19
Capitolo 2: Gli inibitori delle MMPs (MMPIs)
2.1. Considerazioni generali.
Come precedentemente affermato, un’alterazione di uno dei meccanismi di controllo che regolano l’espressione dei livelli fisiologici delle MMPs può essere correlata con serie patologie a carico del sistema nervoso centrale e periferico, del sistema cardiovascolare, muscoloscheletrico, gastrointestinale e non solo. Tali patologie sono caratterizzate da una forte degradazione dei tessuti strutturali da parte di MMPs sovra espresse sia da cellule del tessuto stesso, sia da cellule di natura infiammatoria o tumorale invasiva. Per questo motivo negli ultimi venti anni la ricerca farmaceutica ha sviluppato studi molto importanti nel campo degli inibitori delle MMPs (MMPIs)[12]. Sfortunatamente studi clinici condotti sugli inibitori ad ampio spettro hanno portato a risultati deludenti, specialmente nel campo dell’area terapeutica del cancro. Oggi molti gruppi di ricerca si stanno concentrando sulla scoperta di nuove classi di inibitori, tentando di incrementarne la potenza e soprattutto la selettività nei confronti di specifiche MMPs coinvolte in ogni target patologico[9].
Ripercorrendo le varie strategie che sono state seguite per la progettazione degli MMPIs, il primo approccio è stato quello del design di composti peptidici o peptido-mimetici che mimassero i substrati naturali delle MMPs (approccio substrate-based). A tale scopo sono stati inseriti sullo scheletro delle porzioni (P1, P1’, P2, P2’, P3…) che potessero interagire in modo favorevole con i sottositi dell’enzima (S1, S1’, S2, S2’, S3…) associati ad un gruppo in grado di legare lo Zn+2 catalitico (ZBG) (Fig.2.1)[14].
20
Fig.2.1. Design di MMPIs peptide-based [14].
Comunque, i risultati deludenti ottenuti dagli studi clinici effettuati su questi inibitori
peptide-based hanno aperto intense discussioni su come eliminare gli inconvenienti mostrati dagli inibitori ad ampio spettro, in modo da ottenere nuove classi di composti ad elevata potenza e, soprattutto, elevata selettività verso specifiche MMPs coinvolte nelle patologie di interesse, al fine di minimizzare gli effetti collaterali. Per raggiungere questo obiettivo gli studi si sono concentrati su inibitori non peptidici che fossero selettivi, attivi per somministrazione orale, e dotati di una buona durata d’azione. Le strategie di design seguite sono state prevalentemente due.
La prima è stata il design di MMPIs selettivi ottimizzando le interazioni tra gli inibitori e l’enzima, in modo da ottenere un incremento della potenza (approccio ligand-based). A tale proposito il gruppo di Wittaker affermò che un buon inibitore deve possedere nel suo scaffold fondamentale uno ZBG, almeno un gruppo funzionale che instauri un legame ad idrogeno con lo scheletro dell’enzima, ed una o più catene laterali substrato-simili che stabiliscano delle favorevoli interazioni di Van der Waals con le tasche del sito attivo dell’enzima, come la S1, la S1’ e la S2’. Questo approccio è senza dubbio
21
ancora il più popolare per ottenere potenti MMPIs, anche se oggi le nuove sfide includono lo sviluppo di nuovi inibitori allosterici non leganti lo Zn+2.
La seconda strategia è stata l’utilizzo delle strutture delle MMPs come base per il
design degli inibitori. Infatti la crescente disponibilità di strutture cristallizzate ai raggi X ad alta risoluzione di molti membri di questa famiglia di proteine ha reso possibili approcci di design structure-based, che sono oggi comunemente utilizzati. Nella maggior parte dei casi le strutture 3D attualmente disponibili per le MMPs sono presentate come complessi del loro dominio catalitico con vari inibitori noti peptidici, peptido-mimetici o non peptidici. Inoltre la combinazione con le tecniche di chimica combinatoriale e computazionale ha accelerato lo sviluppo degli inibitori. Tutti questi studi hanno fornito utili informazioni per spiegare i meccanismi delle interazioni chimico-biologiche di alcune MMPs[14].
2.2. ZBGs presenti negli MMPIs.
Tra i vari ZBGs presenti negli MMPIs, i più utilizzati sono stati senza dubbio gli acidi idrossammici. Essi sono dei chelanti che legano lo ione Zn+2 in modo bidentato attraverso i due atomi di ossigeno, e che formano legami ad idrogeno con specifici residui amminoacidici del sito attivo delle MMPs (Fig. 2.2).
22
Fig.2.2. Rappresentazione schematica di un inibitore contenente come ZBG un acido idrossammico
(evidenziato nel riquadro) legato al sito attivo di una MMP [15].
