DEPARTMENT OF BIOLOGICAL AND ECOLOGICAL SCIENCES (DEB)
PhD Course in GENETICS AND CELLULAR BIOLOGY - XXVII cycle
BIO/13
ROLE OF RAB7A AND RILP
IN CELL MIGRATION
PhD student:
Azzurra Margiotta
Coordinator of the PhD course
Tutor
Prof. Giorgio Prantera
Prof. Cecilia Bucci
INDEX
ABSTRACT ... I RIASSUNTO ... II
1. INTRODUCTION ... 1
1.1 Cell migration ... 1
1.2 Actin filaments and formation of protrusions ... 2
1.2.1 Filopodia protrusions and myosin X... 3
1.3 Cell-matrix adhesions ... 4
1.3.1 Integrins ... 6
1.3.2 β1-integrin... 8
1.4 Contraction and retraction ... 9
1.5 Rac, Cdc42 and Rho are important regulators of cell migration ... 9
1.6 Rab GTPases ... 10
1.7 Rab7a ... 12
1.7.1 Rab7a and the Charcot-Marie-Tooth type 2B disease ... 15
2. AIM OF THE PROJECT ... 17
3. MATHERIAL AND METHODS ... 18
3.1 Cryopreservation of E. coli DH5α stocks ... 18
3.2 Competent E. coli cells preparation ... 18
3.3 Bacterial transformation ... 19
3.4 Preparation of plasmidic DNA ... 20
3.4.1 Mini-preparation of DNA ... 20
3.5 Molecular cloning ... 21
3.5.1 Primer design ... 21
3.5.2 Polymerase Chain Reaction (PCR) ... 23
3.5.3 Purification of PCR products ... 23
3.5.4 Digestion of amplified products and acceptor plasmids ... 24
3.5.6 Agarose gel electrophoresis of DNA ... 24
3.5.7 Ligation ... 25
3.5.8 Amplification of the new plasmids ... 25
3.6 Site-directed mutagenesis... 26
3.6.1 Primer design ... 26
3.6.2 PCR reaction ... 27
3.6.3 DpnI digestion ... 27
3.6.4 Amplification of the plasmids ... 28
3.7 Cell culture ... 28
3.8 Mammalian cell transfection ... 29
3.8.1 Transfection of DNA plasmids ... 29
3.8.2 RNA interference ... 30 3.9 Cell lysis ... 32 3.10 SDS-PAGE ... 33 3.11 Western blotting ... 34 3.12 Co-immunoprecipitation assay ... 36 3.13 Immunofluorescence ... 36 3.14 Wound-healing assay ... 37 3.15 Adhesion assay ... 39
3.16 EEA1 and active β1-integrin colocalization assay ... 39
3.17 Rac1 activation assay ... 39
3.18 Live imaging ... 40
3.19 Statistical analyses ... 40
4. RESULTS ... 41
4.1 Rab7a regulates cell migration through Rac1 and vimentin ... 41
4.1.1 Depletion of Rab7a alters cell motility ... 41
4.1.2 Rab7a silencing does not influence Golgi apparatus reorientation ... 44
4.1.3 Rab7a alters filopodia formation ... 45
4.1.4 Rab7a impairs myosin X localization to filopodia tips ... 46
4.1.5 Loss of Rab7a reduces cell adhesion ... 49
4.1.6 Rab7a alters β1-integrin localization and activation ... 51
4.1.7 Rab7a does not impair vinculin abundance ... 56
4.1.8 Loss of Rab7a does not impair Rac1 abundance ... 57
4.1.10 Characterization of the interaction between Rab7a and vimentin ... 61
4.1.10.1 Construction of vimentin deleted mutants ... 62
4.1.10.2 Rab7a interacts with coil1A vimentin domain... 64
4.1.10.3 Vimentin depletion affects Rab7a abundance... 65
4.1.11 Rab7a regulates vimentin filaments orientation during cell migration ... 66
4.1.12 Rab7a is localized in the area of the cell facing the wound during migration ... 68
4.2 Study on the role of Rab7a mutants in cell migration ... 69
4.2.1 Construction of HA-Rab7a mutants ... 69
4.2.2 Expression of HA-Rab7a mutants ... 73
4.2.3 Rab7a mutants expression does not impair cell adhesion ... 74
4.2.4 Rab7a CMT2B mutants behave like the wild type protein in cell adhesion assays ... 76
4.2.5 CMT2B-associated Rab7a mutants do not alter vimentin reorientation during migration ... 78
4.2.6 CMT2B-causing Rab7a mutants are localized in the area of the cell facing the wound during migration ... 81
4.3 Characterization of the effects of RILP on cell migration ... 83
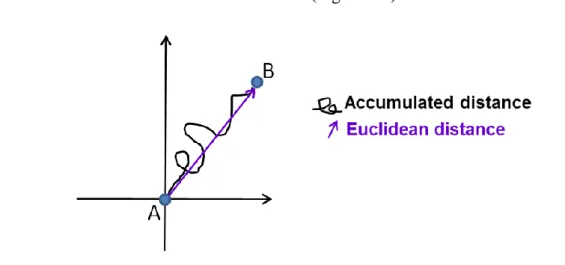
4.3.1 RILP affects cell velocity and accumulated distance of migrating cells ... 83
4.3.2 RILP depletion does not alter Golgi apparatus reorientation ... 86
4.3.3 RILP does not influence microtubule cytoskeleton rearrangements ... 87
4.3.4 RILP modulates cell adhesion ... 88
5. DISCUSSION ... 90
6. APPENDIX ... 101
7. REFERENCES ... 102
I
ABSTRACT
Rab7a is a small GTPase of the Rab family that regulates late stages of endocytosis. RILP is an important downstream effector of Rab7a, which can be recruited to membrane from cytosol by this GTPase. Rab7a and RILP control together several aspects of vesicular trafficking. This PhD work focuses on the role of Rab7a and RILP in cell migration, which is a cellular process characterized by cell polarization, protrusion of the cell membrane, formation of new adhesions, contraction and retraction of the cell body.
First of all, we demonstrated in wound healing assay that Rab7a modulates cell motility affecting cell velocity and directness. Moreover, Rab7a alters the formation of filopodia by mislocalizing myosin X, an actin motor known to promote the formation of these protrusions. Furthermore, we investigated the role of Rab7a in cell adhesion. We evaluated the ability of control and Rab7a-depleted cells to spread out on fibronectin-coated coverslips and we found that Rab7a Rab7a- depleted-cells showed a lower ability to spread while re-expression of active Rab7a recovered partially the function. Interestingly, activation, localization and intracellular trafficking of β1-integrin, a component of cell-matrix adhesions, were impaired after Rab7a silencing. Furthermore, as Rab7a interacts with Rac1 and vimentin, important factors for cell motility, we investigated the role of Rab7a on the regulation of these proteins. Interestingly, we found that Rab7a silencing impairs Rac1 activation state but not its abundance. In addition, Rab7a regulates vimentin filaments reorientation during migration. In fact, Rab7a depletion reduced the percentage of migrating cells with vimentin filaments facing the wound. We made some Rab7a mutants to investigate deeply the role of Rab7a in cell migration but the preliminary data collected did not show any difference in behavior of these proteins compared to wild type Rab7a. Similarly, Rab7a mutants which are responsible of the onset of Charcot-Marie-Tooth type 2B neuropathy did not act differently from the wild type protein in the performed experiments.
In light of the above, we demonstrated that Rab7a is necessary for proper cell migration, being able to modulate several aspects of this process and by regulating several key proteins.
Then, we characterized the role of RILP in cell migration by evaluating its role on cell motility, microtubule cytoskeleton, cell polarization and adhesion. We demonstrated that RILP controls cell velocity and cell adhesion, but not Golgi apparatus reorientation and microtubules reorganization.
The discovery of the involvement of Rab7a and RILP in cell migration could be of importance both in physiological and pathological situations.
II
RIASSUNTO
Rab7a è una piccola GTPasi della famiglia delle proteine Rab e regola gli stadi tardivi dell'endocitosi. RILP è un importante effettore di Rab7a che può essere reclutato dal citosol sulla membrana da questa GTPasi. Rab7a e RILP controllano insieme diversi aspetti del traffico vescicolare. In questo lavoro di dottorato si è studiato il ruolo di Rab7a e RILP nella migrazione cellulare, che è un processo importante in tutti gli organismi ed è caratterizzato dalla polarizzazione cellulare, protrusione della membrana plasmatica, formazione di nuove adesioni, contrazione e retrazione del corpo cellulare.
