso delle cellule staminali embrionali in laboratorio. Inoltre risulterebbe possibile un’applicazione in campo medico in quanto potrebbero essere usate cellule staminali adulte del paziente stesso evitando problemi di rigetto (Wurmser and Gage, 2002).
Fig. 1.3 Plasticità delle cellule staminali adulte.
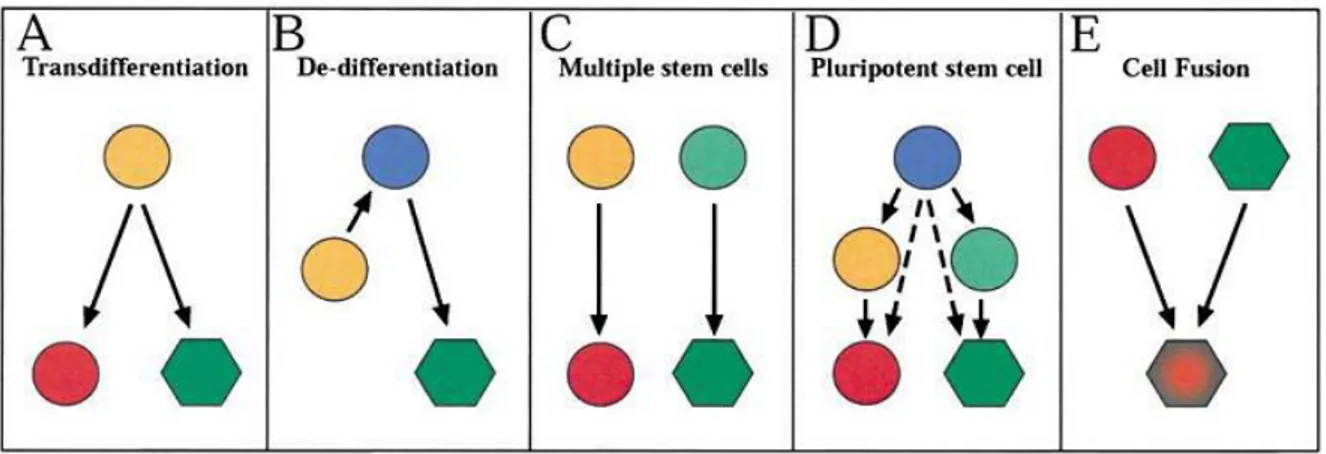
Esistono diversi possibili meccanismi che possono essere alla base della plasticità: transdifferenziazione, de-differenziazione, esistenza di un progenitore pluripotente o trapianto di cellule non omogenee che includono diversi tipi di cellule staminali adulte. La
transdifferenziazione permette di spiegare la generazione di cellule appartenenti a lineages
differenti, tramite l’attivazione di un programma genico fino a quel momento dormiente (Fig.1.4 A). La de-differenziazione (Fig.1.4 B) è un meccanismo che si realizza comunemente negli anfibi, i quali ricostituiscono un arto tagliato grazie alla capacità delle cellule differenziate terminalmente di revertire la propria natura e tornare a una condizione di maggiore potenzialità. Queste cellule comunque non diventano pluripotenti, ma mantengono memoria del tessuto dal quale provengono. Il trapianto di diversi tipi di cellule staminali
adulte (Fig.1.4C) è strettamente collegato al grado di omogeneità delle cellule usate: è
possibile che nel campione di cellule trapiantate siano presenti due o più tipi di cellule staminali adulte. Verosimile appare anche l’esistenza di cellule progenitrici con maggiore
modello di tipo gerarchico (Fig.1.4D). Analisi più accurate, condotte al fine di riprodurre gli eventi di plasticità precedentemente notati, in alcuni casi non hanno avuto successo, mettendo invece in evidenza anche fenomeni di fusione cellulare (Fig.1.4E). La fusione cellulare è un meccanismo che avviene normalmente nella generazione di miotubi scheletrici multinucleati a partire da mioblasti. Inoltre è coinvolta anche in fenomeni patologici come la formazione di tessuto granulomatoso in seguito a infiammazione indotta da corpi estranei (Wurmser and Gage, 2002; Terada et al., 2002; Wagers and Weissman, 2004).
Fig. 1.4 Modelli che potrebbero spiegare la plasticità.
Vista l’ambiguità dei risultati, diventa necessario adottare delle procedure sperimentali più rigorose al fine di escludere eventuali fenomeni di contaminazione o fusione cellulare, analizzando non solo le cellule che compongono la coltura di studio, in modo da ottenere una popolazione più omogenea possibile, ma anche le caratteristiche funzionali delle cellule da esse derivate.
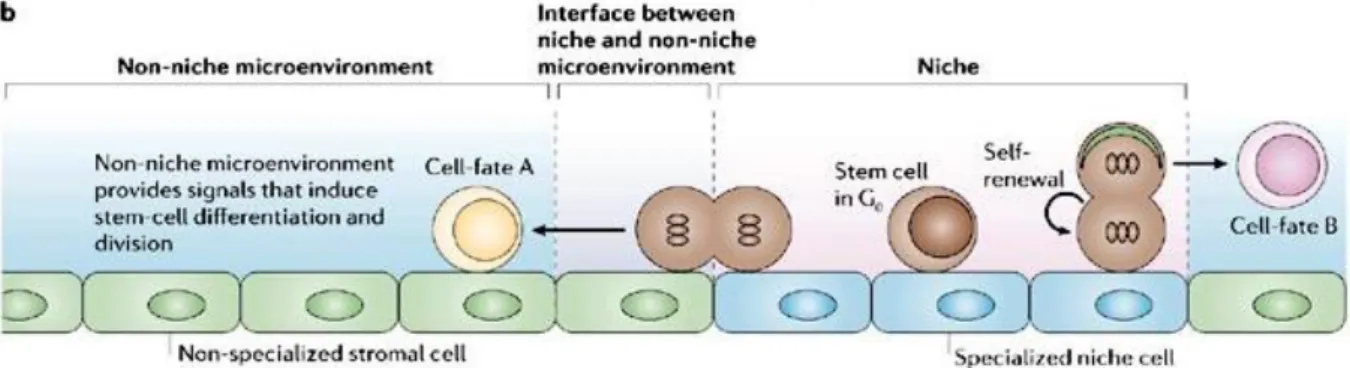
1.4 La nicchia
La capacità delle cellule staminali adulte sia di autorinnovarsi che di differenziare è alla base dell’omeostasi tissutale. Questo processo deve, però, essere finemente regolato in quanto, squilibri, in un senso o nell’altro, porterebbero una carenza di cellule staminali nel relativo pool o una loro eccessiva espansione, condizioni che possono essere entrambe collegate a fenomeni fisiologici o patologici quali l’invecchiamento e il cancro (Yin and Li, 2006). Mantenere questo equilibrio è compito della nicchia, che quindi è uno specifico sito all’interno di un tessuto adulto in cui le cellule staminali risiedono e vengono regolate. La nicchia esplica il suo compito grazie al bilanciamento di due diversi tipi di divisione che la
cellula staminale può compiere: la divisione simmetrica e quella asimmetrica. La divisione
simmetrica permette la formazione di due cellule identiche a partire da una cellula staminale
o un progenitore di partenza: è alla base quindi del self-renew e del mantenimento del pool delle cellule staminali e dei progenitori. La divisione asimmetrica porta invece alla formazione di due cellule figlie diverse: si ritiene che una delle due mantenga le caratteristiche di cellula staminale, mentre l’altra venga indirizzata verso un particolare destino differenziativo. Questo tipo di divisione si può realizzare tramite una diversa distribuzione di determinanti molecolari tra le due cellule figlie o a causa di esposizione delle cellule a microambienti diversi. Nel primo caso si parlerà di asimmetria divisionale, nel secondo di asimmetria ambientale (Wilson and Trumpp, 2006).
