1. INTRODUZIONE
1.1 Polimeri anfifilici
Macromolecole che mostrano affinità per il mezzo acquoso rappresentano una classe di polimeri che comprende dai biopolimeri, i quali mediano i processi vitali, a resine sintetiche di grande importanza commerciale. I polimeri anfifilici sono macromolecole costituite da porzioni idrofobe e da porzioni idrofile, che permettono la loro dispersione in fase acquosa.
1.1.1 Caratteristiche strutturali e proprietà delle dispersioni acquose
La struttura del polimero può derivare da un singolo monomero o da più monomeri. Queste unità possono essere posizionate in modo da dare copolimeri casuali, alternati o aggraffati (Figura 1.1). AABABBBABAABAAABBABBBAAB AAAAAAAAAAAAAAA B B B B B B B B B B B B B B B ABABABABABABABABABABABAB AAAAAABBBBBBAAAAAABBBBBB copolimero alternato copolimero casuale copolimero a blocchi copolimero aggraffato
Figura 1.1 Distribuzione dei monomeri.
(a)
(b)
(c)
Figura 1.2. (a) copolimero lineare; (b) copolimero ramificato;
(c) copolimero ramificato- struttura dendridica.
L’affinità per l’acqua può essere data dall’interazione di segmenti ionici, polari o idrofili che si legano tramite legami a idrogeno, oppure può riguardare la solvatazione di strutture anfifiliche, cioè di polimeri che combinano porzioni idrofile e idrofobe.
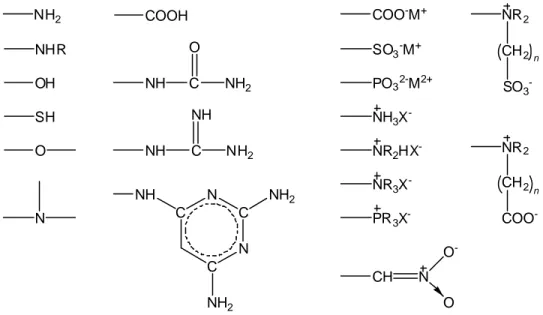
NH2 NHR OH SH O N COOH NH C O NH2 NH C NH NH2 C C N C N NH NH2 NH2 COO-M+ SO3-M+ PO32-M2+ NH3X -NR2HX -NR3X -PR3X -NR2 CH2 SO3 -NR2 CH2 COO -n n CH N O -O
Figura 1.3. Gruppi funzionali che impartiscono idrofilia.
Polimeri anfifilici in acqua possono costituire sospensioni, dispersioni o soluzioni, in funzione dello stato di aggregazione, della idrofilia del polimero e del tipo di miscelazione.1 In mezzo acquoso tali polimeri formano generalmente dispersioni colloidali, costituite da aggregazioni di macromolecole, a causa della incompleta solubilità derivante dalle caratteristiche anfifiliche. Un colloide polimerico può a sua volta avere caratteristiche liofiliche o liofobiche, a seconda della maggiore o,
rispettivamente, minore tendenza del polimero ad “assorbire” acqua dal mezzo sospendente.2 All’aumentare delle caratteristiche idrofile della struttura polimerica le particelle vengono rigonfiate dall’acqua in misura crescente, fino al punto in cui l’affinità per l’acqua rende le macromolecole completamente solubili ed il sistema perde le caratteristiche tipiche di una dispersione colloidale.
In questa tesi verranno presi in considerazione materiali polimerici appartenenti a tre tipologie strutturali caratterizzate da una ampia variabilità delle caratteristiche liofiliche/liofobiche, e quindi della natura delle rispettive dispersioni acquose. Questi sono:
a) copolimeri anfifilici a blocchi, in grado di dar luogo a soluzioni, aggregazioni micellari o particelle strutturate (sfere stratificate di tipo core-corona) di maggiori dimensioni, in funzione della natura dei comonomeri e del peso relativo dei singoli blocchi;
b) polimeri ionomerici (ossia contenenti una frazione non trascurabile ma comunque inferiore al 10-15% di comonomeri ionizzabili), in grado di
autodisperdersi in mezzo acquoso senza l’ausilio di tensioattivi o disperdenti;
c) polimeri sostanzialmente idrofobi contenenti piccole quantità di gruppi funzionali polari o ionizzabili, che possono essere disperdibili in acqua come tali o, più frequentemente, grazie all’ausilio di additivi tensioattivi o disperdenti; questi ultimi sono comunemente noti come lattici polimerici.
Per i colloidi in generale, e i colloidi polimerici in particolare, si può schematizzare il processo di formazione in due modi: (a) per disintegrazione di particelle più grandi oppure (b) per condensazione di particelle più piccole, di solito molecole.
Nel primo caso, se si esclude il caso di alcuni copolimeri a blocchi in grado di autodisperdersi ed assemblarsi in strutture micellari termodinamicamente stabili, il polimero preformato deve essere emulsionato nel diluente e ciò richiede grandi quantità di agenti emulsionanti e di energia meccanica. Polimeri sopra un certo peso molecolare sono inoltre così viscosi che è praticamente impossibile suddividere il materiale in massa, o anche una soluzione del polimero, in piccole particelle. Polimeri a basso peso molecolare, specialmente quelli contenenti gruppi ionici lungo la catena, possono essere abbastanza facilmente emulsionati per formare dispersioni stabili. Per questo motivo tale procedura viene adottata prevalentemente per polimeri ottenuti con processi di polimerizzazione a stadi, che forniscono in genere pesi molecolari moderati e spesso
non possono essere condotti direttamente in mezzo acquoso a causa della nucleofilicità dell’acqua. Ad esempio ionomeri poliuretanici, che hanno siti ionici idrofili tra segmenti di catena idrofobi predominanti, sono autodisperdenti in condizioni favorevoli. Questi prodotti, se opportunamente progettati, possono formare dispersioni stabili in acqua senza l’influenza di forze di separazione e in assenza di disperdenti. Le particelle tendono tuttavia ad avere dimensioni maggiori e a più ampia distribuzione rispetto a quelle delle dispersioni colloidali ottenute per polimerizzazione in emulsione.
I lattici sintetici, o anche semplicemente lattici, sono tuttavia ottenuti più frequentemente per polimerizzazione in emulsione con meccanismo a catena, coinvolgente propagazione radicalica. Comunque in condizioni appropriate avvengono anche meccanismi ionici, sia in mezzo acquoso sia in mezzo non acquoso. Il termine
lattice deriva dal nome dato alla linfa di aspetto lattiginoso, ottenuta da alcune piante
tropicali come l’albero della gomma (Hevea braziliensis), costituita da colloidi di gomme politerpeniche naturali sospese in mezzo acquoso, contenenti piccole quantità di proteine e altre sostanze che agiscono da stabilizzanti colloidali. Le dispersioni polimeriche colloidali sono termodinamicamente instabili ma cineticamente stabilizzate grazie alla presenza di cariche elettrostatiche superficiali che fanno sì che le particelle si respingano mutuamente. Tale repulsione elettrostatica, a volte coadiuvata da un effetto di tipo sterico (in presenza di tensioattivi non ionici) impedisce che le forze attrattive di Van der Waals causino l’aggregazione delle particelle, che porterebbe alla coagulazione del componente polimerico del lattice.
I lattici hanno di solito una bassa viscosità, quasi come quella dell’acqua, benché in alcune condizioni possano essere molto viscosi. La tensione superficiale di questi sistemi può variare da circa 20 mN?m-1 a quella dell’acqua pura, 73 mN?m-1. Le dimensioni delle particelle colloidali variano da circa 10 nm a 1000 nm in diametro e sono generalmente sferiche. Per tale motivo i colloidi polimerici hanno generalmente un aspetto lattiginoso, ma possono essere anche bluastri, traslucidi se le particelle sono sufficientemente piccole da non diffondere la luce visibile. Quando le particelle hanno dimensioni molto uniformi, i colloidi possono mostrare opalescenza.
1.1.2 Applicazioni
I polimeri anfifilici sono utili materiali per una molteplicità di applicazioni che spaziano dalla stabilizzazione di dispersioni colloidali alle applicazioni in campo biomedico. Sono impiegati come modificatori reologici di fluidi semplici e complessi, come tensioattivi polimerici3, come agenti schiumogeni o antischiumogeni4, emulsionanti, agenti solubilizzanti, flocculanti e disperdenti di sospensioni solide5 e quindi come componenti di vernici e inchiostri, in formulazioni di cosmetici6, alimenti, e fluidi per il recupero secondario di petrolio, come veicoli per il rilascio controllato di farmaci.7
I polimeri anfifilici sono interessanti alternative ai tensioattivi in molte applicazioni. Il loro autoassemblaggio varia fortemente con l’architettura del copolimero, tanto che il loro comportamento può essere molto diverso per diverse architetture polimeriche (copolimeri a blocchi, aggraffati e a stella) e solo per i copolimeri a blocchi c’è una stretta rassomiglianza con il comportamento dei tensioattivi.
Analogamente ai tensioattivi a basso peso molecolare, i polimeri anfifilici possono dar luogo ad una varietà di strutture che includono lamelle, sfere, cilindri e micelle.