Innanzitutto, si è dimostrato in esperimenti di wound healing che Rab7a modula la motilità cellulare influenzando la velocità e la direzionalità delle cellule. Inoltre, Rab7a altera la formazione dei filopodi regolando la localizzazione di miosina X, che è una proteina motore responsabile della formazione di queste protrusioni. Successivamente, si è studiato il ruolo di Rab7a nell'adesione cellulare con la fibronectina, una componente della matrice extracellulare. In particolare, si è valutata l'abilità di cellule controllo e silenziate per Rab7a di aderire su vetrini ricoperti da fibronectina. Si è dimostrato che dopo il silenziamento di Rab7a le cellule presentavano una minor abilità nell'aderire al substrato. Tuttavia, la riespressione della proteina Rab7a attiva ha determinato un recupero parziale della funzionalità. Si è riscontrato, inoltre, un'alterazione nell'attivazione, localizzazione e traffico intracellulare dell'integrina β1, una componente delle adesioni cellula-matrice extracellulare, dopo la deplezione di Rab7a. Questa GTPasi interagisce con Rac1 e vimentina, che sono due proteine importanti per la migrazione cellulare. Pertanto, si è studiato il ruolo di Rab7a nella regolazione di queste proteine. Innanzitutto, si è visto che il silenziamento di Rab7a altera lo stato di attivazione di Rac1 ma non la sua abbondanza proteica. Inoltre, Rab7a regola la riorientazione dei filamenti di vimentina durante la migrazione. Infatti, la deplezione di Rab7a ha ridotto la percentuale delle cellule in migrazione che presentavano i filamenti di vimentina orientati nella direzione di migrazione. Successivamente, si sono creati dei mutanti della proteina Rab7a in modo da valutare meglio il ruolo di Rab7a nella migrazione ma i dati preliminari ottenuti non hanno evidenziato alcuna differenza di comportamento tra le proteine mutate e quella wild type. Similiarmente, anche i mutanti di Rab7a che sono responsabili dell'insorgenza della neuropatia di Charcot-Marie-Tooth di tipo 2B non hanno mostrato differenze con la proteina wild type negli esperimenti eseguiti. Alla luce di tutto ciò, si è dimostrato che Rab7a è necessaria per la migrazione cellulare ed è capace di modulare diversi aspetti e proteine chiave di questo processo.
III Successivamente si è analizzato la funzione di RILP nella migrazione cellulare valutando il suo ruolo nella motilità, nella regolazione dei microtubuli, nella polarizzazione e nell'adesione cellulare. Si è, quindi, dimostrato che RILP controlla la velocità e l'adesione cellulare ma non la riorientazione dell'apparato di Golgi e la riorganizzazione dei microtubuli.
La scoperta del coinvolgimento di Rab7a e RILP nella migrazione cellulare potrebbe essere rilevante sia in condizioni fisiologiche che patologiche.
1
INTRODUCTION
1.1 Cell migration
Cell migration is the ability of a cell to actively move from one place to another and it is a relevant process in all organisms. It isn't important only during development but also during the lifetime as it participates in wound repair and immune responses (Etienne-Manneville, 2013; Raftopoulou and Hall, 2004). The general mechanism of cell migration involves the repetition of four basic steps: protrusion of the cell membrane, formation of new adhesions, contraction and retraction of the cell body (Etienne-Manneville, 2013; Vicente-Manzanares et al., 2005).
During migration cells undergo several changes (Figure 1.1). When a cell receives migratory stimuli, several morphological modifications occur leading to the development of a polarized phenotype. First of all, cells are oriented towards the direction of migration presenting a protruding, leading front opposite to a retracting rear edge. Cell polarization refers to the presence of asymmetry within a cell that is generated by spatial differences in a variety of subcellular components. During migration, the polarized phenotype is identified by the extension of dynamic membrane protrusions at the leading edge, the formation and disassembly of focal adhesions (FAs), the reorganization of microtubules, actin and intermediate filaments, and the reorientation of the Golgi apparatus and of the microtubule organizing centre (MTOC) into the direction of the wound (Vicente-Manzanares et al., 2005; Etienne-Manneville, 2013). In particular, proper microtubules organization is essential for the polarized structure. Microtubules are composed of α- and β-tubulin polymers and tubulin subunits are organized in a polar manner, with a fast-growing plus end which radiates toward the cell periphery and a slow-growing minus end which is attached to the MTOC, from which microtubules are generated (Muthuswamy and Xue, 2012). During migration, microtubules are oriented along the axis of migration and play an important role in establishing directionality by regulating actin polymerization and transporting membrane vesicles to the leading edge (Etienne-Manneville, 2013). Moreover, during cell polarization, the reorientation of the nucleus at the rear of migrating cells is required for cell polarization, by promoting correct MTOC localization and thus the establishment of front-rear polarity (Maninová et al., 2013). In addition, other changes, such as in vesicular transport pathways and in gene transcription, occur during migration (Raftopoulou and Hall, 2004; Leduc and Etienne-Manneville, 2015). Cell polarity is essential for directional migration and enables the cell to turn intracellularly generated forces into net cell body translocation (Vicente-Manzanares et al, 2005; Lauffenburger and Horwitz, 1996).
2 All these events are essential for directed migration and several molecules coordinate both in space and time these processes (Vicente-Manzanares et al., 2005).
Figure 1.1 Cellular changes during migration. A polarized migrating cell shows
reorganization of the cytoskeleton, Golgi apparatus and MTOC orientation, and the formation of protrusions and adhesion sites at the leading edge (adapted from Ridley et al., 2003).
1.2 Actin filaments and formation of protrusions
The actin cytoskeleton is an important player in cell migration as its reorganization influences cell shape and polarization, adhesions and contraction of the cell body. Actin filaments can organize into a variety of architectures that are responsible for the regulation of different aspects of cell migration (Figure 1.2). Lamellipodia are broad, flat, sheet-like structures composed of branched actin filaments which push the cell membrane by polymerizing against it. Instead, filopodia are thin, cylindrical, needle-like projections at the front of the cell and are composed of unbranched, bundled actin filaments oriented with their growning ends toward the membrane. Cytoplasmic organelles are excluded from lamellipodia and filopodia that are the main components driving motility (Lauffenburger and Horwitz, 1996; Blanchoin et al., 2014). Contractile structures are represented by transverse arcs, focal adhesion-anchored stress fibers and cell cortex which ensure mechanical integrity and coherent movement of the cell as a whole. Transverse arcs and stress fibers are localized in all the cell except for the lamellipodia/filopodia region. Ventral stress fibers run approximately parallel to the direction of the movement, linking
3 focal adhesion sites, whereas transverse arcs run parallel to the leading edge, just behind the actin network forming the lamellipodia. Finally, cell cortex is a thin layer of actin which coats the plasma membrane at the back and sides of the cell and it is important for cell shape (Blanchoin et al., 2014).
Figure 1.2 Actin filaments structures in migrating cells. Schematic representation of a
migrating cell showing different actin architectures as indicated (adapted from Blanchoin et al., 2014).