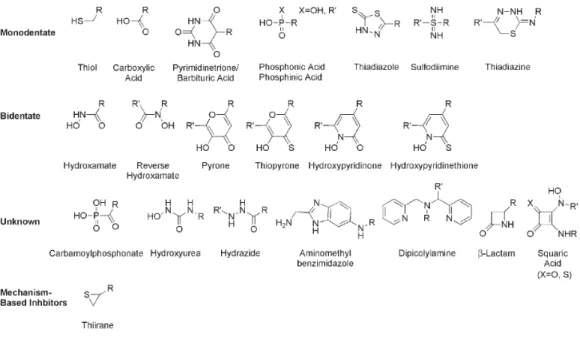
Alcuni inibitori delle MMPs contenenti il gruppo dell’acido idrossammico hanno mostrato in vitro potenza nanomolare ed anche subnanomolare, ma nessuno di essi ha completato con successo gli studi clinici. Ciò è dovuto principalmente al loro basso profilo farmacocinetico, in quanto in vivo possono venire idrolizzati rapidamente ad acidi carbossilici. Inoltre gli acidi idrossammici possono presentare scarsa biodisponibilità orale e mostrare una affinità di legame verso metalli di transizione, come Fe(III), Ni(II), e Cu(II), simile o addirittura maggiore rispetto a quella misurata per lo Zn(II), ostacolando così la selettività per le MMPs nei confronti di altre Metalloproteasi e soprattutto altri metalloenzimi. Queste scoperte hanno messo in luce alcune potenziali limitazioni di questa classe di composti, pertanto sono stati sviluppati altri ZBGs, differenti dall’idrossammato, da inserire nella struttura degli MMPIs[15]. In questo campo un grande contributo è stato dato dalle ricerche di Cohen e coll. In un suo recente studio[15] Cohen ha classificato i vari ZBGs in base al loro meccanismo di interazione con lo Zn+2 catalitico del sito attivo delle MMPs (Fig. 2.3).
23
Fig. 2.3. Strutture di ZBGs classificati in base al binding mode con lo Zn+2 catalitico.
R e R’ rappresentano potenziali siti di attacco per lo scaffold [15].
In generale è stato osservato che gli inibitori contenenti uno ZBG monodentato si mostrano chelanti più deboli rispetto ai bidentati. Dopo l’idrossammato lo ZBG più studiato è stato l’acido carbossilico, prevalentemente perché composti portanti questo gruppo funzionale nella loro struttura sono i precursori sintetici di quelli contenenti un acido idrossammico. Tra i gruppi monodentati, i tioli si sono rivelati molto potenti in
vitro, ma hanno mostrato instabilità in vivo data la potenziale formazione di disolfuri. Per identificare nuovi ZBGs Cohen e Puerta hanno utilizzato vari approcci, uno dei quali è stato quello di sviluppare dei modelli di complessi bioinorganici in grado di mimare il meccanismo di legame dei ligandi col sito attivo delle MMPs. Essi hanno dimostrato che il complesso tris-(pirazolil-)borato dello Zn+2 costituiva un modello adeguato per riprodurre le tre His del sito attivo delle MMPs complessate con lo Zn+2 catalitico, e che l’acido acetoidrossammico formava un complesso che era strutturalmente identico all’ambiente di coordinazione degli acidi idrossammici con lo
24 Zn+2 nel sito attivo delle MMPs (Fig.2.4)[16].
Fig.2.4. Il complesso modello [(TpPh,Me)Zn(acetoidrossammato)] [12].
Questi modelli sono stati usati per valutare il binding mode di nuovi potenziali ZBGs, scegliendo piccoli frammenti che potessero legare lo Zn+2 in modo bidentato (Fig 2.5)[12].
Fig.2.5. Nuovi ZBGs scoperti da Puerta e Cohen [12].
