2.1 Introduzione
In questo capitolo verranno descritte le tecniche di coltura cellulare in quanto, grazie ad esse, è stato possibile ricreare in vitro delle reti neuronali, usate come modello di studio per analizzare i processi di auto-organizzazione del sistema nervoso.
Numerosi studi precedenti a questo lavoro hanno dimostrato che singoli neuroni coltivati in vitro non possono essere considerati come semplici elementi, ma sviluppano connessioni sinaptiche organotopiche, formando una complessa struttura morfologica, e esibiscono una grande varietà di proprietà elettriche simili a quelle osservate in vivo. [1, 2, 3]
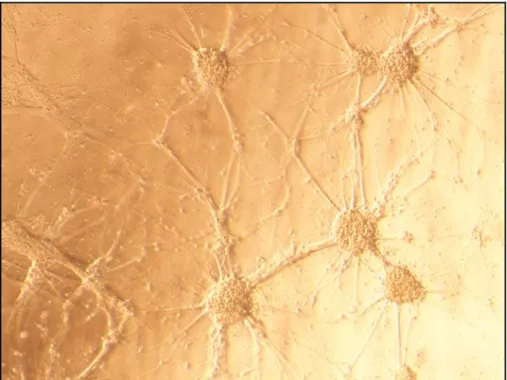
Le reti neurali in vitro ci hanno permesso di realizzare osservazioni ottiche non invasive e quindi di seguire la dinamica della crescita neurale e dell’organizzazione della rete (figura 2.1).
Figura 2.1 Esempio di rete neurale in vitro da noi realizzata
2.2 Cenni sulle colture
Nel 1952 è stata messa in coltura la prima linea cellulare stabilizzata umana, proveniente da un tumore alla cervice: la linea HeLa. Da allora il contributo delle colture cellulari alla ricerca in campo biomedico è stato estremamente significativo, anche per la varietà dei campi d’applicazione considerabili.
I vantaggi offerti dalle colture cellulari sono: 9 semplicità e elevata riproducibilità;
9 possibilità di analisi dei meccanismi cellulari e molecolari del fenomeno in esame;
9 controllo ambientale;
9 economicità e rapidità di risposta; 9 disponibilità.
9 sistemi semplificati rispetto ad un organismo integrato;
9 condizioni di esposizione alle sostanze diverse da quelle in vivo;
9 difficoltà di correlare le concentrazioni in vitro con quelle in vivo;
9 le sostanze inoculate possono interagire con il terreno di coltura.
2.3 Tipi di colture
Le cellule in coltura crescono in apposite fiasche, piastre o provette di plastica contenenti un terreno appropriato, all’interno di un apparecchio denominato incubatore (figura 2.2), in cui l’atmosfera è mantenuta a concentrazioni fisse di CO2, a un’umidità del 90% e ad
una temperatura di 37°C. [4, 5, 6, 7, 8, 9, 10, 11]
Una coltura di cellule in vitro si realizza isolando le cellule da un tessuto e ponendole in presenza di tutti i fattori e i metaboliti necessari alla loro crescita.
L’isolamento delle cellule si ottiene triturando il tessuto e sottoponendo i frammenti all’azione di agenti chelanti come l’EDTA e di enzimi proteolitici come tripsina o collagenasi che degradano la matrice del tessuto.
Le cellule isolate da un tessuto sono molto eterogenee in quanto, oltre alle cellule del parenchima tissutale, sono presenti in varia proporzione anche quelle del sangue, dell’endotelio, del connettivo e altre ancora a seconda del tessuto analizzato.
Parecchi approcci sono usati per separare i diversi tipi cellulari da una sospensione cellulare mista.
Uno sfrutta differenze nelle proprietà fisiche: le cellule più grosse possono essere separate da quelle più piccole e le cellule più dense da quelle meno dense mediante centrifugazione.
Un altro approccio si basa sulla tendenza di alcuni tipi cellulari ad aderire con forza a vetro e plastica, il che permette di separarli da cellule che aderiscono con minor forza.
Un perfezionamento importante di quest’ultima tecnica dipende dalle proprietà di legame specifiche degli anticorpi. Anticorpi che legano specificamente la superficie di un solo tipo cellulare in un tessuto possono essere accoppiati a varie matrici – come collageno, granuli di polissacaridi o plastica – per formare una superficie di affinità a cui possono aderire soltanto le cellule riconosciute dagli anticorpi. Le cellule legate sono poi recuperate mediante blanda agitazione, trattamento con tripsina per digerire le proteine che
mediano l’adesione o, nel caso di una matrice digeribile (come il collageno), degradando la matrice stessa con enzimi (come la collagenasi).
