1. INTRODUZIONE
1.1 Il tumore al polmone
Il tumore al polmone è il risultato della progressiva trasformazione di normali cellule dell’epitelio bronchiale in cellule tumorali maligne, attraverso un processo “multistep” che comporta il lento accumulo di alterazioni genetiche.
Contribuiscono alla cancerogenesi del polmone sia fattori esogeni (fumo di tabacco e inquinamento) sia fattori endogeni (si parla di suscettibilità individuale).
1.2 Istologia del tumore al polmone
Dal punto di vista istologico si distinguono due principali tipi di tumore al polmone: small-cell lung cancer (SCLC) e non small-cell lung cancer (NSCLC) (Muller, 2000; Kaiser, 1992; Parkers, 1996).
SCLC
Rappresenta il 20-25% di tutti i casi di tumore al polmone e si riscontra più frequentemente negli individui di sesso maschile in età avanzata (circa 70 anni di età) (http://www-dep.iarc.fr/).
Si sviluppa generalmente nella parte centrale del polmone sottoforma di piccole cellule fornite di un grande nucleo e scarso citoplasma. E’ caratterizzato da elevata proliferazione cellulare e insorgenza di metastasi anche nei primi stadi della malattia.
NSCLC
E’ la forma di tumore polmonare più diffusa (75-85% dei casi) e se ne distinguono tre sottogruppi istologici: lo squamous-cell carcinoma (SCC), l’adenocarcinoma (AD), e il large-cell lung carcer (LCLC).
SCC rappresenta il 45% dei tumori NSCLC ed è il tipo istologico più frequente tra i Caucasici.
Forma di cancro fortemente associata al fumo di tabacco, si sviluppa solitamente nella parte centrale del polmone e nello stadio più avanzato è possibile osservare ponti intracellulari e necrosi del tessuto (Muller, 2000; Kaiser, 1992).
AD è il tipo di tumore polmonare predominante nelle popolazioni orientali e costituisce il 45% dei NSCLC.
L’adenocarcinoma si sviluppa in genere nelle parti periferiche del polmone e può diffondersi velocemente ad entrambi i lobi (Muller, 2000; Kaiser, 1992; Corrin, 1997). Sono frequenti metastasi precoci al cervello, alla pleura e alle ghiandole renali.
LCLC rappresenta solo il 9% dei tumori NSCLC.
Se fino agli anni ’50 nella popolazione caucasica si registrava un rapporto di 17:1 tra i casi di SCC e i casi di AD, oggi si assiste ad una progressiva diffusione dell’adenocarcinoma e il rapporto si aggira intorno a 2:1 (Djordjevic et al., 1997; Hoffman D. e Hoffman I., 1995). E’ possibile che l’incremento dei casi di AD derivi dall’aumento del consumo di sigarette “light” che rispetto alle sigarette “normali” contengono una minore quantità di nicotina e una maggiore quantità di nitrosammine specifiche del tabacco (TSNAs). In accordo con questa ipotesi è l’osservazione che il trattamento con TSNAs comporti in animali da laboratorio quasi esclusivamente l’insorgenza dell’adenocarcinoma (Hecht, 1999). Inoltre i fumatori di sigarette leggere hanno la tendenza a fumare più intensamente e ad inalare più profondamente, cosicché i cancerogeni possono penetrare a fondo in zone dove è più frequente l’insorgenza dell’AD (Hoffman D. e Hoffman I., 1995; Hoffman D. et al., 1997).
1.3 Epidemiologia del tumore al polmone
Malattia rara fino agli inizi del XX secolo, il tumore al polmone è, ad oggi, la causa di morte più comune per cancro nei paesi industrializzati. Si registrano annualmente nel mondo circa 900000 nuovi casi tra gli uomini e circa 330000 tra le donne, con un tasso di mortalità del 90% entro 5 anni. In entrambi i sessi l’incidenza è bassa sotto i 40 anni e cresce fino ai 70 anni, con una prevalenza tra le classi socio-economiche più basse nelle quali si riscontra il maggior numero dei fumatori (Lung Cancer in: World Cancer Report, IARCPress 2003).
L’associazione tra il cancro al polmone e l’abitudine al fumo è probabilmente tra le relazioni più investigate in epidemiologia: ad un incremento dell’uso di tabacco corrisponde nei 20 anni successivi una crescita di incidenza della patologia, una diminuzione del consumo è invece seguita da una diminuzione dell’incidenza.
La probabilità di insorgenza della malattia cresce con il numero di sigarette fumate al giorno, ma soprattutto con la durata dell’esposizione al fumo, mentre diminuisce in soggetti che abbiano cessato di fumare da almeno 5 anni. Il rischio non scende in modo significativo per i consumatori di sigarette leggere mentre si osserva una relativa diminuzione del rischio tra i fumatori di sigarette con filtro rispetto ai fumatori di sigarette senza filtro. Inoltre i consumatori di tabacco scuro sono soggetti ad un rischio due volte più alto di cancro al polmone rispetto ai fumatori di sigarette “bionde” (Lung Cancer in: World Cancer Report, IARCPress 2003).
E’ inoltre molto forte la correlazione tra esposizione occupazionale e tumore polmonare. Sono stati identificati molti degli agenti che determinano un aumento del rischio, tra questi asbesto e miscele di idrocarburi policiclici aromatici (Boffetta et al., 1995). Il tasso di incidenza di tumore al polmone nel mondo standardizzato per età su 100000 individui è riportato in figura 1. I valori più bassi si riscontrano in Africa e nell’Asia meridionale, mentre i dati più preoccupanti si registrano in America del Nord e in
Europa (Parkin et al., 1997). La percentuale di casi di tumore tra le donne è molto elevata negli USA, in Canada, Danimarca e UK, ma è bassa in paesi come Francia, Giappone e Spagna, nei quali solo recentemente è cresciuta la percentuale di donne fumatrici (Hoffman D. e Hoffman I., 1997).
5
T
u
m
o
re
a
l
p
o
lm
o
n
e
n
e
i
m
a
sc
h
i
T
a
ss
o
d
i
in
c
id
e
n
z
a
s
ta
n
d
a
r
d
iz
za
to
p
e
r
et
à
s
u
1
0
0
0
0
0
i
n
d
iv
id
u
i
1.4 Agenti cancerogeni
Il fumo di tabacco è composto da una fase gassosa e da una particolata. La fase particolata contiene più di 3500 composti dei quali almeno 55 sono stati identificati come cancerogeni per l’uomo (Hoffman D. e Hoffman I., 1997).
Le sostanze chimiche presenti nel tabacco comprendono idrocarburi policiclici aromatici (PAHs), N-nitrosammine, ammine aromatiche, ossido di etilene, 1-3 butadiene ed altre. In particolare le nitrosammine e PAHs risultano essere cancerogeni per l’uomo. La popolazione si trova comunemente esposta ai PAHs attraverso i prodotti del tabacco, la normale alimentazione e la combustione fossile (Hecht, 1999).
Sembrano inoltre essere responsabili del 10-20% di tutti i casi di tumore polmonare agenti cancerogeni come l’asbesto, il cromo esavalente e il benzene, che si trovano negli ambienti di lavoro (Doll R. e Peto R., 1981; IARC Cancer: Causes, Occurence and Control, 1990; IARC Occupational Cancer in Developing Countries, 1994).
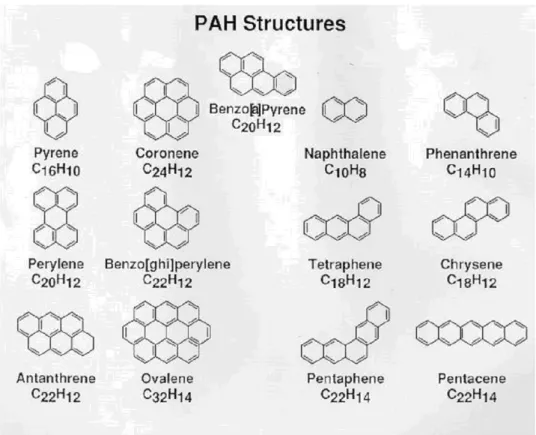
In figura 2 sono rappresentate le strutture molecolari dei principali idrocarburi policiclici aromatici.
Figura 2: struttura dei principali idrocarburi policiclici aromatici
Queste sostanze sono xenobiotici, molecole di natura ciclica che, essendo liposolubili, sono in grado di accumularsi all’interno delle cellule e interferire con le loro funzioni. Il metabolismo di uno xenobiotico avviene in due fasi ad opera di specifici enzimi. Gli enzimi di fase I producono una biotrasformazione alterando la fisionomia fisico-chimica del composto attraverso l’aggiunta o l’eliminazione di gruppi funzionali (funzionalizzazione), in modo che il metabolita sia più idoneo a reagire con gli enzimi di coniugazione della fase II. Le reazioni di fase I, ossidazione, riduzione, idrolisi e idratazione, possono risultare nella formazione di un derivato cancerogeno di una sostanza di per sé inattiva.