Dopo aver determinato la struttura ai raggi X dei complessi, è stata valutata l’attività inibitoria di questi nuovi ZBGs verso la MMP-3, riscontrando un aumento della potenza rispetto all’acido acetoidrossammico preso come riferimento[12]. Come chiaramente intuibile queste valutazioni sono state fatte solo per quanto riguarda lo ZBG senza prendere ancora in esame lo scheletro peptido-mimetico, principale responsabile della potenza di un inibitore. Pertanto è stato poi effettuato uno studio per costruire potenti
25
MMPIs completi che contenessero al loro interno i nuovi ZBGs individuati; tra questi è stato scelto per esempio il maltolo, un derivato pironico, sulla base della nota biocompatibilità e della buona idrosolubilità di questa classe di composti[12]. L’approccio utilizzato è stato di una SAR bioinorganica, le cui fasi sono riassunte nella Fig.2.6. Inizialmente i complessi ZBG-modello sono stati cristallizzati e caratterizzati, poi le coordinate dei frammenti ZBGs sono state superimposte nella struttura cristallizzata della MMP target per generare un iniziale complesso recettoriale. Utilizzando questo complesso ibrido come template, sono stati applicati dei metodi computazionali di docking per individuare frammenti idrofobici che si inserissero nei siti di legame del sito attivo della proteina prossimali allo Zn+2. A questo punto sono stati progettati dei linkers adeguati che connettessero lo ZBG con i frammenti dello scheletro, per ottenere degli MMPIs completi che potessero essere ulteriormente ottimizzati con tecniche convenzionali di Medicinal Chemistry al fine di migliorarne la solubilità ed il profilo farmacocinetico[15, 17].
26
L’approccio appena descritto viene definito Fragment-based lead design (FBLD). Esso presenta vari vantaggi rispetto ai metodi più tradizionali di sviluppo di leads: in primo luogo i siti attivi sono più efficacemente sondati da piccoli frammenti che non siano limitati da costrizioni steriche o da legami ad idrogeno non corretti; inoltre composti a più bassa complessità possono coprire in modo più efficace le diversità chimiche disponibili[17].
2.3. Inibitori chelanti lo zinco diversi dall’idrossammato:
i pirimidin-2, 4, 6-trioni.
I pirimidin-2, 4, 6-trioni, o acidi barbiturici, sono una classe di composti conosciuti da decenni. In passato venivano impiegati come sedativi e ipnotici ma, data la dipendenza e la tolleranza che inducono e la relativa pericolosità che presentano, sono stati sostituiti dalle Benzodiazepine. Oggi continuano ad essere prescritti come antiepilettici e per il trattamento di interventi traumatici alla testa; inoltre sono utilizzati anche in anestesia.
Brandstetter, Grams e al.[18] hanno individuato poi i pirimidin-2,4,6-trioni come una nuova classe di inibitori delle MMPs grazie a studi basati su HTS (high-throughput
screening). La loro attività è stata rilevata soprattutto nei confronti delle Gelatinasi A e B (MMP-2 e MMP-9) ma, per valutare l’inibizione delle Gelatinasi, fu scelto come sistema modello la MMP-8 sia per la disponibilità di informazioni cristallografiche sia perché la sua attività enzimatica è simile nella maggior parte dei casi a quella delle Gelatinasi[18]. Dalle informazioni derivanti dai raggi X di un inibitore pirimidintrionico complessato con una MMP-8 si è osservato che questo mima i substrati peptidici della proteina formando legami a idrogeno con i residui chiave del suo sito attivo e fornisce la
27
possibilità di introdurre gruppi appropriati nella tasca di specificità S1’ e nella S2’[19]. E’ stato osservato che nell’interazione con l’enzima la forma enolica è favorita rispetto a quella chetonica, in quanto, come verrà approfondito in seguito, il protone ossidrilico viene impiegato nella formazione di un legame ad idrogeno con un residuo specifico del sito catalitico[19]. N NH OH O O R2 R1 HN NH O O O R2 R1
Fig.2.1. Equilibrio cheto-enolico della struttura pirimidintrionica.
Per questo motivo è stato preso come modello per la complessazione con la MMP-8 l’inibitore RO200-1770, la cui struttura è riportata nella Fig.2.7.
Fig.2.7. RO200-1770, 2-idrossi-5-fenil-5-N-(4-(2-idrossietil)-piperidil)-4,6-pirimidindione [20]
.
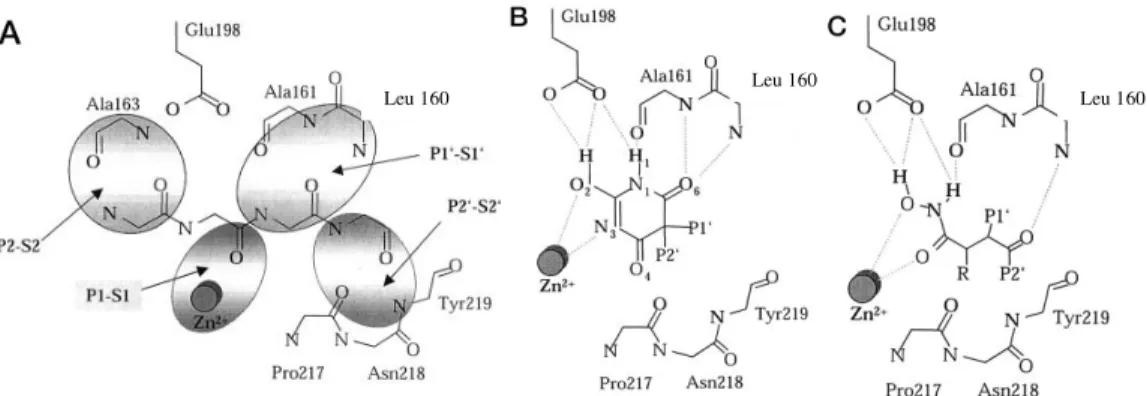
La Fig.2.8 A mostra schematicamente i residui del sito attivo della MMP-8 responsabili del riconoscimento del substrato.