Fig. 1.5 Divisione asimmetrica: asimmetria divisionale e ambientale.
L’esistenza delle nicchie venne per prima ipotizzata nei mammiferi, ma, a causa della loro complessità, studi preliminari vennero effettuati su organismi modello invertebrati come Caenorhabditis elegans e Drosophila melanogaster. In questi organismi è stato possibile individuare cellule staminali e relative nicchie, con una risoluzione a livello di singole cellule. Anche se la situazione che si ritrova nei mammiferi è molto più complessa, si può ipotizzare l’esistenza di meccanismi regolatori comuni (Morrison and Spradling, 2008). Pensare a una nicchia come a una mera struttura fisica è erroneo. Infatti la nicchia esplica la sua funzione anche grazie al coinvolgimento di diverse componenti quali la matrice extracellulare che
contribuisce alla regolazione delle cellule staminali; le segnalazioni paracrine, endocrine e
nervose che mettono in relazione le cellule staminali con l’intero organismo; i prodotti metabolici quali le specie reattive dell’ossigeno (ROS) che sono un indice delle condizioni
del tessuto; le cellule somatiche che ancorano le cellule staminali. Inoltre le nicchie non sono degli elementi statici, ma dinamici: esempi sono forniti dalla formazione de novo di follicoli piliferi in seguito a espressione costitutiva della β-catenina, o dall’ematopoiesi extramidollare in condizioni di stress (Scadden, 2006).
Fig. 1.6 Segnalazioni all’interno della nicchia.
Funzionalmente, si possono distinguere due tipologie di nicchia: di self-renewal e di storage. Nella prima, la cellula staminale va incontro alla divisione simmetrica e/o asimmetrica e può essere semplice o complessa. La nicchia semplice è caratterizzata dalla presenza di un solo tipo di cellule staminali ancorate tramite giunzioni aderenti a specifiche cellule stromali. La nicchia complessa si riscontra laddove la cellula staminale contatta molteplici cellule somatiche, o dove sono presenti più tipologie di cellule staminali. Infine, la nicchia di storage è caratterizzata dall’ospitare cellule staminali quiescenti che vengono attivate solo in caso di danno tissutale (Ohlstein et al., 2004). Ancora non è chiaro se le funzioni vengano svolte da nicchie strutturalmente e fisicamente diverse o se esista in realtà una sola tipologia. Una delle ipotesi è l’esistenza di una nicchia con regioni funzionalmente diverse deputate a compiti specifici. In questa tipologia di nicchia, la regione di storage si trova al centro della struttura, e marginalmente, al confine con la regione esterna alla nicchia, si trovano le regioni in cui si realizzano le divisioni simmetriche e/o asimmetriche. Il tipo di divisione che si realizza
dipende dalla convergenza di segnali provenienti dall’ambiente della nicchia e da quello esterno alla nicchia, e inoltre, è probabile che si realizzi l’asimmetria divisionale così come quella ambientale (Wilson and Trumpp, 2006).
Fig. 1.7 Modello di nicchia.
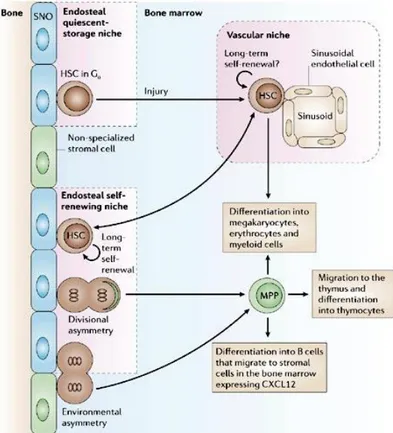
La nicchia meglio caratterizzata è quella delle cellule staminali ematopoietiche (HSCs), ipotizzata per la prima volta nel 1978 da Schofield. Diverse evidenze, usando marker di superficie specifici come le proteine appartenenti alla famiglia SLAM (Kiel et al., 2005), suggeriscono la presenza di due tipologie di nicchie all’interno del midollo osseo: la nicchia
dell’endostio e quella vascolare. Nella prima le HSCs si ritrovano a ridosso degli osteoblasti,
nella seconda invece si ritrovano vicino a cellule endoteliali dei vasi sinusoidali nella parte centrale del midollo osseo. Queste due nicchie sono funzionalmente diverse. La nicchia dell’endostio ospita le Long Term-HSCs (LT-HSC) che sono HSC fondamentalmente quiescenti e in grado di contribuire all’ematopoiesi a lungo termine e che la sostengono per l’intero arco della vita. È stata riscontrata una stretta relazione fra le LT-HSC e gli osteoblasti. Infatti aumentando il numero degli osteoblasti tramite inattivazione condizionale del recettore di tipo 1A per la bone morphogenetic protein (BMPR1A) (Zhang et al., 2003) o tramite overespressione dell’ormone paratiroideo e del relativo recettore (Calvi et al., 2003) si assiste a un aumento del numero degli osteoblasti e, in parallelo, a un aumento delle HSCs. Inoltre un’ablazione condizionale degli osteoblasti grazie all’uso del gene della timidina chinasi, che induce la morte cellulare in risposta al ganciclovir, è associata a una diminuzione sia delle HSCs che delle tipologie cellulari che da esse derivano. Questa situazione viene revertita in seguito a rimozione del ganciclovir (Visnjic et al., 2004). Nella nicchia vascolare sono molto probabilmente localizzate le Short Term-HSC (ST-HSC) che sono in grado di contribuire all’ematopoiesi solo per un breve periodo di tempo. Diversi studi supportano l’ipotesi
secondo la quale la nicchia vascolare in realtà promuova la proliferazione e la differenziazione delle cellule staminali e dei progenitori (Kopp et al., 2005). Questa funzione della nicchia vascolare può essere probabilmente esplicata grazie a un maggior apporto di fattori di crescita e ossigeno, data la vicinanza ai vasi sanguigni (Yin and Li, 2006).
Fig. 1.8 Nicchia delle HSCs.
La nicchia vascolare pare svolgere anche altri ruoli importanti, è cruciale infatti nei processi di homing e di mobilization. Il processo di mobilization prevede il distacco delle HSCs dalla nicchia osteoblastica, il passaggio a quella vascolare e quindi l’attraversamento dell’endotelio per ritrovarsi nel flusso sanguigno, costantemente legate al vaso sanguigno. Il processo di homing è esattamente il contrario della mobilization ed è estremamente importante negli esperimenti di trapianto dove il raggiungimento e l’attecchimento nella nicchia midollare sono requisiti indispensabili per il ripopolamento del pool staminale (Yin and Li, 2006). In questi processi intervengono dei fattori cruciali: tra questi va ricordata la chemochina CXCL12 espressa da diversi tipi di cellule stromali del midollo osseo come gli osteoblasti e le cellule dell’endotelio. Questa chemochina viene riconosciuta dal recettore CXCR4 e induce nelle cellule target motilità, chemotassi, adesione, secrezione di metalloproteasi della matrice e
secrezione di fattori angiogenetici (Wilson and Trumpp, 2006). Anche se non è del tutto conosciuta la segnalazione molecolare che sta alla base della nicchia delle HSCs, sono noti alcuni meccanismi fondamentali. Tra questi sono presenti la segnalazione di Notch, dell’osteopontina, dell’SCF, dell’angiopoietina. Il recettore Notch-1 è espresso da HSCs mentre il suo ligando Jagged1 è espresso da cellule stromali del midollo osseo e da osteoblasti. L’espressione di Jagged1 risulta aumentata negli osteoblasti esposti all’ormone paratiroideo, per questo motivo il concomitante aumento del numero delle HSCs viene in parte attribuito alla via di segnalazione di Notch (Calvi et al., 2003). Un altro meccanismo che pare regolare il numero delle HSCs è la secrezione dell’osteopontina nella matrice cellulare. Questa glicoproteina pare agire come regolatore negativo delle HSCs mantenendole in uno stato di quiescenza (Nilsson et al., 2005). Kit è espresso nelle LT-HSCs. Mutazioni sul locus del gene Kit o del gene SCF compromettono il sistema ematopoietico indicandone il ruolo essenziale (Bernstein et al., 1991). Questa via di segnalazione risulta importante anche per l’adesione delle HSCs agli osteoblasti in quanto vengono attivati VLA4 e VLA5 (Kovach et al., 1995). Tie2 è un recettore tirosina chinasico espresso dalle HSCs e dalle cellule endoteliali, mentre il suo ligando Ang-1 è espresso dagli osteoblasti. Ang-1 favorisce la quiescenza delle HSCs e la loro adesione agli osteoblasti tramite Tie2 (Arai et al., 2004).