1.2.1 Filopodia protrusions and myosin X
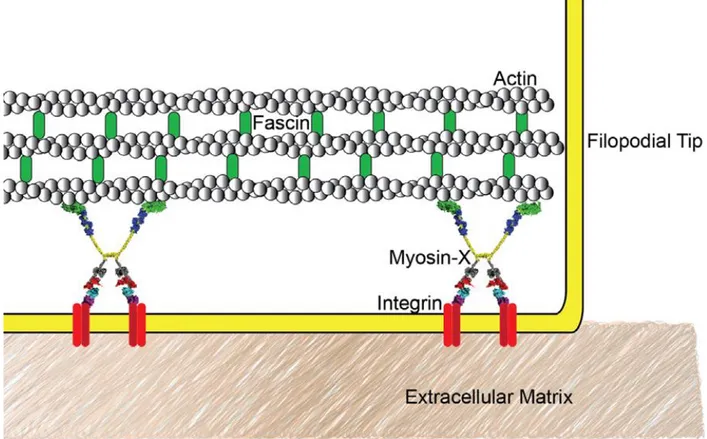
Filopodia are fingerlike processes with a diameter of about 200 nm and length of several µm. They are dynamic structures that can show different modes of movement. The most common motions of filopodia are linear growth and retraction, whereas angular rotations around their anchor point are necessary to "scan" the area of the environment surrounding the cell. Filopodia are important in sensing the cell environment, initiating cell contacts, and transmitting cell-cell signals. Moreover, they exert pulling forces (Blanchoin et al., 2014; Bornschlögl, 2013). The filopodial core is made up of 15-30 tightly packed actin filaments, which can exhibit a dense protein complex at the tip. Different proteins regulate filopodial formation, maintenance and dynamics. Among these, myosin X is a molecular motor that plays an important role in filopodial formation and is responsible for transporting various proteins, including transmembrane receptors, along actin filaments to the tips of filopodia to participate in filopodia maintenance and stabilization, and environment and cue sensing (Bornschlögl, 2013; Jacquemet et al., 2015). In fact, myosin X overexpression induces an increase of the number of filopodia per cell,
4 whereas its silencing decreases endogenous filopodia. Myosin X binds β-integrin receptors through its tail domain. This interaction is hypothesized to allow myosin X to link the internal actin cytoskeleton with the extracellular matrix (Figure 1.3). In support of this, studies of cells expressing myosin X mutants lacking integrin binding have showed decreased filopodial adhesion. Moreover, myosin X over-expression leads to increased cell invasion, whereas its depletion inhibits cancer cell invasion. Myosin X is thought to drive cell invasion by transporting integrins to the tips of filopodia to tether the extra-cellular matrix (ECM). Myosin X undertakes its biological roles when activated by its dimerization (Courson and Cheney, 2015; Jacquemet et
al., 2015).
Figure 1.3 Myosin X localizes to the filopodial tip. Myosin X seems to be the molecular link
between the actin cytoskeleton and integrin-based adhesions to the extracellular matrix in filopodia (Courson and Cheney, 2015).
1.3 Cell-matrix adhesions
Lamellipodia and filopodia are stabilized by adhesions that connect the actin cytoskeleton with the ECM. Cell-matrix adhesions are associated with actin microfilaments in the cytoplasm, whereas they establish tight connection with the substrate in the ECM. Adhesions regulate cell motility by assembling at the leading edge and disassembling at the rear edge in response to
5 extracellular cues. However, adhesions also disassemble at the front of the cell, and their components can be part of new adhesions that will form in the same region. This process is called adhesion turnover (Parsons et al., 2010). Adhesions have been classified basing on their size, stability and localization in several subtypes: nascent adhesions, focal complexes and focal adhesions. They differ from each other in the levels of the protein components, however they seem to be in a continuum of structures rather than distinct adhesion types. The earliest detectable adhesions are the nascent adhesions, small, short-lived adhesions, which have origin in the lamellipodium just behind the leading edge. They can either disassemble in about a minute or mature to larger, dot-like adhesions called focal complexes. Focal complexes are localized at the lamellipodium-lamellum interface, therefore slightly further back from the leading edge. These adhesions are slightly larger in size (~1 µm in diameter) and persist for several minutes. Then, they mature in more stable structures that are referred to as focal adhesions. They are larger and elongated, in fact, their diameter is of about 2 µm whereas their length is of 3-10 µm. Focal adhesions reside at the ends of actin stress fibers (Figure 1.4). As the cell move forward, adhesions are disassembled and re-formed in a more advanced position. Continuous cycles of adhesion and retraction allow the translocation of the cell in the direction of migration (Parsons
et al., 2010).
Figure 1.4 Nascent adhesions, focal complexes and focal adhesions localization sites.
Nascent adhesions form in the lamellipodium. Nascent adhesion maturation gives origin to focal complexes, which are localized at the convergence of the lamellipodium and lamellum (the
6 transition zone). At the contrary, focal adhesions reside at the ends of actin stress fibers (adapted from Parsons et al., 2010).
In addition to nascent adhesions, focal complexes and focal adhesions, fibrillar adhesions have been detected in fibroblasts grown in fibronectin-rich environments for extended time. These adhesions are highly elongated, have a long lifetime and are located towards the center of the cell. However, they are not prominent in rapidly migrating cells (Parsons et al., 2010).
The formation, maturation and disassembly of adhesions are regulated by actin polymerization and actomyosin contraction. The most important components of these adhesions are the adhesive receptor integrins, but several adhesion components have been identified. Among them vinculin is recruited to the cytoplasmic tails of β integrins, through the interaction with talin, an actin-binding protein. Vinculin is important in stabilizing focal adhesions. In fact, cells lacking vinculin have fewer and smaller adhesions, whereas cells over-expressing vinculin show an increased number and size of focal adhesions compared to control cells. Moreover, cell motility is reduced in cells devoid of vinculin (Peng et al., 2011; Parsons et al., 2010).
1.3.1 Integrins
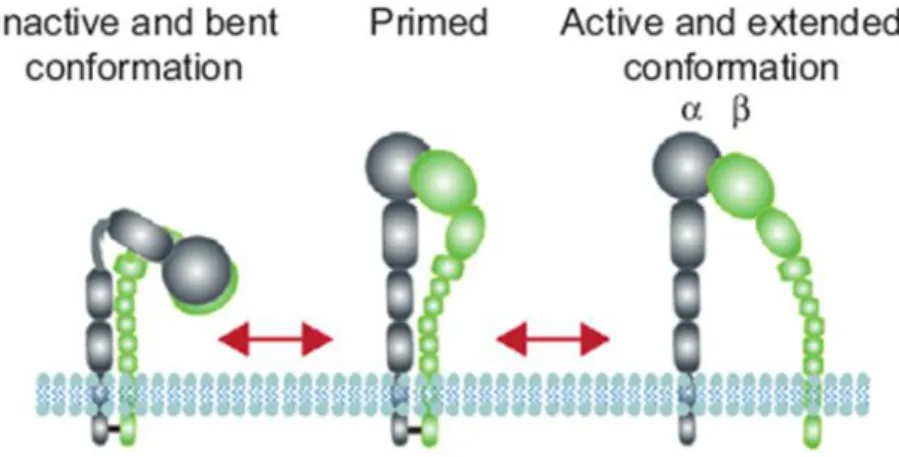
Integrins are a family of transmembrane cell surface heterodimeric molecules that constitute a major class of adhesion receptors. They are expressed in all cell types, erythrocytes excluded. In vertebrates, the assembly of 18 α-integrin and 8 β-integrin subunits within the endoplasmic reticulum leads to the formation of 24 different integrin heterodimers. They undergo further post-translational modification in the Golgi complex and then they localize as large, membrane-spanning proteins, at the cell surface linking ECM to the actin cytoskeleton in cell-matrix adhesions. In particular, their large extracellular domain binds to a variety of matrix ligands, whereas the short intracellular tail domain interacts with a number of adhesion components which mediate the association with actin filaments (De Franceschi et al., 2015). Some integrin subunits appear only in a single heterodimer, while β1 subunit is part of twelve integrins and αV subunit of five (Humphries et al., 2006). Integrins are differently expressed in tissues and exhibit a distinct binding affinity to ligands (Damianovich et al., 1992; Humphries et al., 2006). In addition, changes in the conformation of integrins and their clustering into oligomers can influence the binding of ligands. Integrins can exist in bent "closed" inactive conformation, intermediate extended conformations (also known as "primed") and extended "open" active conformation. These conformations reflect a low ability to bind ligand, an activated, and an activated and ligand-occupied integrin conformers, respectively (Figure 1.5). Instead, the formation of heterooligomers due to the interaction of integrin heterodimers is defined integrin
7 clustering. This event can be induced by binding of an extracellular ligand to different integrin extracellular domains at the same time (referred to as multivalent ligand), by inside-out signals that stimulate the recruitment of protein complexes that bind with several integrin cytoplasmic domains or by free diffusion of integrins in the plane of the membrane. Integrin clustering is important for triggering outside-in signalling, integrin recycling and mechanotransduction by focal complexes and focal adhesions (Shattil et al., 2010).
Figure 1.5 Schematic representation of integrin conformations. (adapted from De Franceschi
et al., 2015).