Gli enzimi di fase II catalizzano invece reazioni biosintetiche che convertono le sostanze in composti a PM più elevato provvisti di gruppi idrofili e per questo più facilmente eliminabili con le urine, la bile ed altri meccanismi. Nelle reazioni di
coniugazione sono coinvolti l’acido glicuronico, i solfati, gli amminoacidi, i gruppi acetile e il glutatione.
Il glutatione ridotto (GSH) ha un ruolo di primaria importanza nei processi di fase II. Attraverso l’azione delle Glutatione-S-transferasi (GSTs), esso interviene nella neutralizzazione di composti elettrofili impedendone gli effetti citotossici.
I coniugati, che si formano ad opera delle GSTs per reazione del sulfidrile nucleofilo del glutatione con i composti contenenti un atomo di carbonio elettrofilo, vengono trasportati fuori dalla cellula e divengono substrati delle Gamma-glutamil-transpeptidasi (GGTP), che, presenti nella porzione esterna della membrana cellulare, provvedono al distacco dell’acido glutammico. La sostanza legata al dipeptide così formatosi ritorna all’interno della cellula dove viene acetilata e definitivamente allontanata dall’organismo per escrezione attraverso le feci e le urine.
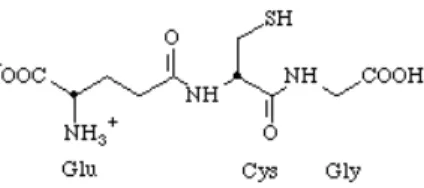
Nella figura 3 sono rappresentate le fasi del meccanismo di detossificazione GSH dipendente.
Glutatione:
Il gruppo SH della cisteina e’ un potente nucleofilo che intercetta substrati elettrofili:
Gli addotti con glutatione non vengono escreti direttamente: subiscono in genere un ulteriore metabolismo con successive perdite dei residui Glu e Gly ed acetilazione del gruppo amminico della cisteina:
Dal metabolismo degli xenobiotici possono essere generati radicali liberi che, grazie alla grandissima capacità di combinarsi con i fosfolipidi di membrana, le proteine recettoriali e/o di struttura e con gli acidi nucleici, possono produrre grave tossicità cellulare.
Uno schema generale del processo di metabolizzazione di uno xenobiotico è riportato in figura 4. XENOBIOTICO penetrazione XENOBIOTICO Metabolismo di Fase I Metabolismo di Fase I METABOLITI Metabolismo di Fase II Metabolismo di Fase II COMPOSTI CONIUGATI escrezione COMPOSTI CONIUGATI
C
C
E
E
L
L
L
L
U
U
L
L
A
A
XENOBIOTICO Reazioni radicaliche Cicli di ossido-riduzione Metabolismo di Fase Metabolismo di FaseII COMPOSTI RADICALICI SPECIE REATTIVE DELL’OSSIGENO METABOLITI ELETTROFILICI P R O T E Z I O N E ENZIMI ENZIMI ANTIANTI--OSSIDANTIOSSIDANTI
Metabolismo Metabolismo di Fase II di Fase II Deteriorazione delle menbrane Perossidazione dei lipidi SCAVENGERS SCAVENGERS Inattivazione di enzimi Danni al DNA altro
Figura 4: biotrasformazione e detossificazione
Qualora i sistemi di detossificazione degli xenobiotici non siano sufficienti all’eliminazione dei composti tossici, le sostanze precancerogene possono subire un’attivazione divenendo cancerogeni. Questi ultimi sono in grado di legarsi
covalentemente agli acidi nucleici con conseguente formazione di addotti. Se i sistemi di riparazione funzionano efficientemente non interviene nessuna variazione nella sequenza del DNA e dunque la replicazione cellulare porta alla formazione di cellule normali. Nel caso in cui gli enzimi della riparazione non funzionino correttamente, invece, possono avvenire mutazioni a livello di oncogeni e di geni soppressori tumorali. Ne deriva instabilità genomica, espressione di geni aberranti e alterazioni della struttura cellulare. Il risultato ultimo è la formazione di cellule tumorali maligne.
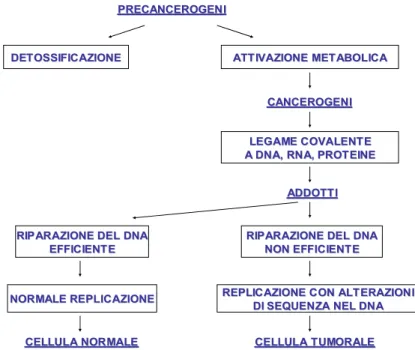
La figura 5 mostra la successione degli eventi che portano alla formazione di cellule tumorali maligne in seguito all’azione di un cancerogeno.
PRECANCEROGENI
PRECANCEROGENI
DETOSSIFICAZIONE
DETOSSIFICAZIONE ATTIVAZIONE METABOLICAATTIVAZIONE METABOLICA
LEGAME COVALENTE
LEGAME COVALENTE
A DNA, RNA, PROTEINE
A DNA, RNA, PROTEINE
ADDOTTI
ADDOTTI
RIPARAZIONE DEL DNA
RIPARAZIONE DEL DNA
EFFICIENTE
EFFICIENTE
RIPARAZIONE DEL DNA
RIPARAZIONE DEL DNA
NON EFFICIENTE NON EFFICIENTE CANCEROGENI CANCEROGENI NORMALE REPLICAZIONE NORMALE REPLICAZIONE CELLULA NORMALE CELLULA NORMALE
REPLICAZIONE CON ALTERAZIONI
REPLICAZIONE CON ALTERAZIONI
DI SEQUENZA NEL DNA
DI SEQUENZA NEL DNA
CELLULA TUMORALE
CELLULA TUMORALE
1.5 Rischio di tumore al polmone in base al sesso
Alcuni dati indicano che il rischio di tumore al polmone è due volte maggiore nelle donne rispetto agli uomini a parità di sigarette fumate al giorno (Risch et al., 1993; Zang et al., 1996).
Le donne sviluppano più comunemente l’adenocarcinoma, mentre negli uomini è più frequente il tumore a cellule squamose (Ernster, 1996). Questo sembra essere dovuto al consumo di differenti tipi di sigarette (Thun et al., 1997), light per le donne e normali per gli uomini.
Recentemente si sta assistendo ad un decremento dei casi di cancro polmonare tra gli uomini, ma non fra le donne (Wingo et al., 1999). Nonostante sia stato ipotizzato che questa differenza fosse determinata da una asimmetria tra i due sessi nella percentuale dei fumatori (Prescott at al., 1998), essa potrebbe originare dal fatto che gli uomini cessano di fumare abbastanza precocemente mentre le donne iniziano, ma pure cessano, più tardi (Wingo et al., 1999).
1.6 Suscettibilità genetica per il cancro al polmone
Il fumo di tabacco è responsabile della maggior parte dei carcinomi polmonari negli uomini e nelle donne. A questo proposito, è chiaro che un calo del consumo di sigarette porterebbe ad una considerevole diminuzione dei casi di malattia nella popolazione. Viste le differenze nella suscettibilità di ogni individuo, un tale sforzo andrebbe però incontro solo ad un limitato successo. Si osserva infatti che soltanto il 15% dei fumatori sviluppa il cancro al polmone entro i 75 anni di età (J.F. Fraumeni Jr., 1975; J.J Mulvihill, 1976; T.A. Sellers et al., 1990; T.A. Sellers, 1992; Peto R. et al., 2000). La differente suscettibilità può essere spiegata attraverso la diversa capacità individuale di metabolizzare i cancerogeni del tabacco (C.C. Harris, 1989), ma può dipendere anche da variazioni che intervengono nei geni soppressori tumorali e negli oncogeni.
I polimorfismi genetici possono consistere in sostituzioni di una singola base (SNPs), delezioni o inserzioni sia di singole basi che di ampie parti di un gene, oppure in amplificazioni di una sequenza genica. Alcuni geni possono essere presenti in una o più varianti alleliche e di queste la maggioranza non comporta di solito nessuna differenza nel fenotipo da persona a persona. Se un polimorfismo causa un’alterazione della normale funzione biologica del gene, si possono avere variazioni nel metabolismo dei precancerogeni, che vengono attivati ma non detossificati. Si creano in questo modo composti elettrofili che sono in grado di legarsi al DNA formando addotti. Se il danno non viene efficientemente riparato prima che inizi la replicazione le cellule figlie accumuleranno mutazioni potenzialmente pericolose.
Ne deriva che i polimorfismi genetici possono essere responsabili delle differenti quantità di cancerogeni che si formano nel polmone di individui che fumano un comparabile numero di sigarette (Shields, 1999). Inoltre polimorfismi nei sistemi di riparazione del DNA (o del controllo del ciclo cellulare) possono portare ad una grande differenza nell’efficienza della riparazione in individui che presentano dosi interne di cancerogeni paragonabili.
L’identificazione di geni polimorfici correlati all’insorgenza del cancro potrebbe rivelarsi uno strumento utile per individuare soggetti ad alto rischio a causa della loro aumentata suscettibilità genetica. Si prospetta dunque la possibilità di attuare programmi di prevenzione per gli individui ad alto rischio e di poter valutare con maggiore accuratezza l’effettivo rischio individuale.