Recentemente è stata, inoltre, avanzata l’ipotesi dell’esistenza di una nicchia unica nel midollo osseo composta dalla coppia HSC-MSC. Infatti una sottopopolazione di cellule staminali mesenchimali (MSCs), selezionate positivamente per la nestina, sono spazialmente associate alle HSC ed esprimono geni necessari per il mantenimento delle stesse. Questo set di geni viene down regolato in seguito a mobilizzazione forzata delle HSC. Inoltre, in vivo, la deplezione delle MSC nestina+ causa una repentina diminuzione del numero delle HSC e una riduzione dell’homing di HSC trapiantate in animali letalmente irradiati (Mendez-Ferrer et al., 2010).
2.0 Cellule staminali mesenchimali (MSCs)
Le cellule staminali mesenchimali (MSCs) sono cellule stromali clonogeniche, dall’aspetto fibroblastoide e capaci di differenziare verso diversi lineages come osso, cartilagine, muscolo, tendine e adipe.
Fig. 2.1 Cellule staminali mesenchimali.
Già dal 1860, Cohnheim aveva ipotizzato la loro esistenza, ma fu Friedenstein, nella seconda metà del 1900, che riuscì a dimostrarla e a isolare le MSCs. La loro fonte principale è il midollo osseo, dove sono presenti con una frequenza di 1 su 10.000 cellule mononucleate. Sono state però ritrovate anche in molti altri tessuti come il tessuto adiposo, liquido amniotico e tessuti fetali e virtualmente, in accordo a dati recenti, dovrebbero essere presenti in tutti i tessuti dell’organismo. Recenti risultati suggeriscono una loro localizzazione perivascolare e una appartenenza delle MSCs alla grande famiglia dei periciti . Infatti, cellule che esprimono markers mesenchimali contemporaneamente sono positive per markers specifici dei periciti, inoltre periciti isolati tramite sorting (CD146+, CD34-, CD45-, CD56-) possono differenziare in vitro in senso osteogenico, adipogenico, condrogenico e miogenico, caratteristica peculiare delle MSCs (da Silva Meirelles et al., 2008).
2.1 Identificazione delle MSCs
Uno dei problemi della biologia delle MSCs consiste nella loro identificazione data la mancanza di markers molecolari univoci. A complicare la situazione si aggiunge il fatto che diversi laboratori isolano queste cellule da numerose sedi con tecniche differenti L’identificazione delle MSCs viene realizzata tramite una combinazione di proprietà fisiche, fenotipiche e funzionali. Un classico metodo per identificare e fare una stima delle MSCs è il saggio delle Colony Forming Unit-Fibroblast (CFU-F) nel quale le singole cellule multipotenti, piastrate a bassa densità, sono in grado di formare una colonia di cellule multipotenti. Le analisi fenotipiche si basano sull’uso di una combinazione di markers di superficie. In particolare, le MSCs risultano negative per CD45 (marker di cellule ematopoietiche), CD14 o CD11 (marker di cellule del sistema immunitario), CD31 (marker di cellule endoteliali e ematopoietiche), e positive per Stro1 (Kolf, Cho and Tuan, 2007), SSEA-4, CD105, CD73, CD4SSEA-4, CD146 e molti altri. Il saggio in vitro che permette di identificare le MSCs si basa sulla loro multipotenzialità e consiste nell’indurre la differenziazione almeno verso tre dei possibili lineages: osteogenico, adipogenico e condrogenico. La differenziazione osteogenica in vitro è realizzata incubando le MSCs confluenti per un periodo di 2-3 settimane con delle specifiche sostanze inducenti, quali Acido Ascorbico, Dexametasone e β-glicerofosfato. Nelle fasi terminali del processo si verifica un aumento dell’espressione di fosfatasi alcalina e le cellule risulteranno positive a colorazioni specifiche come Alizarin Red e von Kossa. La differenziazione adipogenica può essere stimolata in vitro incubando le MSCs con un cocktail di sostanze inducenti che include Insulina, Dexametasone, Isobutilmetilxantina e Indometacina. Le cellule cambiano morfologia, diventando rotondeggianti e accumulano gocce lipidiche in vacuoli citoplasmatici. Inoltre esprimono geni specifici come PPARγ e aP2 e risultano positive alla colorazione con Oil Red. Infine, la differenziazione condrogenica viene indotta centrifugando le MSCs e coltivandole in presenza di Transforming Growth Factor β (TGFβ). Le cellule pertanto producono collagene di tipo2 e risultano positive alla colorazione con toluidina blu (Chamberlain et al., 2007).
Un’altra metodologia che permette di valutare la potenzialità delle MSCs consiste nel loro trapianto in vivo nella regione sotto-cutanea o nelle capsule surrenali, assistendo alla conseguente formazione di osso ectopico (Parekkadan and Milwild, 2010).
Fig. 2.2 Differenziazione in vitro delle MSCs.
Recenti studi, inoltre, hanno permesso di identificare nuove popolazioni con un potenziale proliferativo e differenziativo maggiore se paragonato a quello delle MSCs. Si tratta delle Marrow Isolated Adult Multilineage Inducible Stem Cell (MIAMI), delle Multipotent Adult Progenitor Cells (MAPC) (Reyes et al., 2001; D’Ippolito et al, 2004), che potrebbero essere un gruppo di cellule staminali più primitive e potrebbero rappresentare un progenitore comune di MSCs e HSCs (Giordano et al., 2007); e delle Mesodermal Progenitor Cells (MPCs) (Petrini et al., 2008). In particolare, le MPCs sono state ritrovate nel midollo osseo adulto umano e, in opportune condizioni di coltura, sono capaci di differenziare in vitro in cellule mesenchimali, ma anche in cellule endoteliali. Le MPCs hanno un fenotipo preciso ed esprimono alcuni marcatori di superficie embrionali, non condivisi dalle cellule mesenchimali, e una volta messe in condizioni differenziative, perdono questi marcatori e acquisiscono quelli mesenchimali. Le cellule mesenchimali, invece, non possono essere indotte a revertire in MPCs, confermando la natura più primitiva.
