1.
INTRODUZIONE
1.1
Usi del bis(pinacolato) diboro
Il primo impiego del bis(pinacolato) diboro per la formazione di legami C-B è stato riportato nel 1993 dal gruppo di lavoro di Miyaura. In suddetto lavoro veniva riportata una procedura platino(0)-catalizzata per ottenere una diborazione vicinale di alchini tramite l’impiego appunto del B2Pin2.1
Questa reazione è selettiva nel generare esclusivamente il prodotto cis-diborilato e tollera una vasta gamma di gruppi funzionali e può essere portata avanti sia su alchini terminali che interni (Schema 1).
Schema 1. Diborazione di alchini Pt-catalizzata
R1 R2 B2pin2 Pt(PPh3)4 (3 mol%) DMF / 80°C / 24 h R2 Bpin pinB R1 +
Il B2Pin2 risulta essere particolarmente versatile rispetto agli altri reagenti (alcosso) diboro in quanto sia il B2Pin2 stesso, sia i prodotti di borilazione possono essere maneggiati all’aria e mostrano una elevata stabilità all’idrolisi rendendo più agevoli le procedure di work-up della reazione e eventuali processi di purificazione.
Da questo primo impiego in reazioni di addizione a nucleofili del B2Pin2 , il range dei substrati da impiegare in questa tipologia di razioni è stato ampliato anche agli alcheni, agli 1,3-dieni, agli alleni, ai chetoni α,β- insaturi.2
Schema 2. Diborazione di alcheni Pt-catalizzata
R2 R1 B2pin2 Pt(dba)2 (3 mol%) toluene / 50 °C / 1 h R1 R2 Bpin pinB +
Le reazioni di diborazione di alcheni hanno dato buoni risultati con alcheni terminali variamente sostituiti e con cicloalcheni (per i quali è evidente una stereoselettività syn) impiegando catalizzatori
a base di platino e vari reagenti di diboro. Catalizzatori a base di rodio si sono rivelati invece maggiormente efficaci per eseguire le diborazioni di alcheni interni che con altri catalizzatori sono spesso affette da una reazione collaterale di β−eliminazione. Per quanto riguarda quest’ultima tipologia di reazione Morken e collaboratori sono riusciti a preparare 1,2-dioli sia con selettività
syn/anti che con alta enantioselettività tramite l’ossidazione di diborani ottenuti per diborazione di
alcheni sotto l’influsso di catalizzatori di rodio chirali.3
Schema 3. Generalità sulle diborazioni di alcheni
(Cat)B B(Cat) R Pt(Cod)2 toluene, t.a. R (Cat)B B(Cat) (Pin)B B(Pin) B(Pin) (Pin)B Pt(dba)2 toluene Ph (Cat)B B(Cat) 1 [Rh] 2 H2O2, NaOH Ph OH OH Ph OH OH
La diborazione di alcheni terminali è di particolare interesse perché genera un composto portante due gruppi boronato, uno primario e uno secondario. Si può sfruttare la differenza di questi due gruppi per eseguire una elaborazione selettiva del composto in base alla loro differente reattività. Sempre Morken e collaboratori hanno proposto una procedure utile all’ottenimento di una diborazione enantioselettiva su alcheni terminali (Schema 4). Vari gruppi tollerano le condizioni di reazione, comprese funzionalità alcoliche alliliche protette.4
Schema 4. Diborazione enantioselettiva di alcheni terminali
R 2.5 mol% Pt2(dba)3 6 mol% (R,R)-L B2(Pin)2 (1.05 equiv.) THF H2O2 1) 2) R OH OH O P O O O Et Et Et Et Et Et Et Et (R,R)-L=Al di là di queste diborazioni vicinali Pt-catalizzate di composti idrocarboniosi insaturi il B2Pin2 va incontro a reazioni Pd-catalizzate di coupling con alogenuri, ottimali per l’ottenimento di aril-boronati da impiegare in reazioni di coupling di Suzuki. La reazione è altamente compatibile con i vari gruppi funzionali che si possono trovare sull’anello aromatico. Similmente la reazione può essere eseguita su alchenil-alogenuri. Cambiando il palladio con il rodio o l’iridio nel sistema catalitico si può inoltre ottenere pure aril-boronati a partire da semplici areni o riuscire a compiere la borilazione di alcani lineari (Schema 5).
Schema 5. Borilazione di aril-alogenuri, areni e alcani lineari
. B2pin2 X R Pd cat. (3 mol%) AcOK (3 eq.) / 80 °C Bpin R X = Br, I, OTf + B2pin2 + R1 R3 R2 X X = Br, I, OTf PdCl2(PPh3)2. 2PPh3 ( 3 mol% ) KOPh ( 1.5 eq.) / 50 °C Toluene R1 R3 R2 pinB
B2pin2 + 1 / 2 [ I r C l ( C O E )2 ]2 / d t b p y ( 5 m o l % ) r.t. / 4.5 h Bpin 8 3 % B2pin2 + Cp*Rh(C2H4)2 ( 5 mol %) 150 °C, 5 h Bpin 84%
Per ciò che riguarda la sintesi del B2Pin2 la procedura più classica prevede l’impiego del tetrakis(dimetilammino)diboro con l’alcool pinacolico in presenza di acido cloridrico. Di fatto il vero composto di partenza è il bromoborano; questo viene fatto reagire con dimetilammina in pentano per generare il tris(dimetilammino)borano che a sua volta viene fatto reagire con il bromoborano per ottenere il precursore del tetrakis(dimetilammino)diboro, il bromobis(dimetilammino)borano convertibile appunto nel tetrakis(dimetilammino)diboro per reazione con sodio in toluene. 5
Schema 6. Sintesi del B
2Pin
2a partire dal bromoborano
BBr3 6 Me2NH pentano (Me2N)3B 2 (Me2N)3B + BBr3 pentano (Me2N)2B Br (Me2N)2B Br 2 2 Na
toluene (Me2N)2B B(NMe2)2
(Me2N)2B B(NMe2)2 B B O O O O 2 pinacolo / 4 HCl toluene-etere A B C D
Questa procedura però è afflitta dalla necessità di eseguire accurate procedure di purificazione e dalla bassa resa risultante che si aggira intorno ad un valore del 30%. Una procedura più recente consente di ottenere bis(catecolato)diborani in larga scala e con procedura sintetica one-pot a partire da BX3 (X = Cl, Br) e dall’adeguato catecolo. La procedura prevede di eseguire un homocoupling di alocatecolborani in presenza di un’amalgama di sodio. Di fatto la procedura è, come detto, eseguibile one-pot in quanto si può far reagire un catecolo, sostituito in base alle eventuali necessità, con tribromuro o tricloruro di boro e, in seguito all’evaporazione del solvente e dell’acido
alogenidrico generatosi, si può procedere alla diluizione in toluene e all’addizione dell’amalgama di sodio.
Schema 7. Reazione generale di sintesi di composti bis(catecolato)diboro
O BCl O Na/Hg 90°C toluene 3h O B O B O O
La reazione è stata messa a punto per l’ottenimento di esteri catecolici dell’acido diboronico con rese variabili in base alla sostituzione del sistema catecolico che possono raggiungere anche il 96% come nel caso della sostituzione in meta ad uno degli ossidrili catecolici con un sostituente t-butilico. La procedura però interessa anche la sintesi del B2Pin2 in quanto, nonostante la reazione diretta di riduzione del cloroborano corrispondente sia inefficace, si può comunque ottenerlo tramite una reazione di scambio che prevede l’uso del 4-t-butilcatecolo per la procedura one-pot e l’aggiunta di due equivalenti di alcool pinacolico. Nonostante l’alta conversione ci si imbatte nella difficile separazione del B2Pin2 dal catecolo libero che si forma come prodotto collaterale della reazione. Di fatto l’uso di una base per formare un sale di catecolato per trattamento con base o il tentativo di convertire il catecolo in un acetale, in assenza o in presenza di acido, comporta la degradazione del B2Pin2. Gli autori dello studio suggeriscono di eluire la soluzione in una colonna di alumina con dietiletere per ottenere B2Pin2 puro con una resa leggermente superiore al 46%.6
1.2
Reazioni di sostituzione allilica con B
2pin
2Nello svolgimento della tesi è stato necessario raccogliere informazioni riguardo alle reazioni di sostituzione allilica con lo scopo di mettere appunto una procedura sintetica efficace per compiere il primo step delle reazioni investigate in questo lavoro cioè l’attacco del B2Pin2 a substrati vinil-aziridinici o vinil-ossiranici.