Una delle tecniche più sofisticate di separazione cellulare usa un anticorpo accoppiato ad un colorante fluorescente per marcare cellule specifiche. Le cellule marcate possono essere poi separate da quelle non marcate in un separatore cellulare attivato dalla fluorescenza elettronico. In questo strumento, singole cellule che viaggiano in fila indiana in un flusso sottile passano attraverso un raggio laser e la fluorescenza di ciascuna cellula viene misurata rapidamente. Un ugello vibrante genera minuscole goccioline, la maggior parte delle quali contiene una cellula o nessuna. Alle goccioline che contengono una singola cellula viene data automaticamente una carica positiva o negativa al momento dell’informazione, a seconda che la cellula che contengono sia fluorescente o no; le goccioline sono quindi deviate da un forte campo elettrico in un contenitore appropriato. Gli occasionali aggregati di cellule, rivelati dalla loro aumentata dispersione della luce, sono lasciati privi di carica e sono scartati in un contenitore di rifiuti. Queste macchine possono selezionare accuratamente 1 cellula fluorescente su un pool di 1000 cellule non marcate e separare parecchie migliaia di cellule al secondo.
In alcuni casi si possono usare terreni o condizioni di coltura selettivi che permettono la crescita di alcuni tipi cellulari rispetto ad altri.
2.3.1 Colture primarie e linee cellulari continue
Le colture preparate direttamente da un tessuto si dicono colture
primarie o “colture a termine”, per indicare il fatto che le cellule
isolate da un qualsiasi tessuto animale sono in grado di compiere un numero finito di divisioni cellulari in vitro, dopo le quali vanno incontro a degenerazione e morte. Tale fenomeno avviene indipendentemente dalla presenza di metaboliti appropriati per la crescita e si indica come senescenza.
Nel ciclo di vita di tali cellule sono state identificate tre fasi (figura 2.3):
1. fase iniziale di adattamento;
2. fase logaritmica, in cui il numero di cellule cresce in maniera esponenziale (in presenza delle appropriate condizioni);
3. senescenza e morte.
La senescenza cellulare è un fenomeno controllato a livello genetico e sembra sia dovuto a una riduzione dell’attività dell’enzima teleomerasi, la cui funzione è quella di riparare i teleomeri, brevi sequenze di DNA che si trovano all’estremità del cromosoma. I teleomeri proteggono i cromosomi durante la divisione cellulare e ad ogni duplicazione si accorciano di una certa quantità e, quando la loro lunghezza non è più in grado di proteggere la cellula, essa inizia a riprodursi in modo scorretto generando l’invecchiamento.
In genere il numero di “cicli” che una cellula è in grado di effettuare è inversamente proporzionale all’età dell’animale da cui sono prelevati i tessuti. Non a caso le cellule di derivazione embrionale sono quelle che possono essere mantenute in coltura in vitro più a lungo.
Le colture a termine non permettono di far crescere un numero elevato di cellule e il loro isolamento è una procedura relativamente complessa. Questi limiti hanno indotto i ricercatori ad isolare dei ceppi clonali che fossero capaci di una crescita illimitata: le linee
cellulari continue.
Le linee cellulari continue derivano da singole cellule in cui il programma genetico della senescenza è stato annullato attraverso mutazioni spontanee o indotte. In presenza degli opportuni metaboliti, sono capaci di proliferare in modo continuo e vengono per questo definite immortali.
Le linee cellulari continue sono derivate da colture primarie di tumori (ad esempio HeLa) o da manipolazioni genetiche di colture primarie non tumorali. Tali manipolazioni prevedono l'inserimento
di specifici geni virali o anche la sola propagazione per molti passaggi di una coltura primaria. Le cellule trasformate presentano caratteristiche simili alle cellule cancerose: proliferano in vitro fino a raggiungere una densità maggiore delle cellule normali, risentono poco dell’inibizione da contatto e, spesso, crescono senza legarsi ad alcuna superficie.
2.3.2 Colture aderenti e in sospensione
Le colture si distinguono a seconda che le cellule siano in sospensione o aderenti.
Le cellule di origine emopoietica, che normalmente vivono in un mezzo fluido, crescono in sospensione nel terreno di coltura e sono capaci di moltiplicarsi in vitro senza bisogno di aderire ad un substrato.
Le cellule che in vivo fanno parte di tessuti solidi crescono in vitro aderendo alla superficie delle piastre di coltura. L'adesione alle piastre è una condizione necessaria per la crescita in vitro e si tratta di un fenomeno attivo che, per avvenire, richiede l'interazione di recettori di membrana, le integrine, con le proteine adesive, quali la fibronectina, adsorbite sulla superficie della piastra di coltura.
2.4 Terreni per colture
Le cellule in vitro crescono in presenza di un appropriato terreno di coltura.