2.2 Attività immunoregolatoria e trofica delle MSCs
Evidenze recenti hanno sottolineato un’altra proprietà delle MSCs: la capacità di secernere un ampio spettro di molecole trofiche e immunoregolatorie. In base alle segnalazioni esterne, le MSCs possono esplicare sia un’azione immunosoppressiva che immunostimolatoria. In particolare, MSCs espanse ex-vivo risultano in grado di inibire la proliferazione di cellule T,
cellule B, natural killer e la formazione di cellule presentanti l’antigene. L’inibizione dei linfociti T viene esplicata in seguito alla secrezione di molecole solubili come Trasforming Growth Factor β1 (TGFβ1) e Hepatocyte Growth Factor (HGF) e consiste in una soppressione sia della proliferazione che dell’apoptosi dei linfociti stessi (Di Nicola et al., 2002). Anche nei confronti dei linfociti B è stata dimostrata un’attività immunoregolatoria da parte delle MSCs. In questo caso, non viene bloccata solamente la capacità proliferativa delle cellule, ma anche quella di differenziare verso cellule che producono anticorpi e la chemotassi. Inoltre, pare che proprio un cross-talk fra MSCs e linfociti B sia alla base della secrezione di molecole solubili inibitorie come TGFβ1, HGF, prostaglandine E2 (Corcione et al., 2006). Anche in vivo, le MSCs sembrano svolgere l’azione immunosoppressiva. Questo è stato evidenziato in pazienti con Graft-Versus-Host Disease (GVHD) in cui il trapianto di MSCs, prelevate dal midollo della madre, ha portato il recupero dalla patologia. Inoltre è stato anche evidenziato in modelli sperimentali murini in cui co-trapiantando di MSCs singeniche e cellule ematopoietiche allogeniche, si riscontra un aumento significativo dell’attecchimento a lungo termine del midollo osseo. Questo suggerisce che le MSCs possono indurre una tolleranza nei confronti dell’ospite. Parallelamente all’azione immunosoppressiva, è stata recentemente evidenziata anche un’azione immunostimolatoria (Stagg, 2006).
Le MSCs possono secernere anche un ampio spettro di fattori di crescita e citochine che agiscono sulle cellule limitrofe. Infatti le MSCs offrono alle HSCs non solo un sito di attracco per interazioni cellula-cellula, ma le sostengono tramite la produzione di fattori trofici come Stem Cell Factor (SCF), Granulocyte Colony Stimulating Factor (G-CSF), Macrophage Colony Stimulating Factor (M-CSF) e Interleuchina 6 (IL-6). L’azione trofica viene esplicata dalle MSCs anche in seguito a danno tissutale. In seguito alla produzione di segnali infiammatori, le MSCs vengono reclutate producendo a loro volta fattori trofici che inibiscono l’apoptosi e la cicatrizzazione e stimolano l’angiogenesi e la divisione e differenziamento dei progenitori endogeni. Questo meccanismo d’azione è particolarmente importante in terapia cellualre. Infatti, la somministrazione di MSCs in seguito a infarto infatti è associata a un aumento della produzione di Vascular Endothelial Growth Factor (VEGF), un aumento della densità vascolare e del flusso sanguigno e una diminuzione dell’apoptosi (Caplan and Dennis, 2006).
Grazie a queste caratteristiche, le MSCs assumono una posizione sempre più importante come candidate per la terapia cellulare.
3.0 Adipogenesi
Per lungo periodo si è ritenuto che il tessuto adiposo fosse un tessuto deputato esclusivamente all’ accumulo di energia, ma negli ultimi decenni ne è stata rivalutata l’importanza per due ragioni: innanzitutto, la scoperta della leptina ha elevato il tessuto adiposo a elemento cruciale nell’omeostasi dell’organismo, inoltre la presa di coscienza di una crescente obesità mondiale ha posto l’accento sull’aumento delle patologie e della mortalità ad essa legate. Il tessuto adiposo si ritrova in diversi siti dell’ organismo, generalmente in regioni con tessuto connettivo lasso, ma depositi di grasso si possono ritrovare anche in organi interni (Rosen et al., 2000). Esistono due tipi di tessuto adiposo: tessuto adiposo bianco (White Adipose Tissue: WAT) e tessuto adiposo bruno (Brown Adipose Tissue: BAT). Il BAT possiede una maggiore componente mitocondriale ed esprime quasi tutti i geni espressi dal WAT. E’ caratterizzato dall’espressione di geni specifici come Uncoupling Protein-1 (UCP-1) che permette di generare calore dissipando il gradiente protonico formatosi a cavallo della membrana interna mitocondriale durante la catena respiratoria. In questa maniera l’energia viene dissipata piuttosto che accumulata. In termini fisiologici la funzione del BAT è quella di proteggere dal freddo e dall’obesità. Gli esseri umani possiedono una discreta quantità di BAT durante l’infanzia, ma soltanto in parte di questo viene mantenuto durante la vita adulta. I roditori, invece, mantengono il BAT anche durante la vita adulta. Il WAT è invece principalmente deputato all’accumulo e mobilitazione dell’energia sottoforma di triacilgliceroli, in base alle condizioni metaboliche dell’organismo. Inoltre ha capacità secretoria come dimostrato in seguito alla scoperta di molecole quali la leptina. Quello degli adipociti è uno dei possibili destini delle MSCs. Numerosi studi sono stati condotti per comprendere i meccanismi molecolari alla base della differenziazione adipogenica, tuttavia questo processo non è ancora del tutto chiaro. Si può approssimare affermando che la differenziazione adipogenica è un processo bi-fasico con una prima fase di determinazione e una di differenziazione terminale. La determinazione consiste nel commitment delle cellule staminali verso i progenitori adipogenici. Anche se morfologicamente indistinguibili a causa della mancanza di markers molecolari specifici, queste due tipologie cellulari sono funzionalmente distinte in quanto i pre-adipociti hanno perso la capacità di differenziare verso altri lineages che invece possono essere intrapresi dalle MSCs. Nella differenziazione terminale i pre-adipociti assumono le caratteristiche di adipociti maturi esprimendo geni specifici, come il recettore per l’insulina GLUT4 e la proteina legante gli acidi grassi (adipocyte-selective fatty acid binding protein:
aP2), e assumendo la morfologia caratteristica, con la formazione delle gocce lipidiche citoplasmatiche. La fase terminale è conosciuta più nel dettaglio, in quanto sono disponibili diverse linee cellulari di pre-adipociti che permettono di riprodurre in vitro la situazione fisiologica. Tra queste linee cellulari si ricordano la 3T3-L1 e 3T3-F442A che necessitano di due cicli di mitosi prima di differenziare. Il significato di queste divisioni ancora non è noto, ma molto probabilmente proteine coinvolte nei check-points sono anche implicate nella regolazione della differenziazione adipogenica (Rosen and Mc Dougald, 2006).