1.7 Geni selezionati
In questo lavoro di tesi sono stati presi in considerazione un polimorfismo nel gene Glutatione S-transferasi T1 (GSTT1), uno nel gene Glutatione S-transferasi M1 (GSTM1) e un terzo nel gene TP53. I geni GSTT1 e GSTM1 appartengono alla famiglia
delle glutatione S-transferasi e codificano per enzimi di Fase II, coinvolti nei processi di detossificazione dei metaboliti del tabacco. Il prodotto del gene TP53, invece, è una proteina che agisce da soppressore tumorale, inibendo la proliferazione cellulare e controllando il processo di apoptosi.
Tra i vari polimorfismi di TP53 abbiamo rivolto la nostra attenzione su p53PIN3 che corrisponde ad un’inserzione/duplicazione di 16 pb nell’introne 3, mentre i polimorfismi di GSTT1 e GSTM1 consistono in una delezione in una parte codificante del gene con conseguente assenza di attività enzimatica.
1.8 Geni metabolici
Molti composti chimici devono essere metabolizzati per poter esercitare il loro potenziale cancerogenico e la quantità ultima di cancerogeni prodotti dipende, rispettivamente per l’attivazione metabolica e la detossificazione, dall’azione degli enzimi di Fase I e degli enzimi di Fase II (C.C. Harris, 1989). Tra gli enzimi di Fase I svolgono un ruolo di primaria importanza i citocromo P450 (CYPs) e tra quelli di Fase II gli enzimi glutatione S-transferasi (GSTs) e N-acetl transferasi (NAT) (H. Raunio et al., 1995; F.J. Gonzalez et al., 1991). Per molti di questi enzimi esistono ampie variazioni interindividuali dovute alla variabile risposta all’induzione così come alla trasmissione di geni polimorfici (C.C. Harris, 1989).
GSTs
La fase II della biotrasformazione è caratterizzata dalla coniugazione dei composti endogeni solubili in acqua con substrati lipofilici. Le GSTs catalizzano la coniugazione del glutatione (GSH) con composti elettrofili, con conseguente formazione di complessi meno reattivi e più facilmente eliminabili.
I substrati delle reazioni catalizzate dalle GSTs includono i precancerogeni, come gli idrocarburi policiclici aromatici, i farmaci, tra cui anche il paracetamolo, gli agenti chemioterapici e i radicali liberi generati dallo stress ossidativo (Strange et al., 2001). La famiglia comprende 4 classi di enzimi citosolici: GSTα (GSTA), GSTµ (GSTM), GSTπ (GSTP), GSTθ (GSTT) (Shama C. Buche et al., 2002). Tali enzimi sono differentemente espressi nei vari organi e nelle varie etnie.
GSTπ, uno dei maggiori detossificatori della forma attivata del benzopirene, vede la sua più alta espressione nel polmone (Nakajima T. et al., 1995; Hecht S.S., 1999; Fields et al., 1998) ed è codificato dal gene polimorfico GSTP1, localizzato sul cromosoma 11q13. I polimorfismi di questo gene sono conseguenza di una sostituzione a singolo nucleotide che porta alla sostituzione dell’amminoacido Isoleucina (I105) con l’amminoacido Valina (V105). Il risultato è una ridotta attività enzimatica (Watson et al., 1998; Ali-Osman F. et al., 1997) associata ad un più alto livello di addotti idrofobici nel tessuto polmonare (Ryberg et al., 1997) e ad una dose elevata di idrocarburi policiclici aromatici- DNA addotti nei linfociti umani (Butkiewicz et al., 2000). Il polimorfismo del gene GSTM1 consiste in una delezione nella linea germinale (GSTM1-nullo) che risulta in una totale perdita della funzione enzimatica. Il gene è localizzato sul cromosoma 1p13.3. Una percentuale variabile tra il 20 e il 50% degli individui non esprime l’enzima a causa della delezione omozigotica (Seidgard et al., 1988). La percentuale è più alta nei Caucasici e negli Asiatici piuttosto che negli Africani (Bailey et al., 1998). Evidenze dimostrano che in cellule derivanti da individui GSTM1-nulli sono più frequenti danni al DNA dovuti agli idrocarburi policiclici aromatici e ad altri mutageni (Strange et al., 1999).
Il gene GSTT1 è localizzato sul cromosoma 22 e la non espressione dell’enzima nel 20-60% della popolazione è dovuta anche in questo caso ad una delezione del locus (GSTT1-nullo) (Pamble et al., 1994). Circa il 60% degli Asiatici, il 40% degli Africani e
il 20% dei Caucasici non esprime l’enzima (Strange et al., 1999). Diversi studi hanno rivelato associazione tra il genotipo GSTM1-nullo e il rischio di sviluppare tumore al polmone. Nella popolazione giapponese tale genotipo è correlato al tumore a cellule squamose, mentre non sembra correlato all’adenocarcinoma (Kihara M. et al., 1993). Altri studi su popolazioni caucasiche hanno riportato un’associazione tra GSTM1- nullo e squamous cell carcinoma (Zhong et al., 1991; Pinabasi et al., 2003).
Una meta-analisi di 12 studi caso-controllo (1593 casi e 2135 controlli) mostra un moderato aumento del rischio per tutti i tipi istologici di tumore al polmone in associazione al genotipo GSTM1-nullo, con un Odds Ratio (OR) di 1.41 (intervallo di confidenza CI: 1.23-1.61) (McWilliams et al., 1995). Una più recente meta-analisi di 43 studi, comprendente più di 18000 individui, mostra nuovamente associazione tra il genotipo GSTM1-nullo ed un aumento del rischio di tumore al polmone, con un OR di 1.17 (CI 1.07-1.27) (Simone Benhamou et al., 2002). Uno studio caso-controllo su 446 pazienti e 622 controlli nella popolazione tedesca, successivo di un anno alla meta-analisi sopra citata, mostra invece mancanza di associazione, con un OR di 1.34 (CI: 0.99-1.81) (Joachim Schneider, 2004).
Da un’analisi della letteratura emerge che non tutti gli studi hanno rivelato un’associazione tra GSTT1-nullo e un aumento del rischio di tumore polmonare. Uno studio tedesco di tipo caso-controllo su 446 pazienti affetti da tumore polmonare e 622 controlli mostra assenza di associazione tra il genotipo GSTT1-nullo e aumento del rischio dell’insorgenza di cancro al polmone (Joachim Schneider, 2004). Uno studio successivo di un anno al precedente effettuato su 229 pazienti con tumore polmonare e 197 controlli (entrambi di nazionalità cinese) mostra invece associazione tra il genotipo GSTT1-nullo e un aumento del rischio di insorgenza del tumore polmonare con un OR di 1.69 (95% CI: 1.12-2.56) (Shama C. buch et al., 2002).
1.9 Geni del ciclo cellulare
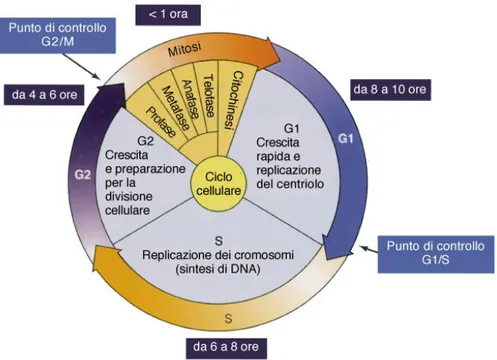
Il ciclo cellulare consta di quattro fasi (G1 di preparazione alla replicazione, S di sintesi, G2 di preparazione alla divisione e M di mitosi) (Figura 6) e la transizione da ciascuna fase del ciclo all’altra è regolata da punti di controllo (checkpoint) a livello dei quali la cellula verifica che ogni processo sia stato completato con successo e che non siano intervenuti danni. I checkpoint sono localizzati tra la fase G1 ed S e alla fine della fase G2 prima della fase M.
Figura 6: schema del ciclo cellulare
Le cellule possono arrestarsi temporaneamente ai checkpoint per rendere possibile la riparazione di un danno cellulare, consentire la dissipazione di segnali cellulari di stress esogeno o permettere l’utilizzo di fattori di crescita, ormoni e nutrienti(Wang et al., 2003). Qualora il danno cellulare risulti irreparabile si attiverà il processo di apoptosi.
Difetti nella regolazione del ciclo cellulare possono risultare in una mutazione genica, un danno cromosomico o un’aneuploidia: ognuno di questi eventi può contribuire alla cancerogenesi. Mutazioni somatiche in importanti geni del ciclo cellulare, come TP53, sono direttamente correlate ai checkpoint cellulari e conseguentemente portano ad aberrazioni cromosomiche, instabilità genomica ed elevato rischio di cancro. Ne deriva che individui con difetti costitutivi nei checkpoint del ciclo cellulare potrebbero essere più suscettibili ai mutageni ed essere sottoposti ad un maggior rischio di cancro.