Integrin signalling is bidirectional. During inside-out signalling, intracellular factors, such as talin, bind to β-integrin tails, leading to conformational changes that increase integrin affinity for extracellular ligands (primed conformation). Inside-out signalling regulates adhesion strength. During outside-in signalling, both the modulation of integrin conformation, due to the binding to extracellular ligands, and the clustering, caused by multivalent ligands, determines intracellular signals that control several biological aspects, such as cell polarity, cytoskeletal structure, gene expression, cell survival and proliferation (Shattil et al., 2010). However, inside-out and outside-in signalloutside-ings are closely loutside-inked. For outside-instance, the primed outside-integroutside-in conformation caused by the binding of intracellular proteins (inside-out signalling) can lead to ligand binding, which in turn causes outside-in signalling (Shattil et al., 2010).
Integrins function can also be regulated by the modulation of their trafficking through the endocytic system. In fact, integrins usually undergo cycles of endocytosis and recycling that control the availability of these receptors at the plasma membrane and therefore their function. Instead, their degradation is less frequent as it has been demonstrated that the half-life of surface integrin is of about 12-24 hours (Paul et al., 2015). Internalization of integrins regulates the turnover of focal adhesions and promotes adhesion formation at the front of migrating cells, thus regulating cell migration. Both inactive and active conformations of integrin heterodimers can be
8 internalised, but the binding of sorting nexins (SNX) 17 and 31 to β-integrins is determinant for their recycling, instead of being degraded (Paul et al., 2015). However, also autophagosomes seem to have a role in degradation of β1 integrins (Tuloup-Minguez et al., 2013). GTPases of the Rab and Arf families seem to be important for regulating integrin trafficking. In particular, it has been demonstrated that Rab5 and Rab21 are important for integrin endocytosis, whereas recycling from early endosomes occurs through the Rab4 short-loop pathway. Instead, perinuclear recycling of integrins is regulated by Rab11 through the long-loop pathway. Finally, Rab25 binds directly to the β1-integrin tail and regulates the trafficking of α5β1 integrin. Rab25 sorts inactive α5β1 integrin for recycling to the cell front, whereas it sorts the active form of this integrin back towards the cell body through late endosomes to chloride intracellular channel 3 (CLIC3)-positive lysosomes where CLIC3 prevents degradation of this integrin and promote its recycling back to the plasma membrane (Paul et al., 2015).
1.3.2 β1-integrin
β1-integrin is a protein of about 130 KDa. Several monoclonal antibodies have been developed to detect conformation-dependent epitopes of this integrin and used in experimental studies (Byron et al., 2009). Therefore, it has been demonstrated that inactive β1-integrin is mostly localized to the plasma membrane and protrusions. In fact, approximately 80% of surface β1-integrin is in the inactive form, whereas only 20% of cell surface β1-β1-integrin is in the active conformation even if this conformation is more clustered than the inactive form. At the contrary, active β1-integrin is predominant in the cytoplasm (Tiwari et al., 2011; Arjonen et al., 2012). In addition to the particular localization, active and inactive β1-integrins show also different endocytosis. In fact, active β1-integrin is internalized together with the ligand and more efficiently than the inactive form. Both active and inactive β1 integrins were found in Rab5-, Rab4a-, Rab21- and Rab11-positive compartments. However, trafficking routes of the two conformations are distinct as only the active β1-integrin can localize to Rab7a-positive compartments, whereas inactive β1-integrin is recycled back through rapid F-actin and Rab4-dependent pathway to ARF6-positive protrusions on the plasma membrane (Figure 1.6) (Arjonen
9
Figure 1.6 Model of trafficking routes of active and inactive β1-integrin. The trafficking
routes of the two conformations of β1-integrin overlap in early endosomes, but show differences in the sequent steps. In fact, inactive β1-integrin is recycled through rapid F-actin and Rab4-dependent pathway to ARF6-positive protrusions on the plasma membrane, whereas the active β1-integrin recycling is less efficient, probably because the ligand, which is internalized together with the receptor, needs to be dissociated prior to receptor recycling. Moreover, only active β1-integrin is found in Rab7a-positive compartments (Arjonen et al., 2012).
1.4 Contraction and retraction
In order to translocate forward, several events must occur in a migrating cell. First of all, protrusive force is needed to extend lamellipodia and filopodia and it is mediated by actin polymerization. In addition, a contractile force is needed to move the cell body forward and it is dependent on the molecular motor protein myosin, by acting on the actin network. This induces contraction in the cell body and tension at adhesion sites, which are used as traction points for migration (Lauffenburger and Horwitz, 1996; Blanchoin et al., 2014). Then, adhesions at the rear are disassembled. Finally the trailing edge, devoid of integrin-cytoskeleton linkages and less supported by the cytoskeleton, retracts, avoiding that the tension rip the cell apart (Lauffenburger and Horwitz, 1996; Vicente-Manzanares et al., 2005).
1.5 Rac, Cdc42 and Rho are important regulators of cell migration
Several intracellular signalling molecules have been implicated in cell migration. In particular, Rac, Rho and Cdc42 GTPases are considered to be key regulator proteins of this process. It seems that they control some aspects of cell migration by integrating their action, but they also regulate specific events independently from each other. For instance, all three GTPases promote the assembly of integrin-based, cell-matrix adhesions and affect, even if in different ways, the microtubule cytoskeleton. Nevertheless, Rac and Cdc42 are required at the front of migrating cells where they regulate the polymerization of actin in the formation of lamellipodial and
10 filopodial protrusions, respectively, whereas Rho modulates the assembly of actomyosin filaments and controls cell body contraction and trailing edge retraction. In addition, Cdc42 is important for defining cell polarity (Raftopoulou and Hall, 2004).
1.6 Rab GTPases
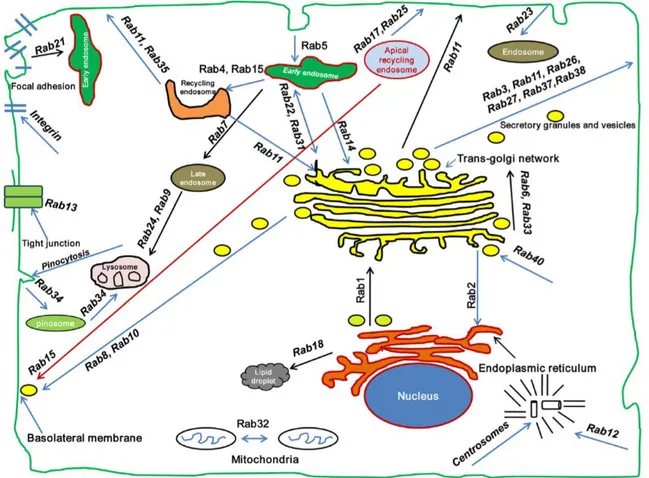
Rab proteins (Ras-related proteins in brain) are small GTPases belonging to the Ras superfamily and regulate the trafficking among the different intracellular compartments (Schwartz et al., 2007). Rab GTPases constitute the largest family of small GTPases with over 70 members in humans. Every Rab protein has a specific localization (Figure 1.7) (Bhuin and Roy, 2014).
Figure 1.7 Rab GTPases subcellular localization. Rab GTPases control membrane trafficking
at various steps and are localized to different compartments (Bhuin and Roy, 2014).
Rab GTPases work as molecular switches cycling between an active form, which binds GTP and is associated with membranes, and an inactive form, GDP-bound, which is citosolic. Active Rab proteins recruit specific effectors through which they regulate vesicle trafficking and fusion. Multiple regulatory proteins are available to modulate Rab proteins activity. Indeed, in the cytosol, prenylated Rabs can be found bound to a GDP-dissociation inhibitor (GDI) which recognizes inactive Rabs and covers up the prenyl groups. In order for a Rab to switch to the active GTP-bound state, the GDI has to be displaced, and this is done by the action of a
GDI-11 displacement factor (GDF). GDFs help Rabs attachment to specific membranes. Some proteins named Guanine nucleotide Exchange Factors (GEFs) catalyze the GDP/GTP exchange. Even though the Rabs have intrinsic hydrolytic activity towards GTP, the process of inactivation is catalyzed by GTPase activating proteins (GAPs), resulting in a renewed association with the GDI and in the recycling of the Rab GTPases back to the cytosol. Therefore, GEFs, GAPs and GDI play a critical role in the regulation of the activity of Rab proteins (Figure 1.8) (Bhuin and Roy, 2014).