Grazie alla loro efficacia di adsorbimento all’interfaccia liquido/solido, possono essere impiegati come additivi per lattici polimerici; tuttavia la maggior parte dei lattici polimerici è costituita da macromolecole esse stesse con caratteristiche anfifiliche, che ne favoriscono la dispersione e stabilizzazione colloidale in un mezzo acquoso anche in assenza di additivi disperdenti. I lattici sintetici hanno a loro volta una vastissima varietà di applicazioni. Lattici che formano film sono impiegati come componenti principali della matrice legante in vernici8, in rivestimenti e impregnanti per pavimenti e substrati porosi9, inchiostri da stampa, adesivi10, sigillanti11 e sempre più in applicazioni dove sostituiscono i tradizionali prodotti a solvente come conseguenza di no rme ambientali più restrittive.
1.2 Lattici da polimerizzazione in emulsione di monomeri vinilici
1.2.1 Aspetti generali
Come accennato in precedenza, dispersioni acquose di particelle polimeriche di dimensioni comprese tra 0,1 e 1 ?m sono dette anche lattici.
A causa delle loro dimensioni, le sospensioni colloidali di tali particelle non sono termodinamicamente stabili, ma possono essere stabilizzate dal punto di vista cinetico grazie alla presenza di opportune funzionalità polari o ioniche (ad esempio derivanti dall’iniziatore o da comonomeri funzionali) o per aggiunta di opportuni agenti emulsionanti, o tensioattivi, che per le loro caratteristiche strutturali riescono a disporsi tra le due fasi, riducendo in questo modo l’energia interfacciale. I tensioattivi sono molecole anfifiliche costituite essenzialmente da una porzione polare idrofila ed una parte idrofoba, che può essere ad esempio una lunga catena alchilica.
I lattici sono in genere preparati tramite polimerizzazione in emulsione, che prevede, nei casi più semplici di processi di polimerizzazione discontinua (batch) o semicontinua, la formazione di una emulsione più o meno stabile del monomero nel mezzo acquoso e la successiva polimerizzazione in presenza di iniziatori radicalici, solitamente solubili in acqua. Comunemente in tale processo vengono aggiunti dei tensioattivi che hanno il doppio ruolo di agenti emulsionanti del monomero in acqua e di stabilizzanti della sospensione delle particelle solide del polimero disperse in acqua al termine della polimerizzazione, ovvero funzionano da disperdenti.
Tuttavia è possibile anche condurre polimerizzazioni in emulsione in assenza di tensioattivi (“soapless”), quando il copolimero abbia caratteristiche anfifiliche derivanti dalla presenza di comonomeri ionizzabili e di altri gruppi ionici, generalmente derivanti dall’iniziatore.
Dispersioni acquose di particelle polimeriche si possono anche ottenere per polimerizzazione in sospensione, nella quale le piccole gocce di monomero, in cui è solubile l’iniziatore, sono sospese in acqua per mezzo di una vigorosa agitazione e grazie alla presenza di stabilizzanti colloidali, generalmente polimerici. In questo caso si ottiene un polimero colloidalmente instabile, che viene isolato dal mezzo di reazione per filtrazione o per centrifugazione. Tale processo dà luogo a sfere generalmente rigide (con temperatura di transizione vetrosa Tg elevata), con diametro che varia tra 0,1 e 2
?m, la cui composizione può essere variata solo in modo limitato. Ne consegue che materiali di questo tipo trovano solo un ristretto numero di applicazioni.
Con il processo in emulsione, invece, si possono ottenere polimeri di composizione desiderata e con una bassa Tg, in grado di formare dei film per semplice deposizione su
un substrato. In seguito ad evaporazione dell’acqua i lattici polimerici possono dar luogo alla formazione di un film oppure di una polvere, a seconda delle caratteristiche del polimero, delle condizioni di essiccamento e della eventuale presenza di additivi, quali coalescenti o plastificanti.
1.2.2 Formazione di films
La constatazione degli effetti deleteri sull’ambiente dei prodotti a base di solventi organici e le normative sempre più restrittive che ne regolano l’impiego ha condotto ad una notevole evoluzione dei rivestimenti derivanti da dispersioni acquose polimeriche. Di conseguenza ciò ha richiesto una migliore comprensione dei processi che governano la trasformazione del lattice polimerico da dispersione acquosa di particelle solide a film polimerico continuo12. Gli studi sui processi di filmazione tendono a migliorare soprattutto le proprietà superficiali, meccaniche, di stabilità all’acqua e di permeabilità dei film stessi.
Per “processo di formazione del film” si intende l’intera sequenza di eventi con i quali, da una dispersione acquosa del polimero si passa ad un film privo di acqua. Il processo globale può essere schematizzato in tre passaggi fra quattro stati come illustrato nella Figura 1.4. Il primo stato corrisponde allo stato bagnato (wet state) del lattice: il polimero solido è disperso in acqua come particelle sferiche, generalmente tra il 15 ed il 50% in peso. Dopo l’evaporazione della maggior parte dell’acqua, si ha la formazione del secondo stato. Esso è descritto come un impaccamento più o meno ordinato di particelle in contatto tra loro con residui di acqua negli spazi interparticellari. In questa fase, mantenendo la temperatura sopra quella di transizione vetrosa Tg, l’evaporazione e le interazio ni intermolecolari si combinano per produrre il
terzo stato in cui le particelle sono deformate in seguito al processo di compattazione, ma ancora distinte tra loro; in questo stato la struttura solida è ancora poco coesa e quindi ancora debole dal punto di vista meccanico. Viene indicata con MFT13 (minimum film- forming temperature) la temperatura minima alla quale si verifica la
transizione tra il terzo ed il quarto stato, ossia la formazione del film; in assenza di dati sperimentali tale temperatura viene considerata di poco superiore alla Tg del polimero.
In quest’ultimo passaggio di formazione del film, l’interdiffusione delle catene polimeriche attraverso i confini interparticellari attenua la distinzione tra le particelle stesse. Il risultato di questo processo finale di coalescenza delle particelle è, in un sistema ideale, un film continuo, isotropo ed uniforme.
Figura 1.4. Rappresentazione schematica dei processi coinvolti nella formazione dei
film
In un sistema reale la situazione è spesso più complessa anche a causa della presenza di ausiliari di polimerizzazione come i sali derivanti dai residui della decomposizione degli iniziatori, i tensioattivi, ed eventuali stabilizzanti etc. che possono influenzare in modo determinante le caratteristiche del film.
Per asciugare il lattice sono necessari da alcune ore a vari giorni in funzione, oltre che della composizione del lattice e della morfologia delle sue particelle, anche delle condizioni ambientali (temperatura14, pressione, umidità14 etc.) e dello spessore del film che si vuole ottenere. Consideriamo ad esempio l’influenza dell’umidità e della composizione del polimero sulla MFT: nel caso di polimeri relativamente idrofobi, la MFT è generalmente prossima alla Tg e la filmazione non è influenzata dall’umidità,
mentre nel caso di polimeri acrilici costituiti da monomeri a basso peso molecolare con caratteristiche idrofile l’umidità tende a diminuire la MFT, anche di alcuni gradi sotto la Tg, a causa dell’effetto plastificante esercitato dalle molecole di acqua che rimangono
assorbite nel film. In alcuni casi, quando la Tg del polimero disperso nel lattice è elevata
rispetto alle necessità imposte dalla sua applicazione, può essere necessario aggiungere appositamente degli additivi plastificanti (in sede di polimerizzazione) o dei coalescenti (a polimerizzazione avvenuta) che hanno il compito di abbassare la MFT del sistema. In particolare, i coalescenti sono dei solventi, a volatilità inferiore o comparabile rispetto a
quella dell’acqua, nei quali il polimero è solubile; essi sono aggiunti per aumentare la velocità di interdiffusione delle catene e, a differenza dei plastificanti, presentano il vantaggio di non essere stabilmente trattenuti dal film polimerico e di non modificarne quindi le proprietà finali.
Nel periodo successivo all’allontanamento dell’acqua e quindi alla formazione del film, spesso si osserva un miglioramento nel tempo sia dell’aspetto che delle proprietà del film stesso15. Questo fenomeno, indicato come further coalescence, è associato all’interdiffusione delle catene polimeriche attraverso la superficie delle particelle16. Esistono prove sperimentali indirette della diffusione interparticellare. Per esempio, essa spiega il miglioramento delle proprietà meccaniche e di permeazione dei film. In molti casi è possibile osservare attraverso la microscopia elettronica a trasmissione (TEM) la scomparsa nel tempo dei contorni delle singole particelle17.
Tuttavia, in alcuni casi, analisi TEM dimostrano chiaramente come la distinzione tra le particelle persista anche in film invecchiati per più di un anno18. Per capire quali siano i fattori che determinano o meno la diffusione interparticellare delle catene polimeriche sono state messe a punto diverse tecniche di osservazione diretta di tale fenomeno. Per esempio, l’interdiffusione in sistemi derivanti dalla miscelazione di due lattici acrilici, uno deuterato e l’altro non deuterato, è stata analizzata mediante scattering neutronico a basso angolo (SANS)19. Ciò ha mostrato come sia la temperatura di formazione del film MFT, sia il grado di reticolazione del polimero ricoprano un ruolo determinante sull’estensione della diffusione interparticellare.