1.2.1 Addizioni coniugate
Inizialmente l’attenzione è stata focalizzata su due procedure piuttosto simili messe appunto dai gruppi di lavoro di Miyaura e Hosomi. Entrambe propongono vie sintetiche per l’attacco del B2Pin2
su chetoni, esteri e nitrili α,β insaturi catalizzate da rame. Durante lo svolgimento del lavoro di tesi ambedue le condizioni di reazione sono state testate su i substrati oggetto della nostra ricerca. Per ciò che riguarda le condizioni proposte da Miyaura la reazione viene eseguita usando il rame quantitativamente (110 mol%) insieme a LiCl e ad una base (nelle condizioni di reazione ottimali è l’acetato di potassio). 6
Schema 8. Procedure di addizione di B
2Pin
2a chetoni esteri e nitrili
α,β
α,β
α,β
α,β
insaturi nelle
condizioni di Miyaura
O B O B O OCuCl / KOAc (1.1 eq) LiCl (1.1 eq) DMF O B O Cu Z Z = COR, CO2R, CN H2O O B O Z
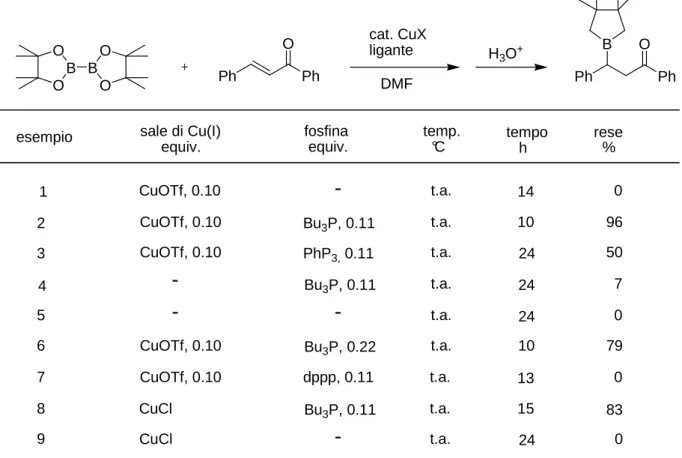
La procedura invece proposta da Hosomi si basa sempre sull’impiego del Cu(I) come promotore della reazione, ma le quantità impiegate sono inferiori (10%). Le condizioni differiscono anche per l’impiego di una fosfina come la tributilfosfina. La reazione messa a punto prevede l’impiego del CuOTf ma ciò non toglie che si possa usare pure il CuCl similmente alla reazione di Miyaura, anche se la reazione così sembra essere più lenta (esempio 8, Tabella 1). 8
La fosfina è necessaria per la reazione e l’impiego di una fosfina meno donativa della tributilfosfina si traduce in un abbattimento della resa mentre l’impiego di una fosfina bidentata riduce la velocità di reazione (esempio 7, Tabella 1). Il ruolo della fosfina è deducibile in base ad alcuni dati forniti dallo stesso Hosomi: in seguito ad una prova eseguita ponendo nell’ambiente di reazione tutti i componenti della reazione, eccezion fatta per il composto carbonilico α,β−insaturo, non si è potuto assistere ad un consumo della specie di diboro impiegata. Questo suggerisce un diverso andamento della reazione rispetto a quanto invece non avvenga nelle reazioni catalizzate da platino nelle quali si assiste al consumo della specie di diboro a causa della formazione di una specie attivata platino-metallica. Non vi sono inoltre evidenze di coordinazione del boro con la fosfina in quanto eccessi della stessa non portano ad un innalzamento della velocità di reazione, anzi la rallentano. Evidentemente la coordinazione tra la fosfina e il rame è più forte rispetto a quella con il boro. Infatti lo spettro 31P NMR non mostra significativi spostamenti del chemical shift del fosforo nella
fosfina in presenza del composto di diboro mentre in presenza del rame si assiste ad un evidente spostamento a campi più bassi. Evidentemente la complessazione fra il ligante e il rame rafforza l’attività catalitica di quest’ ultimo probabilmente tramite la riduzione dell’aggregazione del sale di rame in soluzione.
Tabella 1. Reazione del B
2Pin
2ad un
α,β−
α,β−
α,β−
α,β−
enone catalizzata dal Cu(I)
B B O O O
O Ph Ph
O cat. CuXligante DMF H3O+ Ph Ph O B sale di Cu(I) equiv. fosfina equiv. temp. °C tempo h rese % CuOTf, 0.10 CuOTf, 0.10 CuOTf, 0.10 CuOTf, 0.10 CuOTf, 0.10 CuCl CuCl Bu3P, 0.11 Bu3P, 0.11 Bu3P, 0.22 Bu3P, 0.11 PhP3, 0.11 t.a. t.a. t.a. t.a. t.a. t.a. t.a. t.a. t.a. 14 10 24 24 24 10 13 15 24
--
-0 96 50 7 0 79 0 83 0 1 2 3 4 5 6 7 8 9 dppp, 0.11 esempioAmbedue queste reazioni per l’ottenimento di una β−borazione di composti carbonilici α,β−insaturi presentano delle limitazioni in quanto solo sugli enoni sembrano garantire rese discrete e comunque anche là dove la reazione funziona la procedura è in realtà afflitta dalla necessità di un elevato carico del sistema catalitico, le quali componenti possono essere necessarie da 10 a 110 mol % rispetto al substrato.
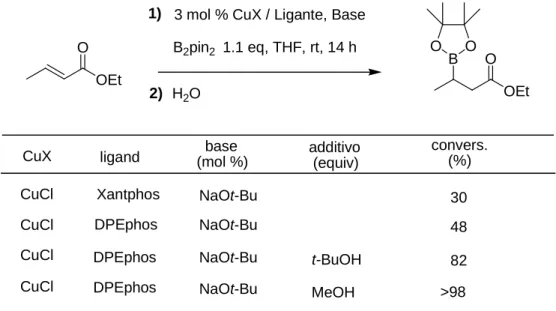
Yun e Al. hanno proposto una procedura analoga a quella di Miyaura che prevede però l’impiego di una componente additiva alcolica che risulta essere determinante nell’incrementare drammaticamente la velocità di reazione. Inoltre viene aggiunto un ligante a base di fosforo. 9
Tabella 2. Aumento della velocità di reazione tramite l’impiego di componenti
alcoliche nella addizione del B
2Pin
2all’(E)-etilcrotonato
3 mol % CuX / Ligante, Base B2pin2 1.1 eq, THF, rt, 14 h 1) 2) H2O OEt OEt O O B O O
CuX ligand base(mol %) additivo (equiv) convers. (%)
CuCl Xantphos NaOt-Bu
CuCl DPEphos NaOt-Bu
NaOt-Bu NaOt-Bu DPEphos DPEphos CuCl CuCl t-BuOH MeOH 30 48 82 >98
Il metanolo sembra essere il più indicato come componente alcolica, probabilmente per la sua maggior capacità di protonare le specie di organo rame generate dall’addizione della specie di boril-rame all’estere.
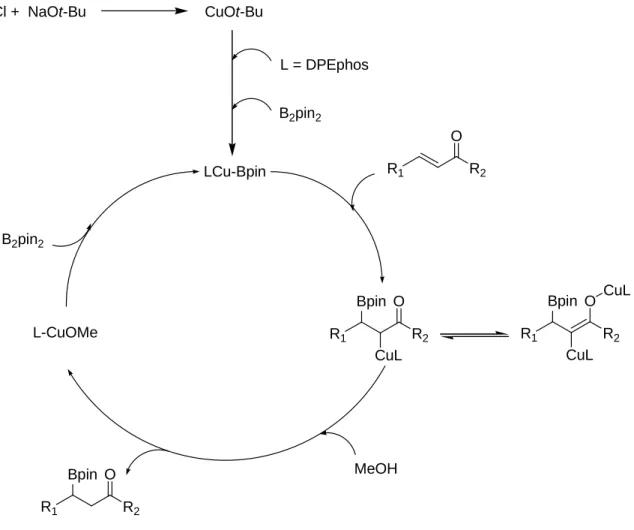
Figura 1. Meccanismo proposto per l’azione del metanolo
CuCl + NaOt-Bu CuOt-Bu
L = DPEphos B2pin2 LCu-Bpin R1 R2 O R1 R2 O Bpin CuL R1 R2 O Bpin CuL CuL L-CuOMe MeOH R1 R2 O Bpin B2pin2
In Figura 1 si può notare come la specie reattiva nella borilazione è un complesso di boril-rame legato alla difosfina. Il MeOH inciderebbe sulla reazione favorendo la protonazione della specie organorame formatasi in situ e rigenerando una specie alcosso-rame in grado di reinserirsi nel ciclo catalitico andando a reagire nuovamente con il B2Pin2.
C’ è la possibilità di accedere a versioni asimmetriche di queste reazioni tramite l’impiego di liganti non racemici. In particolare si ha notizia di risultati ottimali raggiunti nella reazione di borazione nelle condizioni proposte da Yun impiegando (R)-(S)-josiphos e (R)-(S)-NMe2-PPh2-mandyphos.10
Così si possono ottenere boronati in cui lo stereocentro di nuova formazione ha una precisa configurazione. Ciò permette di poter ulteriormente elaborare il composto potendo mantenere l’assetto di tale centro nei prodotti ultimi. Per esempio si può procedere alla ossidazione del carbonio impegnato nel legame C-B ottenendo un alcool non racemico con eccessi enantiomerici piuttosto elevati. Si può prendere in considerazione il composto già visto nella Tabella 2.
Schema 9. Esempio di
β−
β−
β−
β−
borazione di esteri e nitrili
α,β−
α,β−
α,β−
α,β−
insaturi aciclici
O OEt
CuCl 2 mol%, NaOt-Bu 3 mol% (R)-(S)-josiphos 4 mol% MeOH 2 eq THF O OEt Bpin Resa 94% NaBO3 THF/H2O (1:1) O OEt OH ee 90%
Figura 2. (R)-(S)-josiphos e (R)-(S)-NMe2 –PPh2 –mandyphos
P H P CH3 Fe (R)-(S)-josiphos P Fe Ph Ph N H CH3 CH3 P Ph Ph N H H3C H3C (R)-(S)-NMe2 -PPh2 -mandyphos
Le reazioni di Hosomi e Miyaura sono applicabili ache su composti carbonilici α,β−insaturi più sostituiti per accedere a allilboronati trisostituiti. Vi sono dati infatti sull’applicabilità di queste reazioni su acetati derivati da alcool tipicamente ottenibili dalla reazione di Baylis-Hillman e di analoghi composti alcolici portanti funzionalità chetonica o nitrilica al posto della componente esterica.11
La reazione sembra procedere tramite meccanismo SN2’ con riarrangiamento allilico e
elimininazione di acetato. Gli allilboronati ottenuti presentano un alto rapporto E/Z eccezion fatta per gli allilboronati dei nitrili per i quali la prevalenza dell’isomero E risulta meno preponderante.