TP53
Il gene TP53 codifica per una proteina di 53 Kd e 393 amminoacidi. P53 è un fattore di trascrizione strutturalmente costituito da tre domini distinti: un dominio N-terminale di attivazione trascrizionale (TAD), un “core” centrale di legame al DNA (DBD) ed un dominio C-terminale di omo-oligomerizzazione (OD). La struttura tridimensionale della proteina P53 è rappresentata nella figura 7.
Figura 7: struttura della proteina p53.
Core della proteina (verde) legato al DNA (blu scuro). I 6 amminoacidi più frequentemente mutati nel cancro umano sono mostrati in giallo- sono tutti residui importanti per il legami di p53 al DNA. Sfera rossa: atomo di zinco.
La proteina è coinvolta, oltre che nel processo di regolazione del ciclo cellulare, anche nell’induzione del fenomeno di apoptosi. Le modalità con cui P53 programma la morte cellulare non sono del tutto chiare anche se il meccanismo sembra coinvolgere il prodotto del gene BAX. Le mutazioni nel gene TP53 sono molto frequenti in vari tipi di cancro umano. Dei vari polimorfismi del gene TP53, tre sono stati attentamente studiati: il polimorfismo a singolo nucleotide (SNP) G/C al codone 72 (che causa la sostituzione aminoacidica Prolina > Arginina), un’inserzione di 16 bp nell’introne 3 (p53PIN3) ed infine una transizione da G a C nell’introne 6. Svariati studi hanno mostrato
associazione tra il tumore al polmone e il polimorfismo p53 Prolina/Arginina (Wingo et al., 1999; Prescott et al., 1998). Uno studio caso-controllo comprendente 635 pazienti e 635 casi ha esaminato i tre polimorfismi sopra citati del gene TP53 e loro associazione ad un aumento del rischio di insorgenza di tumore polmonare. Gli individui sono stati separati in base alla loro etnia ed il calcolo degli OR è stato eseguito tenendo conto delle differenze etniche. Lo studio ha mostrato associazione con aumento del rischio di tumore polmonare per il polimorfismo nell’introne 3 (polimorfismo di interesse nella tesi) nei caucasici per il genotipo eterozigote W/M con OR aggiustato per età, sesso e fumo di 1.77 (95% CI 1.24-2.52), mentre per il genotipo omozigote con entrambi gli alleli mutati non è stata trovata associazione significativa con un OR aggiustato di 2.37 (95% CI 0.76-7.39). Sempre nello stesso studio è stato analizzato il rischio di tumore al polmone in base all’aplotipo degli individui caucasici ed è risultato che tale rischio è associato in modo significativo a tre aplotipi: (esone4- introne3- introne6) W-W-M (32 pazienti e 21 casi) con un OR aggiustato di 1.78 ( 95% CI 1.02-3.12), W-M-M (22 pazienti e 11 controlli) con un OR aggiustato di 2.33 (95% CI 1.12-4.38) ed infine l’aplotipo M-M-M (102 casi e 70 pazienti) con un OR aggiustato di 1.70 (95% CI 1.23-2.35) (Wu et al., 2002).
1.10 Effetto delle combinazioni genotipiche
L’azione combinata dei geni metabolici è necessaria per il processo di attivazione/detossificazione dei cancerogeni ed è coinvolta nella modificazione del rischio per il cancro.
Da uno studio caso-controllo del 1997 (Randa El-Zein et al., 1997) (54 casi e 50 controlli) è emerso che la combinazione di varie versioni di geni metabolici (CYP2D6, CYP2E1, GSTM1 e GSST1) è fortemente associata al cancro al polmone (i valori di OR per le diverse combinazioni sono compresi nell’intervallo 1.3-14). Il rischio è più alto quando sono coinvolti i geni attivatori (CYP2D6 e CYP2E1).
Lo stesso studio mostra che la durata e l’intensità dell’utilizzo del tabacco, espresse in pacchetti consumati all’anno (pack-years), sono fattori determinanti per l’insorgenza della patologia polmonare. Il rischio relativo di sviluppare il cancro al polmone associato con il consumo di più di 30 pacchetti di sigarette mostra un OR di 6.65 (95% CI 2.3-19.9), mentre ad un consumo di un numero di pacchetti compreso tra 30 e 50 è associato un OR=4.5 (95% CI 1.37-15); il rischio più elevato è apparso associato al consumo di più di 50 pacchetti all’anno con un OR=30 (95% CI 5.7-114) (Randa El-Zein et al., 1997).
In uno studio successivo su 1694 individui caucasici ( 767 casi e 927 controlli) è stata investigata l’associazione di varianti genotipiche combinate per i geni GSTP1, GSTM1 e TP53 con il cancro polmonare. I dati raccolti mettono in evidenza che le combinazioni GSTP1 GG + GSTM1-nullo e GSTP1 GG + TP53 Arg/Pro o Pro/Pro sono associate ad un aumentato rischio di tumore polmonare. Tali combinazioni sembrano promuovere maggiormente la cancerogenesi del polmone in individui di età inferiore ai 55 anni; questo dato è stato verificato eseguendo l’analisi su due sottogruppi, uno comprendente individui aventi almeno 55 anni e uno comprendente individui più giovani (David P. Miller et al., 2002).
L’effetto combinato delle delezioni omozigotiche di GSTM1 e GSTT1 è stato proposto in diversi lavori ma solo uno studio su popolazioni emigranti (Ispaniche e Afro- Americane) ha rivelato un significativo numero di tumori al polmone tra i portatori dei genotipi nulli per i due geni (Schoket et al., 1998).
2. SCOPO DELLA TESI
La peculiarità che differenzia il presente studio da altri citati e’ che la popolazione dei controlli e’ costituita completamente da individui fumatori che, in età tipica, non hanno presentato insorgenza di tumore al polmone. Lo scopo della presente tesi e’ stato di valutare se in un simile contesto i polimorfismi dei geni GSTM1, GSTT1 e TP53 possano essere associati al rischio di sviluppare la neoplasia.
3. MATERIALI E METODI
3.1 Popolazione in studio
La popolazione in studio è costituita da 763 individui di origine norvegese (379 casi e 384 controlli). I pazienti (casi) sono affetti da tumore al polmone di tipo non small cell (NSCLC) diagnosticato e trattato chirurgicamente presso gli ospedali universitari di Oslo e Bergen (Norvegia) tra il 1986 e il 2001. Le informazioni riguardanti il consumo di tabacco, il sesso e l’età sono state reperite tramite la compilazione di un questionario da parte dei pazienti.
Gli individui sani che costituiscono il gruppo dei controlli sono stati reclutati tra 8100 individui di età compresa tra 59-60 e 75-76 anni partecipanti all’Oslo Health Screening 2000-2001 (HUBRO). Di questi, circa 4000 individui hanno accettato di partecipare al progetto, donando un campione di sangue e riempiendo il questionario. Tutti i controlli hanno delle caratteristiche comuni: erano fumatori durante il periodo di campionamento, avevano un’età superiore a 60 anni e fumavano almeno 5 sigarette al giorno. 417 individui sono stati selezionati casualmente dai 4000 di partenza.
Sia i pazienti che i controlli hanno dato l’autorizzazione ad utilizzare i loro campioni di sangue per analisi genetiche; l’approvazione per lo studio è stata data dal comitato etico locale.
3.2 Estrazione del DNA
L’estrazione del DNA è stata effettuata utilizzando il protocollo QIAamp (Qiagen Gmbh, Hilden, Germany) (Figura 8), che si può riassumere brevemente nei seguenti passaggi.
A 200 µl di sangue vengono aggiunti 20 µl di proteasi K (Qiagen), il cui scopo è di degradare le proteine, 8 µl di RNasi (Sigma-Genosys Ltd, Cambridge, UK), per degradare l’RNA, e 200 µl di buffer di lisi (AL), che contiene detergenti che dissolvono i lipidi di membrana.
Le eppendorf (sterili RNAase DNAase free) contenenti la soluzione vengono brevemente centrifugate e incubate in acqua per 10 minuti a 56°C, temperatura ottimale per l’azione dell’enzima RNasi.
Terminata l’incubazione, vengono aggiunti 200 µl di etanolo assoluto così da far precipitare il DNA e separarlo dal solvente.
La soluzione viene applicata su una colonna QIAamp (Qiagen) posta su una eppendorf da 2 ml e si esegue una centrifugazione a 8000 rpm per 1 minuto. In seguito alla centrifugazione il solvente cade sul fondo della eppendorf mentre il DNA rimane adeso alla membrana della colonnina.
Trasferita la colonna in una nuova eppendorf, nella quale si aggiungono 500 µl di un tampone di lavaggio (AW1) (Qiagen), si effettua nuovamente una centrifugazione a 8000 rpm per 1 minuto. Ancora una volta la colonna viene trasferita in una nuova eppendorf nella quale si aggiungono 500 µl di tampone di lavaggio (AW2) (Qiagen). Tramite i due buffer di lavaggio il DNA viene purificato.