Figure 1.8 The Rab cycle. The newly synthesized Rab, in the GDP-bound form, associates with
a Rab escort protein (REP). The REP presents the Rab to a Rab geranylgeranyl transferase (RabGGT), which geranylgeranylates the Rab. The Rab is then recognized by Rab GDP dissociation inhibitor (GDI), which regulates Rab membrane cycle. Targeting of the Rab-GDI complex to specific membranes is mediated by the interaction with a membrane-bound GDI displacement factor (GDF). Then, GDP-bound Rab is converted into the GTP-bound form through the exchange of GDP for GTP. This step is catalyzed by a guanine nucleotide exchange factor (GEF). The GTP-bound "active" Rab interacts with several Rab-specific effector proteins. Active Rab is converted back to the GDP-bound "inactive" form by interacting with a GTPase activating protein (GAP) which catalyzes the hydrolysis of GTP to GDP and the release of an inorganic phosphate (Pi). The Rab is then removed from the membrane by GDI (Stenmark,
2009).
Rab proteins are able to perform multiple roles ranging from transport, sorting, budding, tethering, fusion, motility and recruiting other effector proteins by cycling between the active GTP-bound state and the inactive GDP-bound state (Stenmark, 2009).
Recently, it has been demonstrated that some Rabs, such as Rab5, Rab7b, Rab11 and Rab25, have a role in cell migration (Borg et al., 2014; Caswell et al., 2007; Kessler et al., 2012; Mendoza et al., 2013). Rab5 activation promotes focal adhesion disassembly, migration and invasiveness in tumor cells, whereas Rab7b regulates actin remodeling and influences cell adhesion, polarization and migration (Mendoza et al., 2013; Borg et al., 2014). Rab25 interacts with β1-integrin and controls its localization by regulating integrin-recycling vesicles, while
12 Rab11 promotes cell migration by modulating basal long-distance transport from the rear end to the front of the migrating cell (Caswell et al., 2007; Kessler et al., 2012).
1.7 Rab7a
Rab7a is a small GTPase of the Rab family. It is ubiquitously expressed and localizes mainly to late endosomes. Rab7a regulates vesicular transport to late endosomes and lysosomes in the endocytic pathway, and biogenesis of lysosomes, phagolysosomes and autolysosomes (Bucci et
al., 2000; Harrison et al., 2003; Wang et al., 2011; Hyttinen et al., 2013; Vanlandingham and
Ceresa, 2009). Moreover, it is required for channel trafficking, growth factor-independent survival and apoptosis (Seebohm et al., 2008; Romero Rosales et al., 2009; Edinger et al., 2003). In addition, Rab7a controls the endocytic trafficking of the complex EGF-EGFR, regulating its lysosomal degradation (Ceresa and Bahr, 2006). Further studies have proved that Rab7a controls also the retrograde axonal transport of neurotrophine receptors which are required for neurotrophine signalling, neuronal differentiation, plasticity and survival (Saxena et al., 2005; Deinhardt et al., 2006). Moreover, it has been recently demonstrated that Rab7a interacts and recruits protrudin, a protein which promotes protrusion and neurite outgrowth, on late endosomes compartments (Shirane and Nakayama, 2006; Raiborg, 2015). In addition, Rab7a-dependent lysosomal degradation disturbs the terminal translocation at the final phase of migration of immature neurons during the development of cerebral cortex in vivo (Kawauchi et
al., 2010; Kawauchi, 2011).
Rab7a exerts its functions through the interaction with several effectors (Wang et al., 2011). Some of Rab7a interactors suggest that the functions of this small GTPase are closely connected to cytoskeletal elements.
One of the Rab7a interactors is RILP (Rab-interacting lysosomal protein), an important downstream effector of Rab7a, which can be recruited to membrane from cytosol by Rab7a to regulate late endosome and lysosome traffic, phagosome and autophagosome maturation and signalling receptor degradation (Cantalupo et al., 2001; Jordens et al., 2001; Harrison et al., 2003; Progida et al., 2007). RILP is required also for the formation of multivesicular bodies (MVB) (Progida et al., 2007). RILP is a protein of 401 amino acids and contains two coiled-coil domains (aminoacids 140-180 and 245-280), whereas the truncated form of RILP (amino acids 207-401), named RILP-C33, retains the second half of the protein but lacks the N-terminal domain (Cantalupo et al, 2001). The first half of RILP is required for dynein/dynactin motor recruitment, whereas its C-terminal domain binds to activated Rab7a on late endosomes and lysosomes, therefore both RILP and RILP-C33 interact with the GTP bound form of Rab7a, but
13 only RILP binds to the dynein/dynactin complex (Jordens et al., 2001). Overexpression of RILP induces late endosome/lysosome enlargement and clustering to the perinuclear region by regulating microtubule minus-end directed transport through the interaction with the dynactin/dynein complex. Instead, cells over-expressing RILP-C33 show dispersed lysosomes (Cantalupo et al., 2001; Jordens et al., 2001). ORP1L (OSBP (oxysterol-binding protein) related protein) is another important interactor of Rab7a which modulates dynein motor recruitment by Rab7a-RILP-dynactin complex (Wang et al., 2011). Moreover, Rab7a interacts with FYCO1 (FYVE and coiled-coil domain containing 1), which controls plus-end directed transport of autophagosomes (Pankiv et al., 2010). Therefore, Rab7a controls vesicles movement along microtubule tracks by interacting with different effectors.
A connection between Rab7a and actin filaments comes from the interaction between Rab7a and Rac1, which is a fundamental regulator of actin cytoskeleton (Sun et al., 2005; Bosco et al., 2009). In fact, it has been demonstrated that Rab7a interacts and colocalizes with Rac1 at the fusion zone of the ruffled border in bone-resorbing osteoclasts where Rab7a is involved in the formation of late endosomal-like compartments localized in the plasma membrane. This interaction may mediate late endosomal transport between microtubules and microfilaments in order to regulate ruffled border formation in osteoclasts (Sun et al., 2005). A functional link of the Rac1-Rab7a association has been proved by Frasa and colleagues. In fact, they demonstrated that active Rac1 is responsible for the recruitment of its effector Armus which, in turn, deactivates Rab7a and that the integration of Rac1 and Rab7a activities regulates E-cadherin turnover and stability of cell-cell contacts (Frasa et al., 2010). Finally, coordination between Rab7a and Rac1 functions seems to be important in integrating autophagy with intracellular trafficking and signalling (Carroll et al., 2013).
Rab7a regulates also intermediate filament (IF) proteins. In fact, Rab7a directly interacts with vimentin (Cogli et al., 2013a). Vimentin is one of the most abundant type III IF protein and it is mainly expressed in mesenchymal cells. This IF protein contains an N-terminal head region, a central rod domain and a C-terminal tail (Figure 1.9). The rod domain is a highly conserved α-helical region that is composed by four α-helical segments (1A, 1B, 2A, 2B), divided by three non-helical linker regions (L1, L1-2, L2). Vimentin filaments assembly occurs through the association of two monomers forming a dimer, which in turn associates in an antiparallel manner with another dimer, creating a tetramer, which is the fundamental subunit of IFs. Multiple tetramers assemble to form an elongated filament. IFs undergo extensive reorganization upon phosphorylation. In fact, this post-translational modification controls vimentin assembly and disassembly (Figure 1.10) (Dave and Bayless, 2014).
14
Figure 1.9 Structure of vimentin monomer. The structure of vimentin consists of an
N-terminal "head" domain, a C-N-terminal "tail" domain, and a central rod domain. Three linkers referred to as L1, L12 and L2 divide the rod domain in coil1A, coil1B, coil2A and coil2B (adapted from Tang, 2008).
Figure 1.10 Phosphorylation-mediated disassembly of vimentin. Vimentin depolymerization
is triggered by the action of several protein kinases (adapted from Dave and Bayless, 2014). Several phosphorylation sites are located in the N-terminal head domain of vimentin and phosphorylation mediated by protein kinases on these sites determines disassembly of the IFs, as phosphorylation of the head region increases the distance between two head groups of a dimer, altering tetramer ability to assemble into a filament (Dave and Bayless, 2014). Rab7a regulates vimentin filament assembly. In fact, overexpression of Rab7a increases vimentin phosphorylation and causes redistribution of vimentin in the soluble fraction. Accordingly, Rab7a-depletion induces a decreased phosphorylation of vimentin head domain, thus increasing the amount of filamentous vimentin (Figure 1.11). Rab7a is able to interact with both soluble and filamentous vimentin (Cogli et al., 2013a).