Schema 10. Ottenimento di allilboronati trisostituiti
R1 OR
O
R2 OAc
CuCl, LiCl, KOAc
DMF OR O B2pin2 Bpin R2 R1
rapporti E/Z elevati
Ph
O OAc
CuCl, LiCl, KOAc
DMF O B2pin2 Bpin Ph E:Z = >95:<5 CN OAc
CuCl, LiCl, KOAc,
DMF B2pin2 CN Bpin CN Bpin E:Z = 33:67
Risulta comunque ovvio che nell’ambito delle reazioni di addizione coniugata di composti di diboro non ci si può esclusivamente ridurre all’impiego di sistemi catalitici a base di rame, essendo note procedure analoghe a base di platino, rodio e nichel. In realtà si può pure prescindere dall’impiego di sistemi catalitici a base di metalli (la cui applicabilità è ristretta a substrati aciclici e che generalmente non permettono di formare centri quaternari portanti un atomo di boro). Una valida alternativa all’impiego di catalizzatori metallici può essere rappresentata dall’impiego di carbeni N-eterociclici (NHC) che possono, in qualità di nucleofili, associarsi al boro elettrofilo portando ad un allentamento del legame B-B del B2Pin2 aumentandone la reattività (Schema 11). 12
Figura 3. Esempi di NHC carbeni
MesN NMes Cl MesN NMes Cl ArN NAr Cl CyN NCy BF4
Schema 11. Modello proposto per l’attivazione del B
2Pin
2e per la successiva
addizione coniugata ad un enone
O B O O B O Nuc: B O O O B O Nuc legame B-B polarizzato O O B(pin) (pin)B Nuc O B(pin) B(pin) Nuc O B(pin) B(pin)
La validità del meccanismo proposto è comprovata da una serie di dati sperimentali ricavati dall’analisi del complesso formato tra B2Pin2 e l’NHC: la carica parziale atomica indica che il carattere elettrofilo degli atomi di boro è smorzato nel complesso mentre la densità elettronica del legame B-B risulta superiore verso l’atomo di boro non legato. Quindi il legame B-B risulta polarizzato e allentato visti anche i dati sulla sua lunghezza che risulta aumentata. Pure la spettroscopia NMR conforta l’ipotesi del meccanismo proposta in quanto i dati spettrali del 11B
indicano la scomparsa dell’univoco segnale avente δ pari a 30.1 tipico del B2Pin2 per lasciar spazio a due segnali distinti a campi alti con δ pari a 4.5 e 6.3. Questo sta a indicare come entrambe gli atomi di boro risultino schermati e come siano ora differenti fra loro essendo ora uno sp3e l’altro
sp2.
Quindi è possibile con gli NHC effettuare l’addizione coniugata del B2Pin2 su substrati ciclici come il cicloesenone, anche nel caso in cui i substrati portino un sostituente sul carbonio da attaccare, con la possibilità di ottenere anche carboni quaternari boro-sostituiti. Inoltre va sottolineata la maggiore tolleranza di questo sistema catalitico verso alcuni gruppi che rendono invece difficoltosa la reazione con catalizzatori metallici come il rame (per esempio gli alchini).
Schema 12. Addizione coniugata del B
2Pin
2al cicloesenone catalizzata da NHC
O CyN NCy BF4 10 mol% NaOt-Bu 10 mol% B2pin2 1.1 eq THF, 22 °C, 12 h; O B(pin) Resa >98% Work-up acquoso O Me CyN NCy BF4 2.5 mol% NaOt-Bu 2.5 mol% B2pin2 1.1 eq THF, 22 °C, 6h O Me B(pin) >98 % conv, 96 % resa1.2.2. Sostituzioni alliliche
Le reazioni di sostituzione allilica vere e proprie sono un’ altra categoria di reazioni che è stata oggetto di particolare attenzione durante lo svolgimento del lavoro di tesi. Un gruppo di ricerca molto attivo in questo settore è quello di Szabò. In particolare da questo gruppo provengono dati relativi all’impiego di catalizzatori pincer del palladio (Figura 4) per l’esecuzione di sostituzioni alliliche con reagenti di diboro su ciclopropani, allil-acetati e (particolarmente interessanti per noi) vinil-aziridine (Schema 13).13
Figura 4. Catalizzatori a “pinza” del palladio
Pd Me2N NMe2 L Pd PhSe SePh L Pd Ph2P PPh2 L 1 2 3Schema 13. Reazioni di sostituzione allilica su ciclopropani, allil-acetati e
vinil-aziridine in presenza di catalizzatori a “pinza” del Pd
E' E N Ar Ts R OAc [B(OH)2]2 [1]cat Q B(OH)2
Gli acidi allilboronici che si formano sono generalmente instabili e fortemente reattivi. Ciò, associato alla loro decomposizione in soluzione concentrata o se portati a secco, può limitare le possibilità di accedere a questi composti. Comunque si possono ottenere dati in spettroscopia NMR impiegando il DMSO come solvente. Una alternativa per evitare di affrontare questa serie di problematiche può essere rappresentata dalla conversione diretta dell’acido allilboronico nel più stabile organotrifluoroborato corrispondente tramite trattamento con KHF2.
L’utilità dell’impiego di questi sistemi catalitici rispetto ad altri comunque impiegati nelle reazioni di sostituzione allilica con Pd2(dba)3 e Pd(PPh3)4 è che sono elettronricchi rispetto a questi ultimi: per questa ragione sono meno inclini a dare luogo a transmetallazione con specie allilmetalliche tramite formazione di complessi η3 e di complessi di bis-allilpalladio (v. Schema 14) che sono invece la causa della formazione di prodotti collaterali nelle reazioni aventi luogo sotto l’influsso di catalizzatori del Pd(0).
Schema 14. Meccanismo di formazione di co-prodotti indesiderati nella reazione di
sostituzione allilica di allilacetati con B
2Pin
2B O O R R R Pd R Pd L L R R
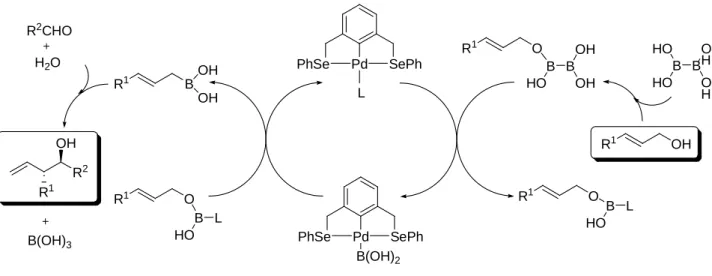
Una procedura simile è impiegabile come metodo efficace per la conversione di alcool allilici in specie allilmetalliche utili poi nei procedimenti di allilazione.14 Può infatti risultare vantaggioso rivolgersi verso procedure che implichino l’uso dell’acido diboronico piuttosto che alle classiche reazioni implicanti l’uso di acidi di Lewis e/o di reagenti organometallici riducenti, entrambe categorie di composti che possono dare problemi di tolleranza per certi gruppi funzionali. Gli acidi diboronici sono infatti reattivi non riducenti e dotati di scarsa acidità di Lewis e perciò sono estremamente compatibili con molti gruppi funzionali. Si può così effettuare una agevole reazione di sostituzione su alcool allici in seguito alla quale si può decidere di isolare l’acido allilboronico generato. In questo caso è necessario però convertirlo in prodotti più proni alle procedure di purificazione (per esempio il corrispondente estere), oppure procedere a reazioni di allilazione preferibilmente one-pot dato che c’è la possibilità di eseguirle in questa modalità così da evitare le procedure di work-up della reazione che potrebbero dare problemi all’allilboronato generato. Nella reazione si impiega una quantità catalitica di acido p-toluensolfonico dato che ciò accelera sia la formazione dell’allilboronato sia la reazione di allilazione. Sempre al gruppo di lavoro di Szabò dobbiamo la presenza di dati su reazioni di questo tipo con allilazione di aldeidi.
Schema 15. Procedura one-pot di generazione di acidi allilboronici seguita da
allilazione di aldeidi
R2CHO [B(OH)2]2 Pd Cl SePh PhSe cat SO3H cat R1 OH HO R1 R2 OH R1 singolo regio- e stereoisomero OH R1 R2CHO [B(OH)2]2 Pd Cl SePh PhSe cat SO3H cat R1 H OH R2 singolo stereoisomeroCome si può apprezzare nello Schema 15, la reazione presenta la favorevole caratteristica di essere, oltre che altamente regioselettiva, pure altamente stereoselettiva in quanto segue la intrinseca diastereoselettività tipica delle reazioni di allilazione con allilboronati. La reazione procede pure su substrati ciclici come si può sempre apprezzare dallo schema con la produzione del diasteroisomero
syn essendo partiti da un doppio legame con configurazione Z. La reazione può essere eseguita
impiegando il B2Pin2 piuttosto che l’acido diboronico (visto che quest’ ultimo non è commercialmente disponibile quanto il B2Pin2).15 La reazione però così risulta inefficace a causa della minore reattività del B2Pin2. Si può ovviare a questo inconveniente sfruttando l’alta tolleranza all’umidità di questa reazione e quindi aggiungendo 8 equivalenti di acqua ogni equivalente di alcool allilico al fine di provvedere ad una idrolisi in situ del B2Pin2 ad acido boronico, idrolisi probabilmente catalizzata dallo stesso acido p-toluensolfonico già implicato nella catalisi degli altri steps della reazione (un aumento della quantità di acido impiegata da 5 a 20 mol %).