Eseguita un’ulteriore centrifugazione, a 14000 rpm per 3 minuti, la colonnina viene trasferita in una nuova eppendorf da 1.5 ml e si aggiungono 200 µl del tampone di eluizione (AE) (Qiagen). Il tutto viene lasciato per 5-10 minuti a temperatura ambiente. La funzione del buffer AE è quella di staccare il DNA dalla membrana della colonna. Si effettua un’ultima centrifugazione a 8000 rpm per 5 minuti e si ottiene il DNA purificato.
Figura 8: estrazione del DNA con il protocollo Qiagen
3.3 Genotipizzazione: PCR MULTIPLEX
Per effettuare la genotipizzazione è stata utilizzata una PCR MULTIPLEX che ha consentito di analizzare per ogni campione di DNA, nella stessa miscela di reazione, i polimorfismi dei tre geni GSTT1, GSTM1, TP53, utilizzando come controllo positivo il gene CYP2A6.
Il DNA dei casi e dei controlli è stato randommizzato sulle piastre da PCR in modo tale da equilibrare il numero dei casi e dei controlli. In questo modo è stato possibile minimizzare il più possibile eventuali “bias” dovuti al campionamento, che potrebbero mascherare l’eventuale associazione tra il genotipo ed il rischio di tumore.
Il protocollo di amplificazione prevede l’impiego della polimerasi termostabile Hot Fire (Solis Biodyne, Tartu, Estonia) e di quattro coppie di primer (tabella 1), una per ognuno degli ampliconi presenti nei quattro geni sopra citati. I primer utilizzati hanno una lunghezza compresa tra le 15 e le 25 paia di basi.
Tabella 1: primer utilizzati per la genotipizzazione dei polimorfismi dei 4 geni CYP2A6, GSTT1, GSTM1 e TP53
Gene Primer forward Primer reverse Lunghezza
del prodotto di PCR CYP2A6 5’AGCGGAACTGTTTCGGAGA 3’ 5’TGTGAGACATCAGAGACAACTTTC 3’ 570 bp GSTT1 5’ CAACAATCCATCCCCAGTC 3’ 5’TCACCGGATCATGGCCAGCA 3’ 233 bp
GSTM1 5’ GCTTCACGTGTTATGAAGGTTC 3’ 5’ CAAAGAGAAAGGAGGATGGG 3’ 189 bp
TP53 5’ TGCTCTTGTCTTTCAGACTTCCT 3’ 5’ GAGCAGTCAGAGGACCAGGTC 3’ 114/130 bp
Di seguito è riportato il protocollo di amplificazione. Il primo ciclo viene effettuato a 95°C per 15 minuti in modo da denaturare la maggior quantità di DNA possibile. La seconda fase è costituita da 20 cicli, divisi ognuno in tre passaggi, il primo dei quali (95°C per 1 minuto) ha ancora lo scopo di denaturare il DNA. Il secondo passaggio (68°C per 30 secondi) consente l’appaiamento dei primer. Durante tale passaggio la temperatura si abbassa di un grado ad ogni ciclo (“touch-down PCR”) fino a raggiungere un valore di 48°C al 20° ciclo. Partendo da una temperatura poco permissiva e abbassandola di 1 grado ad ogni ciclo vengono raggiunte le tm ottimali per
ciascun primer e si garantisce così un’amplificazione specifica delle sequenze bersaglio. Il terzo passaggio della seconda fase (72°C per 1.5 minuti) consente la sintesi del DNA. La terza fase è anch’essa costituita da 20 cicli, divisi ognuno in tre passaggi: il primo a 95°C per 1 minuto per la denaturazione, il secondo a 51°C per 30 secondi per l’appaiamento dei primer. In questo caso si opera alla temperatura molto permissiva di 51°C che garantisce un’alta resa. Il terzo passaggio, a 72°C per 1.5 minuti, garantisce la polimerizzazione.
L’ultimo ciclo viene effettuato a 72°C per 10 minuti; in seguito la temperatura si abbassa fino al valore di mantenimento (4°C).
Una volta terminata la PCR, si procede con una corsa elettroforetica su gel d’agarosio contenente bromuro d’etidio, un composto genotossico capace d’intercalarsi tra le basi del DNA, che permette di evidenziare le bande dei frammenti tramite l’utilizzo di un transilluminatore a raggi ultravioletti .
Nella pratica viene preparato separatamente un mix di primer che viene poi utilizzato nel mix di PCR. L’allestimento dei due mix può essere così schematizzato:
mix di primer: si preparano 8 µl per campione di una mix contenente i primer forward e reverse dei geni CYP2A6, GSTT1, GSTM1 e TP53 in rapporto 1:1:2:1 (concentrati 10 µM), più H2O per portare a volume
mix di PCR: in una eppendorf, contenente 20 ng di DNA secco, si inseriscono per campione 2 µl di buffer 10x (Solis Biodyne), 2 µl di dNTPs (2 mM) (Invitrogen), 2.4 µl di MgCl2 (25 mM) (Solis Biodyne), 8 µl di mix di primer, 0,2 µl di HOT FIRE 5 U/µl (Solis Biodyne) e H2O per raggiungere un volume finale di 20 µl .
3.4 Messa a punto del protocollo
La concentrazione del magnesio è senza dubbio il fattore più critico della PCR, condizionando l’attività della polimerasi e l’ibridazione dei primer ed aumentando la temperatura di denaturazione del DNA. Ad ogni prova sono dunque state variate le concentrazioni di MgCl2 ed il rapporto tra le concentrazioni delle coppie di primer. Bilanciare le concentrazioni dei primer è importante perché, amplificando contemporaneamente i tre geni più il controllo positivo, alcuni di essi possono essere amplificati a discapito di altri che non risultano così visibili su gel dopo corsa elettroforetica.
La prima prova è stata effettuata con:
un mix di primer (miscela in cui si trovano tutti i primer per tutti i geni di interesse) in rapporto 1: 1: 1 :1 (CYP2A6: GSTT1: GSTM1: TP53).
Una volta preparata la mix di primer sono state preparate tre distinte mix di PCR: 1) prima mix preparata con una concentrazione finale di MgCl2 1.5 mM
buffer 10x (Solis Biodyne Tartu Estonia) 2 µl dNTP (2 mM) ( Invitrogen, Carlsbod, California) 2 µl
MgCl2 (25 mM) (Solis Biodyne) 1,2 µl
Mix di primer 8 µl
HOT Start DNA polymerase 5 U/µl (Solis Biodyne). 0,4 µl
DNA 1 µl
H2O 5,4 µl
2) secondo mix preparato con una concentrazione di MgCl2 3 mM
buffer 10x (Solis Biodyne) 2 µl
dNTP (2 mM) ( Invitrogen) 2 µl
MgCl2 (25 mM) (Solis Biodyne) 2,4 µl
Mix di primer 8 µl
HOT Start DNA polymerase 5 U/µl (Solis Biodyne). 0,4 µl
DNA 1 µl
H2O 4,2 µl
Volume finale 20 µl
3) terzo mix preparato con una concentrazione di MgCl2 6 mM
buffer 10x (Solis Biodyne) 2 µl
dNTP (2 mM) ( Invitrogen) 2 µl
MgCl2 (25 mM) (Solis Biodyne) 4,8 µl
Mix di primer 8 µl
HOT Start DNA polymerase 5 U/µl (Solis Biodyne). 0,4 µl
DNA 1 µl
H2O 1,8 µl
Volume totale 20 µl
Una volta terminati i cicli sono stati analizzati i risultati dei prodotti di PCR tramite elettroforesi su gel di agarosio al 3%: le ultime due mix sono state ritenute più produttive della prima.
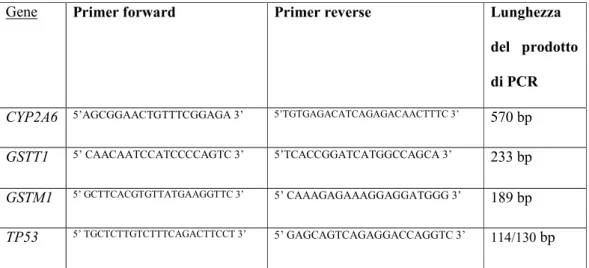
Utilizzando il rapporto 1: 1: 1 :1 (CYP2A6: GSTT1: GSTM1: TP53) fra i primer l’amplificazione del controllo positivo CYP2A6 risultava però essere eccessivamente forte, mentre i geni GSTM1 e TP53 risultavano debolmente amplificati (Figura 9).
Figura 9: Prodotti di PCR corsi su gel di agorosio e analizzati al transilluminatore: l’amplificazione di CYP2A6 è eccessivamente forte rispetto a quella dei geni GSTM1 e TP53.
Al fine di ottimizzare la produttività della reazione sono state condotte ulteriori prove modificando i rapporti fra i primer, ovvero raddoppiando le quantita dei primer di GSTT1 e quadruplicando i primer di GSTM1 e TP53, giungendo così al nuovo rapporto 1: 2: 4: 4 (CYP2A6: GSTT1: GSTM1: TP53).