15
Figure 1.11 Rab7a regulates vimentin assembly/disassembly. Rab7a induces vimentin
phosphorylation at the head domain causing vimentin disassembly (Cogli et al., 2013a).
In addition to mechanical stabilization of cells, vimentin functions include the regulation of cell adhesion, motility and signalling. Moreover, altered actin cytoskeleton and spatial organization of focal adhesion proteins were observed in fibroblasts lacking vimentin (Dave and Bayless, 2014).
Finally, Rab7a interacts and regulates the assembly of peripherin, another IF protein that shares the same structure and assembly of vimentin (Cogli et al., 2013b).
1.7.1 Rab7a and the Charcot-Marie-Tooth type 2B disease
Mutations in the RAB7A gene have been identified as causative of the Charcot-Marie-Tooth type 2B (CMT2B) disease. The CMT2B disease is an autosomal dominant axonal neuropathy which is clinically characterized by prominent sensory loss, marked distal weakness, normal or near-normal nerve conduction velocities and frequent foot ulcers due to deformities. The disease
16 onset is generally in the second or third decade of life (Bucci and De Luca, 2012). The CMT2B-associated Rab7a mutations target very conserved amino acid residues in the Rab7a protein. In particular five are the amino acidic substitutions responsible for the onset of the neuropathy: L129F, K157N, N161T, N161I, V162M (Meggouh et al., 2006; Houlden et al., 2004; Verhoeven
et al., 2003; Wang et al., 2014). GTPases have a GTP-binding and hydrolysis site which is
characterized by four conserved motifs that in the three-dimensional structure of the protein form a pocket able to accommodate the nucleotide and Mg2+. In silico analysis of CMT2B-disease Rab7a mutated proteins predicted important conformational changes (Bucci and De Luca, 2012). CMT2B-associated Rab7a mutants have been biochemically and functionally characterized. It has been demonstrated that CMT2B mutants exhibited higher nucleotide exchange rates, hydrolyzed GTP slower than the wild-type protein and bound prevalently GTP (Spinosa et al., 2008; De Luca et al., 2008). Nevertheless, CMT2B-associated mutants were able to bind the Rab7a effector RILP and to rescue Rab7a function after silencing, suggesting that activated forms of Rab7a were responsible for the onset of the neuropathy (Spinosa et al., 2008; De Luca
et al., 2008). Moreover, CMT2B-disease Rab7a mutants inhibited neurite outgrowth in PC12
cells and in Neuro2A cells as proved by the impaired up-regulation of growth-associated protein 43 (GAP43) and of the nuclear neuronal differentiation marker NeuN, respectively. This effect was similar to that induced by the constitutively active Rab7a mutant, confirming that active Rab7a mutants are responsible for CMT2B (Cogli et al., 2010). The inhibition of neurite outgrowth was probably due to the phosphorylation of nerve growth factore receptor TrkA by CMT2B-associated Rab7a mutants. In fact, the mutant proteins significantly enhanced the phosphorylation of TrkA in response to brief NGF stimulation, affecting Erk1/2 downstream signalling pathway (BasuRay et al., 2010). In addition, CMT2B-disease Rab7a mutants interacts stronger with peripherin and vimentin than the wild type protein and affect more the assembly of these IF than the wild type protein (Cogli et al., 2013a; Cogli et al., 2013b).
17
AIM OF THE PROJECT
Cell migration is an important event during the lifetime of all organisms and it is characterized by cell polarization, protrusion of the cell membrane, formation of new adhesions, contraction and retraction of the cell body. All these events are essential for migration and are coordinated by several proteins.
Rab7a, a small GTPase of the Rab family, localizes to late endosomes and lysosomes and regulates vesicular transport to these compartments. Among Rab7 effector proteins, there are Rac1, a key regulator of actin cytoskeleton and a fundamental player in cell migration, and vimentin, an intermediate filament protein, which is important in cell adhesion and motility. Another Rab7a interactor is RILP that regulates, together with Rab7a, late endosomal and lysosomal trafficking controlling Rab7-positive vesicle movement along microtubule tracks. Given the link between Rab7a and RILP with cytoskeleton components and in particular with key proteins in cell migration, the aim of the project is to clarify the role of Rab7a and RILP in cell migration by studying several aspects of this process, such as motility, formation of protrusions, adhesion with components of the extracellular matrix, cell polarization and cytoskeleton rearrangements. For this purpose, we have used three different approaches: (i) modulating expression of Rab7a to study its role in the various steps of cell migration; (ii) expressing constitutively active, dominant negative and CMT2B-causing Rab7a mutant proteins to study the impact of these mutants on cell migration; (iii) modulating expression of RILP to establish its role in cell migration.
18
MATHERIAL AND METHODS
3.1 Cryopreservation of E. coli DH5α stocks
In a tube, 0,4 ml of a sterile 50% glycerol solution was added to 0,6 ml of a E. coli DH5α bacterial culture cultivated overnight (O/N) in LB medium to give a final concentration of 20% glycerol. The tube was put in liquid nitrogen before being stored at -80°C. To revive the bacteria, the surface of the culture was rapidly scraped using a sterile inoculation loop and streaked onto a LB agar plate which was incubated O/N at 37°C.
3.2 Competent E. coli cells preparation
E. coli competent cells have been made by using the Inoue method (Inoue et al., 1990). This
protocol differs from other procedures in that the bacterial culture is grown at 18°C rather than the conventional 37°C and this leads to a higher efficiency of transformation. The lower temperature likely affects the composition or the physical characteristics of bacterial membranes synthesized at 18°C that are more favourable for uptake of DNA. The term "Transformation Efficiency" is defined as the number of colony-forming units (cfu) produced by 1 µg of plasmid DNA and it is used as a parameter to evaluate the quality of the competent E. coli cells preparation.
Protocol: E. coli DH5α cells from a glycerol stock solution were plated onto a LB agar plate and
incubated O/N at 37°C. A single bacterial colony was then aseptically transferred to a 10 ml tube containing 5 ml of sterile LB medium and placed O/N in a shaking incubator (37°C, 250 rpm). 1 ml was then used to inoculate a 1 L shake flask containing 250 ml of SOB medium and placed O/N on a shaker at a temperature of 18°C to achieve an OD600 = 0,6. When the appropriate OD600
value was reached, the culture was put on ice for 10 minutes, and kept cold for the rest of the procedure. The culture was then centrifuged for 10 minutes (2500g, 4°C). The supernatant was discarded and the pellet resuspended in 80 ml of ice-cold TB solution. The resuspension was left on ice for 10 minutes before being centrifuged again as described above. The supernatant was discarded and the pellet gently resuspended in 20 ml of TB solution. 1,4 ml of 100% DMSO was then added to give a final concentration of 7% DMSO (which is important in protecting the cells upon freezing). The solution was left on the ice for 10 minutes. Aliquots of 500µl were made into microcentrifuge tubes and immediately put into liquid nitrogen until frozen. The tubes were then stored at -80°C for later use.
To establish the transformation efficiency of the competent cells, a control transformation reaction, using a known amount of circular plasmid DNA, was performed.
19 Reagents: SOB medium Bacto Tryptone 20 g/L Yeast Extract 5 g/L NaCl 10 mM KCl 2,5 mM MgCl2 10 mM MgSO4 10 mM
Adjust pH to 6.7-7.0 with NaOH SOC medium SOB Glucose 20 mM TB solution Pipes 10 mM MnCl2 55 mM CaCl2 15 mM KCl 250 mM
Adjust pH to 6.7 with 5N KOH prior to adding the MnCl2. The solution was filter-sterilised and
stored at 4°C for immediate use.
3.3 Bacterial transformation
Transformation is a technique by which bacterial cells take up naked DNA more readily than by natural mechanisms. Transformation of bacteria with plasmids is important not only for studies in bacteria but also because bacteria are used as the means for replicating plasmids. Because of this, also plasmids designed for use in mammalian cells carry both a bacterial origin of replication and an antibiotic resistance gene to be used as a selectable marker in bacteria.