28
Fig. 2.8. A Rappresentazione schematica dei residui chiave del sito attivo della MMP-8 per il legame col
substrato. B Rappresentazione schematica dell’interazione del 2-idrossi-pirimidindione col sito attivo della MMP-8. C Rappresentazione schematica dell’interazione di un inibitore della classe degli acidi idrossammici col sito attivo della MMP-8 [20].
Il confronto con la Fig.2.8 B, nella quale viene rappresentato il legame del
RO200-1770, illustra come l’inibitore mantenga le interazioni più importanti con i residui del
sito attivo. Lo Zn+2 catalitico è coordinato con N3 e O2 dell’anello barbiturico. O2 occupa la stessa posizione assunta dalla molecola d’acqua catalitica durante l’idrolisi dei substrati peptidici, perciò la parziale carica negativa che vi si forma è stabilizzata dall’adiacente Glu198, rafforzando così il suo legame con lo Zn+2 catalitico. La forma enolica risulta pertanto favorita rispetto a quella chetonica. Inoltre gli atomi polari H1-N1-C6=O6 dell’anello barbiturico mimano le interazioni della porzione P1 del substrato con la tasca S1. N1-H1 ammidico forma un legame ad idrogeno con il carbonile dell’Ala161 ed il chetone C6=O6 è stabilizzato dagli atomi di azoto ammidico di Leu160 e Ala161. Il chetone C4=O4 sembra contribuire in modo sfavorevole all’energia di legame con la proteina in quanto causa l’insorgere di interazioni repulsive con l’ossigeno carbonilico della Pro217; per prevenire tali interazioni si verificano dei cambiamenti conformazionali nell’enzima, ovvero il legame peptidico Pro217-Asn218 viene ruotato di
Leu 160
29
circa 100°, e questo comporta un costo a livello energetico. Diversamente dalle interazioni polari mediate dall’anello pirimidintrionico, le interazioni mediate dagli anelli fenilico e piperidinico sono prevalentemente idrofobiche e coinvolgono le tasche S1’ e S2’, rispettivamente. L’interazione più importante con la tasca S1’ è la parziale sovrapposizione tra l’anello fenilico e l’His197 che si instaura ad una distanza ottimale di 3,6Ǻ (Fig.2.9).
Fig.2.9 Dettagli dell’interazione tra l’anello fenilico e His197[20].
Tale His è importante anche perché chela lo Zn+2 catalitico, assieme all’His201 e all’His207. Le interazioni idrofobiche dell’anello piperidinico si instaurano con la parte inferiore della tasca S2’ mediante i residui Pro217-Asn218-Tyr219, e con la parte superiore grazie ai residui Gly158-Ile159-Leu160. La Leu160 inoltre separa le tasche S1’ e S2’.
Sebbene identificati come potenti inibitori delle MMPs grazie ad un programma di screening, i composti pirimidintrionici mostrano sorprendenti analogie con i ben più caratterizzati idrossammati e acidi malonici per quanto riguarda il binding mode. La Fig.2.8 C illustra che la geometria di chelazione dello Zn+2 catalitico da parte dell’idrossammato, è mantenuta anche dal barbiturico attraverso il suo N3 che sostituisce il gruppo chetonico dell’idrossammato stesso. Inoltre l’interazione degli atomi N1-H1 e O6 del barbiturico con i residui Leu160 e Ala161 dello scheletro della
30
proteina è analogo a quello dell’idrossammato, con la sottile differenza che nel primo O6 forma un’interazione addizionale con l’azoto ammidico della Ala
161
, potendo così stabilizzare una carica negativa maggiore. Queste scoperte comportano sia aspetti positivi che negativi; infatti se da un lato la somiglianza strutturale per entrambe le classi di inibitori fa sì che si possano applicare le conoscenze ed i criteri di ottimizzazione in modo reciproco, dall’altro tale somiglianza può anche indicare una limitazione nel trovare inibitori specifici per le varie MMPs. La presenza di una simile geometria di chelazione in classi di composti indipendentemente identificati e strutturalmente non correlati indica che il legame con lo Zn+2 segue un criterio piuttosto universale che domina sulle caratteristiche di legame. Di conseguenza molti, se non la maggior parte degli inibitori delle proteasi chelanti lo Zn+2 mostrano un motivo di legame pressoché invariato e presentano un basso profilo di selettività, almeno nelle fasi precedenti all’ottimizzazione.