Una volta preparata la mix di primer sono state allestite due diverse mix di PCR, la prima con una concentrazione finale di MgCl2 3 mM e la seconda con una concentrazione finale di MgCl2 6 mM
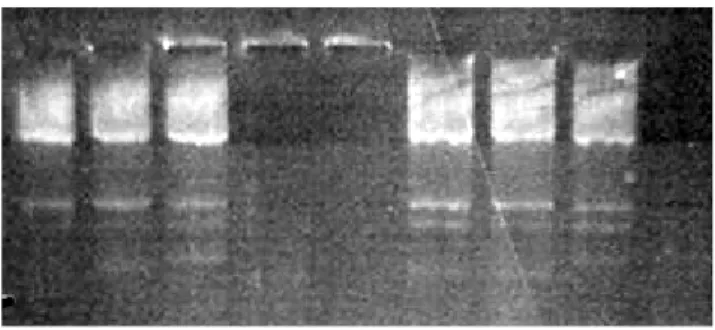
Dall’analisi dei prodotti di PCR (Figura 10) la prima mix è risultata essere la più efficiente, tuttavia il controllo positivo CYP2A6 risultava ancora essere troppo accentuato sul gel.
Figura 10: Prodotti di PCR corsi su gel di agarosio e analizzati al tranilluminatore: il controllo positivo CYP2A6 risulta essere ancora troppo accentuato sul gel.
Sono state allora fatte numerose prove variando il rapporto dei primer nella mix di primer (1: 4: 8: 8; 1: 2: 8: 8; 1: 1: 6: 4; 1: 1: 2: 1) (CYP2A6: GSTT1: GSTM1: TP53) ma mantenendo costanti le quantità dei composti della mix. Il rapporto tra i primer 1: 1: 2: 1 (CYP2A6: GSTT1: GSTM1: TP53) si è rivelato il più adeguato; tuttavia ogni volta che i primer vengono cambiati, la mix di primer deve essere bilanciata nuovamente. Come ultima prova è stato ridotto il quantitativo della polimerasi Hot Fire (Solis Biodyne) passando da 0.4 µl a 0.2 µl, quello che è stato rilevato è che questo quantitativo non variava la produttività della reazione.
3.5 Preparazione del gel di agarosio
Viene allestita una soluzione al 3% d’agarosio in TBE 0.5Χ con aggiunta di bromuro d’etidio 0.1 X.
3.6 Corsa elettroforetica
La corsa elettroforetica viene effettuata in una apposita vasca (Figura 11) a 130 V per 40 minuti al fine di verificare la presenza o assenza delle bande di DNA amplificato tramite l’esposizione del gel al transilluminatore (Figura 11). Grazie all’utilizzo di un marker (indicatore del peso molecolare), è possibile stimare la lunghezza delle bande
ottenute sul gel e quindi verificare se esse siano specifiche o siano il risultato di un’amplificazione aspecifica.
Figura 11: vasca elettroforetica.
3.7 Interpretazione dei risultati
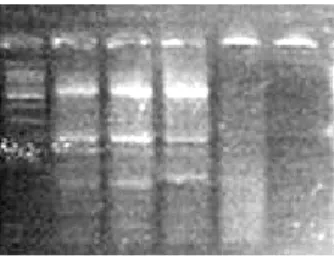
Una volta avvenuta la corsa elettroforetica il gel viene analizzato mediante un transilluminatore a raggi ultravioletti che permette di evidenziare le bande corrispondenti agli ampliconi (figura 12).
Un campione può dare su gel un massimo di quattro bande (a meno che non siano stati amplificati aspecifici): la banda a maggior peso molecolare costituisce il controllo positivo CYP2A6 di 570 paia di basi (pb), la banda di GSTT1 (seconda banda su gel) può essere presente con una lunghezza di 233 paia di basi (pb) oppure può essere totalmente assente (indicando la presenza del polimorfismo GSTT1 nullo). La banda di 189 paia di basi (pb) rappresenta GSTM1 e anch’essa può non essere presente in caso di polimorfismo GSTM1 nullo. Infine l’ultima banda rappresenta TP53 e può essere presente come un’unica banda di 114 paia di basi (bp) (si tratta dell’omozigote avente
entrambi gli alleli wild type), un’unica banda di 130 paia di basi (bp) (si tratta dell’omozigote avente entrambi gli alleli polimorfici), oppure come doppia banda (una di 114 paia di basi e 130 paia di basi), in tal caso si tratta dell’eterozigote avente sia l’allele polimorfico che l’allele wild type.
Figura 12: gel di agarosio riportante le bande dei geni GSTT1, GSTM1, TP53 ed il controllo positivo al transilluminatore.
T P5 3 ,1 6 bp repeats
(1 1 4 /13 0 bp) G S T T 1 (2 3 3 bp) C on trollo pos itiv o
C Y P2 A 6 (5 70 bp)
G S T M 1 (1 8 9 bp )
3.8 Analisi statistica
La popolazione dei controlli e dei casi sono state scelte in modo tale che fossero appaiate per frequenza per diverse variabili (tabella 2) fra cui, l’età, il sesso e l’abitudine al fumo (caratterizzata da tre parametri: sigarette medie al giorno, sigarette medie all’anno e pack-years). In questo modo è stato possibile minimizzare il più possibile eventuali “bias” dovuti al campionamento, che avrebbero potuto mascherare l’eventuale associazione tra il genotipo ed il rischio di tumore.
Tabella 2: caratteristiche dei pazienti affetti da tumore polmonare (casi) e dei controlli (individui sani).
a
DS, deviazione standard; b test di Wilcoxon non parametrico per due campioni indipendenti c test X2 . d numero medio di pacchetti fumati al giorno, moltiplicato per il numero di anni che l’individuo ha fumato (tenendo conto anche di eventuali interruzioni)
L’equilibrio di Hardy-Weinberg è stato calcolato per ciascun polimorfismo tramite il test X2 nei controlli. I valori di P riportati sono significativi per P <0.05. Di tutti i geni esaminati sono state calcolate le frequenze genotipiche. Le frequenze alleliche calcolate
Parametri
Pazienti con tumore polmonare (casi) n = 379 Controlli n = 384 Età (anni ± DSa) Sesso Maschi Femmine Abitudini al fumo
Sigarette medie al giorno ± DS Sigarette medie all’anno ± DS Pack-years mediod ± DS 63.2 ±±± 10.2 ± 114 25 15.6 ±±± 8.3 ± 40.4 ±±± 12.1 ± 31.1 ±±± 17.7 ± 63.5 ±±±± 7.1 101 70 14.8 ±±±± 6.3 42.3 ±±±± 7.9 31.6 ±±±± 15.1 P=0.31b P=0.92c P=0.61b P=0.17b P=0.16b
sono poi state confrontate nelle due popolazioni in esame. Le associazioni tra le varianti alleliche e la patologia sono state stimate tramite un’analisi di regressione logistica multivariata, utilizzando il programma statistico STATA (versione 8.0), calcolando gli odds ratios (ORs) e gli intervalli di confidenza (CIs) ad essi associati. L’analisi statistica per il polimorfismo del gene TP53 è stata effettuata utilizzando il modello recessivo (utilizzando come gruppo di riferimento l’omozigote con entrambi gli alleli mutati).
4. RISULTATI
In questo lavoro di tesi è stata analizzata l’eventuale associazione tra i polimorfismi dei geni GSTT1 (GSTT1 nullo), GSTM1 (GSTM1 nullo) e TP53 (p53PIN3) e il rischio di cancro al polmone tra i fumatori. In uno studio caso-controllo sono stati genotipizzati 763 individui di origine norvegese (379 casi e 384 controlli).
I polimorfismi di GSTT1 e GSTM1 sono stati scelti come oggetto del nostro studio alla luce dell’importante ruolo svolto dai due geni nei processi di detossificazione degli xenobiotici. Per quanto riguarda il gene TP53, il polimorfismo p53PIN3 è stato poco studiato e proprio per questo risulta interessante ricercare una sua possibile implicazione nella cancerogenesi del polmone.
4.1 Geni GSTT1 e GSTM1
Le varianti alleliche GSTM1 nullo e GSTT1 nullo non sono in grado di codificare per l’enzima funzionante a causa di una delezione in una porzione codificante del gene. Le frequenze genotipiche osservate per questi due geni sono riportate nelle tabelle 3 e 4.
Tabella 3: frequenze genotipiche del gene GSTM1 GSTM1 NUMERO % Genotipo nullo
391
55,54
Genotipo positivo313
44,46
TOTALI704
100
Tabella 4: frequenze genotipiche del gene GSTT1
L’associazione tra il polimorfismo di ciascun gene e il rischio di insorgenza di tumore polmonare è stata valutata tramite un’analisi di regressione logistica multivariata. Gli OR presentati sono sia OR grezzi che OR aggiustati per età, sesso ed abitudine al fumo (Tabella 5 ) e non mostrano associazione per nessuno dei due geni.