Protocol: Competent DH5α cells were thawed on ice. Cells were mixed gently by flicking the
bottom of the tube with finger and 200µl of cells were aliquoted for each transformation into 1,5 ml tubes. Then 10-100 ng DNA (for preparation of DNA solution to be used in mammalian cells) or 5-10µl DNA (in case of ligation products) were added to bacterial cells. Tubes were incubated on ice for 30 minutes and each transformation was then subjected to heat shock by placing the tube into a 42°C water bath for 45 seconds and subsequently on ice for 2 minutes. 800µl of SOC were added to each tube which was then placed in 37°C shaker for 1 hour. This outgrowth step allowed the bacteria to generate the antibiotic resistance proteins encoded on the plasmid so that they will be able to grow once plated on the antibiotic-containing agar plate. Indeed, after 1 hour of incubation, 200µl of each transformation are plated on LB-agar containing ampicillin (75
20 μg/ml) or kanamycin (30 μg/ml), depending on the resistance that the plasmid conferred to bacteria. Plates were incubated inverted at 37°C O/N to let the transformed colony appear.
Reagents:
LB medium Bacto Tryptone 10 g/L
Yeast Extract 5 g/L NaCl 10 g/L Add distilled water to final volume.
LB-agar medium Bacto Tryptone 10 g/L Yeast Extract 5 g/L NaCl 10 g/L Agar 15 g/L Add distilled water to final volume.
3.4 Preparation of plasmidic DNA
A plasmidic preparation is a method used to extract and purify plasmidic DNA. Many methods have been developed to purify plasmidic DNA from bacteria. These methods invariably involve three steps: growth of bacteria transformed with the plasmid, harvesting and lysis of the bacteria, and purification of plasmid DNA. Kits are available from varying manufacturers to purify plasmidic DNA, which are named by size of bacterial culture and corresponding plasmid yield. In increasing order, there are the miniprep, midiprep, maxiprep, megaprep and gigaprep. We used the QIAGEN Plasmid Maxi Kit (for maxi-preparation of plasmidic DNA) and the Wizard Plus Midipreps DNA Purification System from Promega (for midi-preparation of plasmidic DNA) according to the manufacturer's instructions in order to obtain DNA solutions to be used in mammalian cells. Mini-preparation of DNA was performed according to the "Boiling prep" method as described below. This is a rapid method for small-scale isolation of plasmidic DNA from bacteria. Mini-preparations were used in the process of molecular cloning to analyze bacterial clones.
3.4.1 Mini-preparation of DNA
Protocol: after transformation of bacteria with plasmid, a single colony growth on the plate was
touched gently with an inoculation loop and bacteria were transferred to a 10 ml tube containing 5 ml of sterile LB medium containing the appropriate antibiotic. The tube was then placed O/N in a shaking incubator (37°C, 250 rpm). 1,5 ml of culture was then centrifuged for 2 minutes (13000 rpm, 4°C) and the supernatant was discarded. 50µl of 25% sucrose (prepared in distilled
21 water) were added. The sample was then swirled for 30 seconds and 300µl of M-STET solution were added. The sample was swirled for 10 seconds and 25µl of 10 mg/ml lysozime (dissolved in TE buffer) was added. The tube was put at 100°C for 45 seconds and rapidly in ice. Then it was centrifuged for 15 minutes (13000 rpm, 4°C) and pellet was removed. 40µl of 3M sodium acetate (pH 5.2) and 270 µl of isopropanol were added to the supernatant, which was mixed for inversion. After 1 minute of incubation at room temperature the sample was centrifuged for 15 minutes (13000 rpm, room temperature) and the supernatant was removed. Pellet was washed with 250 µl of 70% cold ethanol and then air-dried. Pellet was finally resuspended in 40µl of 100µg/ml RNAsiA solution (dissolved in TE buffer).
Reagents: STET solution Triton X-100 5% EDTA (pH 8.0) 50 mM Tris-HCl (pH 8.0) 50 mM Sucrose 80 g/L TE buffer Tris (pH 7.5) 10 mM EDTA (pH 8.0) 1 mM
3.5 Molecular cloning
Molecular cloning is a set of experimental methods in molecular biology that are used to assemble recombinant DNA molecules and replicate them, using in vivo systems, for further processing. The essential steps of molecular cloning are the production of the nucleic acid fragment of interest, ligation (incorporating the fragment in an appropriate vector), transformation (incorporating the vector system/fragment of interest into an appropriate host), selection (obtaining hosts with the correct vector system/fragment of interest), amplification (manipulating the host to create multiple copies of the vector/fragment of interest), and isolation (isolate the vector/fragment of interest).
3.5.1 Primer design
All the plasmids generated in this study contain fragments that were PCR-amplified from either pcDNA3 2xHA-tagged Rab7a wt or pCMV6-ENTRY Myc-tagged vimentin wt plasmids. DNA
22 fragments were amplified using the primers (DNA oligonucleotides) indicated in Table 3.1 and designed according to the following criteria: primers length should be of 18-30 nucleotides, they should contain a sequence of 16-18 nucleotides complementary to the target region, the GC content should be between 40% and 60% with the 3' of the primer ending in C or G to promote binding, the melting temperature should be similar for pairs of primers and around 60°C-65°C, primers should be designed so that self-complementation and formation of secondary structures are avoided. All oligonucleotides contained restriction sites on the ends to allow easier downstream cloning. Restriction sites for AsiSI or EcoRV enzymes were included in forward primers used for vimentin and Rab7a amplifications, respectively. Restriction sites for MluI or NotI enzymes were included in reverse primers used for vimentin and Rab7a amplifications, respectively. Insertion of the vimentin and Rab7a fragments will occur at the same restriction sites situated on the acceptor plasmids.
DNA
fragment Primers (Forward/Reverse) Template
Vimentin 1-256 5'-TCTGCCGCCGCGATCGCCATGTCCACCAGG-3' pCMV6-ENTRY Myc-tagged vimentin wt 5'-CGTACGCGTGATTTGGACATGCTGTTC-3' Vimentin 92-256 5'-ATAGCGATCGCCATGATCAACACCGAG-3' pCMV6-ENTRY Myc-tagged vimentin wt 5'-CGTACGCGTGATTTGGACATGCTGTTC-3' Vimentin 256-411 5'-ATAGCGATCGCCATGATCGATGTG-3' pCMV6-ENTRY Myc-tagged vimentin wt 5'-CGTACGCGTAATCCTGCTCTCCTC-3' Vimentin 256-466 5'-ATAGCGATCGCCATGATCGATGTG-3' pCMV6-ENTRY Myc-tagged vimentin wt 5'-CCGCGTACGCGTTTCAAGGTCATC-3' Vimentin 1-141 5'-TCTGCCGCCGCGATCGCCATGTCCACCAGG-3' pCMV6-ENTRY Myc-tagged vimentin wt 5'-CTGATTACGCGTTTGGCCCTTGAGCTGCTC-3' Rab7a 11-207 5'-CCACGGGATATCCATGGTTATCATCCTGGGAGATTCTGGAG-3' pcDNA3 2xHA-tagged Rab7a wt 5'-ATAAGAATGCGGCCGCTCACTGCCCCTTCAGCAACTGC-3' Rab7a 1-176 5'-CGGGATATCCATGACCTCTAGGAAGAAAG-3' pcDNA3 2xHA-tagged Rab7a wt 5'-ATAAGAATGCGGCCGCTCACTGTTTAAGTGCATTCCT-3'
Table 3.1 List of primers used to amplify DNA fragments. DNA fragments produced, the
23
3.5.2 Polymerase Chain Reaction (PCR)
DNA fragments were PCR-amplified from plasmidic DNA as indicated in Table 3.1 by using
Taq polymerase. This DNA polymerase assembled a new DNA strand from nucleotides by using
single-stranded DNA as a template and primers which are required for initiation of DNA synthesis. Cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA generated new DNA. As PCR progressed, the new DNA was itself used as a template for replication so that DNA was exponentially amplified.
The mixture was prepared as follows:
Reaction buffer 1X DMSO 0-5% Forward primer 2 pM Reverse primer 2 pM dNTPs 200 µM Template DNA 40 µg Taq DNA polymerase 0,05U/µl
H2O up to 25 µl
DMSO was added as additive for the amplification of vimentin fragments in order to aid in the denaturing of the GC-rich template.