Più recentemente Winnik e coll. hanno approfonditamente indagato il fenomeno della further coalescence attraverso misure di fluorescenza sfruttando il fenomeno del trasferimento energetico non radiativo (NRET, non radiative energy transfer)20,21 tra specie fluorescenti con caratteristiche rispettivamente di donatore ed accettore. In tali lavori sono stati sintetizzati due lattici acrilici marcati con indicatori di fluorescenza (1-2 % mol) diversi: un donatore D (generalmente un derivato fenantrenico) ed un accettore A (generalmente un derivato antracenico). I due lattici sono stati poi mescolati ed il processo di formazione del film è stato studiato mediante analisi di trasferimento energetico non radiativo dal donatore all’accettore, evidenziabile grazie a tecniche rapide di irraggiamento pulsato ed acquisizione delle curve di decadimento della fluorescenza (Figura 1.5). Inizialmente, mentre il film si sta asciugando, si osserva un trasferimento energetico abbastanza piccolo, ad indicare che le particelle mantengono la
loro individualità. Al trascorrere del tempo e mantenendo la temperatura sopra la Tg del
polimero, si osserva un incremento del trasferimento energetico, ossia un decadimento più rapido della fluorescenza, a conferma dell’avvenuta interdiffusione delle catene polimeriche attraverso i contorni delle particelle e quindi dell’avvicinamento di D ed A.
Figura 1.5. Schematizzazione dei processi coinvolti nell’analisi NRET
La versatilità mostrata dalle tecniche di fluorescenza nello studio dei processi di coalescenza dei film ha permesso di investigare anche sistemi più complessi quali, ad esempio, lattici funzionalizzati aventi morfologia core-shell in cui i markers fluorescenti sono segregati in regioni specifiche delle particelle polimeriche22.
1.3 Dispersioni acquose poliuretaniche
1.3.1 Aspetti generali
Nel Paragrafo 1.1.1 si era accennato al fatto che lattici polimerici si possono ottenere anche a partire da polimeri preformati, o in seguito a reazioni di poliaddizione a stadi condotte direttamente nel mezzo acquoso come nel caso di alcuni silossani. Una classe importante di dispersioni acquose polimeriche è costituita da lattici poliuretanici. Nei processi convenzionali di sintesi in massa o in solvente i poliuretani si ottengono mediante reazioni di poliaddizione tra isocianati polifunzionali e macroglicoli, ossia reagenti che contengono due o più funzionalità ossidriliche. La reattività dei poliisocianati verso i composti con caratteristiche nucleofile verrà discussa più in dettaglio nel Paragrafo 1.1.3. Può essere anche utilizzata una combinazione di
Emissione di fluorescenza dalla specie D
Energy Transfer non radiativo da D ad A Diffusione polimerica interparticellare Deformazione/coalescenza A A D D A A A A D
macrodioli ed estensori amminici o diolici a catena corta (Figura 1.6 A). Nel primo caso si formano omopolimeri lineari, mentre nel secondo si ha la formazione di copolimeri lineari segmentati (Figura 1.6 B).
OCN R' NCO HO R OH HO CH2 x OH H2N CH2 y NH2 diisocianato macroglicole estensori RO C O NH R' NH C O O n polimero lineare RO C O NH R' NH C O O CH2 O C O NH R' NH C O O n RO C O NH R' NH C O NH CH2 NH C O NH R' NH C O O n x y
copolimero lineare segmentato
(A) (B)
Figura 1.6. Struttura generale (A) dei monomeri impiegati; (B) dei poliuretani e
poliuretani-uree ottenuti nella sintesi.
All’aume ntare del grado di funzionalità dei monomeri di partenza (f >2) si ottengono, oltre ad i polimeri lineari già citati, anche polimeri ramificati e reticolati.
Nella catena macromolecolare è possibile quindi individuare domini rigidi in corrispondenza dei legami uretanici o ureici e domini flessibili in corrispondenza dei macroglicoli.
I poliuretani lineari sono materiali termoplastici, con buone proprietà chimico-fisiche, meccaniche e di processabilità, e sono quindi importanti materie prime per la produzione di rivestimenti, vernici, adesivi e impregnanti. In genere, tuttavia, le formulazioni impiegate portano alla formazione di prodotti reticolati con migliori caratteristiche meccaniche, ma maggiori limitazioni in fase applicativa, essendo spesso costituite da sistemi bicomponenti reattivi.
Nella sintesi possono essere utilizzati isocianati aromatici, alifatici o cicloalifatici, mentre i macroglicoli sono generalmente di natura polieterea o poliesterea, con pesi molecolari variabili.
Dal punto di vista applicativo i poliuretani mostrano una elevata versatilità: a partire da essi si possono produrre, infatti, materiali con proprietà anche molto diverse tra loro (elastomeri e materiali rigidi). Sono quindi molteplici le possibilità di impiego: dagli adesivi, alle schiume, sia rigide che flessibili, dall’industria automobilistica a quella tessile, dai rivestimenti (coatings), alle vernici per legno e metalli.
1.3.2 Ionomeri poliuretanici
Le dispersioni acquose poliuretaniche, analogamente ai lattici acrilici, sono sistemi binari nei quali le particelle di polimero sono disperse in un mezzo acquoso continuo23,24. A differenza degli analoghi sistemi in solvente, tuttavia, la reattività dei gruppi isocianato con l’acqua ne limita fortemente la formulazione in sistemi bicomponenti. Le particelle devono quindi essere costituite da polimero preformato, con al più qualche gruppo funzionale reattivo utilizzabile per reazioni di reticolazione. Inoltre per disperdere nel mezzo acquoso un polimero, per sua natura insolubile in acqua, è necessaria la presenza di un emulsionante, che può essere esterno, ossia un tensioattivo aggiunto in fase di formulazione, oppure interno. In quest’ultimo caso vengono utilizzati nella polimerizzazione dei monomeri aventi gruppi idrofili, o facilmente trasformabili in gruppi idrofili per reazione successiva alla fase di polimerizzazione25.
Si possono così produrre sia poliuretani non ionomerici, contenenti segmenti flessibili idrofili, ottenuti, ad esempio, usando un poli(etilenglicole) come macrodiolo, sia poliuretani ionomerici, che sono i più utilizzati. Questi ultimi sono caratterizzati dalla presenza di gruppi ionici legati covalentemente alla catena principale, i quali favoriscono l’autoemulsione del polimero e permettono di ottenere dispersioni molto stabili, con particelle aventi dimensioni anche molto piccole (0,01?0,2 ?m).
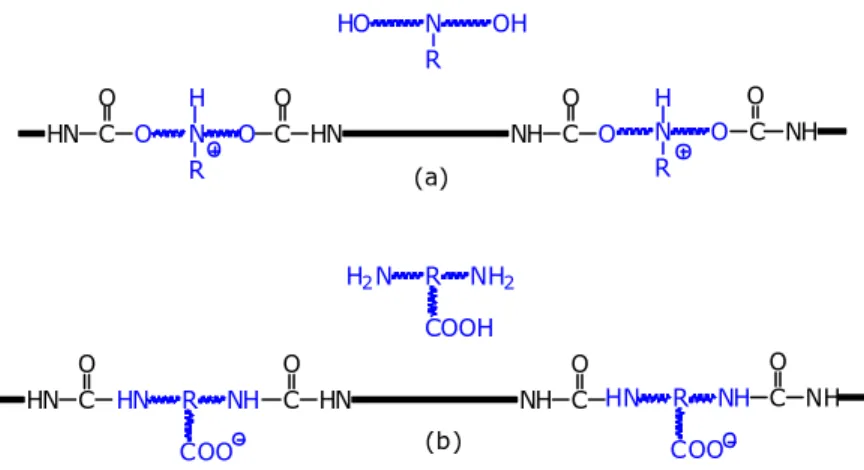
I poliuretani ionomerici possono essere ottenuti sia in forma cationica sia in forma
anionica. I primi contengono gruppi amminici terziari alchilati o protonati, mentre nei
secondi sono comunemente presenti gruppi carbossilato o solfonato. (Figura 1.7)
Un metodo per preparare uretani cationici è un adattamento della reazione di Menschutkin alla chimica dei poliuretani ed è stata chiamata poliaddizione
diammina. Se uno di questi componenti contiene un lungo segmento polietereo si ottiene uno ionomero. Questi ionomeri sono stati anche chiamati ioneni.
Figura 1.7. Ionomero poliuretanico cationico (a) ed anionico (b).
O CO NH N N Br Br N N N N + =
= segmento derivante dal diisocianato
Schema 1.1. Poliuretani cationici da poliaddizione quaternarizzante.