I terreni base disponibili in commercio contengono tutti i componenti nutritivi necessari alla crescita delle cellule, quali:
9 aminoacidi (essenziali e non essenziali), necessari per la sintesi delle proteine;
9 vitamine, che agiscono come catalizzatori o come substrati per facilitare o controllare alcune funzioni metaboliche;
9 zuccheri (generalmente glucosio), che rappresentano la principale fonte di energia o di carbonio per le biosintesi;
9 sali inorganici, essenziali per la crescita delle cellule e il mantenimento delle funzioni cellulari. Forniscono gli ioni sotto forma di sodio, magnesio, potassio, calcio, fosfato, cloruro, solfato e bicarbonato e agiscono anche come tampone per proteggere le cellule dalle fluttuazioni del pH dovute alle variazioni ambientali e ai prodotti del catabolismo.
I più comuni terreni base di coltura sono: ¾ il MEM: Minimum Essential Medium;
¾ il DMEM: Dulbecco’s Modification of MEM; ¾ l’RPMI: Roswell Park Memorial Institute.
Essi differiscono tra loro per il contenuto in amminoacidi e sali e per la concentrazione di glucosio.
La composizione esatta dei singoli terreni ed il tipo di terreno adatto per una data linea cellulare vengono di solito specificati dalla ditta produttrice.
Per la crescita, le cellule richiedono un valore di pH del mezzo compreso tra 7.2 e 7.4.
Per mantenere costante il pH, si usa in genere un terreno contenente un'elevata quantità di bicarbonato di sodio (NaHCO3), che funziona
da sistema tampone. In soluzione acquosa il bicarbonato si dissocia, determinando il rilascio di anidride carbonica (CO2) nell'incubatore
e l'aumento del pH nella fase acquosa. La concentrazione di CO2
della fase gassosa all'interno dell'incubatore tende a controbilanciare questo aumento, mantenendo così una ragionevole acidità nel terreno colturale.
Il pH del terreno dipende generalmente dalla percentuale di CO2
nell'incubatore. Un aumento nella pressione atmosferica di CO2
porta ad un abbassamento del pH che invece aumenta quando c'è una fuoriuscita di anidride carbonica. La concentrazione di CO2
richiesta può andare da 0,5% a 8,5% in funzione del contenuto di NaHCO3 nel terreno utilizzato e del pH desiderato (figura 2.4).
Figura 2.4 Relazione tra la concentrazione di NaHCO3 e il pH.
La linea solida rappresenta il 10% di CO2,, la tratteggiata il 5% di CO2.
Ad esempio, a 10 mM di NaHCO3, il pH è 6.91 per il 10% di CO2
Per avere un’indicazione visiva del pH, i terreni vengono addizionati di rosso fenolo, un indicatore che ha un colore rosso-arancio a pH 7.3, vira al giallo-rosso-arancio a pH acido e al rosso-viola a pH alcalino. In contatto con la CO2 dell’atmosfera, il terreno
tenderà ad alcalinizzare, diventando violaceo e quindi non più utilizzabile, perché tossico per le cellule. Con il proliferare della crescita cellulare, in incubatore al 5% di CO2, il terreno tenderà al
giallo a causa dell’acidificazione prodotta dal metabolismo cellulare. Se le piastre tenute in incubatore, invece, virano al violaceo, significa che la regolazione della CO2 del macchinario è
errata oppure che le cellule stanno morendo (non sono metabolicamente attive).
Il terreno base una volta preparato viene conservato a 4°C e, prima di essere utilizzato, in cappa sterile, viene completato con altri tre componenti instabili nel range di temperatura tra +2 e +8 °C, e perciò non contenuti in esso:
1. glutammina; 2. antibiotici; 3. siero.
La glutammina è un amminoacido essenziale molto labile, pertanto se il terreno è conservato per più di 15 giorni è necessario aggiungerla nuovamente fresca prima dell’uso.
Gli antibiotici vengono usati a scopo preventivo per evitare contaminazioni batteriche. Di norma si usano penicillina e streptomicina.
Il siero è la frazione di sangue che rimane dopo la coagulazione e la centrifugazione delle componenti cellulari e rappresenta il
supplemento più comune delle colture cellulari. È una miscela complessa di proteine ed elementi fondamentali per la crescita in vitro della maggior parte delle cellule, che non sopravviverebbero con il solo terreno base. Oltre a contenere fattori di crescita quali PDGF, EGF, IGF ecc., e almeno due fattori di adesione (fibronectina e vitronectina), il siero contiene anche altri elementi importanti per la crescita cellulare, quali la trasferrina, essenziale per il metabolismo del ferro, l'albumina, capace di veicolare vitamine e lipidi poco solubili nel mezzo acquoso, colesterolo, acidi grassi e glucocorticoidi, ed elementi minerali in tracce (Cu, Zn, Co, Mo, Va, Se) che funzionano come cofattori per alcuni enzimi cellulari.