3.1 Adipogenesi: meccanismi molecolari
La differenziazione adipogenica prevede l’attivazione di set genici specifici necessari per il raggiungimento dello stadio di adipocita maturo. Un ruolo centrale viene svolto dal recettore nucleare Peroxisome Proliferator- Activated Receptor γ (PPARγ) e dai membri della famiglia CCAAT-Enhancer- Binding Proteins (C/EBPs). PPARγ è un membro della superfamiglia dei recettori nucleari attivati da ormoni. Questi, per legarsi al DNA e attivare la trascrizione dei geni targets, devono formare un dimero e legarsi a un ligando specifico. PPARγ formerà un eterodimero con il recettore per l’acido retinoico (RXR), ancora ignoto è però il suo ligando endogeno. Infatti non è ancora stata trovata una molecola fisiologica che si leghi con un’affinità paragonabile a quella degli altri recettori nucleari. Esistono due isoforme di PPARγ (PPARγ1 e PPARγ2) generate in seguito a trascrizione a partire da due promotori diversi e a splicing alternativo. PPARγ2 risulta più lunga dell’altra grazie a 30 aa all’ammino-terminale ed è la forma predominante negli adipociti; PPARγ1 è espressa invece anche in altri tipi di cellule, come macrofagi, pneumociti, epitelio della mammella, della prostata e del colon (Rosen et al., 2000). PPARγ è stato collegato allo sviluppo degli adipociti grazie ad esperimenti di acquisto e perdita di funzione. Infatti, l’espressione ectopica di PPARγ in cellule somatiche, come i fibroblasti riesce a indurre una forte adipogenesi (Tontonoz et al., 1994). Inoltre PPARγ può promuovere la transdifferenziazione di miociti in adipociti quando coespresso con C/EBPα (Hu et al., 1995). Con questi esperimenti si dimostra che PPARγ è sufficiente per indurre l’adipogenesi, per dimostrare che è anche necessario sono stati effettuati studi di perdita di funzione. Tuttavia i topi knockout muoiono allo stadio E10- 10.5 a causa di compromissioni della placenta (Barak et al., 1999). Il problema è stato ovviato grazie all’uso di due approcci differenti. Una stategia prevedeva la produzione di blastocisti chimeriche ottenute dall’unione di ES wild-type e PPARγ-/- (Rosen et al, 1999), un’altra
consisteva nella produzione di blastocisti con ES PPARγ-/- e cellule tetraploidi wild-type destinate alla formazione degli annessi extra-embrionali, compresa la placenta (Barak et al., 1999). In entrambi gli studi si è evidenziata la mancata formazione di WAT e una notevole riduzione del BAT. Sono stati condotti anche degli studi volti a chiarire il ruolo delle due diverse isoforme di PPARγ, ottenendo però dati contrastanti. In uno studio, in seguito a eliminazione delle due isoforme sulla linea 3T3-L1, è stata indotta l’espressione ectopica di PPARγ1 o di PPARγ2. L’espressione di quest’ultimo portava un recupero dell’adipogenesi, evento invece che non si realizzava con l’espressione di PPARγ1 (Ren et al., 2002). In un altro lavoro, si dimostra invece che entrambe le isoforme riescono a recuperare l’adipogenesi in fibroblasti PPARγ-/-, anche se con un’efficienza diversa (Mueller et al., 2002). Questi ultimi risultati hanno trovato conferma in esperimenti condotti su topi PPARγ2 knockout. In una linea di topi knock-out, si assisteva a una diminuzione del tessuto adiposo, ma non a una sua totale scomparsa e i fibroblasti embrionali (MEF), derivati da essi, avevano un’alterata adipogenesi (Zhang et al., 2004). In un’altra linea di topi, si è sviluppata resistenza all’insulina, ma una corretta formazione del tessuto adiposo. Questi risultati suggeriscono che PPARγ2 non è indispensabile per la differenziazione adipogenica. PPARγ risulta di notevole importanza anche nel mantenimento dello stato differenziato. L’espressione di una forma dominante negativa in adipociti maturi derivati da 3T3-L1 porta infatti una de-differenziazione e una perdita delle gocce lipidiche e dei markers specifici (Tamori et al., 2002). Altri geni coinvolti nell’adipogenesi appartengono alla famiglia delle proteine C/EBPs, una classe di fattori di trascrizione che possiedono il dominio cerniera di leucine. I membri più conosciuti sono C/EBPα, C/EBPβ, C/EBPγ, C/EBPδ e CHOP: i primi tre attivano l’adipogenesi, gli ultimi due la reprimono. Analisi condotte sulle linee cellulari di preadipociti indotte a differenziare mostrano che l’espressione di C/EBPβ e C/EBPδ è precoce e aumenta durante la differenziazione, CHOP è soppresso, mentre l’espressione C/EBPα viene indotta più tardivamente. Questi dati suggeriscono una fine regolazione reciproca fra i diversi elementi della famiglia (Rosen et al., 2000). Studi su topi doppi knockout C/EBPβ -/- e C/EBPδ -/- mostrano una riduzione del tessuto adiposo, ma negli adipociti presenti si riscontra comunque una normale espressione dei geni da loro indotti: C/EBPα e PPARγ. Questo risultato contrasta con dati ottenuti su MEFs C/EBPβ -/- e C/EBPδ -/- nei quali non si rileva espressione di C/EBPα e PPARγ. In vivo, quindi, si può ipotizzare la presenza di altri meccanismi che vicariano C/EBPβ e C/EBPδ, garantendo la maturazione degli adipociti
anche in loro assenza (Tanaka et al., 1997). Un ruolo più determinante è invece svolto da C/EBPα. Infatti topi knockout per questo gene muoiono subito dopo la nascita e per mantenerli in vita è necessario recuperarne l’espressione epatica. In questi topi è del tutto assente il WAT, mentre il BAT è normale. L’espressione ectopica di C/EBPβ non permette il recupero totale dei topi knockout, infatti si ha un recupero delle normali funzioni epatiche, ma una ridotta formazione del WAT. Quindi C/EBPα non può essere sostituito dagli altri membri della famiglia e possiede un ruolo fondamentale nella formazione del WAT. Nonostante l’importanza dei loro ruoli, le proteine C/EBPs non possono esplicare la loro funzione in assenza di PPARγ, infatti, ad esempio, C/EBPα non può garantire l’adipogenesi in fibroblasti PPARγ -/- (Rosen et al., 2002). L’espressione di PPARγ in cellule C/EBPα -/- mostra invece una corretta espressione genica e deposizione lipidica, ma viene sviluppata un’insensibilità all’insulina. Questi esperimenti suggeriscono il ruolo centrale rivestito da PPARγ nell’adipogenesi. Nella differenziazione adipogenica agiscono anche altri fattori come i membri della famiglia Kruppel Like Factor (KLF). Tra questi ricordiamo KLF5 e KLF15: il primo è indotto da C/EBPβ e C/EBPδ, che hanno elementi di risposta sul suo promotore, e agisce attivando PPARγ in concerto con i fattori C/EBPs (Oishi et al. 2005); il secondo promuove la differenziazione e l’espressione del recettore per il glucosio sensibile all’insulina GLUT4 (Gray et al., 2002). Altri membri della famiglia KLF agiscono inibendo la differenziazione adipogenica (Rosen et al., 2006). Tra i tanti altri fattori conosciuti che hanno effetti positivi sull’adipogenesi ricordiamo anche Krox20, che induce l’espressione di C/EBPβ, e la proteina legante l’elemento di risposta all’ cAMP (CREB). In questa maniera si può ipotizzare una cascata trascrizionale che vede una competizione iniziale fra fattori attivatori e repressori dell’adipogenesi. Quando prevalgono i fattori attivatori, tra i quali Krox20, si ha l’attivazione della cascata con il coinvolgimento iniziale di C/EBPβ e C/EBPδ. Questi vanno a stimolare, insieme a fattori appartenenti alla famiglia KLF e altri attivatori, C/EBPα e PPARγ, che si auto-sostengono e spingono in avanti la differenziazione.
Fig. 3.1 Meccanismi molecolari alla base dell’adipogenesi.