Questa tipologia di reazione è estendibile anche alla allilazione di chetoni oltre che di aldeidi previa aggiunta di InI che funziona da catalizzatore attivando l’acido allilboronico nelle reazioni di allilazione.
Per ciò che riguarda le considerazioni meccanicistiche sulla reazione tandem one-pot di allilazione di composti carbonilici con acidi allil-boronici generati da alcool allilici, si può ritenere che tutto inizi con la attivazione del gruppo ossidrilico dell’alcool allilico con formazione di un estere diboronico. A questo punto da questo composto che porta ancora il legame B-B, il gruppo boronato è trasferito dal diboronato al catalizzatore generando un complesso η1. Questo gruppo boronato viene a sua volta trasferito in una reazione di sostituzione dell’ossidrile attivato dell’alcool allilico dando come prodotti l’acido allilboronico corrispondente e acido borico. A questo punto si è formata la specie in grado di allilare l’aldeide spontaneamente (anche se una certa catalisi acida può coadiuvare questa reazione). Può risultare interessante notare come in questo processo il palladio non compia nessuno shift tra lo stato di ossidazione 0 e lo stato +2 ma rimanga sempre +2.
Figura 5. Meccanismo proposto per l’allilazione di aldeidi con acidi allilboronici
generati da alcool allilici
Pd PhSe SePh Pd PhSe SePh B(OH)2 L R1 O B B OH OH HO R1 O B L HO R1 OH B B O H O H HO HO R1 O B L HO R1 B OH OH R2 R1 OH B(OH)3 R2CHO H2O
Le reazioni di sostituzione allilica con reagenti di diboro possono essere effettuate anche con l’impiego di sistemi catalitici a base di rame. Scegliendo poi opportuni liganti chirali si può accedere a versioni asimmetriche di queste reazioni. Vari dati sono disponibili su reazioni di sostituzione allilica con B2Pin2 su carbonati allilici con l’impiego di Cu(I) come catalizzatore in presenza di un ligante a base di fosforo e di una base.16
Schema 16. Reazione asimmetrica di carbonati allilici con B
2Pin
2in presenza di
complessi del rame(I) con ligante chirale
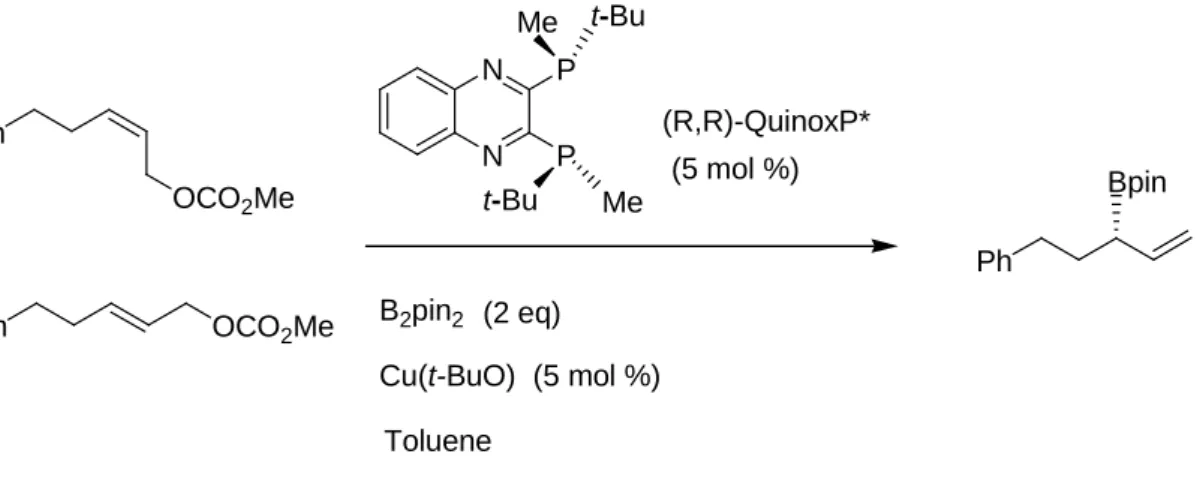
Ph OCO2Me Ph OCO2Me N N P P t-Bu Me t-Bu Me (R,R)-QuinoxP* B2pin2 (5 mol %) Cu(t-BuO) (5 mol %) (2 eq) Ph Bpin Toluene
L’enantioselettività della reazione è influenzata dalla configurazione del doppio legame nell’allilcarbonato in quanto sull’isomero Z la reazione riportata nello Schema 16 dà luogo ad un eccesso enantiomerico del 96% l’isomero E porta solo al 44% di ee.
Figura 6. Meccanismo proposto
Cu P P O t-Bu Cu P P Bpin R OCO2Me C C H H OCO2Me R Cu P P C C H H Cu P P pinB O CO2Me B2pin2 pinB OMe R -CO2 pinB R H A B C D
Per quanto riguarda il possibile meccanismo della reazione illustrato in Figura 6 il composto di alcossirame A reagisce con il B2Pin2 per generare il composto di borilrame B. Quest’ ultimo va a formare il complesso π Cu-alchene C. L’addizione del legame B-Cu sul doppio legame C-C porta all’intermedio β−borilalchilrame D. Effetti stereo-elettronici sono causa della completa regioselettività. Il legame σ Cu-Cβ è infatti stabilizzato da interazioni con il σ∗ Cα-O. Infine
l’intermedio D va incontro a β-alcossi eliminazione per dare l’α-allilboronato chirale e una specie rame-carbonato che va a rigenerare poi il composto A.
Figura 7. Ragioni della stereoselettività
N N P P C C Cu pinB H N N P P C C Cu pinB R H OCO2Me OCO2Me R H H favorito
1.3
Reazioni catalizzate da nichel
Nell’ambito del lavoro di tesi, di fronte a risultati insoddisfacenti di vari sistemi catalitici basati sul rame, si è resa necessaria una ricerca volta a individuare l’eventuale possibilità di impiegare sistemi catalitici di altro tipo nelle reazioni di sostituzione allilica con B2Pin2. Tra i vari sistemi catalitici quindi presi in considerazione il nichel ha suscitato particolare interesse e si è poi rivelato essere la chiave necessaria per sbloccare la situazione di empasse inizialmente venutasi a creare.
Un gruppo di lavoro particolarmente attivo nello sviluppo di procedure Ni-catalizzate implicanti l’impiego di reagenti del boro è il gruppo del professor Oshima. Una parte dei dati da questo prodotti è riservata ad alchilazioni nichel-catalizzate di aldeidi con trialchilborani.17 Generalmente i reagenti di alchilboro non reagiscono con aldeidi a causa della loro scarsa reattività; se però si tratta una aldeide con trietilborano in presenza di cesio carbonato e di quantità catalitiche di Ni(cod)2 e di P(t-Bu)3 in toluene a temperatura ambiente per ventiquattro ore si può ottenere il prodotto alchilato in buone rese. I trialchilborani possono essere preparati per idroborazione di alcheni.
Schema 17. Alchilazione di aldeidi con alchilborani preparati tramite idroborazione
n-Bu
BH3
.
SMe2THF, 0 °C, 3 h
(n-C6H13)3B
cat. Ni(cod)2 / P(t-Bu)3
Cs2CO3, PhCHO
toluene, r.t., 24 h Ph
OH
n-C6H13
89 %
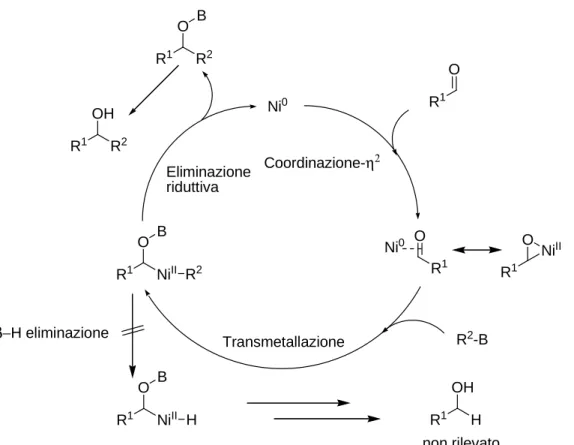
Per ciò che riguarda il meccanismo di reazione si ritiene che inizialmente una specie del nichel(0) vada a generare con l’aldeide un complesso η2. In seguito la transmetallazione con il trialchilborano
(o con il borato formatosi per azione del cesio carbonato) genera un intermedio che va incontro ad una eliminazione riduttiva promossa dalla P(t-Bu)3 per generare un alcool e restituire la specie di nichel(0) da cui si era originata la reazione. La reazione procede senza la formazione del possibile alcool primario prodotto collaterale della possibile β-eliminazione di idruro che si potrebbe avere come reazione competitiva della eliminazione riduttiva.
Figura 8. Meccanismo proposto
Ni0 R1 O Ni0 R1 O NiII R1 O NiII B R2 R1 O R2 B R1 OH R2 R1 O NiII B H R1 OH H Coordinazione-η2 R1 O Transmetallazione R2-B Eliminazione riduttiva β−H eliminazione non rilevato
Il cesio carbonato è fondamentale nella reazione e un tentativo di sostituzione con potassio carbonato o con acqua comporta un abbattimento netto delle rese della reazione. Risulta però interessante come sostituendo il toluene con l’acqua come solvente si riesca (a volumi maggiori rispetto a quelli impiegati di toluene) ad ottenere rese alte anche senza impiegare il cesio carbonato e tutto ciò nonostante il trialchilborano impiegato nelle prove (il tri-n-butilborano) sia insolubile in acqua così come il catalizzatore a base di nichel.