In alternativa, poliammonio-poliuretani possono essere sintetizzati preparando inizialmente un poliuretano contenente atomi di azoto terziari e poi quaternarizzando gli atomi di azoto un secondo stadio (Schema 1.2). Partendo da prepolimeri NCO terminati basati su polieteri, si possono così ottenere poliuretani segmentati quaternari. Similmente, poliuretani cationici con gruppi solfonio terziari possono essere preparati quando il glicole di partenza è un bis-2-idrossietil solfuro (Schema 1.3). La porzione ionica, o il suo precursore, può anche essere il diisocianato o parte di un polieterediolo a catena lunga. N R OH HO H2N R NH2 COOH HN NH C C O O HN R NH COO C O NH HN R NH COO C O HN HN NH C C O O O O N R O C O NH N R O C O HN H H (a) (b)
OCN NCO OCN NCO HO N OH HO N OH HO + OH + OCN NCO N N A A N N
Schema 1.2. Poliuretani cationici da poliaddizione NCO e seguente
quaternarizzazione o neutralizzazione. HO CH22 S CH22 OH O CH22 S CH2 2 O CH2 2 S CH22 O CH2 S 2 OCN NCO S S S S S S S S n (CH3)2SO4 = segmento polietereo Schema 1.3
Metodi speciali per l’introduzione di centri ionici, sia anionici che cationici, sono infine le reazioni di poliuretani o poliuree segmentati ad alto peso molecolare con composti ciclici come sultoni, lattoni o anidridi in presenza di basi forti. La reazione può avvenire con un gruppo amminico secondario e i prodotti ottenuti con i sultoni sono ammoniosolfonati zwitterionici, che possono essere convertiti in ionomeri cationici per quaternarizzazione e in ionomeri anionici per reazione con basi (Schema 1.4).
N N H H NH O O O N CO CH2 CH2 COO K + KOH NH + O SO2 NH ( CH2)3 SO3 N ( CH2)3 SO3H N (CH2)3 SO3 K CH3 (CH3)2SO4 KOH
Schema 1.4. Successiva introduzione di gruppi ionici in un poliuretano contenente
gruppi amminici secondari.
NH CO NH O SO2 NH CO NH NH CO N CH23 SO3 HN O O NH CO N CH2 CH2 COOH (C2H5)3N + + NH CO NH NH CO N CH2 SO3Na CH2O, NaHSO3 Schema 1.5
Una procedura di maggiore interesse pratico è la modificazione, sempre con l’impiego di sultoni o lattoni, di poliuretani già formati contenenti gruppi ureici. Ionomeri possono essere anche preparati per solfometilazione o amminometilazione. (Schema 1.5).
Per ottenere poliuretani anionici, tuttavia, la procedura più comune è basata sull’impiego di dioli che portano un acido carbossilico (Schema 1.6) o un gruppo solfonato. Nel caso dell’introduzione di gruppi carbossilici, questi ultimi vengono neutralizzati in una fase successiva, ad esempio con ammine terziarie.
In questa reazione deve essere fatta attenzione in modo da evitare la reazione tra i gruppi NCO e i gruppi COOH che causa indesiderate ramificazioni e reticolazione. Per questo si preferiscono composti con gruppi COOH stericamente ingombrati, come acido
? ,? -dimetilolpropionico. Con altri acidi, come l’acido tartarico, la neutralizzazione
dovrebbe avvenire immediatamente dopo l’addizione dell’acido.
N HN OCN NCO + OCN NCO HO OH COOH HO + OH OCN NCO COOH COO
Schema 1.6. Poliuretani anionici con gruppi carbossilato.
I gruppi solfonato sono di solito inseriti attraverso un diamminoalcansolfonato, poiché questi composti sono solubili in acqua e la reazione con i prepolimeri NCO terminati non è ostacolata dall’acqua (Schema 1.7).
OCN NCO SO3 Na H2N NH SO3 Na NH CO NH = in H2O
Questo metodo di produrre dispersioni poliuretaniche senza un agente emulsionante non è limitato a prepolimeri poliuretanici terminati con gruppi NCO. Analoghe trasformazioni con incorporazione dei centri ionici sono possibili anche nelle macromolecole ormai prive di centri isocianato reattivi. Tuttavia le caratteristiche applicative tipiche dei poliuretani, per i quali la procedura più comune porta alla formazione di una struttura reticolata a partire da prepolimeri o estensori di catena multifunzionali, rende spesso impraticabile o poco efficace il processo di trasfo rmazione del polimero finale in ionomero.
In generale il tipo e la quantità dei gruppi ionici influenza non solo la disperdibilità e la dimensione delle particelle, ma anche le proprietà fisiche, meccaniche e di adesione, la resistenza ai solventi e le proprietà filmanti.
Anionomeri poliuretanici che contengono gruppi solfonati o carbossilati incorporati sono predominanti. I policarbossilati forniscono buon carattere idrofobo mentre i polisolfonati danno dispersioni con stabilità eccellente anche in condizioni sfavorevoli.
Al contrario, le dispersioni cationiche non trovano al momento un largo impiego. Dispersioni acquose di microcapsule con pareti preparate da poliisocianati e sali di guanidinio sono state studiate per possibili usi come additivi speciali per impieghi tipografici (carta copiativa)26, o nei processi conciari27.
1.3.3 Procedure sintetiche
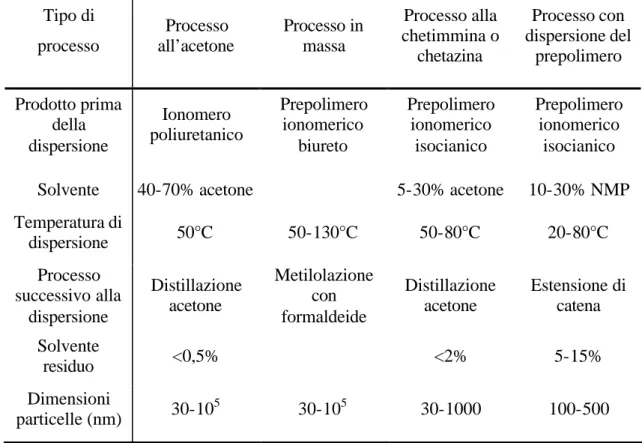
I principali metodi utilizzati per la sintesi di dispersioni acquose poliuretaniche secondo lo schema di reazione convenzionale (che prevede una reazione di addizione tra un eccesso di diisocianato ed un macrodiolo per formare un prepolimero terminato con la funzionalità isocianica, a cui segue un’estensione di catena attraverso la poliaddizione del prepolimero con dioli o diammine24,28) sono quattro: processo all’acetone, processo in massa, processo alla chetimmina o alla chetazina e processo con dispersione del prepolimero.
Il processo all’acetone è stato uno dei primi metodi che furono messi a punto verso la fine degli anni sessanta, nel quale l’intera polimerizzazione è effettuata in solvente; il
polimero viene poi disperso in acqua, ed infine il solvente allontanato sotto vuoto (Schema 1.8).
SO3 Na
soluzione in a cetone
aggiunta di acqua precipitazione di una dispersione
distillazione dell'acetone
dispersione in acqua
Schema 1.8
Nel processo in massa, la sintesi è effettuata in assenza di solventi e ad una temperatura superiore a 130°C, facendo reagire il prepolimero contenente terminazioni isocianato con ammoniaca od urea. I gruppi terminali di tipo urea o biureto così formati (vedi Paragrafo 1.3.4) sono sufficientemente idrofili da permettere la successiva dispersione del polimero in acqua.
Il processo alla chetimmina e quello alla chetazina prevedono invece l’estensione di catena in acqua, utilizzando come estensori rispettivamente diammine o idrazine bloccate, quali le chetimmine o le chetazine; la deprotezione, realizzata dall’acqua al momento della dispersione, avviene quindi contemporaneamente all’estensione di catena.
Successivamente si è cercato di sviluppare un processo che consentisse di diminuire la quantità di solvente utilizzato e con il processo con dispersione del prepolimero29, attualmente quello di maggior impiego, si è giunti ad operare con quantità minime di solvente, dell’ordine del 2?3%.
In questo caso la sintesi del prepolimero viene effettuata in massa, oppure in solvente ad altissima concentrazione, a caldo (50°C?80°C), in presenza del macrodiolo, del diisocianato e di un comonomero idrofilo o comunque contenente una funzionalità in grado di essere trasforma ta in gruppo idrofilo successivamente alla polimerizzazione, come descritto nel paragrafo precedente. Ad esempio, la neutralizzazione di gruppi funzionali carbossilici e/o solfonici o la quaternarizzazione di gruppi amminici terziari
può essere effettuata sia prima che durante la dispersione in acqua, operando quindi sul prepolimero NCO terminato o sul polimero finale.
In genere lo stadio finale di estensione di catena è realizzato contemporaneamente alla dispersione in acqua, gocciolando lentamente una soluzione acquosa dell’estensore di catena nella miscela di reazione contenente il prepolimero neutralizzato, ossia già contenente gruppi funzionali idrofili.
In Tabella 1.1 sono riportati alcuni parametri di confronto tra i metodi sopra descritti per la sintesi di dispersioni acquose poliuretaniche.
Tabella 1.1. Confronto tra i metodi di preparazione di dispersioni acquose
poliuretaniche. Tipo di processo Processo all’acetone Processo in massa Processo alla chetimmina o chetazina Processo con dispersione del prepolimero Prodotto prima della dispersione Ionomero poliuretanico Prepolimero ionomerico biureto Prepolimero ionomerico isocianico Prepolimero ionomerico isocianico
Solvente 40-70% acetone 5-30% acetone 10-30% NMP
Temperatura di dispersione 50°C 50-130°C 50-80°C 20-80°C Processo successivo alla dispersione Distillazione acetone Metilolazione con formaldeide Distillazione acetone Estensione di catena Solvente residuo <0,5% <2% 5-15% Dimensioni particelle (nm) 30-10 5 30-105 30-1000 100-500
Dieterich e coll. hanno proposto, per la formazione di dispersioni acquose, ed in particolare per il processo all’acetone, quattro stadi principali, ai quali possono essere tuttavia ricondotti tutti i processi di sintesi precedentemente descritti.