3.2 Adipogenesi: segnali extracellulari
L’attivazione dei pathways specifici che portano all’adipogenesi è il risultato dell’integrazione di segnalazioni extracellulari e intracellulari. Esistono diverse segnalazioni che in vitro modulano la differenziazione adipogenica, ma non tutte sono state confermate anche in vivo. Alcune vie hanno degli effetti inibitori: è il caso della segnalazione delle proteine Wnt che, legandosi al recettore 7TM Frizzled e al co-recettore LPR, attivano una via di segnalazione che si conclude con la stabilizzazione della β-catenina. Questa trasloca quindi nel nucleo dove interagisce con i fattori di trascrizione TCF/LEF, normalmente presenti sul promotori di geni target ma bloccati da co-repressori come Groucho. Il legame della β-catenina determina lo spostamento di Groucho e la regolazione dei geni bersaglio, portando, come risultato finale, l’inibizione dell’adipogenesi, bloccando l’espressione di PPARγ e di C/EBPα. Quest’azione inibitrice ad opera delle proteine Wnt è confermata da studi in cui la distruzione della via da esse attivata porta una maggiore differenziazione adipogenica. D’altra parte, l’espressione costitutiva di alcuni membri della famiglia, come Wnt10b, inibisce l’adipogenesi. Un’altra via che regola negativamente la differenziazione adipogenica è quella di Hedgehog. Queste proteine determinano l’attivazione di una via di segnale che termina con il coinvolgimento delle proteine GATA e di fattori di trascrizione appartenenti alla famiglia GLI e l’inibizione della differenziazione adipogenica. Sovraespressione di Sonic hedgehog porta infatti un blocco della differenziazione adipogenica nelle linee 3T3-L1. Le vie di segnalazione della superfamiglia TGFβ hanno degli effetti più complessi in quanto alcuni membri attivano l’adipogenesi, mentre altri la inibiscono. La superfamiglia TGFβ comprende tre famiglie: TGFβ, attivine e BMP. Le proteine appartenenti alla superfamiglia TGFβ si
legano a recettori serina/treonina chinasi e attivano una via di segnalazione fosforilando e attivando le proteine Smad. TGFβ fosforila Smad2 o Smad3, BMP agiscono su Smad1, Smad5 o Smad8. Una volta fosforilata, Smad si dissocia dal recettore e si lega a Smad4 con la quale trasloca nel nucleo e determina la regolazione dei geni targets. Le proteine Smad attivate da alcuni membri della famiglia TGFβ , una volta traslocate nel nucleo, si legano alle C/EBPs e causano l’inibizione della trascrizione di PPARγ. La miostatina (un membro della famiglia TGFβ) può invece sia attivare che inibire l’adipogenesi in vitro, in base al tipo di cellule e alle condizioni di coltura. Alcuni membri della famiglia delle BMP (BMP2 e BMP4) invece attivano il programma adipogenico agendo su un Schnurri-2 il quale, traslocando nel nucleo, crea uno scaffold per specifiche proteine SMAD e C/EBPs sul promotore di PPARγ. Tra le vie che attivano la differenziazione adipogenica ricordiamo quella di Notch. Una via coinvolta nella differenziazione adipogenica è quella delle chinasi attivate da mitogeni (MAPK). La loro attivazione nelle fasi precoci determina una necessaria fase di proliferazione, nelle fasi terminali causa invece una fosforilazione di PPARγ con conseguente inibizione della differenziazione. Quindi risulta necessaria una loro regolazione tramite l’azione di MAPK fosfatasi. Una segnalazione molto importante è quella mediata dall’insulina. Questa, nella prima fase della differenziazione, si lega ai recettori per Insulin Growth Factor-1 (IGF-1) e attiva le Insulin Receptor Substrate proteins (IRS). Esistono diversi tipologie di IRS e loro mutazioni hanno effetti più o meno gravi sull’adipogenesi. Le vie attivate dal legame dell’insulina con il suo recettore sono molteplici e si concludono con l’attivazione del programma adipogenico (Rosen and McDougald, 2006).
3.3 Cross-regolazione del destino delle cellule mesenchimali
Le MSCs possono intraprendere diversi destini differenziativi. La scelta di un destino piuttosto che un altro è finemente regolata, e, una volta presa, i fattori che la attivano contemporaneamente inibiscono le altre vie. Questo è vero anche per la decisione fra adipogenesi e osteogenesi. La comprensione del meccanismo che sta alla base di questa scelta risulta, quindi, di fondamentale importanza anche in campo clinico in quanto permetterebbe lo sviluppo di terapie mirate volte a limitare gli effetti dovuti a eventuali squilibri. L’alterazione del bilanciamento fra la differenziazione osteogenica e quella adipogenica delle MSCs è infatti presente in diverse patologie umane. Per esempio, l’invecchiamento e l’uso di corticosteroidi portano un aumento dell’adipogenesi nel midollo osseo e una diminuzione della formazione dell’osso, mentre in pazienti con iperplasia ossea si riscontra un incremento dell’ossificazione anche in posizione ectopica, come all’interno del tessuto adiposo. Diverse sono le vie di segnalazione coinvolte: primi fra tutti i master genes di ciascun destino (PPARγ e Runx2), le Bone Morphogenetic Proteins (BMPs), le proteine Wnt, Hedgehogs, la segnalazione Notch/Delta, le Fibroblast Growth Factor (FGF), l’Insulina, l’Insulin-like Growth Factor (IGF).
Diversi dati, ottenuti sia tramite analisi in vitro che in vivo, sostengono il ruolo inibitorio di PPARγ nel processo osteogenico, il suo effetto non è però tutto o nulla, bensì dipende dal ligando utilizzato per la stimolazione. Attivatori potenti come il rosiglitazone riescono infatti a inibire totalmente l’osteogenesi bloccando fattori chiave per questo processo come Runx2, OSX e Dlx5. Stimolazione di PPARγ con un ligando a bassa affinità, come il troglitazone, non mostra alcuna inibizione dell’osteogenesi. A conferma di questi dati, è stato evidenziato che una down-regolazione di PPARγ porta una diminuzione della massa grassa e un aumento dell’osteogenesi in vitro e della massa trabecolare in vivo.
Come detto precedentemente, la segnalazione mediata dalle BMP stimola l’adipogenesi. Queste hanno però una duplice azione in quanto stimolano anche l’osteogenesi agendo su OSX in maniera dipendente o indipendente da Runx2. Ciò è possibile grazie al legame delle BMP a recettori diversi, che quindi favoriscono una via piuttosto che un’altra: il recettore BMP-IA trasduce un segnale adipogenico, mentre BMP-IB uno osteogenico. Inoltre, pare che le BMP abbiano un effetto dose-dipendente in quanto cellule staminali embrionali vengono indotte verso un destino adipogenico quando trattate con una bassa concentrazione di BMP-2, mentre intraprendono un cammino osteogenico a concentrazioni elevate.
Fig.3.3 Duplice ruolo della segnalazione delle BMPs.
La segnalazione di Wnt, come detto precedentemente, ha un’azione inibitoria nei confronti dell’adipogenesi, in quanto va a bloccare l’espressione di PPARγ e C/EBPα, in vitro. Conferma di ciò viene da esperimenti di distruzione della β-catenina in cui si assiste a una differenziazione spontanea in adipociti sia in vitro che in vivo. La via di Wnt, oltre a inibire l’adipogenesi, attiva l’osteogenesi. Questa relazione è suggerita dall’analisi di pseudo-gliomi, dove si assiste a un notevole incremento o una diminuzione della massa ossea in base alla presenza di mutazioni con acquisto o perdita di funzione del co-recettore LPR5. Inoltre, la distruzione mirata della β-catenina in vivo causa una totale mancanza dello scheletro, mentre una sua stabilizzazione induce l’espressione di marcatori osteogenici precoci in linee di cellule staminali.