Una procedura molto simile a quella appena vista può essere impiegata per effettuare una addizione 1,4 con trialchilborani su esteri α,β−insaturi.
Schema 18. Addizione 1-4 di trietilborano nichel catalizzata
R1 OR2 O Et3B 3 eq Ni(cod)2 (8 mol%) P(t-Bu)3 (19 mol%) Cs2CO3 (3 eq) toluene, r.t., 17-24 h R1 OR2 O Et
Visto che l’acidità di Lewis per i trialchilborani è simile a quella del B2Pin2 è plausibile aspettarsi pure che nelle stesse condizioni appena viste anche il B2Pin2 potesse dar luogo ad addizioni 1,4 coniugate. Le condizioni di reazione sono state messe appunto eseguendo una serie di prove per la
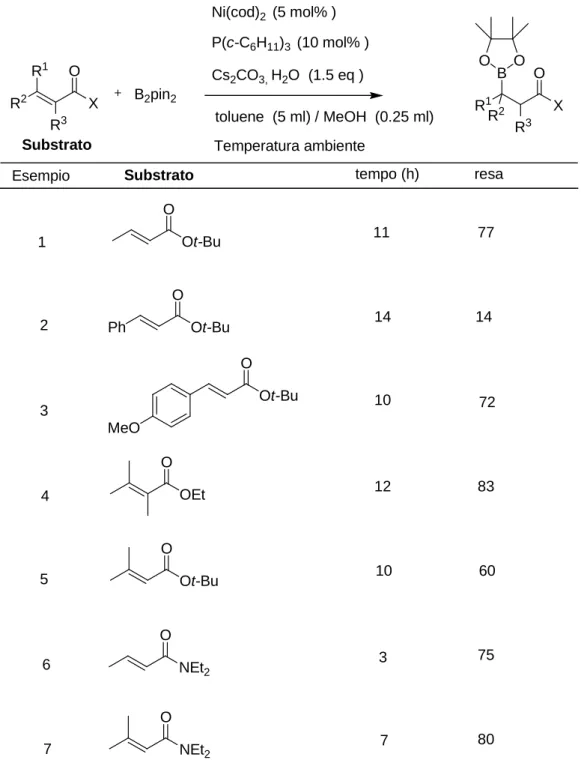
β-borazione dell’etil e del t-butil-crotonato.18 Le condizioni di reazione migliori prevedono l’impiego del metanolo come cosolvente del toluene e l’impiego di cesio carbonato e acqua. Sulla base di queste osservazioni è si sono state poi effettuate prove su vari esteri e ammidi (che reagiscono più velocemente dei corrispondenti esteri vedi esempio 6 e 7 Tabella 3) ottenendo risultati soddisfacenti anche con esteri β,β di sostituiti, tri- e tetra sostituiti.
Tabella 3.
β−
β−
β−
β−
borazione di esteri e ammidi
α,β−
α,β−
α,β−
α,β−
insaturi con B
2pin
2 R2 X R1 R3 O B2pin2 Ni(cod)2 (5 mol% ) P(c-C6H11)3 (10 mol% ) Cs2CO3, H2O (1.5 eq ) toluene (5 ml) / MeOH (0.25 ml) Temperatura ambiente R1 X B R2 R3 O O O SubstratoSubstrato tempo (h) resa
Ot-Bu O 11 77 Ph Ot-Bu O 14 14 Ot-Bu O MeO 10 72 O Ot-Bu 10 60 O OEt 12 83 NEt2 O 3 75 NEt2 O 7 80 Esempio 1 2 3 4 5 6 7
Per ciò che riguarda il meccanismo della reazione questo inizia, similmente a quanto già visto nella alchilazione di aldeidi con alchilborani, con la formazione di un complesso η2tra una specie di nichel(0) e il substrato. Il B2Pin2 si coordina tramite il boro con il gruppo carbonilico dell’estere o dell’ammide. L’acidità di Lewis del boro promuove la formazione di un complesso η3del nichel
con l’estere, seguita da una transmetallazione prima e da eliminazione riduttiva poi. Il metanolo provvede poi a protonare la specie di enol borinato formatasi per convertirla nel prodotto finale.
Figura 9. Meccanismo proposto
Ni X R2 R1 R3 O X R2 R1 R3 O Ni B B X R2 R1 R3 O B B Ni X R2 R1 R3 O B Ni B R2 X R1 R3 O R1 X R2 R3 O O B O R1 X O R3 R2 B protonolisi B B B = O B O coordinazione η2 coordinazione η3 transmetallazione eliminazione riduttiva
Sempre lo stesso gruppo di lavoro ha pubblicato uno studio in cui la stessa tipologia di reazione finora vista veniva estesa all’apertura di vinilciclopropani lineari.19 Questo lavoro si è rivelato essere particolarmente interessante in quanto verteva su sistemi chimicamente più simili a quelli da noi studiati durante il lavoro sperimentale. Quindi Oshima ha raccolto una serie di dati sulla reattività di questi sistemi in base alla diversa sostituzione sul ciclo: le reazioni sono risultate più efficaci e stereoselettive verso i prodotti isomeri E e Z (a favore dell’isomero E) nel caso di sostituzione con gruppi elettronattrattori, con una maggior richiesta di carico nel sistema catalitico
all’aumentare delle dimensioni di tali gruppi. Risulta interessante per considerazioni meccanicistiche notare come la sostituzione con un unico gruppo elettronattrattore in cis rispetto al gruppo vinilico porta a rese maggiori rispetto alla stessa sostituzione ma con il gruppo elettronattrattore in trans.
Tabella 4. Apertura di vinilciclopropani con B
2pin
2E2 E1 B2pin2 (1.5 eq) Ni(cod)2 ( x mol%) P(c-C5H9)3 (3x mol%) K3PO4. 3H2O (2.25 eq) toluene / MeOH = 30:1 25 °C, 10 h E2 E1 O B O E1 E2 x resa E/Z CO2Et CO2Et 5 84 94:6 CO2Me CO2Me 2 73 91:9 CO2tBu CO2tBu 7.5 66 95:5 CO2tBu CO2tBu H 5 85 91:9 H 10 44 85:15
Per quanto riguarda il meccanismo di reazione si ritiene che il ciclopropano sia attivato dall’acidità di Lewis del boro per poi andare incontro all’addizione ossidativa con il nichel(0) a dare un complesso π-allil(ossa-π-allil)nichel. La transmetallazione dà un complesso π−allilnichel portante una funzionalità enolato di boro. Questo complesso va incontro a eliminazione riduttiva e successiva protonazione dell’enolato di boro del boronato allilico generatosi dando infine il boronato allilico finale.
Figura 10. Meccanismo proposto
EtO OEt O O B B Ni0 EtO EtO O O Ni B B EtO O EtO O B Ni B eliminazione riduttiva addizione ossidativa transmetallazione protonolisi EtO EtO O O B O OIl fatto che, come prima accennato, la sostituzione con un gruppo elettronattrattore in cis al gruppo vinilico porti a rese più alte della stessa sostituzione in trans porta a credere che la configurazione
cis permetta una coordinazione bidentata del nichel che poi va facilmente incontro ad addizione
ossidativa dando l’intermedio chiave della reazione direttamente o via ossanichelcicloottadiene. Nel caso della configurazione trans il complesso bidentato non si forma e l’addizione ossidativa risulta lenta sia diretta, sia tramite l’ossanichelcicloesadiene. Ciò comporta un aumento nello sviluppo di prodotti di reazioni collaterali.
Oshima impiega i boronati allilici prodotti (sia isolati, sia con procedura one-pot) per compiere allilazioni su composti carbonilici. La procedura da esso proposta prevede di impiegare quantità catalitiche di triflato rameico, verosimilmente come attivatore del composto carbonilico, ma non ci è possibile sapere se effettivamente il suo impiego sia fondamentale per far avvenire l’ allilazione.
Schema 19. Impiego dei boronati ottenuti per apertura di vinilciclopropani con
B
2pin
2per reazioni di allilazione di composti carbonilici
E E O B O E = CO2Et (E/Z = 94:6) PhCHO (1.2 eq) Cu(OTf)2 (10 m ol%) toluene 25°C 91%, anti / syn = 95:5 E EPh OH Ni(cod)2 ( x mol %) P(c-C5H9)3 (3x mol %) K3PO4 3H2O (2.25 eq) toluene / MeOH = 30:1 25 °C, 10 h E E PhCHO (1.2 eq) Cu(OTf)2 (10 m ol%) toluene 25°C 79%, anti / syn = 94:6 E E O B O one-pot B2pin2 (E/Z = 94:6)
1.4
Reazioni di allilazione di composti carbonilici.
I dati su questa tipologia di reazione sono piuttosto abbondanti essendo l’impiego dei boronati allilici come composti allilanti ormai conosciuto da tempo e dettagliatamente approfondito per ciò che riguarda la sua intrinseca diastereoselettività.