Utilizzando la struttura cristallografica della MMP-8 complessata col composto lead
RO200-1770 sono stati sintetizzati e testati molti altri inibitori (Fig.2.10).
31
Nella tabella 2.1 sono riportate le attività di inibizione di tali composti nei confronti di alcune MMPs espresse come IC50 [nM].
Tab.2.1 Profili di inibizione di alcune MMPs (IC50 [nM]) [20].
Il composto lead RO200-1770 mostra una capacità di inibizione micromolare non specifica, fatta eccezione per la MMP-3 verso la quale l’affinità di legame diminuisce di circa dieci volte. Per facilitare la sintesi, la piperidina del composto lead è stata sostituita con un suo isostero, una piperazina, ottenendo la molecola RO204-1924. Essa ha mostrato una diminuzione piuttosto uniforme nell’affinità di legame, che può essere razionalizzata con un maggiore dispendio energetico di desolvatazione per legare l’anello piperazinico. I composti I-COL043 e RO206-0027 rappresentano i risultati di due approcci incrociati per ottimizzare rispettivamente le interazioni P1’-S1’ e P2’-S2’; per ognuno dei due è stato registrato, rispetto al lead, un aumento di circa 10 volte della capacità di inibizione verso le 8, -2, -9 e -3, mentre per quanto riguarda la MMP-1 e la MTMMP-1-MMP è l’affinità di legame è peggiorata o rimasta pressoché invariata. Le ottimizzazioni di P1’ e P2’ di I-COL043 e RO206-0027 sono state combinate nel composto RO206-0032, ed i dati dimostrano l’esistenza di un effetto additivo del
32
contributo delle due porzioni alla capacità di inibizione; l’incremento dell’affinità di legame alla MMP-3 risulta meno marcato, mentre per quanto riguarda la MMP-1 si assiste ad una media, piuttosto che ad una somma, degli effetti dei due composti[20]. In uno studio successivo ancora Grams, Brandstetter e al. hanno proseguito l’ottimizzazione del composto RO206-0032, che da qui in avanti verrà chiamato 3[18].
Ottimizzazione del residuo P1’
Il processo di ottimizzazione del residuo P1’ è stato effettuato mantenendo fissa la porzione 4-(4-nitro-fenil)-piperazinica come promettente residuo P2’. I risultati degli studi di SAR sono riassunti nella Tab.2.2.
33
Sostituendo il residuo ottilico di 3 con uno fenossi-fenilico (9), bifenilico (10) o 4-butossi-fenilico (11) è stato ottenuto un miglioramento dell’attività in media di due volte. Tutti i composti 3 e 911 interagiscono molto bene con la tasca S1’ delle MMP8, -2 e -9. Comunque il tentativo di instaurare qualche legame ad idrogeno ulteriore con lo scheletro degli amminoacidi chiave del sito attivo si è rivelato fallimentare. Sono stati sintetizzati inoltre derivati portanti un’ammide, un ossidrile o un gruppo etereo sulla loro catena (12, 13, 14), ma i risultati sono stati peggiori. Studi strutturali delle due Gelatinasi hanno portato a supporre che la S1’ dovesse essere considerata più come un tunnel che come una tasca; pertanto si è cercato di migliorare ulteriormente l’affinità di legame del composto 9 introducendo un altro piccolo sostituente nella posizione 4- dell’anello ossifenilico (Tab.2.3).
34
Tab.2.3 SAR di vari composti a struttura 5-(4-fenossi-fenil)-2,4,6-pirimidintrionica [18].
35
composto RO204-1924, ottenendo il composto 15 del quale poi sono stati sintetizzati il bromo- (16), cloro- (17), metil- (18) e metossi- (19) derivato; tutti questi composti hanno mostrato un’attività maggiore di 15 nei confronti sia delle Gelatinasi che della MMP-8. Il composto più attivo su tutti e tre questi enzimi è stato il 16.
Ottimizzazione del residuo P2’
Facendo riferimento ancora alla Tab.2.3 si osserva come la sostituzione del gruppo nitro del composto 9 con un gruppo solfonamidico (20) provochi un incremento di tre volte della capacità di inibizione verso la MMP-2. In ogni modo, poiché un ulteriore aumento del peso molecolare e della lipofilia potrebbe presentare problemi nelle successive fasi di sviluppo del farmaco, è stato deciso di riconsiderare come residuo P2’ l’originario gruppo idrossietil-piperazinico, variando la lunghezza della catena alifatica.