Anche la segnalazione delle proteine appartenenti alla famiglia Hedgehogs sono coinvolte nell’inibizione dell’adipogenesi e nella concomitante stimolazione dell’osteogenesi, tramite aumento dell’espressione di Runx2 e Alp. Inoltre Sonic hedgehogs agisce insieme a BMP-2 potenziando la differenziazione ostogenica. Recenti studi hanno dimostrato il coinvolgimento di un ulteriore fattore nella regolazione del destino delle cellule staminali mesenchimali: si tratta della proteina TAZ, un fattore di trascrizione con dominio di legame PDZ. TAZ pare che interagisca direttamente con i master genes dell’adipogenesi e dell’osteogenesi (PPARγ e Runx2, rispettivamente) causando un’inibizione della prima e un’attivazione della seconda. In particolare, è stato visto che TAZ è incrementato in seguito a stimolazione della linea murina di mioblasti C2C12 con BMP-2 e che a sua volta causa un incremento dell’espressione dell’osteocalcina, marker terminale dell’osteogenesi, tramite l’azione su Runx2. Inoltre, un
diminuzione della segnalazione di TAZ comporta una compromissione dell’osteogenesi, come dimostrato sia da esperimenti in vitro tramite l’uso di siRNA, che in vivo su embrioni di zebrafish trattati con morfolino. Oltre a stimolare l’osteogenesi, TAZ determina un’inibizione di PPARγ in seguito al legame diretto sulle sequenze consensus presenti nei suoi geni targets, come evidenziato da esperimenti di co-immunoprecipitazione sulla linea di pre-adipociti 3T3-L1 (Hong et al., 2005). Esistono anche delle segnalazioni che hanno effetti positivi su entrambi i processi differenziativi: è il caso della segnalazione delle FGF, insulina e IGF (Muruganandan, Roman and Sinal, 2009). Nella scelta fra i due diversi destini sembrano essere coinvolte anche alcune caratteristiche strutturali come la morfologia cellulare e la tensione citoscheletrica. hMSC soggette a una forte tensione citoscheletrica tendono più facilmente a un destino osteogenico. Inoltre mutazioni a livello del gene per la Guanine Nucleotide- Binding Protein Stimulatin Activity polipeptide 1 (GNAS1) sono state riscontrate in pazienti con eteroplasia ossea che porta alla formazione di osso ectopico. Questo suggerisce che anche proteine G sono coinvolte nella decisione del destino delle MSCs (Nuttall and Gimble, 2004).
4.0 Kit
L’oncogene virale v-kit è stato identificato per la prima volta nel 1986, come gene trasformante dell’Hardy- Zuckerman 4 feline sarcoma virus. Solo successivamente venne anche identificato il suo omologo cellulare c-kit e venne mappato a livello del locus W. Di questo locus erano conosciute già da tempo mutazioni con fenotipi caratteristici come difetti di pigmentazione, anemia e ridotta fertilità. Era stato quindi ipotizzato che il relativo prodotto genico fosse coinvolto nella melanogenesi, nell’ematopoiesi e nella gametogenesi. Mutazioni sul locus Sl generavano un fenotipo analogo, suggerendo una relazione tra i due prodotti genici. Solo con studi successivi si capì che i loci codificavano per una coppia recettore/ligando. Esistono diverse mutazioni del locus W con diversi gradi di gravità. Una delle prime evidenziate fu appunto la mutazione W in cui il recettore Kit è assente nella cellula a causa di una delezione di 78 amminoacidi a livello del dominio transmembrana. Topi omozigoti per questa mutazione muoiono in utero o nel periodo perinatale. Sono state riscontrate anche altre mutazioni a carico del gene Kit: si tratta per lo più di mutazioni puntiformi che aboliscono l’attività tirosina-chinasica, tra queste ricordiamo la mutazione Wv
. Topi omozigoti per questa mutazione sono vitali, ma manifestano una forte anemia, sono bianchi e sterili (Nocka et al., 1990). Esperimenti dell’ultimo decennio hanno mostrato come topi mutanti W, oltre alle alterazioni macroscopiche a carico delle cellule ematopoietiche, germinali e dei melanoblasti, mostrino delle anomalie in altri sistemi in cui la via di Kit è attiva. In questi topi mutanti infatti sono state trovate alterazioni dell’attività delle cellule interstiziali del Cajal (Huizinga et al., 1995) sia deficit dell’apprendimento e della memoria spaziale. Le linee di topi che recano mutazioni a carico del recettore Kit rappresentano quindi un ottimo modello di studio per indagarne il ruolo anche in quei sistemi in cui precedentemente non era stato evidenziato un fenotipo macroscopico. Nell’uomo, alterazioni dell’espressione e funzione del recettore sono state associate a diverse patologie. La perdita di funzione di Kit è stata riscontrata nel piebaldismo e in diverse forme di tumore come melanoma e cancro alla tiroide. Un acquisto di funzione è stato associato ad altre forme tumorali come il tumore gastrointestinale (GISTs) e la leucemia mieloide acuta. Infine, alterazioni della via di segnalazione, con la produzione da parte del tumore sia del ligando che del recettore, sono state riscontrate nel carcinoma del polmone a piccole cellule e nel neuroblastoma (Rönnstrand, 2004). Visto il coinvolgimento in patologie purtroppo molto
diffuse, lo studio del gene Kit risulta di particolare importanza, al fine di comprendere appieno il suo meccanismo d’azione e poter sviluppare farmaci mirati.
4.1 Struttura e isoforme del recettore Kit
Kit, anche conosciuto come CD117, è un membro della famiglia dei recettori tirosin-chinasi di tipo III. A questa famiglia appartengono anche altri recettori come c-fms e il Platelet-Derived Growth Factor (PDGF) receptor. Questi recettori possiedono una porzione extracellulare con 5 domini immunoglobulinici, una regione transmembrana e un dominio citoplasmatico con l’attività tirosin-chinasica. I 3 domini Ig più distali sono deputati al riconoscimento del ligando specifico, in questo caso lo Stem Cell Factor (SCF), il 4° dominio è utile per la dimerizzazione, mentre il 5° dominio ha una funzione sconosciuta. Il dominio tirosin-chinasico è bipartito a causa della presenza di una regione intermedia che separa il dominio di legame all’ATP dal dominio di fosforilazione.
Fig. 4.1 Struttura del recettore Kit.
Questo recettore è codificato da un gene di 21 esoni, in una regione del DNA di 81 Kb che mappa sul cromosoma 5 nel topo e sul cromosoma 4 nell’uomo. La proteina è di 145- 150 KDa ed è glicosilata. Esistono due isoforme di Kit nel topo e quattro nell’uomo, prodotte in seguito a splicing alternativo, inoltre esiste una forma solubile generata in seguito a taglio
proteolitico. Le isoforme, presenti sia nel topo che nell’uomo, sono caratterizzate dalla presenza o assenza di un tetrapeptide (Gly Asn Asn Lys) nella regione extracellulare. Questa isoforma è generata in seguito all’uso alternativo di un sito donatore di splicing. Studi volti a chiarire le differenze funzionali tra queste due isoforme hanno dimostrato che la versione priva del tetrapeptide, pur avendo la stessa affinità per SCF, risponde in maniera più forte e repentina. Un’altra variante, presente solo nell’uomo e non nel topo, è caratterizzata dalla presenza o meno di una singola serina nella regione interchinasica, in seguito all’uso alternativo di un sito accettore di splicing. Inoltre è presente anche una versione troncata espressa solo nelle cellule germinali post-meiotiche del testicolo. Questa forma possiede esclusivamente il secondo dominio chinasico Kit solubile (Kits) è normalmente presente nel siero ed è prodotto in seguito a taglio proteolitico del recettore. Si può ipotizzare che la produzione di una versione solubile sia un meccanismo di regolazione, infatti Kits lega SCF con alta affinità e ne previene il legame con Kit di membrana. Esistono anche altri meccanismi di regolazione di Kit sia a livello trascrizionale che post-traduzionale: infatti, in seguito al legame con l’SCF, il recettore viene internalizzato tramite vescicole ricoperte di clatrina e degradato in seguito a ubiquitinizzazione (Broudy, 1997; Ashman, 1999 ; Rönnstrand, 2004).