I. La viscosità subisce una prima drastica diminuzione, in seguito all’iniziale aggiunta di acqua alla soluzione in solvente. Ciò è stato attribuito alla minore
efficacia delle associazioni ioniche, che si verificano dopo la salificazione dei gruppi ionici sulla macromolecola, a causa della presenza dell’acqua.
II. L’ulteriore aggiunta di acqua provoca una diminuzione della concentrazione del solvente organico, il cui effetto solvatante sui segmenti idrofobici del polimero risulta notevolmente ridotto. Di conseguenza, i segmenti idrofobici tendono ad aggregarsi, causando un incremento della viscosità del sistema. III. In seguito, la precipitazione di aggregati e la separazione di una fase dispersa
rendono il sistema opalescente e torbido.
IV. Infine la formazione di micelle sferiche, con i gruppi ionici rivolti verso la superficie ed i segmenti idrofobici verso l’interno, causa un definitivo crollo della viscosità ed un notevole aumento della torbidità. In questo stadio il sistema è descrivibile come una fase continua acquosa ed una fase discontinua organica. Rimuovendo il solvente per distillazione è possibile ottenere la dispersione totalmente acquosa.
1.3.4 Diisocianati
Reattività generale
Per la sintesi di polimeri uretanici sono di norma utilizzati sia diisocianati alifatici che aromatici.
Nella sintesi di dispersioni acquose poliuretaniche sono maggiormente impiegati isocianati alifatici, rispetto agli aromatici, poiché hanno una tossicità inferiore e minore reattività nei confronti dell’acqua. Tale reazione porta infatti alla formazione di anidride carbonica ed ammine, a loro volta reattive verso il gruppo isocianato e quindi in grado di alterare il bilancio stechiometrico della polimerizzazione.
La reattività degli isocianati con l’acqua ha una notevole rilevanza nella preparazione di dispersioni acquose. Sia il meccanismo della reazione che la natura dei prodotti dipendono dal rapporto tra le concentrazioni di acqua e isocianato30,31. Per alti rapporti CH2O/CNCO, e a pH neutro, l’acqua si addiziona all’isocianato (Schema 1.9), per dare un intermedio acido carbammico (ii), che si decompone spontaneamente in un’ammina primaria ed anidride carbonica. L’ammina liberata reagisce prontamente con l’acido
carbammico intermedio, per dare un sale di carbammato (iii), stabile in acqua a basse temperature. A temperature superiori, il sale di carbammato si decompone, formando anidride carbonica ed un’ammina, che si addiziona ad un secondo gruppo isocianato, generando un derivato ureico (iv).
R N C O + H2O R N H C O OH R NH2 R N C O R N H C O O R NH3 R N H C O N H R R N C O - CO2 - CO2 ? (i) (ii) (iii) (iv) Schema 1.9
In presenza di un eccesso di isocianato, ovvero per bassi rapporti CH2O/CNCO, la reazione con acqua non è spontanea a temperatura ambiente, probabilmente a causa della scarsa solubilità in acqua dell’isocianato. Solventi polari, come eteri del glicole etilenico o ammidi, favoriscono la reazione aumentando la solubilità del substrato. In generale la reazione è catalizzata da ammine terziarie e composti metallici.
Dalle strutture di risonanza del gruppo isocianato (Schema 1.10) si evidenzia l’elevato carattere elettrofilo del carbonio isocianico, che risulta molto reattivo nei confronti dell’addizione nucleofila sul doppio legame C=N di composti insaturi o aventi idrogeni acidi.
Schema 1.10
Nel caso di isocianati aromatici, la delocalizzazione del doppietto elettronico dell’atomo di azoto sull’anello aromatico conferisce all’atomo di carbonio isocianico un maggior carattere elettrofilo. Negli isocianati alifatici, invece, il carattere
elettrondonatore dei gruppi alchilici incrementa la densità elettronica sull’atomo di azoto, rendendo l’atomo di carbonio isocianico meno elettrofilo. Di conseguenza, sebbene la reattività sia influenzata dalla natura dei sostituenti, dall’ingombro sterico e dal tipo di catalizzatore utilizzato, gli isocianati alifatici risultano meno reattivi degli isocianati aromatici e quindi più adatti all’impiego per sintesi e formulazioni in presenza di acqua.
Sintesi di prepolimeri di-isocianici per reazione con alcoli.
Polimeri uretanici vengono spesso preparati a partire da macromeri ottenuti per poliaddizione tra diisocianati e macrodioli, ossia oligomeri con gruppi alcolici terminali su catene polieteree o poliesteree. L’addizione nucleofila di funzionalità ossidriliche sugli isocianati (N- idro-C-alcossi addizione), che genera carbammati o uretani
sostituiti, può essere catalizzata da trietilammina, cloruro stannico, cloruro fe rrico,
2-etilesanoato ferrico e composti organostagno quali dibutilstagno di-(2-2-etilesanoato) e ottoato di stagno.
Con alcoli primari e secondari la reazione procede velocemente, anche in assenza di catalizzatori, a temperature di 50÷100°C. Alcoli terzia ri e fenoli reagiscono più lentamente; inoltre i carbammati derivanti da alcoli terziari, ad alta temperatura, si decompongono facilmente dando ammine, anidride carbonica ed olefine (Schema 1.11).
Schema 1.11
In funzione del rapporto stechiometrico tra diolo (DO) e diisocianato (DI), nell’intervallo 1? DI/DO ? 2 si può regolare il peso molecolare del prepolimero risultante, che avrà terminazioni isocianato.
R N C O OH N C H R O O ? R NH 2 CO2
Estensione di catena per reazione con ammine, uretani e uree.
Lo stadio finale di estensione della catena polimerica ed eventuale reticolazione (se si usano monomeri con funzionalità superiore a 2) comporta generalmente la formazione di derivati ureici per addizione di ammine ad isocianati. Questa è una reazione solitamente esotermica, veloce e quantitativa la cui velocità è fortemente influenzata dalla basicità dell’ammina.
Generalmente, le ammine reagiscono con gli isocianati più velocemente degli alcoli. Ammine alifatiche primarie e secondarie ed ammine aromatiche primarie reagiscono velocemente a temperature di 0-25°C ed in assenza di catalizzatori. Diminuendo la basicità dell’ammina, come nel caso di ammine difenil-sostituite, decresce la velocità della reazione di addizione.
Gli isocianati, a temperature di 100-140°C, possono reagire anche con i gruppi NH
uretanici ed ureici dei prepolimeri stessi (Schema 1.12), formando rispettivamente allofanati (v) e biureti (vi).
N R N C C O O N R N C C O O R' N H C O OR'' C N R' C O OR'' O N H R OCN R' N H C O N R'' H C N R' C O N O N H R OCN H R'' + + (v) (vi) Schema 1.12
Tali reazioni comportano la trasformazione di macromolecole lineari in strutture ramificate o reticolate, e comunque di più alto peso molecolare, per ulteriore reazione con una seconda molecola di isocianato. Durante la sintesi di dispersioni acquose poliuretaniche, ad esempio nel processo in massa, possono essere raggiunte le temperature a cui risulta favorita la formazione di allofanati e biureti; ciò può comportare sia la variazione dei rapporti stechiometrici tra monomero diisocianico e macroglicole, sia, come si è detto, la reticolazione del polimero.
Altre reazioni.
Tra le reazioni di un certo interesse per la sintesi di poliuretani in dispersione acquosa si possono citare inoltre la reazione con acidi carbossilici, con formazione di N-carbossianidridi (vii) termicamente instabili (Schema 1.13) che si decompongono, a caldo o in presenza di catalizzatori, ad ammidi ed anidride carbonica.
La decomposizione del derivato (vii) è fortemente dipendente sia dalle strutture dell’isocianato e dell’acido carbossilico, sia dalle condizioni di reazione. Generalmente la temperatura a cui avviene la decomposizione è di 80-180°C.
Processi di reticolazione possono aver luogo anche per trimerizzazione degli isocianati; in particolare con gli isocianati alifatici è frequente la ciclotrimerizzazione ad esaidro-s-triazintrioni 1,3,5-trisostituiti o isocianurati (viii), in opportune condizioni di reazione31,32 (Schema 1.14).
Schema 1.13
Schema 1.14
Macroglicoli
Come accennato in precedenza, la sintesi di polimeri uretanici prevede spesso un primo stadio di poliaddizione di un diisocianato con un diolo, che è solitamente costituito da catene polieteree o poliesteree terminate con gruppi ossidrilici. Tali componenti sono comunemente detti macroglicoli o macrodioli e la loro struttura
R N C O R1 COOH R N C O H C R1 O O R1 N C OR2 H O CO2 ? ? Cat (vii) 2 R N C O Base N N C O O R R R N C O N N N O O O R R R (viii)
chimica e peso molecolare (tipicamente da poche centinaia ad alcune migliaia) sono determinanti per le proprietà finali del polimero24,33.