I primi studi su questa categoria di reazioni erano stati eseguiti su reazioni di allilazione in cui l’agente allilante era l’estere pinacolico dell’acido crotilboronico.20 La scelta di impiegare l’estere
era giustificata dalla necessità di evitare il fenomeno dello shift 1,3 metallotropico. Questo è un fenomeno che affligge alcune specie allilmetalliche nelle quali si osserva una instabilità configurazionale del doppio legame che isomerizza da E a Z e viceversa, rendendo di fatto improponibile una qualsivoglia considerazione sulla determinazione di un cammino stereoselettivo per la reazione impartito dalla configurazione del composto allilico. Tutto ciò è dovuto al fatto che alcune specie metalliche hanno una bassa barriera energetica per l’interconversione del loro legame con l’allile da η1a η3 e ciò dà possibilità alla componente metallica di compiere lo shift
metallotropico tra le posizioni 1 e 3 del gruppo allilico. Così si creano le condizioni per l’isomerizzazione del doppio legame (nonché una serie di problematiche sulla regioselettività). Dei vari composti di allilboro quelli risultati più resistenti allo riarrangiamento borotropico sono quelli in cui il boro è legato ad atomi di ossigeno o di azoto, per cui si sono prestati bene esteri e ammidi dell’acido crotilboronico.
Schema 20. Meccanismo di isomerizzazione tramite shift 1,3 metallotropico
M Y Y M M Y Y M M Y
Può risultare interessante osservare le vie sintetiche utilizzate al tempo delle ricerche di Hoffmann (1982) per la sintesi dei derivati dell’acido crotilboronico con configurazione ben determinata: la sintesi veniva eseguita facendo reagire il crotilmagnesio bromuro con clorobis(dimetilammino)borano, ottenendo una miscela di ammidi dell’acido metilallilboronico. Per isomerizzazione con ZnBr2 a 120 °C veniva ottenuta una miscela di ammidi dell’acido crotilboronico E/Z che venivano infine separate per distillazione. Dalle ammidi si poteva passare agli esteri tramite trattamento con un 1,2 diolo come il pinacolo. Gli esteri possono essere ottenuti direttamente per reazione dei composti di crotilpotassio con FB(OCH3)2 e successiva transesterificazione per ottenere il composto voluto.
Schema 21. Sintesi di E e Z crotilboronato
MgCl ClB NMe2 NMe2 B Me2N NMe2 B NMe2 NMe2 B NMe2 NMe2 ZnBr2 B O O HO HO B O O n-BuLi KOt-Bu -78 °C K FB(OCH3)2 HO HO B O O K n-BuLi KOt-Bu -78 °C FB(OCH3)2 HO HO B O OGli studi eseguiti su questi composti hanno rivelato come le reazioni che implicano l’allilazione di composti carbonilici con specie di allilboro (ma il discorso è valido pure per altre componenti metalliche diverse dal boro), siano strettamente correlate alle reazioni di addizione aldolica coinvolgente enolati metallici. Innanzitutto si riscontra in suddette allilazioni una intrinseca diastereoselettività del tutto analoga alla diastereoselettività semplice della addizione aldolica con enolati metallici. Si può infatti apprezzare come gli allilboronati E tendano a generare dopo allilazione di una aldeide esclusivamente la coppia di enantiomeri con configurazione relativa dei centri chirali di neoformazione anti, così come gli allilboronati Z nella stessa reazione diano la coppia di enantiomeri syn. Tutto ciò quindi ricorda molto come gli enolati metallici Z reagiscano
nella addizione aldolica generando aldoli syn piuttosto che anti i quali sono invece i prodotti prediletti degli enolati E.
Schema 22. Diastereoselettività della reazione aldolica e della allilazione di aldeidi a
confronto
R H O Y OM R1 R R1 OM Y O R R1 OM Y O R O H B O O B O O R OH R OH R OH R OH Reazione aldolica Allilazione di aldeidiLa similitudine nella selettività non è limitabile al semplice esito della reazione ma si estende anche al meccanismo implicato nella generazione della selettività. Infatti si ritiene che, similmente a quanto avviene nella reazione aldolica in presenza di enolati metallici dove si sviluppano gli stati di transizione ciclici di tipo Zimmerman-Traxler, sia plausibile aspettarsi che le ragioni della selettività vadano ricercate nella presenza di stati di transizione ciclici a sei termini. Nelle condizioni cinetiche in cui vengono svolte le reazioni infatti saranno le stabilità relative di questi stati di transizione a determinare quale prodotto si formerà. Perciò risulta evidente come la differente configurazione negli allilboronati comporti differenti interazioni dei sostituenti nello stato di transizione ciclico e quindi differenti livelli energetici tra stati di transizione diastereoisomeri.
Schema 23. Stati di transizione ciclici nelle reazioni di allilazione di aldeidi con
boronati allilici
O B R H L L Si Re unlike H R O H O B H R L L Re Re like R OH R OH enantiomero enantiomero interazione 1,3 diassialeNello schema è stato presentato l’esempio dell’allilboronato Z. Si può notare come lo stato di
transizione che condurrebbe alla coppia di enantiomeri anti sia afflitto dalla presenza di una
sfavorevole interazione 1,3 diassiale tra uno dei sostituenti del boro e il gruppo R dell’aldeide. Ciò generalmente prevale sulla sfavorevole interazione gauche che si ha nello stato di transizione che
porta agli enantiomeri cis. Ovviamente non ci possiamo aspettare una altrettanto efficace selettività
se al posto di una aldeide si usasse un chetone in quanto in questo caso verrebbe meno un discriminante importante come la differenza di dimensioni che c’è tra il semplice idrogeno e un qualsiasi altro generico gruppo R, riscontrabile quando si tratta di aldeidi. Un'altra caratteristica importante per la selettività sembra essere la lunghezza del legame ossigeno-metallo nello stato di transizione. In particolare le interazioni tra il gruppo R e il gruppo legato al metallo sono massimizzate tanto più il legame ossigeno-metallo è corto. In questo il boro (1.4 Ǻ) risulta essere migliore rispetto ad altri metalli come il silicio, il titanio o lo stagno. Infine anche la grandezza dei gruppi legati al boro possono incidere sulla diastereoselettività.
Già negli studi di Hoffmann si fanno considerazioni sull’impiego di allilboronati portanti un sostituente in α al boro e in forma non racemica. Questi composti venivano ottenuti secondo, una
procedura proposta da Matteson (Schema 24), inserendo un ausiliario chirale sul boro e poi sfruttando l’induzione asimmetrica da questo impartita per eseguire reazioni di alchilazione enantioselettive.
Schema 24. Procedura di Matteson
H3C Cl2HC B O O C6H11 C6H11 CH3Li ZnCl2 B O O C6H11 C6H11 Cl Li B O O C6H11 C6H11 CH3
La reazione di questi composti non racemici con aldeidi achirali porta nel caso di assenza di sostituenti sul C1 alla possibile formazione di composti epimeri in quanto differiscono e per la configurazione del centro chirale neoformato e per la configurazione del doppio legame. Non è dato in assoluto di conoscere a priori se ci sarà una prevalenza e eventualmente quale dei due epimeri prevarrà. Infatti come si può vedere nellao Schema 25 per ciò che riguarda considerazioni in termini di effetti sterici (considerando un sostituente R1 alchilico non polare) entrambe gli stati di
transizioni sono esposti a sfavorevoli interazioni: da una parte lo stato di transizione 4 è affetto da interazioni tra il sostituente R1 e la componente alcolica dell’estere boronico, la cui dimensione può
quindi incidere sulla selettività della reazione, dall’altra lo stato di transizione 5 presenta sfavorevoli interazioni alliliche a causa della disposizione pseudo-assiale del gruppo R1.21
Schema 25. Reazione di allilboronati portanti un centro chirale in
αααα
al boro con
aldeidi
B OR OR R1 R2 H O B O R1 OR OR R2 O B OR OR R2 H R1 R2 R 1 OH R2 OH R1 4 5Un esempio di selettività per uno dei due stati di transizione è il caso dell’estere pinacolico dell’acido Z-pentenilboronico. Qui la presenza di un metile in posizione allilica al gruppo in α al boro fa si che le sfavorevoli interazioni alliliche nello stato di transizione 5 siano determinanti nel favorire il cammino passante per lo stato di transizione 2.
Schema 26. Selettività nella reazione dell’estere pinacolico dell’acido
Z-pentenilboronico con aldeidi
B O O O B H3C R O O H3C B O O O CH3 CH3 R R OH R OH 6 7 RCHO
In studi più recenti sono state fornite delle alternative al metodo di sintesi degli allilboronati
α−sostituiti non racemici prima presentato (secondo Matteson). Un approccio sintetico più recente prevede l’impiego del 3-cloropropenilboronato come substrato su cui eseguire una alchilazione allilica con un reattivo di Grignard, rame catalizzata sotto l’influenza di fosforoammiditi chirali.
Schema 27. Metodo alternativo per la sintesi di allilboronati portanti un centro
chirale in
αααα
all’atomo di boro in forma non racemica
Cl B OR OR R 1MgBr Cu IL* SN2 B OR OR R1
Dai dati sperimentali si deduce che le condizioni più favorevoli per la reazione sono l’impiego del diclorometano come solvente e di etilmagnesio bromuro come nucleofilo. Sulla base di queste condizioni sono state testate varie fosforoammiditi e vari boronati, differenti fra loro per la componente alcolica con cui erano esterificati, per poter verificare quali composti dessero una miglior risposta in termini e di enantioselettività, e di rapporto SN2 e SN2’. Le condizioni ottimizzate sono riportate nello Schema seguente.