Il siero di uso più comune nelle colture cellulari è il siero fetale di bovino (FBS).
2.5 Colture di cellule aderenti
Tutte le linee derivate da cellule di origine nervosa, epiteliale o mesenchimale crescono aderenti alle piastre di coltura.
Le cellule aderenti crescono fino ad occupare l’intera superficie disponibile: a questo stadio si dicono confluenti.
A confluenza la crescita si arresta e le cellule, per evitare che muoiano, devono essere staccate e trasferite in nuove piastre.
Per il trasferimento è necessario staccare le cellule dal fondo della piastra con una soluzione di EDTA e tripsina. L’EDTA chela il Ca2+ e il Mg2+, indispensabili per l’adesione, mentre la tripsina degrada le proteine della matrice che mantengono le cellule aderenti al substrato.
Avvenuto il distacco, l’azione dell’EDTA e della tripsina viene neutralizzata dall’aggiunta di nuovo mezzo di coltura che contiene cationi bivalenti in eccesso ed inibitori della tripsina (contenuti nel siero).
Le cellule vengono quindi contate, seminate in nuove piastre e fatte crescere per il tempo necessario.
2.5.1 Piastre per coltura
Le piastre per colture cellulari sono generalmente in polistirene. Il polistirene è un materiale rigido, duro e fragile, con superficie lucida. Presenta una buona resistenza chimica a soluzioni acquose, ma limitata resistenza ai solventi.
La caratteristica che lo ha reso ideale per l’uso in “tissue culture” è la totale mancanza di tossicità sulle cellule in coltura. In più, la sua notevole trasparenza consente un’osservazione diretta delle cellule al microscopio.
La superficie delle piastre è trattata chimicamente in modo da renderla idrofila e carica negativamente. Il polistirene è, in tal modo, capace di legare in modo stabile, anche se non covalente, i fattori di adesione presenti nel siero. [12]
Le piastre si possono distinguere in: fiasche;
capsule di Petri; piastre multiwell.
Figura 2.5 Piastre da coltura
Fiasche
Le fiasche sono contenitori ad imboccatura stretta, chiuse con tappo a vite.
Ne esistono di diverse dimensioni: 25, 75 e 150 cm2.
Mediante un trattamento adatto della superficie interna, il fondo della fiasca fornisce una superficie di crescita ottimale per le più diverse colture dipendenti da una matrice. Contemporaneamente, le superfici laterali e superiore, non trattate, permettono di ridurre le perdite di tessuto cellulare durante la fase di distacco, grazie alla riduzione dell’aderenza del tessuto al di fuori della zona di crescita. La speciale filettatura impedisce lo spanamento della stessa, permettendo un uso multiplo del tappo.
La superficie frontale, inclinata sotto l’apertura, limita il pericolo di traboccamento del liquido e permette uno svuotamento senza residui.
Bisogna stare attenti a non superare il volume massimo consigliato per evitare che il terreno esca fuori dall’apertura della fiasca quando questa è posizionata orizzontalmente.
Per facilitare la conta delle cellule al microscopio, in alcuni tipi di fiasche per colture di cellule aderenti, la superficie trattata è dotata di retinatura esterna e alcuni modelli hanno la graduazione stampata da entrambi i lati.
Le fiasche sono molto utili nel caso in cui le cellule debbano essere portate fuori dal laboratorio, ma sono meno comode per il trasferimento delle colture a causa della relativa difficoltà di accesso attraverso l’imboccatura.
Capsule Petri
Le capsule Petri sono di facile utilizzo in tutte le manipolazioni sperimentali successive alla semina e sono più economiche delle fiasche.
Le più usate sono quelle dal diametro di 5 e 9 cm, con area, rispettivamente, di circa 20 e 60 cm2.
Il design delle capsule per colture cellulari prevede la presenza di un anello zigrinato, che ne permette un’impugnatura facile e sicura. Inoltre, la struttura discontinua che caratterizza tale anello sulla parte superiore, riduce la formazione della condensa e l’eventuale adesione tra piastra e piastra.
Solamente la base della piastra, totalmente piatta, è trattata internamente per la crescita cellulare.
Le capsule vengono sterilizzate tramite raggi gamma e vengono poi confezionate in sacchetti con facile apertura di tipo “peel-off”.
Piastre multiwell
Le piastre multiwell sono costituite da diversi pozzetti, rotondi o rettangolari, con fondo a U oppure piatto.
Esistono piastre a 4, 6, 8, 12, 24 e 48 pozzetti, i cui bordi sono leggermente più alti del livello della piastra per ridurre il rischio di cross contaminazione.
Il trattamento a cui è sottoposto il fondo dei pozzetti fa sì che si ottenga una superficie di crescita ottimale per le cellule più diverse. Le superfici laterali interne non sono trattate.