I glicoli a basso peso molecolare usati più comunemente, l’1,4-butandiolo e l’1,6-esandiolo, danno luogo alla formazione di segmenti rigidi e fragili all’interno del polimero, a causa della elevata frazione di gruppi uretanici. Macroglicoli a catena lunga, con pesi molecolari intorno a 1000-6000 uma, producono poliuretani più flessibili, caratterizzati da una migliore affinità per l’acqua.
I macrodioli impiegati più comunemente sono di tipo polietereo o poliestereo. In generale i polieteri sono meno viscosi e più facilmente processabili dei poliesteri. Mentre i segmenti polieterei conferiscono una maggiore stabilità idrolitica al polimero, i segmenti poliesterei favoriscono le interazioni polari ed i legami a idrogeno tra le catene macromolecolari. Ciò determina un orientamento delle catene, quindi un irrigidimento del sistema, che acquista una elevata resistenza meccanica.
1.3.5 Struttura e proprietà dei polimeri poliuretano -ureici e dei film da essi derivati
La stabilità delle dispersioni acquose poliuretaniche, ottenute per lo più con una delle procedure descritte nel Paragrafo 1.3.2, è assicurata dalla formazione di un doppio strato elettrico tra i gruppi ionici covalentemente legati al polimero ed i loro controioni, presenti nella fase acquosa. L’interazione elettrostatica repulsiva tra i gusci carichi delle particelle ne ostacola la tendenza alla coalescenza, contribuendo alla stabilizzazione della dispersione acquosa. La stabilità di una dispersione è peraltro condizionata anche dalle dimensioni delle particelle che la costituiscono. In particolare, maggiore è la dimensione delle particelle e maggiore è la loro tendenza a sedimentare, ovvero minore è la stabilità colloidale del sistema. Particelle più piccole, invece, formano dispersioni più stabili ed hanno inoltre una migliore capacità filmante, grazie alla loro elevata energia superficiale. .
Come già discusso nel Paragrafo 1.2.2 il processo di formazione di un film polimerico da dispersioni acquose è molto complesso. Le prestazioni di un coating, ossia un rivestimento protettivo costituito da un legante polimerico, dipendono in larga misura dalla facilità di formazione del film, oltre che dalle proprietà chimiche e
meccaniche del materiale polimerico. È noto come la capacità filmante di polimeri in solvente sia superiore rispetto ai corrispondenti sistemi acquosi. Questi ultimi, se non formulati correttamente, possono più facilmente dare luogo a film con microfratture e altri difetti superficiali che, lasciando penetrare acqua, rendono più fragile e meno duraturo il rivestimento, oltre a causare la perdita di efficacia protettiva.
I poliuretani sono copolimeri segmentati, contenenti sequenze alternate di segmenti flessibili e rigidi, derivanti rispettivamente dal macrodiolo e dal diisocianato29f,34 (Figura 1.8). Allo stato solido l’incompatibilità tra tali sequenze genera una caratteristica struttura con microseparazione di fase.I principali fattori che determinano la formazione delle microfasi sono: la formazione di legami a idrogeno tra i gruppi uretanici ed ureici ed i gruppi carbossilici presenti sul polimero, l’interazione elettrostatica tra i gruppi ionici e, infine, la possibile cristallizzazione sia dei segmenti rigidi sia di quelli flessibili. Inoltre vi possono essere parziali miscelazioni di fase, in seguito ad interazioni tra i domini rigidi e flessibili, mediante legame a idrogeno con formazione di estese zone interfacciali.
Segmento rigido Segmento flessibile Diisocianato Macrodiolo Legame uretanico
Figura 1.8. Segmenti rigidi e flessibili in un polimero uretanico.
Si ritiene che le proprietà dei film di ionomeri poliuretanici siano determinate sia dal comportamento delle microfasi sia dal loro carattere ionico.
Infatti, nel caso di ionomeri poliuretanici, la presenza dei gruppi ionici, introdotti per rendere possibile l’autoemulsione del polimero, porta ad una reticolazione fisica
derivante dalla separazione dei gruppi ionici stessi in microdomini che è responsabile di alcune delle proprietà di tali ionomeri29a,b.
Va tuttavia considerato che polimeri molto idrofili, con pesi molecolari e Tg bassi,
danno luogo a film di qualità ottima per quanto riguarda struttura e morfologia, ma scarsa per le proprietà richieste ad un rivestimento. Un film così ottenuto risulta infatti appiccicoso e poco resistente all’umidità (polimero idrofilo). L’ottimizzazione del processo di formazione di un film polimerico a partire da una dispersione acquosa richiede quindi un compromesso tra buona capacità filmante e proprietà richieste per il rivestimento.
1.3.6 Proprietà biocide
L’attacco di polimeri da parte di batteri, funghi e altri microrganismi si manifesta attraverso la perdita di proprietà meccaniche, degradazione della superficie, decolorazione, macchie e alterazioni delle caratteristiche estetiche e di altre proprietà35 a causa di reazioni di varia natura, derivanti dall’impiego della materia organica come alimento dei microrganismi stessi o dall’attacco chimico, generalmente di tipo idrolitico, causato dai metaboliti prodotti dai microrganismi.
Particolarmente sensibili sono le emulsioni acquose, le dispersioni ed i materiali con una o più componenti di origine naturale.36
Nel caso in cui il materiale polimerico sia il costituente di un rivestimento superficiale (ad esempio protettivo, vernice, prodotto di rifinizione) l’aggiunta di un biocida in fase di formulazione è il modo più consueto per prevenire la colonizzazione dei polimeri da parte dei microrganismi. Nel caso in cui la specie ad attività biocida non sia legata chimicamente al polimero, il suo rilascio può essere un pericolo per l’ambiente, e la protezione è limitata nel tempo. Fissare il biocida sullo scheletro polimerico attraverso un legame idrolizzabile provoca soltanto un miglior controllo della durata ed efficacia dell’effetto biocida, ma non risolve il problema della tossicità.
I sali di ammonio quaternari (QAS), ed in particolare quelli con un sostituente a lunga catena alchilica avente almeno otto atomi di carbonio, sono noti da molto tempo come biocidi attivi in acqua. A basse concentrazioni i QAS commerciali sono batteriostatici, fungicidi, algistatici, sporostatici e tubercolostatici; alle medie
concentrazioni i QAS sono battericidi, fungicidi, algicidi e virucidi contro i virus lipofili, mentre risultano scarsamente attivi contro virus idrofili, anche ad alte concentrazioni37. È noto che i QAS esercitano la loro attività biocida per interazione con la parete cellulare dei batteri.38
La rottura della membrana citoplasmatica, seguita dal rapido rilascio di ioni K+ e altri costituenti citoplasmatici è lo stadio cruciale nell’azione biocida di queste specie anfifiliche cationiche. Il meccanismo di azione non è noto con precisione, tuttavia l’importanza della loro interazione con le specie cariche negativamente presenti nella membrana, come i fosfolipidi acidi e le proteine di membrana, è già stata riportata in letteratura. Nella ricerca di biocidi non liscivianti, non pericolosi per l’ambiente, sono stati quindi studiati prodotti biocidi costituiti da QAS legati covalentemente ad un polimero. Polimeri solubili che portano QAS sono stati trovati essere biocidi più attivi dei monomeri corrispondenti, fatto che può essere spiegato dalla maggiore densità di carica locale sul polimero che rende l’interazione con la parete cellulare più efficace. A causa dell’alta densità di carica, i polielettroliti sono capaci di interagire fortemente con cariche opposte. I policationi interagiscono quindi più fortemente con la membrana citoplasmatica di quanto facciano i corrispondenti cationi monomerici; se questo fosse l’unico fattore da prendere in considerazione i policationi potrebbero possedere attività antimicrobiche più elevate dei cationi monomerici.
Nel caso di polimeri reticolati insolubili i risultati riportati in letteratura sono tuttavia variabili.
La sequenza degli avvenimenti elementari nell’azione biocida dei sali d’ammonio quaternari può essere schematizzata come segue:
1. adsorbimento sulla superficie della cellula batterica 2. diffusione attraverso la parete cellulare
3. assorbimento sulla membrana citoplasmatica 4. rottura della membrana citoplasmatica 5. perdita dei costituenti citoplasmatici 6. morte della cellula.
È ben noto che la superficie della cellula batterica è di solito carica negativamente, come evidenziato dalla sua suscettibilità all’elettroforesi. L’adsorbimento di policationi sulla superficie colloidale carica negativamente è ovviamente facilitata dall’effetto cooperativo rispetto ai cationi monomerici. È quindi ragionevole assumere che nei
processi elementari la cinetica del primo stadio sia molto aumentata per i polimeri in confronto a quella per i monomeri.
Per quanto riguarda il secondo stadio, va considerato che c’è molta differenza nella struttura delle pareti cellulari tra batteri Gram-positivi e Gram-negativi. I primi hanno una struttura della parete cellulare semplice; fuori dalla membrana citoplasmatica c’è solo uno strato rigido di peptidoglicano che dota le cellule batteriche delle ben note forme di cocchi, bacilli, ecc. Lo strato di peptidoglicano, benché relativamente spesso, è composto da reticoli porosi, che permettono a molecole di dimensioni non eccessivamente grandi di penetrare all’interno della cellula senza difficoltà. Infatti anche i policatio ni con pesi molecolari relativamente bassi si ritiene possano diffondere facilmente attraverso le pareti della cellula dei batteri Gram-positivi. D’altra parte, i batteri Gram- negativi hanno pareti cellulari molto più complesse, comprendenti una seconda me mbrana fuori dello strato di peptidoglicano, chiamata membrana esterna, la cui struttura è simile a quella della membrana citoplasmatica. A causa della struttura a doppio strato, la membrana esterna è una potenziale barriera contro molecole esterne ad alto peso molecolare. Generalmente , più alto è il peso molecolare e più difficile è la diffusione attraverso la parete cellulare, in particolare nei batteri Gram- negativi.