Schema 28. Condizione ottimali per l’ottenimento di allilboronati portanti un centro
chirale in
α
α
α
α
all’atomo di boro in forma non racemica
B Cl EtMgBr O O P N OMe OMe S CO 2Cu CH2Cl2 - 78 °C 5.5 mol % 5.0 mol % 8 B Et ee 95,5 % 9
Visti i migliori risultati ottenuti con l’impiego del composto 8 rispetto al corrispondente estere pinacolico, su di esso è stata messa appunto una reazione di allilazione one-pot a bassa temperatura catalizzata da un acido di Lewis. Usare il boronato 8 pare dare dei vantaggi nella selettività E/Z rispetto al corrispondente estere pinacolico grazie alla riduzione delle interazioni di non legame il gruppo etile pseudoequatoriale e il gruppo legato al boro quando si forma lo stato di transizione. Ovviamente la sintesi degli allilboronati in forma non racemica non è da relegare al solo ottenimento di reagenti utili ad allilazioni stereoselettive in quanto i prodotti ottenuti sono facilmente elaborabili. Per esempio con workup ossidativo si può convertirli in alcoli allilici chirali. C’ è inoltre la possibilità di convertire gli allilboronati chirali in allil-trifluoroborati, agenti allilanti efficaci poiché si convertono in-situ in allil-difluoroborani altamente reattivi. La preparazione di questi sali a partire dal corrispondente pinacolato è semplice, quantitativa e non è caratterizzata da racemizzazione. Il valore aggiunto di questi reattivi come allilanti è la capacità di reagire non solo con le aldeidi ma anche con i chetoni.
Schema 29. Impiego degli organotrifluoroborati per l’allilazione di chetoni
B Et O O KHF2 ( 6 equiv ) acetone/ H2O BF3K Et ( 64 % ) R1 R2 O BF3.
OEt2 ( 2 equiv ) CH2Cl2 - 78 °C, 14 h R1 Et R2 HO R1 = Ph, R2 = H R1 = Ph, R2 = Me ( 70%, 2.6:1 E/Z, 87% ee ) ( 95%, 2.5:1 E/Z, 85% ee )Sulla reazione di allilazione dei chetoni non si è infatti ancora raggiunta la amplia conoscenza sviluppata invece sulle aldeidi. Vi sono comunque lavori recenti tesi a colmare questa lacuna. Sono infatti ora accessibili dati sulla possibilità di eseguire allilazioni asimmetriche su chetoni con allilboronati.22 La procedura si basa sull’impiego di una componente catalitica costituita da un diolo
chirale. La scelta del diolo non era ovviamente casuale: il suo impiego era stato determinato dalla pregressa conoscenza della capacità della reazione di allilazione di essere accelerata dalla presenza di un acido di Lewis o da un acido forte di BrØnsted e dalla facilità con cui i dialcossiborani aciclici vanno incontro a scambio di ligante. Un diolo chirale con adeguate caratteristiche avrebbe potuto perciò agire sia da acido di BrØnsted che da ligante chirale sull’allilboronato. Si riporta sinteticamente in Tabella 5 una serie di esperimenti con diversi dioli, solventi, e condizioni di reazione nella reazione dell’acetofenone con due diversi boronati, uno ciclico e uno no.
Tabella 5. Ottimizzazione dei reagenti e delle condizioni di reazione per l’allilazione di
chetoni con allilboronati
Ph CH3 O B OR OR 15 mol % Catalizzatore 1 R = i -Pr 2 R = C(CH3)2C(CH3)2 Ph OH H3C
cataliz. boronato temp. (°C) solvente resa % er
1 0 PhCH3 13 3 1 1 0 0 PhCH3 PhCH3 54 50:50 4a 35 72:28 1 0 PhCH3 4b 82 83:17 4b 1 - 35 PhCH3 47 95:5 4b 1 - 25 PhCF3 89 94.5:5.5 4b 1 - 35 PhCH3 : PhCF3 1 : 3 83 97:3 4b 2 - 35 PhCH3 : PhCF3 1 : 3 0 esempio 1 2 3 4 5 6 7 8 O O OH Ph Ph OH Ph Ph (+ )-TADDOL 3 X X OH OH 4a X = H (S )-BINOL 4b X = Br
Dalla Tabella 5 si può dedurre come una prima prova con (+)-TADDOL porti a rese più alte rispetto alla prova in bianco priva di catalizzatore. La enantioselettività non è però rilevabile impiegando questo catalizzatore. L’impiego dell’(S)-BINOL non porta a grandi rese ma migliora molto l’enantioselettività. La sostituzione del BINOL a posizioni 3,3’ con un atomo di bromo migliora fortemente sia la resa che la selettività. Un ulteriore raffinamento della selettività della reazione si ottiene lavorando a basse temperature. Si assiste però pure ad un calo della velocità della reazione. Passando dal toluene all’α,α,α−trifluorotoluene si riesce a risolvere il problema delle basse rese a
basse temperature. Per poter raggiungere i –35 °C c’ è la necessità di impiegare una miscela dei due solventi testati. Si arriva così alle migliori condizioni di reazione ottenute nell’ambito di questo screening di liganti, reattivi e solventi. Da notare come la stessa reazione che è efficace con il boronato 1 è del tutto inattiva se si impiega il boronato ciclico 2 probabilmente a causa della stabilità del boronato ciclico.
Per ciò che riguarda le considerazioni meccanicistiche vari dati indicano la formazione di un complesso in cui il catalizzatore risulta associato al boronato. Innanzitutto tramite l’1H NMR è stato
osservato un rapido scambio per il gruppo isopropossilico legato al boro. La cinetica della reazione è del primo ordine rispetto al catalizzatore ed è stato osservato un effetto non lineare del rapporto enantiomerico del catalizzatore sulla selettività della reazione. Il meccanismo proposto prevede che una volta formatosi il complesso diolo-boronato l’ossidrile libero del diolo attivi l’allilboronato tramite la formazione di un legame a idrogeno con l’ossigeno del gruppo isopropossilico. La presenza del ligante chirale nello stato di transizione impartisce enantioselettività alla reazione.
Figura 11. Possibile stato di transizione
Br O B O O H O
In questo caso si è visto come il catalizzatore sia stato scelto in base anche alla capacità di comportarsi da acido di BrØnsted. Questa scelta era stata operata in quanto è nota la attività catalitica che composti dotati di caratteristiche acide possono esercitare sulla reazione di allilazione di aldeidi con boronati allilici. La catalisi acida per queste reazioni è relativamente recente come approccio essendo stato descritto da Miyaura nel 2002. Nei suoi studi si evidenzia come la reazione di allilazione su varie aldeidi eseguita con l’estere pinacolico dell’acido propenilboronico ad una temperatura di – 78 °C, sia fortemente accelerata dalla presenza di un acido di Lewis.23 Le rese
riportate nello schema vanno correlate con le rese in assenza di catalizzatore che a –78 °C sono nulle.
Tabella 6. Effetti della catalisi acida
aldeide AlCl3 Sc(OTf)3
rese 4-CF3C6H4CHO 80 (16 h) 69 (16 h) PhCHO 88 (16 h) 80 (16 h) 4-MeC6H4CHO 47 (24 h) 62 (24 h) 4-MeOC6H4CHO 82 (36 h) 84 (36 h) n-C10H21CHO 54 (16 h) 73 (16 h) c-C6H11CHO 48 (16 h) 74 (16 h) (E)-PhCH CHCHO 69 (24 h) 74 (24 h) R1CHO R3 BPin R2 cat. L.A. R1 OH R2 R3 * *
L’impiego delle bassa temperature e della catalisi acida rafforza la già insita differenza di reattività tra aldeidi elettronpovere (più reattive) e elettronricche (meno reattive), tanto da rappresentare un metodo per far reagire selettivamente una aldeide rispetto ad un’ altra nell’ambiente di reazione in base alle differenti caratteristiche elettroniche.
Schema 30. Sfruttamento della differenza di reattività tra aldeidi a bassa temperatura
sotto catalisi acida
4-CF3C6H4CHO 4-MeOC6H4CHO Bpin
4-CF3C6H4
OH
4-MeOC6H4
OH
senza catal. temp. ambiente 89 % (81:19)
AlCl3 (10 mol % / -78 °C) 57 % (99:1)
Sc(OTf)3 (10 mol % / -78 °C) 72 % (99:1)
toluene 16 h
Una osservazione importante è che la catalisi acida non influisce sulla struttura dello stato di transizione che rimane ciclico del tipo Zimmerman-Traxler. Ciò è ipotizzabile in quanto le caratteristiche di diastereoselettività come quelle di regioselettivtà della reazione vengono mantenute inalterate.
Una volta citate quindi una serie di applicazioni degli allilboronati come agenti allilanti per aldeidi e chetoni ci rimane da focalizzare l’attenzione sui composti azometinici. Infatti per noi è stato importante trovare dati in letteratura da cui ottenere delle linee guida per la messa a punto delle condizioni di reazione per poter, con successo, espandere l’impiego degli allilboronati da noi ottenuti, appunto anche alle allilazioni di immine. Particolarmente illuminante è stato un articolo pubblicato dal gruppo di lavoro di Shu Kobayashi in quanto qui si riferiva di α− amminoallilazioni di aldeidi in presenza di ammoniaca.24 Questo articolo veniva infatti incontro alle nostre necessità
di ottenere una procedura pratica per l’allilazione di immine non N-protette, in quanto le N-protette avevano dato risultati insoddisfacenti.
In questo articolo viene descritta una procedura tre-componenti che prevede di miscelare a –10° C l’aldeide in ammoniaca liquida ed etanolo (nel quale l’ammoniaca ha un’ alta solubilità) per due ore dopodiché alla miscela si aggiunge gocciolandolo l’allilboronato. La reazione dopo altre 3 ore viene lasciata andare a temperatura ambiente e viene eseguito il work-up. (questa procedura viene all’evenienza modificata per particolari classi di aldeidi come riportato nella tabella seguente). Il fatto che generalmente si usi un eccesso di ammoniaca e che ciò possa alterare l’equilibrio tra ammoniaca/aldeide e azometina fa pensare che il primo dei due meccanismi sia il più probabile. La sostituzione dell’ammoniaca con una ammina come la benzilammina comporta il blocco della reazione nonostante si assista comunque alla formazione dell’immina.