4.2 Kit: trasduzione del segnale
L’attivazione del recettore Kit avviene in seguito a legame con il suo ligando specifico: l’SCF. Si assiste quindi alla formazione di un omodimero e all’autofosforilazione a livello di tirosine specifiche. La presenza delle tirosine fosforilate permette il legame di proteine con domini Src homology 2 (SH2) o phospo-tyrosine binding domains (PTB). Studi recenti hanno dimostrato che, in assenza della segnalazione dell’SCF, l’autofosforilazione di Kit viene inibita grazie a uno specifico dominio citoplasmatico giustapposto alla membrana. Questo dominio pare esplicare la sua attività inibitoria, grazie all’acquisizione di una conformazione a α-elica che gli consente di bloccare fisicamente il primo dominio chinasico. La fosforilazione in seguito al legame dell’SCF porta un cambiamento conformazionale permettendo la liberazione del dominio chinasico e la formazione di docking sites per proteine adattatrici. Mutazioni a livello di questa regione portano attivazione costitutiva del recettore e sono state riscontrate nei tumori gastrointestinali (GISTs) (Chan et al., 2003; Ma et al., 1999).
Fig. 4.2 Schema di azione del dominio auto-inibitorio del recettore Kit.
In seguito all’autofosforilazione di Kit e al legame delle proteine adattatrici, vengono indotte diverse vie di segnalazione: Ras/Erk; PhosphoInositol3Kinase (PI3K); PhospholipaseCγ (PLC γ); JAK/STAT.
La via Ras/Erk è generalmente associata alla divisione e sopravvivenza cellulare. Una volta creati i siti di attracco, si lega la proteina adattatrice Grb2 tramite il dominio SH2. Questa proteina recluta Sos che facilita lo scambio GDP/GTP in proteine G come Ras. Quando legata con il GTP, Ras si trova nella sua forma attiva e a sua volta attiva Raf-1 che è una serina-treonina chinasi. Raf-1 fosforila, attivando, Mek1 e 2, e questi ultimi a loro volta fosforilano e attivano Erk 1 e 2 che traslocano nel nucleo e si legano sui promotori dei loro geni targets. PI3K causa la fosforilazione del fosfatidilinositolo-4,5-bisfosfato (PIP2) producendo il
fosfatidilinositolo-3,4,5-trisfosfato (PIP3). Quest’ultimo permette il reclutamento e
l’attivazione di proteine contenenti domini di pleckstrin homology (PH), come l’Akt che può quindi fosforilare i suoi bersagli. Questa via di segnalazione è associata alla divisione mitotica, alla differenziazione, sopravvivenza, adesione, secrezione e riorganizzazione dell’actina citoscheletrica. PLCγ agisce su PIP2 producendo diacilglicerolo (DAG) e
inositolo-1,4,5-trisfosfato (IP3). DAG rimane associato alla membrana attivando la chinasi PKC, IP3 migra
verso il reticolo endoplasmatico dove innesca il rilascio di Ca+ all’interno del citoplasma. La via di PLCγ agisce sulla proliferazione, chemiotassi e adesione, i risultati sul suo coinvolgimento risultano però controversi. Dal recettore Kit viene attivata anche la via JAK/STAT. In particolare Kit fosforila in maniera rapida e transiente JAK2, che risulta costitutivamente legata al recettore. JAK2 attivato va a fosforilare le proteine STAT (STAT1, STAT3, STAT5A e STAT5B). STAT, una volta fosforilato, dimerizza e trasloca nel nucleo dove attiva la trascrizione di geni targets che agiscono sul controllo della proliferazione e dell’apoptosi (Rönnstrand, 2004). Tutte le vie di segnalazione che partono dal recettore Kit
hanno quindi un’azione sinergica in quanto agiscono sul controllo dell’adesione e chemiotassi, della proliferazione e dell’apoptosi.
Fig. 4.3 Cascata del segnale attivata dal legame dell’SCF con Kit.
Infine, esistono diversi meccanismi deputati allo spegnimento della segnalazione di Kit:
l’attivazione di tirosin-fosfatasi, come SHP-1, che, rimuovendo i gruppi fosfati dalle tirosine, riportano il recettore alle condizioni di riposo;
l’internalizzazione del recettore tramite vescicole di clatrina, seguita da ubiquitinizzazione e degradazione proteica;
produzione della forma solubile in seguito a taglio proteolitico della regione extracellulare.
4.3 Espressione di Kit: uno stem cell gene
Kit risulta coinvolto in diversi tipi di cellule staminali, agendo probabilmente con meccanismi comuni. La sua segnalazione, infatti, è coinvolta nella migrazione, proliferazione e sopravvivenza di cellule staminali e precursori ematopoietici, cellule staminali cardiache, cellule germinali primordiali (PGC), cellule staminali neurali dell’occhio e melanoblasti. L’espressione di Kit è inoltre stata riscontrata anche a livello di cellule interstiziali del Cajal, dove risulta indispensabile per il loro sviluppo post-natale, la loro proliferazione e la loro attività pacemaker (Klüppel et al., 1998). Al fine di indagare a fondo l’azione di Kit, risulta interessante individuare in quale momento dello sviluppo e in quali cellule è espresso. Questo
è stato possibile grazie alla produzione di topi knockin in cui il gene LacZ è inserito nel primo esone del gene Kit. Esperimenti condotti su cellule in cui entrambi gli alleli portavano LacZ hanno dimostrato che, in assenza di Kit, la migrazione, la proliferazione e la sopravvivenza sono compromesse nelle PGC a partire da E9.5, nei melanoblasti a partire da E11 e nei precursori ematopoietici da E11-E12 in poi. L’espressione di Kit è stata riscontrata anche nelle cellule interstiziali di Cajal, ma qui non pare essere fondamentale durante lo sviluppo embrionale (Bernex et al., 1996). Nell’organismo adulto, Kit è espresso da circa il 70% delle cellule ematopoietiche CD34+, inclusi i progenitori ematopoietici e le LT-HSCs. Studi effettuati con anticorpi bloccanti hanno dimostrato che Kit è fondamentale nella formazione della componente mieloide, ma non risulta necessario per quella linfoide. L’espressione di Kit inoltre subisce un calo durante la differenziazione delle filiere ematopoietiche, ad eccezione dei mastociti nei quali è mantenuta un’elevata attività. La generazione di altre linee transgeniche ha permesso di studiare la regolazione dell’espressione di Kit nelle cellule ematopoietiche e nelle PGC. In questo caso i topi transgenici sono stati ottenuti ponendo il gene per la Green Fluorescent Protein (GFP) sotto il controllo di alcune sequenze regolatorie del gene Kit. A livello del promotore e del primo introne del gene Kit, sono stati identificati 6 siti ipersensibili (HS) all’azione della DnasiI. Sono stati creati 3 diversi costrutti: nel primo costrutto la GFP è clonata a valle del promotore e del sito HS1, il secondo costrutto contiene anche il sito HS2, mentre nel terzo sono presenti tutti e sei i siti.
Fig. 4.4 Costrutti Kit/GFP.
Analisi dell’espressione del transgene hanno mostrato che il primo costrutto è sufficiente a guidare l’espressione nelle PGCs, ma fallisce negli altri sistemi. Il costrutto 2, invece,