Quindi i polimeri sono potenzialmente più attivi degli analoghi composti a basso peso in termini di adsorbimento sulla membrana citoplasmatica e dell’interazione con la membrana seguita dalla sua rottura; sono tuttavia svantaggiati nella diffusione attraverso le pareti della cellula, specialmente per i batteri Gram- negativi. Di conseguenza, l’efficacia dei polimeri, potenzialmente maggiore di quella dei QAS a basso peso molecolare, è di fatto regolata dalle proprietà di trasporto e/o diffusione attraverso la parete cellulare.
Figura 1.9 Bersagli potenziali per biocidi.
[Denyer, S. P.; Stewart, G. S. A. B. International Biodeterioration & Biodegradation 41, 261, 1998]
È ben noto che le membrane citoplasmatiche dei batteri sono composte da fosfolipidi e proteine di membrana. I maggiori componenti dei fosfolipidi sono la fosfatidiletanolammina (PE), zwitterionica, e il fosfatidilglicerolo (PG), acido. Ad esempio, in Escherichia coli, PE e PG costituiscono rispettivamente il 75 e il 25% dei lipidi. In Staphylococcus aureus, il PG acido e il suo dimero cardiolipina raggiungono il 61% dei fosfolipidi totali. Policationi con gruppi biguanidinici in catena, che sono disinfettanti polimerici altamente attivi, hanno una forte interazione con il PG acido, mentre l’interazione con fosfolipidi neutri è scarsa.
I composti di guanidina agiscono in base allo stesso meccanismo dei QAS36. Sali di poli(esametilene guanidina) e sali di poli(alchilene biguanidina), agenti antimicrobici ben noti aventi una bassa tossicità, sono ampiamente utilizzati per trattamenti dell’acqua, ad esempio per piscine e negli stabilimenti termali39.
Da quando fu riportato nel 1930 che guanidine sostituite hanno proprietà antimicrobiche, i derivati guanidinici con attività antibatterica e antifungina sono stati studiati come disinfettanti in medicina, agenti di protezione delle colture e antisettici per prodotti industriali, alimenti e altri materiali di uso quotidiano. Tra i derivati guanidinici con attività antimicrobica, i policondensati di sali di guanidinio o biguanidinio con diammine hanno guadagnato importanza come antisettici per cosmetici e per il tessile40. Le biguanidine polimeriche prodotte da ICI American Inc. sotto il nome commerciale di Arlagard E e Cosmocil CQ, sono noti biocidi con un ampio spettro di attività
antimicrobica e sono usati come antisettici per cosmetici e per tessuti-non-tessuti in impieghi monouso (ad esempio salviette disinfettanti). Derivati guanidinici termostabili sono stati anche impiegati come additivi per fibre sintetiche; in questo caso tali derivati devono infatti essere miscelati col polimero prima della estrusione. Gli additivi antimicrobici dispersi all’interno della matrice polimerica durante lo stadio di fusione migrano continuamente verso la superficie della fibra sintetica, fornendo una efficace attività antimicrobica effettiva anche dopo molto tempo. La maggior parte degli antimicrobici guanidinici commerciali è tuttavia solubile in acqua. Questo comporta da un lato una agevole applicazione per semplice impregnazione dei substrati tessili, ma rappresenta anche un punto di debolezza in quanto il trattamento biocida è scarsamente stabile al lavaggio. La inalterabilità antimicrobica a ripetuti lavaggi può essere aumentata diminuendo la solubilità in acqua dei derivati guanidinici, ad esempio incorporandoli in una struttura polimerica scarsamente o per nulla idrosolubile.
Benché le diammine siano ampiamente usate come estensori di catena nella sintesi di poliuretani, come riportato nel Paragrafo 1.3.1, lo stesso non si può dire per i derivati della guanidina che non è una diammina ordinaria, quanto piuttosto un derivato imminico dell’urea. La guanidina è una base organica forte (il pKa relativo alla
dissociazione acida della guanidina protonata è pari a 13,641) per cui in pratica al posto della guanidina stessa come estensori di catena possono essere al più usati sali di guanidinio42. Questi ultimi sono commercialmente disponibili in questa forma, in quanto i composti guanidinici liberi non sono stabili allo stoccaggio.
Di fatto, come accennato già nel Paragrafo 1.3.2, l’impiego di sali di guanidinio come tali nella fase di estensione di catena durante la sintesi di poliuretani è stata riportata una sola volta in letteratura26,27.
1.4 Copolimeri anfifilici a blocchi
1.4.1 Aspetti generali
I copolimeri a blocchi hanno molte proprietà e applicazioni utili, ma la loro applicazione a livello industriale è stata finora limitata a causa di una varietà di fattori, legati prevalentemente al tipo di processo necessario per la loro sintesi43. Infatti, fino alla metà degli anni ’80 i copolimeri a blocchi potevano essere ottenuti essenzialmente
solo per polimerizzazione ionica. In anni recenti le polimerizzazioni radicaliche controllate come la polimerizzazione radicalica a trasferimento atomico (ATRP), la polimerizzazione mediata da nitrossido e la polimerizzazione a trasferimento di catena a addizione- frammentazione reversibile (RAFT) hanno considerevolmente ampliato le opportunità per la sintesi di copolimeri a blocchi. Questa espansione è principalmente dovuta alla possibilità di impiego di molti monomeri vinilici, non polimerizzabili se non per via radicalica, oltre che al processo di polimerizzazione che può essere condotto anche in condizioni blande, usando monomeri funzionali, in presenza di acqua anche come solvente e in condizioni non particolarmente rigorose di purezza dei reagenti e solventi, a differenza di quanto richiesto dalle polimerizzazioni ioniche.
Recentemente sono stati riportati diversi studi sulla sintesi tramite ATRP di copolimeri anfifilici a blocchi e sulle loro proprietà in soluzione44.
Questo tipo di copolimeri può subire transizioni morfologiche o di fase a seguito di semplici variazioni di pH45, di temperatura, di forza ionica o della natura del solvente46 e sono frequentemente impiegati come disperdenti47 per pigmenti o cariche inorganiche, oppure come tensioattivi disperdenti per polimerizzazioni in emulsione. Contrariamente ai tensioattivi e agli stabilizzanti convenzionali, i copolimeri a blocchi possono essere sintetizzati e modificati in maniera opportuna per applicazioni specifiche, e le loro proprietà, derivanti dalla composizione o dal peso molecolare dei singoli blocchi, possono essere modificate per ottenere l’effetto desiderato. Inoltre l’ampia varietà di monomeri idrofili e idrofobi commercialmente disponibili e polimerizzabili tramite tecniche radicaliche controllate consente di ampliare notevolmente il numero di possibili combinazioni di polimeri anfifilici che possono essere preparati rispetto a quelli finora ottenibili con metodi di polimerizzazione ionica.
Copolimeri anfifilici a blocchi possono mostrare comportamenti altamente differenziati in un mezzo acquoso. La morfologia degli aggregati nelle dispersioni acquose di copolimeri anfifilici a blocchi è controllata principalmente da un bilancio di forze che coinvolge tre parametri45: lo stiramento (deformazione) dei blocchi idrofobi che formano generalmente il nucleo di aggregati a simmetria sferica, l’interazione repulsiva tra le catene idrofile della corona (ossia del guscio esterno di strutture cosiddette “core-corona”), e la tensione interfacciale all’interfaccia nucleo-corona. Così molti fattori possono influenzare le morfologie finali degli aggregati a causa dei loro effetti sui tre parametri. Tra questi fattori, i più importanti sono la natura del blocco che
forma il nucleo e del blocco che forma la corona, la composizione del copolimero, la natura del solvente comune, e la presenza e la natura di additivi, in particolare se specie ioniche.
La natura del blocco che forma il nucleo influenza la morfologia dell’aggregato principalmente attraverso il suo effetto sulla tensione interfacciale all’interfaccia nucleo-corona, che aumenta con l’aumento dell’idrofobicità del blocco che costituisce il nucleo. La lunghezza del blocco che forma la corona influenza la morfologia dell’aggregato attraverso il suo effetto sull’interazione repulsiva tra le catene della corona.
I copolimeri a blocchi in un solvente selettivo per uno dei blocchi, formano micelle o aggregati come risultato dell’associazione dei blocchi insolubili46. Le micelle sono stabilizzate in soluzione per mezzo delle interazioni dei blocchi solubili con le molecole di solvente. In dipendenza dalla composizione dei copolimeri a blocchi, si possono distinguere micelle a stella e micelle a spazzola (crew-cut). Benché non ci sia un confine netto tra queste due classi di aggregati, il primo è di solito costituito da copolimeri a blocchi nei quali i blocchi che formano la corona sono molto più lunghi dei blocchi che formano il nucleo, mentre il secondo è costituito da copolimeri nei quali i blocchi che formano il nucleo sono più lunghi. La presenza di gruppi ionici o ionizzabili in uno dei blocchi, come nel caso di gruppi acidi nel blocco costituente la corona di particelle disperse in un mezzo acquoso, ha un effetto molto importante e complicato sul comportamento di aggregazione attraverso la sua influenza sull’interazione repulsiva tra le catene della corona.