Per assicurare corretti orientamento della piastra e posizionamento del coperchio, su piastra e coperchio sono presenti uno o due angoli smussati. Le zone zigrinate sui lati delle piastre e la forma del coperchio permettono di maneggiare con sicurezza o di separare il coperchio della piastra con facilità. Queste caratteristiche impediscono che in caso di piastre impilate, condizione che si presenta quando lo spazio a disposizione nell’incubatore non è sufficiente, vi sia aderenza tra il coperchio di una piastra e il fondo della successiva.
La configurazione delle piastre assicura un’ottima ventilazione delle colture, grazie alla regolazione degli scambi gassosi tra pozzetto e ambiente; ciò riduce l’evaporazione dei terreni di coltura, senza alterare la diffusione di CO2.
2.5.2 Scongelamento e semina delle cellule
Prima di iniziare la procedura occorre portare il terreno di coltura a 37°C, immergendolo nel bagnetto termostatato.
Se necessario, il fondo della piastra deve essere ricoperto con il gel e questa deve essere poi mantenuta nell’incubatore per circa 15-20 minuti.
La procedura deve essere la più rapida possibile e comprende diversi passi:
1. prendere la vails contenente le cellule dal congelatore ed immergerla nel bagnetto termostatato a 37°C fino al completo scongelamento;
2. pulire l’esterno della vails con alcool assoluto;
3. sotto cappa sterile aprire la vails e trasferirne il contenuto, con una pipetta, in una falcon sterile in cui erano stati preventivamente inseriti 3 ml di mezzo completo;
4. centrifugare per 5 minuti a 900 giri/minuto per eliminare il crioprotettore (DMSO);
5. aspirare il surnatante;
6. aggiungere terreno fresco e spipettare in modo da separare e risospendere le cellule nella provetta;
7. seminare le cellule nella piastra dalla quale, durante la centrifuga, era stato aspirato il gel in eccesso.
2.5.3 Efficienza di semina
Generalmente le cellule, al di sotto di una certa densità, non crescono in modo efficiente e vanno incontro a morte.
Come regola generale non è prudente scendere sotto una densità pari a 104 cellule/cm2.
2.5.4 Tempo di duplicazione
La popolazione cellulare cresce in modo esponenziale: il numero di cellule si raddoppia ad ogni duplicazione.
Il tempo impiegato per ogni duplicazione è caratteristico di ogni linea cellulare, ma può essere modificato entro certi limiti dalle condizioni di coltura, ad esempio dal tipo di terreno e di siero.
Il tempo necessario alla duplicazione è di circa 20-24 ore per le cellule animali e 24-30 ore per quelle umane.
Conoscendo il tempo di duplicazione di una popolazione, è possibile calcolare il tempo necessario affinché le colture giungano a confluenza.
2.5.5 Tecnica di tripsnizzazione I reagenti necessari sono:
9 PBS (tampone fosfato a pH neutro); 9 tripsina;
9 terreno completo.
La procedura comprende diverse fasi:
1. controllare la piastra al microscopio per accertarsi dello stato di salute delle cellule e del raggiungimento della confluenza;
2. aprire la piastra sotto cappa sterile e aspirare completamente il terreno di coltura;
3. fare due risciacqui con PBS al fine di eliminare completamente eventuali cellule morte e residui di terreno;
4. aggiungere una quantità nota di soluzione tripsina/EDTA (circa 2 ml per una piastra da 25 ml);
5. riporre la piastra in incubatore per circa tre minuti e, trascorso tale tempo, osservare al microscopio che la soluzione si sia distribuita in modo omogeneo e che le cellule si siano completamente staccate. E’ possibile favorire il distacco dando dei colpi laterali alla piastra;
6. bloccare la tripsina con un quantitativo di mezzo pari e pipettare varie volte scaricando il liquido in punti diversi della piastra in modo da risospendere bene tutte le cellule;
7. trasferire la sospensione in una provetta e centrifugare per 5 minuti a 900 giri/minuto;
8. aspirare il surnatante;
9. aggiungere terreno fresco e spipettare per separare le cellule; 10. distribuire nelle nuove piastre in cui, se necessario, era stato preventivamente messo il gel e aspirato quello in eccesso.
Tutte le operazioni devono essere svolte con una certa rapidità, in modo da tenere le cellule fuori dall’incubatore il minor tempo possibile.
2.5.6 Congelamento di cellule
Le cellule che vengono congelate devono essere in fase logaritmica di crescita o devono aver appena raggiunto la confluenza.
Il congelamento mantiene le cellule vive in completa quiescenza per anni. Tramite congelamento, quindi, si può costituire uno stock di cellule che mantengono le caratteristiche fisiologiche e biochimiche delle cellule di partenza.