The following PCR profile was used:
First denaturation step 94 °C 7 minutes Denaturation step 94 °C 1 minute
30 cycles Annealing step 50 °C 30 seconds
Elongation step 72 °C 45 seconds Final elongation step 72 °C 7 minutes
End 4 °C ∞
3.5.3 Purification of PCR products
Qiaquick PCR purification kit (Qiagen) and GenElute Gel Extraction kit (Sigma) were used according to the manufacturer’s instructions for PCR product purification and to extract DNA fragments from agarose gels in order to remove primers, salts, agarose, ethidium bromide and other impurities. The PCR purification kit was preferred when no aspecific PCR products were
24 generated during the amplification reaction, otherwise samples were loaded on agarose gel and Gel Extraction kit was used by gel-extracting bands of the expected size.
3.5.4 Digestion of amplified products and acceptor plasmids
In order to clone the DNA fragments in the proper plasmids, compatible ends on both fragments and plasmids (pcDNA3 2xHA-tagged Rab7a wt; pCMV6-ENTRY Myc-tagged vimentin wt) were created by digesting them with appropriate restriction endonucleases.
Protocol: 1 g of DNA was digested with 3 U of each enzyme (AsiSi and MluI for cloning of
vimentin mutants, whereas EcoRV and NotI for cloning of Rab7a deleted mutants) in the buffer suggested by the manufacturer for 2 hours at 37°C. The reaction volume was 10 times the volume of enzymes used to dilute the glycerol of the enzyme storage buffer. Therefore, Rab7a wt and vimentin wt fragments were removed from pcDNA3 2xHA-tagged Rab7a wt and pCMV6-ENTRY Myc-tagged vimentin wt, respectively. At the end of the digestion step, enzymes activity was heat-inactivated.
3.5.5 Gel extraction and purification
GenElute Gel Extraction kit (Sigma) was used according to the manufacturer’s instructions for digestion products purification by extracting DNA fragments of the expected size from the agarose gel were they were loaded and removing salts, agarose, ethidium bromide and other impurities.
3.5.6 Agarose gel electrophoresis of DNA
Conventional agarose gel electrophoresis was used to analyze DNA fragments. The electrophoretic mobility of DNA fragments mainly depends on the fragment size and to a lesser extent on the conformation of the DNA, type and concentration of agarose used as well as applied voltage and electrophoresis buffer used.
Protocol: gels containing 1% (w/v) of agarose in TAE buffer were used for the separation of
DNA fragments. Ethidium bromide, an intercalating agent, was added to gels to the final concentration of 0,5 μg/ml for later visualization of the DNA bands. Before the loading, DNA samples were mixed with 1/6 volume of 6x loading buffer. 1 Kb DNA Ladder (Cat. 15615-024, Invitrogen) was used as a molecular weight standard. The electrophoresis was performed in TAE buffer. Finally, ethidium bromide-DNA complexes were visualized under UV transilluminator due to their bright red-orange fluorescence.
25 Reagents: Loading buffer Bromophenol blue 0,5% Xylene cyanol 0,5% Glycerol 30% Ladder 1 Kb DNA Ladder (1 µg/µl) 100 µl Loading buffer 150 µl TE buffer 643,5 µl 5M NaCl 6,5 µl TAE Tris 2 M
Acetic acid glacial 5,71% 0,5M EDTA (pH 8.0) 50 mM
3.5.7 Ligation
For cloning of DNA inserts into plasmids, a ligation reaction was set up for each vimentin and Rab7a mutant. Suitable controls without inserts, to check the amount of undigested or single-site digested plasmid, were included.
Protocol: amount of the used acceptor plasmid DNA varied from 25 to 50 ng. For the ligation
reaction, plasmid and insert DNA were generally taken in the molar ratio 1:10. The amount of insert to use, having a certain amount of vector, is given by the formula:
The ligation reaction was performed in a 20 µl final volume comprising plasmid and insert DNA, ligation buffer and T4 DNA Ligase (1U, Invitrogen). Ligation was carried out O/N at 16°C. The ligation reaction was then used for bacterial transformation.
3.5.8 Amplification of the new plasmids
Competent bacterial cells were transformed with the products of ligation in order to amplify the new plasmids generated. Several bacterial colonies from each sample were then singularly transferred to different 10 ml tubes containing 5 ml of sterile LB medium each and placed O/N in a shaking incubator (37°C, 250 rpm). Both LB-agar and LB medium contained the appropriate antibiotics to select the transformed bacteria. These cell cultures were used for mini-preparation of plasmidic DNA, which were then screened by digestion and analyzed by agarose gel
26 electrophoresis to identify positive bacterial colonies with the plasmids of interest containing the desired inserts. Plasmids that corresponded to the expected size after digestion with the enzymes used for cloning the insert inside the plasmid were sequenced to ensure an absence of PCR-introduced mutations. Finally, maxi-preparation or midi-preparations of plasmidic DNA was performed.
3.6 Site-directed mutagenesis
In vitro site-directed mutagenesis is an invaluable technique for creating plasmid modifications and studying protein structure-function relationships. This technique was used to make point mutations in a DNA sequence and switch amino acids in the relative protein. Pfu DNA Polymerase, synthetic oligonucleotide primers containing the desired mutation and a temperature cycler were used.
3.6.1 Primer design
The plasmid used as a template for site-directed mutagenesis was pcDNA3 2xHA-tagged Rab7a wt. In order to create pcDNA3 2xHA-tagged plasmids containing a set of nucleotides coding for mutated Rab7a proteins, pairs of complementary oligonucleotides containing the desired mutation, flanked by unmodified nucleotide sequence, were designed (Table 3.2).
Rab7a mutants to generate Primers (Forward/Reverse)
Rab7a N30A 5'-CACTCATGAACCAGTATGTGGCCAAGAAATTCAGTAATCAG-3' 5'-CTGATTACTGAATTTCTTGGCCACATACTGGTTCATGAGTG-3' Rab7a S34I 5'-GTGAACAAGAAATTCATTAATCAGTACAAAGC-3’ 5’-GCTTTGTACTGATTAATGAATTTCTTGTTCAC-3’ Rab7a S111G 5’-CTCATCCAGGCCAGGCCCCGGGATCC-3’ 5’-GGATCCCGGGGCCTGGCCTGGATGAG-3’ Rab7a N117A 5’-GCCAGTCCCCGGGATCCTGAAGCCTTCCCTTTCGTTGTGTTGGG-3’ 5’-CCCAACACAACGAAAGGGAAGGCTTCAGGATCCCGGGGACTGGC-3’ Rab7a K175E 5’-GCAAGGAATGCACTTGAACAGGAAACAGAGG-3’ 5’-CCTCTGTTTCCTGTTCAAGTGCATTCCTTGC-3’
Table 3.2 List of primers used to generate plasmids coding for Rab7a mutated proteins.
Rab7a mutants and primers used to generate them in the site-directed mutagenesis experiments are listed. Codons that contains point mutations (underlined) and that code for different amino acids are highlighted in yellow.
27
3.6.2 PCR reaction
The procedure utilized a supercoiled double-stranded DNA plasmid with an insert of interest (pcDNA3 2xHA-tagged Rab7a wt) and two synthetic oligonucleotide primers containing the desired mutation. The oligonucleotide primers, each complementary to opposite strands of the plasmid, were extended during temperature cycling by using Pfu DNA polymerase, which replicated both plasmid strands with high fidelity. Incorporation of the oligonucleotide primers generated a mutated plasmid containing staggered nicks.
The mixture was prepared as follows:
Reaction buffer 1X Primer #1 2,5 ng/µl Primer #2 2,5 ng/µl dNTPs 50 µM Template DNA 0,2 ng/µl Pfu DNA polymerase 0,05U/µl H2O up to 50 µl
The following PCR profile was used:
First denaturation step 95 °C 1 minute Denaturation step 95 °C 50 seconds
18 cycles Annealing step 60 °C 50 seconds
Elongation step 68 °C 1 minute/Kb of plasmid length Final elongation step 68 °C 7 minutes
End 4 °C ∞
3.6.3 DpnI digestion
Following temperature cycling, the PCR product was treated with DpnI endonuclease, which is specific for methylated and hemimethylated DNA. It was used to digest the parental DNA template, which had the nucleotides sequence coding for the wild type protein, and to select for mutation-containing synthesized DNA. In fact, the plasmid used as a template was dam methylated given that it was isolated by E. coli DH5α strain as indicated in paragraph 3.3.