Tabella 5: OR grezzi e aggiustati per età ed intervalli di confidenza per i geni GSTT1 e GSTM1
a Odds ratio aggiustato per età, sesso ed abitudine al fumo
GSTT1 NUMERO %
Genotipo nullo
129
18,32
Genotipo positivo
575
81,68
TOTALI
704
100
Gene/polimorfismo Odds ratio grezzo
(95% CI) Odds ratioa (95% CI)
GSTT1/GSTT1 nullo
1.00 (0.67-1.50) 1.11 (0.61-2.04) GSTM1/GSTM1 nullo0.89 (0.64-1.23) 0.97 (0.59-1.60)
4.2 Gene TP53
Le frequenze genotipiche trovate per il gene TP53 sono riportate in tabella 6.
Tabella 6: frequenze genotipiche del gene TP53
L’associazione tra il polimorfismo del gene TP53 e il rischio di insorgenza di tumore polmonare è stata valutata tramite un’analisi di regressione logistica multivariata. Sono presentati sia gli OR grezzi sia gli OR aggiustati per età, sesso ed abitudine al fumo (Tabella 7). Nonostante i dati suggeriscano un lieve aumento del rischio l’associazione non appare significativa.
Tabella 7: OR grezzo e aggiustato per età ed intervallo di confidenza per il gene TP53
a
Odds ratio aggiustato per età, sesso ed abitudine al fumo
TP53 NUMERO % W/W
535
75,35
W/M147
20,71
M/M28
3,94
TOTALI710
100
Gene/polimorfismo Odds ratio grezzo (95% CI)
Odds ratioa (95% CI)
5.
DISCUSSIONE
In questo lavoro è stata analizzata la possibile associazione tra le varianti alleliche GSTT1-nullo, GSTM1-nullo e P53PIN3 e la suscettibilità individuale a sviluppare il tumore polmonare del tipo istologico NSCLC nei fumatori.
L’analisi di regressione logistica multivariata non ha evidenziato nessun aumento del rischio per i polimorfismi dei geni GSTT1 e GSTM1, nonostante essi siano coinvolti nei processi di detossificazione delle sostanze cancerogene.
Il risultato da noi ottenuto appare in accordo con quello di una recente metanalisi, comprendente 130 studi per un totale di 23.452 casi e 30.397 controlli, nella quale sono state studiate cinque varianti comuni nei quattro geni GSTM1, GSTT1, GSTP1 e GSTM3 (Ye Z. et al., 2006). Tale analisi conclude che la relazione dei genotipi GSTM1-nullo e GSTT1-nullo con il rischio di cancro al polmone sia incerta, soprattutto alla luce della totale mancanza di associazione che si riscontra in letteratura negli studi più ampi. Anche il nostro studio, seppur differenziandosi per aver studiato l’associazione esclusivamente tra i fumatori, sembra in linea con quanto osservato negli studi precedentemente riportati in letteratura.
Anche per la variante genotipica p53PIN3 dal nostro studio non è emersa nessuna associazione con il rischio di insorgenza di tumore polmonare. Tuttavia, il risultato da noi ottenuto mostra un trend molto simile a quello di uno studio caso-controllo più ampio (2.238 casi e 2.289 controlli) dove e’ stata trovata un’associazione statisticamente significativa (Hung et al., 2006). Nei norvegesi la frequenza degli omozigoti varianti (la categoria probabilmente a rischio) si aggira attorno al 4%, quindi (secondo Breslow NE, Storer BE. General relative risk functions for case-control studies. Am J Epidemiol. 1985 Jul;122(1):149-62.) il nostro studio non aveva la
numerosita’ del campione adeguata per rilevare rischi inferiori a 2.2, considerando una potenza statistica dell’80%).
Considerando i risultati del nostro studio, in relazione a quelli riportati da Hung et al. e Wu et al., e’ ragionevole ritenere che i polimorfismi intronici di TP53 giochino un ruolo nell’influenzare la suscettibilita’ al tumore polmonare in situazioni di stress genotossico. Precedentemente e’ stato mostrato che la variante genotipica p53PIN3 e’ associata ad una riduzione dei livelli di mRNA (F. Gemignani et al., 2004), e’ pertanto probabile che i polimorfismi intronici abbiano un ruolo nel modulare la regolazione e/o l’espressione del gene. Essendo P53 essenziale nel prevenire la proliferazione di cellule danneggiate, una sua eventuale disregolazione potrebbe facilitare il processo cancerogenetico.
E finalmente è il momento di dire Grazie…
dal cuore, per primi, a mamma e a papà, perché grazie a loro oggi discuto
questa tesi davanti a tutte le persone a cui tengo. Grazie per avermi
“supportata e sopportata”, rendendo il mio percorso così felice.
Grazie a Monica e a Giovanni, per avermi tanto coccolata e per essere
sempre così presenti anche adesso che sono grande.
Il grazie più dolce è però per i piccoli Francesca, Matteo e Giulia, per aver
reso più colorate le mie giornate di studio scarabocchiando tutti i miei
quaderni!!
Grazie mille a Lara, Melania e Valentina, amiche insostituibili, per le
interminabili chiacchierate e le grandi risate, per l’impegno e le
soddisfazioni condivise e per i dispiaceri superati insieme.
Un grazie grande così è per Lucia e Davide, amici da sempre, e per
sempre, nonostante la lontananza e le diverse strade intraprese.
Grazie a tutti i ragazzi del laboratorio e al Dott. Stefano Landi, per aver
contribuito al raggiungimento del mio obiettivo. Soprattutto grazie a
Daniele, che mi ha seguita in prima persona comprendendo anche le
piccole difficoltà.
Un grazie speciale a Katy, che è stata la migliore compagna di lavoro oltre
che una splendida amica.
Infine, grazie a tutte le persone che mi hanno aiutata e sostenuta!!
Grazie, grazie, grazie!!!
Formattato: Inglese (Regno Unito)
BIBLIOGRAFIA
Ali-Osman F., Ankade O., Antoun G., Mao J. X., Buolamwini J. (1997), Molecular cloning, characterization and expression in Escherichia coli of full-lenght cDNAs of three human glutathione S-transferase π gene variants. Evidence for differential catalytic activity of the encoded proteins., J. Biol. Chem. 272, pp. 10004-10012.
Bailey, L. R., Roodi, N., Verrier, C. S., Yee, C. J., Dupont, W. D. and Parl, F. F. (1998), Breast cancer risk and CYP1A1, GSTM1 and GSTT1 polymorphisms: evidence of a lack of association in Caucasian and African Americans. Cancer Res. 58, pp. 65-70.
Boffetta, P., Kogevinas, M., Simonato, L., Wilbourn, J., Saracci, R. (1995) Int J Occup Environ Health. 1(4): 315-325.
Breslow NE, Storer BE. General relative risk functions for case-control studies. Am J Epidemiol. 1985 Jul;122(1):149-62.
Butkiewicz D., Grzybowska E., Phillips D. H., Hemminki K., Chorazy M. (2000), Polymorfisms of the GSTP1 and GSTM1 genes and PAH-DNA adducts in human mononuclear white blood cells. Environ. Mol. Mutagen. 35, pp. 99-105.
C.C. Harris (1989), Iterindividual variation among humans in carcinogen metabolism, DNA adduct formaion and DNA repair. Carcinogenesis 10, pp. 1563-1566.
Corrin, B., Otto, W.R., Wright, N.A. Stem cells in the lung. In Pathology of lung tumors. Churchill Livingstone, New York, (1997).
David P. Miller, Geoffrey Liu, Immaculata De Vivo, Thomas J. Lymch, John C. Wain, Li Su and David C. Christiani (2002), Combinations of the Variant Genotypes of GSTP1, GSTM1 and p53 are associated with an Increased Lung Cancer Risk. Cancer Res. 62, pp. 2819-2823.
Djordjevic, M.V., Hoffmann, D., Hoffmann, I. (1997) I. Prev Med 26,435-440.
Doll, R., Peto, R. (1981) J Natl Cancer Inst 66, 1191-1308.
Ernster, V. L. (1996) Annu. Rev. Public Health. 17, 97 - 114.
F. J. Gonzalez, C. L. Crespi and H. V. Gelboin (1991), DNA-expressed human cytocrome P450s: a new age of molecular toxicology and human risk assessment. Mutation Res. 247, pp.113-127.
Federica Gemignani, Victor Moreno, Stefano Landi, Norman Moullan, Amelie Chabrier, Sara Gutierrez-Enriquez, Janet Hall, Elisabeth Guino, Miguel Angel Peinado, Gabriel Capella, Federico Canzian (2004), Oncogene, 23, 1954-1956.
Fields W. R., Morrow C. S., Doss A. J., Sundberg K., Jernstrom B., Townsend A. J. (1998), Overexpression of stably transfected human glutathione S-transferase P1-1 protects against DNA damage by benzo(a)pyrene dio-epoxide in human T47D cell. Mol. Pharmacol. 54, pp. 298-304.