Il congelamento richiede i seguenti reagenti: 9 PBS;
9 tripsina/EDTA; 9 terreno completo 9 soluzione di DMSO. I passi della procedura sono: 1. fare due risciacqui di PBS;
2. aggiungere la tripsina e riporre la piastra nell’incubatore per 3-5 minuti, in modo da distaccare le cellule;
3. bloccare la tripsina con un quantitativo di mezzo pari ;
4. trasferire la sospensione in una falcon e centrifugare per 5 minuti a 900 giri/minuto;
5. aspirare il surnatante, stando attenti a non aspirare le cellule; 6. aggiungere il crioconservante, ovvero il DMSO, e spipettare per risospendere le cellule adagiate sul fondo dalla forza centrifuga; 7. trasferire la soluzione nelle vials, piccole provette adatte al congelamento delle cellule (1 ml per vials);
8. chiudere accuratamente il tappo della vails e sigillarlo esternamente con del parafilm;
9. porre il tutto nel frigorifero termostatato a -83°C.
La velocità con cui il campione viene congelato è molto importante in quanto congelamenti troppo rapidi portano alla formazione di cristalli di ghiaccio all’interno delle cellule. Questi, al momento dello scongelamento, provocano lisi della membrana plasmatica. Il processo deve perciò essere lento, così da favorire la formazione di cristalli di ghiaccio al di fuori della cellula.
2.5.7 Fissazione e colorazione
Le cellule possono essere fissate e colorate al fine di consentire un’analisi più facile e accurata.
Per la fissazione è possibile impiegare due diversi tipi di reagenti: 9 aldeidi (formaldeide);
9 alcool al 70%.
Esiste, inoltre, anche una svariata gamma di coloranti cellulari, fluorescenti e non fluorescenti, vitali e non vitali, specifici per il DNA o per particolari organelli, oppure in grado di marcare l’intera cellula.
Ad esempio, il cristal violet è un colorante non fluorescente in grado di marcare interamente solo le cellule vitali.
Le fasi della procedura di colorazione con cristal violet sono: 1. aspirare il mezzo;
2. fare 2 risciacqui con PBS;
3. sotto cappa chimica, trattare i campioni con una soluzione di formaldeide al 10% per 10 minuti, così da fissare le cellule nelle loro posizioni,
4. eliminare l’eccesso di formaldeide, 5. risciacquare con PBS;
6. trattare con una soluzione di cristal violet per 1-2 minuti e quindi aspirarlo;
7. eseguire vari lavaggi con acqua distillata per eliminarne l’eccesso;
2.5.8 Conta cellulare
Esistono diversi metodi per contare le cellule, tra questi uno dei più semplici ed economici è l’uso dell’emocitometro o camera di
Burker.
Questa è costituita da un vetro spesso, in cui è ricavata una camera capillare. La parete superiore della camera è costituita da un vetrino bloccato da due graffe laterali. Al microscopio diventano evidenti una serie di linee ortogonali tra loro, che definiscono una serie di aree e, quindi, di volumi.
La procedura da seguire è la seguente:
1. preparare la sospensione cellulare eliminando gli aggregati cellulari mediante trattamento enzimatico o chimico (es. tripsina, EDTA);
2. utilizzando una pipetta Pasteur, trasferire una piccola quantità di sospensione cellulare in entrambe le camere del vetrino (con il coprioggetto già montato sopra le camere), permettendo il riempimento per capillarità;
3. partendo dalla prima camera contare le cellule nel quadrato centrale da 1 mm e nei quattro quadrati da 1 mm agli angoli (figura 2.6). Per ciascun quadrato non si contano le cellule che toccano la linea centrale del perimetro in basso e a destra;
4. ripetere la procedura per la seconda camera.
Se si contano più di 500 cellule nei 10 quadrati, aggiustare la sospensione cellulare in modo opportuno.
Figura 2.6 Quadrato centrale della camera di Burker
Conta: ciascun quadrato dell’emocitometro con il coprioggetto in
posizione ha un volume di 0.1 mm3 o di 10-4 cm3. Essendo 1 cm3 equivalente a 1 ml, la concentrazione di cellule in 1 ml è calcolato con la seguente formula:
cellule per ml = conteggio medio per quadrato x fdil x 104
dove fdil è il fattore di diluizione.
Un’altra tecnica utilizzata è quella del visual counting. Le fasi sono: 1. fissare e colorare le cellule;
2. fotografare 4 0 5 punti del campione sotto microscopio;
3. eseguire al computer una conta visiva del numero di cellule in ciascuna foto.
Facendo una media dei risultati ottenuti è possibile stimare il numero totale di cellule sul campione.
2.6 Sterilizzazione
La sterilizzazione è la prima procedura da eseguire negli esperimenti che necessitano di materiale biologico per la loro realizzazione. [13]
Le tecniche di sterilizzazione più comuni usate nelle colture cellulari sono:
raggi UV; autoclave.