Tabella 7.
αααα
-amminoallilazione di aldeidi
p-NO2C6H4CHO p-BrC6H4CHO p-MeOC6H4CHO o-HOC6H4CHO (pyridin-2-il)CHO (tiofen-2-il)CHO (E)-PhCH CHCHO Ph(CH2)2CHO PhCHO PhCHO c-C6H11CHO ( )-PhCHMeCHO HO2CCHOH H.
2O 1 2 3 4 5 6 7 8 9 10 11 12 13 A A A A A A A A B B C C C 84 3 80 96 92 91 76 85 77 75 78 80 69 8 <1 5 4 <1 nr 12 3 3 4 4 RCHO metodo resa 1 2 R H O B O O R R NH2 OH nr quantA: l'aldeide (0.5 mmol) in ammoniaca viene lasciata agitare a -10 °C per 2 h. Alla soluzione si aggiunge l'allilboronato (1.2 eq) goccia a goccia. La miscela viene lasciata agitare a -10 °C per 3 h e poi a temp eratura ambiente per 1 h prima del work-up.
B: l'aldeide (0.5 mmol) in ammoniaca acquosa al 28-30% wt (ca 20 eq) e etanolo, viene lasciata agitare a temperatura ambiente per 30 min. Alla soluzione si aggiunge l'allilboronato (1.2 eq). La miscela viene lasciata agitare a temperatura ambiente per 2 h.
C:l'allilboronato (1.2 eq) in ammoniaca liquida ed etanolo viene lasciato agitare a temperatura ambiente per 2 h. L'aldeide (0.5 mmol) in etanolo viene aggiunta alla soluzione e la miscela viene lasciata agitare per 2 h a temperatura ambiente prima del work-up.
La reazione di (Z)- o (E)-crotilboronato porta alla solita selettività syn/anti apprezzata per le aldeidi. Un esempio di sfruttamento di questa selettività si può vedere nella sintesi della Alloisoleucina, peptide non comune reperibile in certi antibiotici peptidici. Si può vedere in Schema 31 come la reazione possa pure essere applicata nell’ambito di sintesi enantioselettive.
Schema 31. Diastereoselettività nella allilazione di immine con allilboronati
R H O B O O B O O R R NH2 NH2 R = Ph p-NO2C6H4 metodo C (v. Tabella 8 ) R = Ph : 85%, >99% syn R = p-NO2C6H4 90%, >99% syn R = Ph : 79%, >93% anti R = p-NO2C6H4 92%, >92% antiSchema 32. Sintesi dell’alloisoleucina
HO2CCHO H
.
2O (1.5 equiv, >99% Z) B O O metodo C -10 °C 3 h H2 Pd/C r.t. 12 h ( 91%, in 2 stadi ) HO O NH2 alloisoleucina >99% syn2. RISULTATI E DISCUSSIONE
2.1 Reazioni di apertura borilativa di vinil-epossidi e vinil-aziridine
Il lavoro di tesi è stato concepito partendo dalla convinzione che si potesse ottenere una apertura borilativa di vinil-epossidi e vinil-aziridine. Questa nostra idea era sostenuta dall’abbondanza di dati presenti in letteratura riguardo a procedure rame catalizzate per la formazione di legami C-B tramite il trattamento di composti portanti doppi legami α,β−insaturi o di substrati allilici con B2Pin2. Inoltre un ulteriore appoggio per intraprendere questo lavoro era dato dalla presenza di procedure Ni-catalizzate per l’apertura borilativa di vinilciclopropani, substrati per molti aspetti analoghi a quelli verso i quali ci apprestavamo a rivolgerci.
2.1.1 Reazioni catalizzate da rame
Le prime reazioni da noi eseguite sono state improntate alle condizioni proposte nei lavori di Hosomi e Miyaura e Yun descritte nella parte introduttiva. Inizialmente il sistema catalitico verso il quale ci siamo orientati era quindi basato sul rame. In particolare si è deciso di cominciare il lavoro testando le condizioni proposte da Yun secondo le quali è previsto l’impiego di un sale di rame in quantita catalitiche, di una base, di un ligante a base di fosforo e di una componente alcolica additiva (MeOH nella fattispecie). Il solvente in questo caso è il THF. Il primo substrato da noi utilizzato è stata un’aziridina vinilica ciclica a cinque termini.
Schema 1. Prima reazione di apertura borilativa eseguita
N Ts
CuCl 10 mol% / LiOtBu 10 mol % BINAP 10 mol% B2Pin2 THF/ MeOH (4 eq) NHTs PinB Resa 23% 1 2
La reazione ha avuto successo (anche se con rese basse) e dopo aver eseguito una purificazione tramite flash cromatografia, si è potuto isolare con resa del 23 % il prodotto di apertura in cui il boro ha dato vita ad un addizione di tipo 1,4. Si può notare come l’ attacco del B2Pin2 porti selettivamente alla formazione del prodotto anti. Ciò è stato confermato dalla bassa differenza di chimica shift tra i segnali dei due protoni metilenici del prodotto: i due segnali dati da questi protoni diastereotopici cadono infatti sotto forma di multipletti uno a 2.15-2.29 ppm e l’altro a 1.67-1.75 ppm, con una differenza di circa 0.50 ppm tra di loro in accordo con quanto riportato in letteratura per composti analoghi.25 Va considerata come prova della stereochimica anti i valori delle costanti
di accoppiamento per questi protoni: un caso in cui è stato possibile individuare con precisione il valore di queste costanti è il caso del composto 10 (lo si vedrà tra i composti ottenuti nel capitolo 1.2.1 anche se non sarà possibile purificarli), per il quale i valori delle costanti di accoppiamento per uno dei protoni metilenici sono risultate essere: 3.2, 8.8, 13.7 Hz. Inoltre una ulteriore conferma di questa stereochimica relativa è derivata anche dalle successive prove sui prodotti di allilazione ottenuti da questi boronati e che a loro volta hanno mostrato una determinata stereochimica relativa che non sarebbe stata possibile se questi boronati allilici non fossero stati trans. Di queste prove si parlerà più approfonditamente nel capitolo 2.5.
In seguito a questo risultato abbiamo cercato di capire se effettivamente le condizioni proposte da Yun erano necessarie oppure se era possibile rivolgersi alle più semplici condizioni proposte da Miyaura che prevedevano l’impiego di una sale di rame in quantità stechiometriche e di usare come solvente la DMF (Tabella 1 esempio 4). Come si può notare in Tabella 1 le modifiche da noi applicate sono state inspirate dalle condizioni proposte da Yun, cioè si è provata la variazione del solvente con THF e l’inserimento o della componente alcolica o del ligante a base di fosforo (esempi 2 e 3).
Tabella 1. Variazioni delle condizioni di reazione
NTs B2Pin2 CuCl / Base Solvente NHTs PinBCondizioni Solvente Additivo Ligante
CuCl 110 mol% / tBuOLi 10 mol% CuCl 110 mol% / tBuOLi 10 mol% CuCl 110 mol% / tBuOLi 10 mol% CuCl 110 mol% / AcOK 110 mol% LiCl 110 mol% DMF THF THF DMF -BINAP rac. -MeOH 1 2 Esempio 1 2 3 4
Tutte queste prove hanno dato risultati non soddisfacenti portando a miscele complesse nelle quali il prodotto sembra essere presente in quantità irrilevanti. Visti i deludenti risultati ottenuti applicando le condizioni proposte da Miyaura o simili ad esse, ci siamo dedicati all’indagine dell’efficienza di quelle proposte invece da Hosomi. A tale fine è stata eseguita una reazione “pilota” per vedere se si poteva ottenere rese migliori rispetto alle condizioni proposte da Yun.
Schema 2. Reazione di apertura borilativa secondo le condizioni di Hosomi
1 N Ts 2 Cu(OTf) 10 mol% Bu3P 11 mol% B2Pin2 Toluene NHTs PinB Resa 19 %
Come si può vedere le principali differenze tra le condizioni di reazione proposte da Yun e quelle di Hosomi sono la mancanza in quest’ultime della base e del metanolo.
La reazione procede effettivamente con rese paragonabili a quelle ottenute in condizione di Yun, ma sempre con limitazioni importanti per ciò che riguarda la resa anche se ciò stride con l’aspetto dello spettro NMR del grezzo che lasciava presagire rese migliori. Si è testata pure la DMF come solvente ma si è ottenuto solo un peggioramento delle rese.
Si sono quindi adottate le condizioni di Yun su substrati alternativi alla aziridina 1, in particolare si sono impiegati gli epossidi ciclici 3,4,5, e l’aziridina lineare 6, ma tutte le reazioni hanno portato a miscele complesse o al recupero del substrato praticamente intonso.
Tabella 2. Reazione secondo Yun su vari substrati
Resa Substrato O O O Ph N Ts Recuperato solo substrato Recuperato solo substrato Miscela complessa Miscela complessa
Esempio Tempo di reaz.
1 2 3 4 20 h 20 h 1 h 4 h 3 4 5 6
Visto i deludenti risultati nel cercare di ampliare la rosa dei substrati impiegabili applicando le condizioni di Yun, si è allora eseguito uno screening di substrati applicando le condizioni di Hosomi che si erano comunque rivelate pressappoco valide come le condizioni di Yun.