Le micelle a stella di solito assumono forma sferica perché le interazio ni repulsive tra le catene della corona sono forti, a causa della densità relativamente alta delle catene della corona sulla superficie del nucleo.
In confronto con le micelle a stella, gli aggregati sferici a spazzola di solito hanno numeri di aggregazione, ossia numero di macromolecole presenti in ogni singola particella, molto più grandi. Ciò è dovuto al fatto che l’area occupata per catena della corona diminuisce come diminuisce la lunghezza dei blocchi che formano la corona. A causa del grande numero di aggregazione, lo stiramento delle catene idrofobe nei nuclei diventa più importante nel limitare la crescita della dimensione del nucleo. In particolare, la minimizzazione dell’energia interfacciale è bilanciata non solo da un
incremento della repulsione delle catene intercorona, ma anche da un incremento nello stiramento della catena dei blocchi che formano il nucleo.
Poiché la frazione dei blocchi insolubili nei copolimeri è grande, gli aggregati a spazzola sono convenzionalmente preparati dissolvendo per prima cosa il copolimero in un solvente comune per entrambi i blocchi. In seguito si aggiunge lentamente acqua deionizzata così che i blocchi idrofobi iniziano ad associarsi per formare le micelle. Nei primi stadi di micellizzazione, i nuclei idrofobi degli aggregati sono di solito altamente rigonfiati dal solvente comune. La velocità dello scambio di catena del polimero tra gli aggregati può essere molto veloce. Come il contenuto di acqua aumenta, la concentrazione degli unimeri decresce, e il solvente comune è gradualmente rimosso dal nucleo delle micelle, con conseguente riduzione della mobilità delle catene. Questo processo è analogo a quello di congelare le strutture diminuendo la temperatura nei copolimeri a blocchi in massa o in miscele parzialmente compatibili tra un copolimero a blocchi e l’omopolimero di uno dei due blocchi.
I copolimeri anfifilici a blocchi con una più alta frazione di natura idrofila possono essere utilizzati come disperdenti ad alta efficacia.
Quando l’azione disperdente debba essere più moderata, ad esempio nel caso della stabilizzazione temporanea di malte cementizie ad opera di additivi superfluidificanti, copolimeri costituiti da un blocco di poli(acido acrilico), o metacrilico, e un secondo blocco di poli(PEG acrilato), ossia di unità di acido acrilico (o metacrilico) esterificate con alcoli polietossilati di lunghezza variabile (da 4 a 20 unità etileneossido), per i quali sono prevedibili caratteristiche moderatamente anfifiliche, potrebbero mostrare proprietà assai diverse dai prodotti attualmente impiegati, costituiti da copolimeri di composizione chimica analoga, ma a distribuzione dei comonomeri prevalentemente casuale.
1.4.2 Polimerizzazione radicalica a trasferimento atomico
Come accennato nel paragrafo precedente, copolimeri a blocchi possono essere ottenuti tramite polimerizzazione radicalica controllata (CRP) di monomeri vinilici. La peculiarità di tali processi di polimerizzazione, e quindi la loro analogia con le vere e proprie polimerizzazioni viventi (che procedono generalmente con meccanismi di tipo
ionico, di coordinazione o ad apertura d’anello), è la sostanziale soppressione delle reazioni irreversibili di trasferimento e di terminazione di catena48.
Tutti i metodi CPR sono basati sull’instaurarsi di un rapido equilibrio dinamico tra una minima quantità di radicali liberi in crescita ed una grande maggioranza di “macromolecole dormienti”. Queste ultime possono essere terminate da alogenuri alchilici nel caso della polimerizzazione radicalica a trasferimento atomico (ATRP), da tioesteri o ditiocarbonati nei processi a trasferimento di catena ad addizione-frammentazione reversibile (RAFT), da alcossiammine nella polimerizzazione mediata da nitrossido (NMP), o potenzialmente anche da specie organometalliche. I radicali liberi sono generati per via termica (NMP), con una reazione catalizzata (ATRP) o reversibilmente tramite reazioni di scambio degenerativo con specie dormienti (RAFT).
Caratteristica comune dei processi CRP è la presenza di stadi di attivazione e disattivazione (con costanti di velocità kact e kdeact , Schema 1.15), benché nella RAFT lo
schema possa essere formalmente semplificato al solo processo di scambio con la costante di velocità apparente kexch.
Pn Pn X Pn X Pn X Pn+m Pm Pm kact kdeact + kp+ M X + Y kact kdeact Pn Pn+m Pm + kp+ M X-Y kt kt + Pn+m Pm kp + M kt Pn Pn+m Pm + kp + M kt X-Pm kexch kact kact k deact kdeact Pn X Pm (1) (3) (2)
I radicali liberi generati propagano e terminano (con costanti di velocità kp e kt) come
in una polimerizzazione radicalica convenzionale. Benché la terminazione non sia formalmente soppressa, in condizioni appropriate il suo contributo può divenire trascurabile; in questo caso la polimerizzazione radicalica procederà in modo sostanzialmente analogo a quello prevedibile per un sistema vivente.
Tra i vari processi sopra descritti la ATRP è stata oggetto di un numero crescente di studi sin dalla sua scoperta, a metà degli anni ‘90. Questo perchè la ATRP, illustrata più in dettaglio nello Schema 1.16, ha mostrato una notevole flessibilità di impiego, essendo adattabile ad una ampia gamma di monomeri vinilici, anche di tipo difunzionale, sia in assenza che in presenza di solventi organici o acquosi.
R-X + Mt n -Y / Legante monomero terminazione R + X-Mtn+1-Y / Legante kact kdeact kp kt
Schema 1.16 ATRP catalizzata da metalli di transizione.
Nel processo ATRP i radicali propaganti sono generati attraverso un processo redox reversibile catalizzato da un complesso di un metallo di transizione (Mtn-Y/Legando,
dove Y può essere un altro legando o il controione). Quest’ultimo, nel caso più comune, subisce una alogenazione ossidativa in presenza di un alogenuro alchilico attivato, ossia di un “radicale dormiente” R-X che può essere l’iniziatore o una catena polimerica propagante. Scegliendo opportunamente le condizioni di reazione (temperatura, solvente), la natura e la concentrazione del sistema catalitico in funzione della natura, e quindi della reattività, del monomero, è possibile modificare il rapporto kact/kdeact e
quindi le concentrazioni allo stato stazionario dei macroradicali nelle forme dormiente (alogenata) e attiva (radicale libero propagante) in modo da mantenere molto più bassa la concentrazione di quest’ultima. In questo modo vengono minimizzate le reazioni di terminazione (kt), che sono processi bimolecolari di accoppiamento o
disproporzionamento di radicali. Inoltre la possibilità di scambio di alogeno, con conseguente modifica della reattività del sistema, può consentire una efficace riattivazione della propagazione in processi a più stadi di copolimerizzazione a blocchi,
anche nel caso in cui il secondo blocco debba essere costituito da monomeri poco reattivi.
Tipicamente non più del 5% delle catene di polimero in crescita totali terminano durante l’iniziale breve stato non stazionario della polimerizzazione. Una polimerizzazione ATRP condotta in modo corretto comporta quindi la formazione di una frazione modesta di catene terminate irreversibilmente, a fronte di una crescita uniforme delle catene restanti. Ne consegue che con tale processo è in genere possibile sintetizzare copolimeri a blocchi per polimerizzazione sequenziale di comonomeri diversi, ottenendo macromolecole costituite da blocchi di peso molecolare predeterminabile.
1.5 Scopo della tesi
Le caratteristiche dei polimeri anfifilici a distribuzione casuale, a blocchi o segmentati sono tali da consentire la loro dispersione in mezzo acquoso senza l’utilizzo di emulsionanti tradizionali.
In questo lavoro di tesi si intendeva avviare una serie di studi esplorativi volti alla preparazione e caratterizzazione di polimeri anfifilici le cui diverse caratteristiche composizionali e strutturali li rendessero adatti a diverse finalità applicative. In particolare si volevano studiare sistemi macromolecolari a diverso contenuto di gruppi o comonomeri idrofili, e quindi caratterizzati da un diverso comportamento in mezzo acquoso. I sistemi selezionati erano i seguenti:
i. ionomeri uretanici;
ii. copolimeri a blocchi a diverso grado di idrofilia;
iii. polimeri in dispersione acquosa (lattici) con gruppi funzionali ionizzabili. Gli ionomeri uretanici rappresentavano una prima tipologia di copolimeri anfifilici di particolare interesse, in quanto ottenibili a partire da prepolimeri poliuretanici per introduzione di estensori di catena costituiti da derivati poliammino-guanidinici difunzionali, potenzialmente in grado di conferire al polimero finale sia caratteristiche autodisperdenti in acqua che proprietà biocide o biostatiche. Entrambe queste caratteristiche delle poliuretano- uree funzionalizzate con gruppi guanidinici salificati