La sterilizzazione con UV viene eseguita generalmente prima di effettuare il trattamento con le proteine di adesione.
L’autoclave, utilizzato comunemente in laboratorio per sterilizzare materiale di vario tipo, è un recipiente cilindrico verticale in acciaio, con pareti robuste. Ne esistono, comunque, anche orizzontali con camera cilindrica o quadrangolare a capacità variabile, tipicamente da 40 a 140 litri. E’ munito di un coperchio a chiusura ermetica provvisto di valvola di sicurezza e di indicatore per il controllo della pressione. La sterilizzazione si ottiene sottoponendo il materiale al vapore saturo sotto pressione alla temperatura di 121° per 20 minuti.
2.7 Prevenzione delle contaminazioni
Le contaminazioni microbiche possono essere causate da batteri, miceti o virus.
I segni precoci di inquinamento batterico sono riscontrabili nell’intorbidamento e nell’acidificazione del terreno di coltura. I batteri hanno una dimensione al limite del microscopio ottico e
appaiono a contrasto di fase come una minuta sabbiolina sulle cellule.
Nella contaminazione da miceti il tappeto cellulare resiste più a lungo, ma si ricopre, in tutto o in parte, di lunghe formazioni filamentose dall’aspetto inconfondibile. La contaminazione da miceti può derivare da linee cellulari infette o dall’operatore e rappresenta un problema rilevante per i laboratori, per la facilità di trasmissione da un ceppo cellulare all’altro attraverso l’ambiente di lavoro.
Le infezioni da virus sono difficilmente riconoscibili.
La forma più subdola di contaminazione è dovuta ai micoplasmi. La presenza di tali agenti può passare del tutto inosservata all’operatore e può provocare cambiamenti profondi nel metabolismo e nelle proprietà del ceppo cellulare.
Le contaminazioni microbiche rappresentano per le colture cellulari un grave problema, in quanto non esistono trattamenti sicuri per combatterle in modo risolutivo. [14, 15, 16]
E’ perciò necessario prevenirle, attuando tutti gli accorgimenti possibili.
La prima regola da seguire è quella di verificare con estrema cura la sterilità delle soluzioni e dei terreni utilizzati. Tuttavia, anche utilizzando soluzioni rigorosamente controllate, può capitare di contaminare una coltura cellulare.
Le infezioni da batteri vengono prevenute con l’uso di penicillina e streptomicina nel mezzo di coltura, quelle da miceti con l’uso di anfotericina B, anche se il suo uso è limitato, in quanto gli antimicotici presentano spesso una certa tossicità per le cellule.
Non esistono sistemi di prevenzioni delle contaminazioni virali o da micoplasmi.
Vi sono delle norme pratiche per evitare le infezioni microbiologiche che riguardano l’ambiente, la strumentazione, l’operatore:
destinare il laboratorio solo alle colture cellulari;
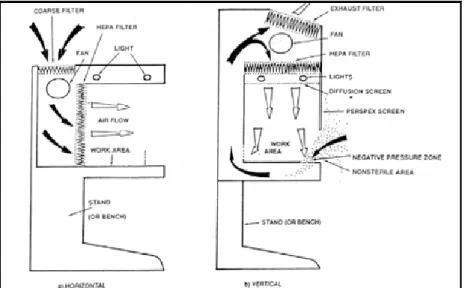
operare sempre sotto cappa a flusso laminare (figura 2.7);
Figura 2.7 Schema di una cappa a flusso laminare
tutto il materiale usato, sia di vetro che di plastica, deve essere sterile;
utilizzare solo pipettatori elettrici, evitando nel modo più assoluto di pipettare soluzioni e terreni con la bocca;
pulire bene la cappa a inizio e fine lavoro e controllarne periodicamente i filtri;
l’operatore deve sempre lavarsi le mani con sapone disinfettante e indossare i guanti prima di iniziare a lavorare con le cellule.
2.8 Cellule nervose in coltura
Quasi tutti i tipi di tessuti, quando dissociati in una sospensione di singole cellule e piazzati in una piastra vanno incontro alla serie di eventi precedentemente descritta.
La situazione con i neuroni è abbastanza differente. [17]
Quando le cellule dal cervello embrionico vengono dissociate e messe in coltura, i neuroni che hanno completato la divisione in situ estendono i processi e diventano elettricamente attivi. Ma se il tessuto è rimosso durante la neurogenesi, è raro osservare cellule che si dividano in coltura e acquisiscano successivamente il fenotipo neurale.