Il composto migliore si è rivelato il 21, sebbene anche le molecole 22 e 23 si mantengano nello stesso range di valori. Di questi tre composti è stato preparato il cloro-derivato nella posizione 4- dell’anello ossifenilico, ottenendo i composti 24-26, ma non è stato riscontrato un considerevole aumento dell’attività. I composti 27 e 28, in cui è stato eliminato il residuo P2’, sono stati sintetizzati per valutare il contributo di tale porzione all’energia di legame ed è stato osservato che il rapporto tra le loro attività di inibizione per le MMP-2 e -9 è simile a quello tra i composti 15 e 17; il rapporto di inibizione verso la MMP-2 è 2.2 per i composti sostituiti e 2.5 per i composti senza il residuo P2’, mentre verso la MMP-9 è 5.6 e 6.5, rispettivamente. Comunque, non tutte le coppie di composti corrispondenti cloro-sostituiti e non mostrano quest’effetto: per esempio il rapporto di inibizione tra i composti 23 e 27 è 0.5 verso la MMP-2 e 2.6 verso la MMP-9. La differenza rispetto ai dati prima riportati potrebbe essere
36
razionalizzata con un sottile riarrangiamento spaziale di uno di questi due composti rispetto all’altro.
Confronto con altri tipi di MMPIs
Il composto 10 (RO-28-2653) è stato confrontato con altri inibitori delle MMPs già ben studiati: Batimastat, AG3340 (Prinomastat) e BAY12-9566.
MMP-1 MMP-2 MMP-3 MMP-8 MMP-9 MMP-14 MMP-16
10 RO-28-2653 16000 10 1800 15 12 10 23
BAY12-9566 16000 40 2000 14 1200 1850 2000
AG3340 26 2.4 95 2.4 1 12 19
Batimastat 25 32 67 27 23 19 29
Tab.2.4 Confronto tra le IC50 [nM] di RO-28-2653 e di altri inibitori [18].
Come si può vedere dalla Tab.2.4 il rapporto tra le attività di inibizione mostrate su MMP-2 e MMP-1 è 1600 per RO-28-2653, 400 per BAY12-9566, 11 per AG3340 e 1 per Batimastat, mentre per quanto riguarda MMP-9 e MMP-1 è 1300 per
RO-28-2653, 13 per BAY12-9566, 26 per AG3340 e 1 per Batimastat. Da questi risultati si
capisce che il composto pirimidintrionico risulta essere il più selettivo nei confronti dei
targets desiderati, le due Gelatinasi.
L’attività antitumorale di RO-28-2653 è stata studiata da Maquoi et al.[9]. Questo inibitore ha mostrato buona capacità di ridurre in vivo la crescita di tumori indotti per inoculazione di linee cellulari che producono MMPs. Inoltre esso ha ridotto la vascolarizzazione tumorale e bloccato l’angiogenesi in un test effettuato sull’anello aortico di ratto.
37
I composti a struttura pirimidintrionica sono stati realizzati non solo come inibitori delle Gelatinasi, ma anche della MMP-13, pioché questo enzima risulta essere primariamente coinvolto in una delle più comuni patologie che interessano il tessuto connettivo: l’osteoartrite (OA). Questa malattia è caratterizzata da una progressiva perdita della cartilagine articolare che può portare ad un complesso meccanismo di degradazione dei componenti della ECM, primo tra tutti il collagene di tipo II cartilagine-specifico. La sua distruzione è un processo irreversibile, considerato di cruciale importanza nella perdita dell’integrità strutturale e funzionale della cartilagine. Tra le varie MMPs la Collagenasi-3 (MMP-13) è specificamente espressa nella cartilagine umana di pazienti affetti da OA, mentre non è stata trovata nei normali tessuti cartilaginei adulti. Per di più, questo enzima è quello che mostra una più alta attività protesica nei confronti del collagene di tipo II. Per questi motivi, è stato supposto che una inibizione selettiva della MMP-13 potesse risultare molto promettente nel trattamento della OA, cercando in più di minimizzare gli effetti collaterali dose- e durata-dipendenti a carico del sistema muscolo-scheletrico (MSS) rilevati negli studi clinici degli MMPI a largo spettro[14]. A tale proposito, è interessante citare uno studio condotto da Soong-Hoon Kim et al.[21]. Lo scopo del lavoro era quello di progettare una serie di potenti inibitori per la MMP-13 diversi dagli acidi idrossammici, per ovviare ai problemi di basso profilo farmacocinetico (scarso assorbimento e rapido metabolismo) mostrato da questa classe di composti. Il drug design è stato condotto con un approccio structure-based partendo da un acido idrossammico di nota potenza, il composto 1 (RS-130830), di cui erano disponibili i raggi X nel complesso con la MMP-13.