H. Raunio, K. Husgufvel-Pursiainen, S. Anttila, E. Hietanen, A. Hirvonen and O. Pelkonen (1995), Diagnosis of polymorfisms in carcinogen-activating and inactivating enzymes and cancer susceptibility- a review. Gene 159, pp. 113-121.
Hecht S.S. (1999), Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 91, pp. 1194-210.
Hoffmann, D., Hoffmann, I. (1995) I.Cancer Treat Res. 72,1-42.
Hoffmann, D., Hoffmann, I. (1997) J. Toxicol. Environ. Health. 50, 307 - 364.
Hoffmann ,D., Djordjevic, M.V., Hoffmann, I. (1997) Prev Med 26:427-434.
Hung RJ, Boffetta P, Canzian F, Moullan N, Szeszenia-Dabrowska N, Zaridze D, Lissowska J, Rudnai P, Fabianova E, Mates D, Foretova L, Janout V, Bencko V, Chabrier A, Landi S, Gemignani F, Hall J, Brennan P. (2006), Sequence Variants in Cell Cycle Control Pathway, X-ray Exposure, and Lung Cancer Risk: A Multicenter Case-Control Study in Central Europe. Cancer Res.66(16):8280-6.
IARC (International Agency for Research on Cancer) Cancer: (1990) Causes, Occurrence and Control.
IARC (International Agency for Research on Cancer) (1994) Occupational Cancer in Developing Countries.
J.J Mulvihill (1976), Host factors in human lun tumors: an example of ecogenetics in oncology. J. Natl. Cancer Inst. 57, pp 3-7.
J.F. Fraumeni, Jr. (1975) Respiratory carcinogenesis: an epidemiological appraisal. J. Natl. Cancer Inst. 55, pp. 1039-1046.
Joachim Schneider, Ulrike Bernges, Monika Philipp, Hans-Joachim Woitowitz (2004), Cancer Letters 208, pp. 65-74.
Kayser, K. Analytical Lung Pathology. Springer-Verlag, (1992) Berlin, Heidelberg, New York.
Kihara, M., Kihara, M., Noda, K., Okamoto, N. (1993). Cancer Lett. 71, pp. 151-155.
Lung Cancer In: World Cancer Report, IARCPress 2003, Lyon France pp182-187.
Muller, K. M. Die Bronchialschleimhaut und das Bronkialkarzinom. (2000) Atemweg, Lungenerkrankungen. (370-374).
McWilliams, J. E., Sanderson, B. J,. Harris, E. L., Richert-Boe, K. E., Henner, W. D. (1995), Cancer Epidemiol. Biomarkers Prev. 4, pp. 589-594.
Nakajima T., Elovaara E., Anttila S., Hirvonen A., Camus A. M., Hayes J. D., Ketterer B., Vainio H. (1995), Expression of polymorfisms of glutathione S-transferase in human lungs: risk factors in smoking-related lung cancer. Carcinogenesis (Lond.) 16, pp.707-711.
Pamble, S., Schroeder, K. R., Spencer, S. R., Meyer, D. J., Hallier, E., Bolt, H. M., Ketterer, B. and Tayler, J. B. (1994). Human glutathione S-transferase theta (GSTT1): cDNA cloning and caracterization of a genetic polymorphism. Biochem. J. 300 (Part. 1), pp. 271-276.
Parkers, W. R. Occupational Lung Disorders. (1996) Butterworths, London 529 pp.
Parkin, D. M., Whelan, S. L., Ferlay, J., Raymond, L., Young, J. Eds (1997) Cancer Incidence in Five Continents, Vol. VII (IARC Scientific publication No.143 and IARC Cancerbase No.2), Lyon, IARCPress.
Peto, R., Darby, S., Deo, H., Silcocks, P., Whitley, E., Doll, R. (2000), Br. Med. J. 321, pp. 323-329.
Pinabasi, H., Silig, Y., Cetinkaya, O., Seyfikli, Z., Pinabasi, E. (2003), Cancer Genet. Cytogenet. 146, pp. 125-129.
Prescott, E., Osler, M., Andersen, P. K, Hein, H. O, Borch-Johnsen, K., Lange, P., Schnohr, P., Vestbo, J. (1998) Int. J. Epidemiol. 27, 27-32.
Randa El-Zein, Joseph B. Zwischenberger, Thomas G. Wood, Scherif Z. Abdel-Rahman, Chad Brekelbaum and William W. Au (1997), Combined genetic polymorfism and risk for development of lung cancer. Mut. Res./ Fund. and mol. mech. Of mutag., vol. 381, issue 2, pp. 189-200.
Risch, H. A, Howe, G. R, Jain, M., Burch, J. D., Holowaty, E.J., Miller, A.B. (1993) Am. J. Epidemiol. 138, 281 - 293.
Risch, H. A, Howe, G. R, Jain, M., Burch, J. D, Holowaty, E. J., Miller, A.B. (1994), Science 263, pp. 1206 -1208.
Ryberg D., Skaugh V., Hewer A., Phillips D. H., Harries L. W., Wolf C. R., Ogreid D., Ulvik A., Vu P., Haugen A. (1997), Genotypes of Glutathione transferase M1 and P1 and their significance for lung DNA adduct levels and cancer risk. Carcinogenesis (Lond.) 18, pp. 1285-1289.
Seidgard, J., Vorachek, W. R., Pero, R. W. and Pearson, W. R. (1988), Hereditary differences in the expression of the glutathione S- transferase activity on trans-stilbene oxid are due to a gene deletion. Proc. Natl. Acad. Sci. USA 85, pp. 7293-7297.
Shama C. Buch, Perin N. Notani e Rajani A. Bhisey (2002). Carcinogenesis, vol. no. 5, 803-807.
Shields, P. G.. (1999) Ann. Oncol,10 (Suppl.5): S7-S11.
Schoket, B., Philips, D. H., Kostic, S. and Vincze, I. (1998), Smoking-associated bulky DNA adducts in brinchial tissue related to CYP1A1 MspI and GSTM1 genotypes in lung patients. Carcinogenesis 19, pp. 841-846.
Simone Benhamou, Won Jin Lee, Anna-karin Alexandrie, Paolo Moffetta e altri (2002), Carcinogenesis, vol 23 no.8 ,1343-1350.
Strange, R.C. and Fryer, A. A. (1999), The glutathione S-transferase: influence of polymorphisms on cancer susceptibility. In: Metabolic Polymorphisms and
Susceptibility to Cancer. (Vineis, P., Malats, N., Lang, M., D’errico, A., Caporaso, N., Cuzick, J. And Boffetta, P., eds.). IARC Scientific Pubblication, No. 148, Lyon, France, pp. 231-249.
Strange, R.C., Spiteri, M. A., Ramachandran, S. and Fryer, A. A. (2001), Glutatione S-transferasi family of enzymes. Mut. Res. 482, pp. 21-26.
Thun, M. J, Lally, C. A, Flannery, J. T., Calle, E. E, Flanders, W. D., Heath, Jr C. W. (1997) J. Natl. Cancer Inst. 89,1580 -1586.
T.A. Sellers, J.E. Baily- Wilson, R.C. Elston, A.F. Wilson, G.Z. Elston, W.L. Ooi and H. Rotchild (1990), Evidence of Mendelian inheritance in the pathogenesis of lung cancer. J. Natl. Cancer Inst. 82, pp. 1272-1279.
T.A Sellers, J.D. Potter, J.E. Baily- Wilson, S.S. Rich, H. Rotschild and R.C. Eltson (1992), Lung cancer detection and prevention evidence for an interaction between smoking and genetic predisposition. Cancer Res. (Suppl.) 52, pp. 2694s-2698s.
Wang, Y., Liang, D., Spitz, M. R., Zhang, K., Dong, Q., Amos, C. I., Wu, X. (2003d), Cancer 98, pp. 1701-1706.
Watson M. A., Stewart R. K., Smith G. B., Massey T. E., Bell D. A. (1998), Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis (Lond.) 19, pp. 275-280.
Wingo, P. A, Ries, L. A, Giovino, G. A, Miller, D. S, Rosenberg, H.M., Shopland, D. R., Thun, M. J., Edwards, B. K. (1999) J. Natl. Cancer Inst. 91, 675-690.
Wu X, Zhao H, Amos CI, Shete S, Makan N, Hong WK, Kadlubar FF, Spitz MR. p53 Genotypes and Haplotypes Associated With Lung Cancer Susceptibility and Ethnicity. J Natl Cancer Inst. 2002 May 1;94(9):681-90.
Ye Z., Song H., Higgins JP, Pharoah P, Danesch J. (2006), Five glutathione s-transferase gene variants in 23,452 cases of lung cancer and 30,397 controls: meta-analysis of 130 studies. PLoS Med. 3(4):e91.
Zang, E. A., Wynder, E. L. (1996) J. Natl. Cancer Inst. 88, 183-192.
Zhong, S., Howie, A. F, Ketterer, B., Taylor, J., Hayes, J. D, Beckett, G. J., Wathen, C. G, Wolf, C. R, Spurr, N. K. (1991). Carcinogenesis. 12, pp. 1533-1537.