I neuroni nelle colture primarie non si dividono ma, sotto condizioni favorevoli, possono essere mantenuti in coltura per diverse settimane, durante le quali sviluppano assoni e dendriti, stabiliscono sinapsi, formando una densa rete, e esprimono i recettori e i canali ionici caratteristici delle corrispondenti cellule in situ. Molto spesso sviluppano una considerevole attività elettrica spontanea, includendo i potenziali sinaptici che sommano per produrre i potenziali d’azione. Quando i neuroni sono co-coltivati con le cellule di Schwann o con gli oligodendrociti, gli assoni diventano melinati.
Inoltre, i neuroni primari mantengono le loro identità individuali, probabilmente perché, generalmente, quando vengono messi in
coltura, sono post-mitotici e hanno compiuto la loro differenziazione. Durante i primi giorni di coltura, prima che la rete diventi troppo complicata, singoli neuroni possono essere osservati nella loro interezza e questo permette la diretta osservazione della crescita degli assoni e del loro modo di diramarsi, [18, 19] e anche precise analisi o modifiche di tali eventi quali misura della tensione e della forza adesiva generate dai singoli coni di crescita [20, 21] o taglio dei neuriti in posizioni ben precise. [22, 23]
In contrasto con le colture primarie, le linee cellulari nervose possono invece crescere e essere sottoposte a numerosi passaggi, fino ad ottenere il numero di cellule desiderato. Queste poi possono essere indotte a differenziarsi da una grande varietà di fattori, cessandone la divisione e determinando l’estensione dei processi e l’espressione di diversi geni specifici dei neuroni. Tali cellule sono elettricamente attive, sintetizzano trasmettitori o peptidi e esprimono i recettori per i neurotrasmettitori.
Questi vantaggi devono essere pesati con il fatto che, comunque, non tutti gli aspetti importanti della differenziazione neurale possono essere espressi dalle linee immortalizzate.
Bibliografia
1. S.M. Potter - Prog. Brain Res 130, 49 (2001)
2. Orit Shefi, Ido Golding, Ronen Segev, Eshel Ben Jacob, Amir Ayali, - Morphological characterization of in vitro neuronal
networks, Physical review 66, 021905 (2002)
3. Orit Shefi, Amir Harel, Dmitri B. Chklovskii, Eshel Ben Jacob, Amir Ayali, - Biophysical constraints on neuronal branching, Neurocomputing 58-60 (2004), p.p. 487-495
4. Tesi di laurea in Chimica e Tecnologia Farmaceutiche di Delia Venanzoni “Organizzazione e differenziazione di neuroblastomi
SH-SY5Y su substrati polimerici microstrutturati: potenziali applicazioni nel campo dell’ingegneria dei tessuti nervosa retinici”, 2002
5. G.J Todaro, H.J. Green - Cell. Biol. 1963, p.p. 7-299
6. S. Aaranson, G.J. Todaro - Cell. Physiol. 1968, p.p. 72-141
7. J. Folkman, A. Moscona – Nature, 1978, p.p. 273-345
9. D. Barnes, G. Sato “Cell” 1980, p.p. 22-649
10. C.J. Avers “Biologia molecolare della cellula” ed. Zanichelli
11. S. Guenzi “Efficienza e sicurezza dei prodotti per la crescita
di cellule eucariote: sieri, terreni e reagenti per colture cellulari”
12. P. De Filippi, G. Tarone, “Colture cellulari – tecniche di
base: i manuali delle scuole”, Scuola Superiore di Oncologia e
Scienze Biomediche
13. M. Amorosa, “Principi di tecnica farmaceutica”, ed. L. Tinarelli
14. M. L. Macy, “Tissue culture association manual” 1980, p.p. 5-1151
15. M. F. Barile, R.T. Acton, J.D. Lynn, “In cell culture and its
application”, eds Academic Press London and New York 1977
16. G. J. McGarrity Adv – “Cell culture” 1982, p.p. 2-99
17. Gary Banker and Kimberly Goslin, editors, “Culturing Nerve
18. D. Bray, - Branching patterns of individual sympathetic
neurons in colture, Cell Biol. 56 (1973), p.p. 702-712
19. C. G. Dotti, C. A. Sullivan and G. A. Bunker, - The
establishment of polarità by hippocampal neurons in colture,
Neuroscience 8 (1988), p.p. 1454-1468
20. P. Lamoreux, R. E. Buxbaum and S. R. Heidemann, - Direct
evidence that growth cones pull, Nature 340 (1989), p.p.
159-162
21. J. Zheng, R. E. Buxbaum and S. R. Heidmann, -
Measurements of growth cones adhesion to culture surfaces by micromanipulations, Cell. Biol. 127 (1994), p.p. 2049-2060
22. G. Shaw and D. Bray, - Movement and extension of isolated
growth cones, Exp. Cell. Res. 104 (1977), p.p. 55-62
23. K. Goslin and G. Banker – Experimental observations on the
development of polarity by hippocampal neurons in culture,