38
Fig.2.11 Caratteristiche di design per un inibitore selettivo della MMP-13
diverso dall’acido idrossammico [21].
Come illustrato nella Fig.2.11 l’anello barbiturico sostituisce la funzione acido idrossammico come chelante dello Zn+2 catalitico dell’enzima; inoltre è stato introdotto un linker che conferisse alla struttura sia rigidità, mimando la conformazione di legame assunta da 1, sia un elemento di novità. Infine la porzione arilica è stata mantenuta ed è stata oggetto di studi di SAR. E’ stato osservato da predizioni di modeling che lo stato conformazionale del composto spiro-barbiturico 4a risulta essere molto vicino a quello del composto 1 nella sua interazione col sito attivo della MMP-13 (Fig.2.12).
Fig.2.12 Interazione del composto 4a (grigio) con la MMP-13 e sua
sovrapposizione con l’inibitore 1 (viola) [21]. (MMP-13 Ki=0.52 nM)
39
E’ interessante notare anche come l’ossigeno carbonilico dell’anello lattamico si sovrapponga bene con uno dei due atomi di ossigeno del solfone nell’inibitore 1. Gli studi di SAR condotti hanno portato alla sintesi di una serie di composti che sono stati testati sulla MMP-13; quelli più attivi sono riportati nella Tab.2.5.
Tab.2.5 SAR di alcuni inibitori progettati [21].
Dalla tabella si osserva che in generale tali composti presentano una migliore selettività per la MMP-13 rispetto quella riportata per 1, comunque non c’è sostanziale differenza di selettività rispetto alla MMP-2 e -9 [21].
Attualmente l’unica molecola a struttura pirimidintrionica che ha mostrato una rilevante differenza di selettività nei confronti delle due Gelatinasi è stata individuata da Breyholz
et al.[22] (Fig.2.13), in quanto presenta una IC50 per la MMP-9 di 26 nM, mentre la IC50 per la MMP-2 è di 1 nM. NH O N H O O R R= N N O Br
40
Sempre per quanto riguarda l’obiettivo di trovare inibitori della MMP-13 terapeuticamente utili e con uno ZBG diverso dall’idrossammato, il gruppo di Reiter[23] ha identificato tramite prove di screening il composto pirimidintrionico 1 come un debole inibitore della MMP-13 (Fig.2.14). Studi di molecular modeling hanno evidenziato che l’etere arilico pendente occupava la tasca S1’ dell’enzima, mentre il gruppo metilico era proiettato in una regione altamente esposta a solvente. Partendo da questi presupposti era stato ipotizzato che, sostituendo il cloro in posizione 4- dell’anello aromatico con un ulteriore etere arilico 4-sostituito, si potesse ottenere un incremento della potenza di inibizione sfruttando al meglio la struttura profonda e stretta della tasca S1’. Questo cambiamento, unito alla contemporanea estensione a residuo butilico della catena in posizione 5- dell’anello barbiturico, ha portato ad un sostanziale aumento della potenza (composto 2). L’eccessiva lipofilia è stata ridotta sostituendo il gruppo butilico con uno etossietilico (composto 3) (clogP 2: 5.05, clogP 3: 3.11).
Fig.2.14 Lead e suo sviluppo iniziale [23].
I raggi X del complesso cristallizzato del composto 3 con la MMP-13 hanno confermato le predizioni di modeling con l’etere arilossiarilico nella tasca S1’ (Fig.2.15).
41
Fig.2.15. Raggi X del complesso cristallizzato i 3 con la MMP-13 [23].
Inoltre è importante sottolineare come l’anello pirimidintrionico si leghi allo Zn+2 catalitico del sito attivo nella sua forma enolica e come una molecola d’acqua venga evidentemente sostituita dall’ossigeno etereo della catena in C5, il che potrebbe spiegare il modesto incremento di potenza osservato da 2 a 3.
Nel tentativo di ottenere inibitori molto selettivi della MMP-13 sulla MMP-14, il composto 3 è stato ulteriormente modificato con studi di SAR ottimizzando il sostituente in posizione 4- dell’anello aromatico terminale. Il composto più interessante è risultato il 29, portante in tale posizione un ossadiazolo (Fig.2.16).
42
Fig.2.16 Inibitore 29 e sua attività su MMP-13 e MMP-14 [23].
Studi in vivo sui ratti hanno evidenziato che il composto 29 presenta moderata emivita (t1/2 = 4.0 h), bassa clearance (0.63 mL/min/kg), basso volume di distribuzione (0.20 L/kg) ed eccellente assorbimento orale (~100%)[23].
In uno studio successivo[24] ancora il gruppo di Reiter ha ulteriormente ottimizzato il composto 29 tramite studi di SAR riguardanti sia la catena in C5 sia l’eterociclo a cinque termini opportunamente sostituito, con lo scopo di ottenere inibitori selettivi per la MMP-13 nei confronti della MMP-2, -8, e -12. Tra tutti i composti sintetizzati quello più interessante è stato il 34 (Fig.2.17).
Fig.2.17
29
43
Data la sua notevole selettività il composto 34 è stato scelto per verificare l’ipotesi che una inibizione selettiva della MMP-13 eliminasse gli effetti collaterali di MSS osservati con altri inibitori. Per questo motivo 34 è stato testato nel modello di fibroplasia nel ratto, utilizzato per simulare la MSS nell’uomo. Dal test è emerso che, nonostante la somministrazione di dosi ripetute ben al di sopra della IC50 per la MMP-13, non è stata osservata fibroplasia. Inoltre è stata riscontrata protezione per il collagene di tipo II nella cartilagine articolare e sollievo per i pazienti affetti da OA[24].
Successivamente, Freeman-Cook et al. hanno individuato in uno studio parallelo[25] una serie di composti spiro-pirimidintrionici analoghi di quelli sintetizzati da Reiter.
Tab.2.6 Confronto delle attività di composti etossietilici legati tramite ossigeno (serie A) con quella di
44
Dai dati riportati nella Tab.2.6 risulta che nei composti ciclici spiro-pirrolidinici si assiste ad un modesto incremento della potenza verso la MMP-13 (5-10 volte) rispetto agli analoghi legati tramite ossigeno. In generale gli alti livelli di selettività verso la MMP-8 e -12 dei composti della serie A vengono mantenuti anche per quelli della serie B. Questo non deve meravigliare, in quanto le interazioni stabilite dal residuo P1’ con la tasca S1’, responsabile della selettività, rimangono invariate in entrambe le serie di composti. Purtroppo però questo non vale per la MMP-2, verso la quale la selettività risulta in qualche modo compromessa negli analoghi spiro-ciclici. In molti casi (composti 14, 16, 20) il rapporto di selettività cade al di sotto del valore soglia desiderato (100 volte). Nel gruppo dei 4-fenil-ossazoli la SAR ha suggerito che la selettività per la MMP-2 si mantiene al di sopra del valore soglia solo per i composti
para-sostituiti. Nel gruppo dei benzoimidazoli (composti 22, 24, 26) il linker spiro-ciclico conferisce ancora un aumento della potenza di 5-10 volte verso la MMP-13, ma il profilo di selettività verso le altre MMPs è diverso: infatti se da un lato non è molto difficile raggiungere alti livelli di selettività per la MMP-2 e -8, dall’altro lo è per quanto riguarda la MMP-12 (analogamente a quanto succede nei corrispondenti composti aciclici 21, 23, 25). Date la selettività e la potenza osservate, il composto 12 è stato sottoposto a studi di farmacocinetica ed ha mostrato un eccellente profilo (volume de distribuzione = 1.3 L/kg, clearance = 3.9 mL/min/kg, t1/2 = 4.5h). Inoltre il composto
12 si è mostrato molto efficiente nel prevenire in vitro la degradazione della cartilagine
bovina nasale indotta da MMP-13 con una IC50 = 12 nM. Sono stati condotti anche dei test in vivo di iniezione intraarticolare di MMP-13 nella cartilagine del ginocchio di un criceto, nei quali il composto 12 ha mostrato una capacità di inibizione superiore all’80% alla dose di 10 mg/kg per os). Date queste caratteristiche positive il composto
45
12 è stato scelto per testare l’ipotesi che un inibitore selettivo della MMP-13 potesse
eliminare l’insorgere di effetti collaterali di MSS. A tale scopo è stato testato nel modello di fibroplasia nel ratto per 14 giorni a dosi molto elevate e , sebbene non abbia prodotto tossicità acuta, sono stati osservati segnali pre-clinici di MSS. Queste scoperte sono state sufficienti a determinare l’interruzione degli studi pre-clinici su questi analoghi[25].
Studi più recenti sono orientati verso l’impiego degli inibitori delle MMPs a struttura barbiturica in diagnostica, utilizzandoli come radiotraccianti in vivo per la visualizzazione non invasiva di MMPs attivate, utilizzando tecniche di scintigrafia come PET e SPECT[22, 26].