DOI: 10.1002/ejoc.201000762
Solving the Puzzling Absolute Configuration Determination of a Flexible
Molecule by Vibrational and Electronic Circular Dichroism Spectroscopies and
DFT Calculations: The Case Study of a Chiral 2,2
⬘-Dinitro-2,2⬘-biaziridine
Sergio Abbate,*
[a,b]Alessia Ciogli,
[c]Stefania Fioravanti,
[d]Francesco Gasparrini,
[c]Giovanna Longhi,*
[a,b]Lucio Pellacani,
[d]Egon Rizzato,
[e]Domenico Spinelli,
[e]and
Paolo A. Tardella
[d]Keywords: Configuration determination / Circular dichroism / Density functional calculations / Conformation analysis / Biaziridines
The absolute configuration of a recently synthesized race-mate of 2,2⬘-dinitro-2,2⬘-biaziridine (2a), a possible catalyst for asymmetric synthesis, has been determined by vibrational circular dichroism (VCD) spectroscopy in the mid-IR region and DFT calculations. Electronic circular dichroism (ECD) spectra have been obtained and Time-Dependent DFT (TD-DFT) calculations have been performed and found to be in
Introduction
Recently, by a direct aza-MIRC (Michael-initiated ring closure) reaction promoted by CaO on nosyloxycarbamates (NsONHCO2R⬘; Ns = 4-NO2C6H4SO2, R⬘ = Et, Bn), the
relevant (⫾)-2,2⬘-dinitro-2,2⬘-biaziridines (2) were obtained by us[1]by starting from some
(E,E)-1,4-dialkyl-2,3-dinitro-1,3-butadienes (1; Scheme 1). Compounds 2, well decorated with interesting functional groups (aziridine ring, nitro, and carboxylate groups) and designed to furnish quite a few possible chemical modifications, represent versatile building blocks that are able to open the way to a variety of new compounds that could be useful as catalysts for asymmetric syntheses or as pharmacologically active substrates.
[a] Dipartimento di Scienze Biomediche e Biotecnologie, Università di Brescia,
Viale Europa 11, 25123 Brescia, Italy Fax: +39-030-3701157
E-mail: [email protected] [email protected] [b] CNISM,
Via della Vasca Navale 84, 00146 Roma, Italy
[c] Dipartimento di Chimica e Tecnologie del Farmaco, Università degli Studi “La Sapienza”,
Piazzale A. Moro 5, 00185 Roma, Italy [d] Dipartimento di Chimica, Università degli Studi
“La Sapienza”,
Piazzale A. Moro 5, 00185 Roma, Italy
[e] Dipartimento di Chimica “G. Ciamician”, Università degli Studi di Bologna,
Via Selmi 2, 40126 Bologna, Italy
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.201000762.
View this journal online at
agreement with the conclusions from VCD and DFT. A de-tailed conformational analysis, for approximately 300 geome-tries, has been carried out, allowing us to find evidence for the most populated conformers chiefly contributing to VCD and ECD spectra. The proposed absolute configuration and prevalent conformers are in correspondence with an equal configuration of the nitrogen atoms of the two aziridine rings.
Scheme 1. Formation of only one racemate out of ten possible stereoisomers of 2 (one enantiomer is depicted).
Examining the structures of biaziridines 2, one can ob-serve that they contain four stereogenic carbon atoms (stable at room temperature) that are related by symmetry in pairs. Also, aziridine nitrogen atoms are stereogenic, but in a dynamic fashion. Of course we will consider nitrogen inversion when discussing the conformational mobility. Taking into account just asymmetric carbon atoms, sym-metry considerations reduce the total number of stereoiso-mers from the possible sixteen (24) to only ten, four
cou-ples of enantiomers (2R,2⬘R,3R,3⬘R/2S,2⬘S,3S,3⬘S and 2R,2⬘R,3S,3⬘S/2S,2⬘S,3R,3⬘R with a C2 symmetry axis, as
well as 2R,2⬘R,3R,3⬘S/2S,2⬘S,3S,3⬘R and 2R,2⬘S,3R,3⬘R/ 2S,2⬘R,3S,3⬘S with no C2 symmetry axis) and two meso
forms (2R,2⬘S,3R,3⬘S = 2S,2⬘R,3S,3⬘R and 2R,2⬘S,3S,3⬘R = 2S,2⬘R,3R,3⬘S).
Quite unexpectedly, the HPLC analyses (carried out with UV, CD, and ELSD detectors simultaneously) of the reac-tion mixtures have shown that the aziridinareac-tion happens to give essentially only a pair (two enantiomers) out of the ten possible stereoisomers of the 2,2⬘-biaziridines 2.
Examina-tion of the 1H NMR spectra of the obtained 2,2
⬘-biazirid-ines has shown that the signals of the two aziridine protons are not coincident but are split apart by approximately δ = 0.45 ppm (i.e., they are not chemically equivalent) thus allowing us to exclude the two couples containing the C2
symmetry axis. Therefore, the obtained racemate must be one of the couples 2R,2⬘R,3R,3⬘S/2S,2⬘S,3S,3⬘R or 2R,2⬘S,3R,3⬘R/2S,2⬘R,3S,3⬘S. Moreover, by the action of sodium iodide, 2a (R = Me, R⬘ = Et) gave a rearrangement with ring enlargement furnishing the relevant 5,5 ⬘-dimethyl-4,4⬘,5,5⬘-tetrahydro-3,3⬘-bi(1,2,4-oxadiazole) 2,2⬘-dioxides (3a)[2]still as a single racemate through an S
N2–SN2
dom-ino ring-opening/ring-closure reaction with complete stere-oselectivity[1](Scheme 2).
Scheme 2. Ring expansion of 2a to 3a (only one enantiomer is de-picted for 3a).
This last result allows us to deduce that the two surviving chiral centers must be homochiral (the heterochiral combi-nation should give rise to a meso-oxadiazolidine) and that the major isolated couple of 2a must be the racemate 2R,2⬘S,3R,3⬘R/2S,2⬘R,3S,3⬘S (Figure 1).
Figure 1. Structure of (2R,2⬘S,3R,3⬘R)-2a.
The problem of configurational assignment of the two eluted enantiomers, 2R,2⬘S,3R,3⬘R versus 2S,2⬘R,3S,3⬘S, of racemate 2a is solved by first measuring their chiroptical properties and comparing them to the corresponding calcu-lated data on a chosen configuration. The advantage of using the vibrational circular dichroism (VCD) technique is in the fact that usually many bands with different signs are observed in the mid-IR region, and reliable VCD spectra calculations can be performed, as documented by well-set-tled literature relevant for this work.[3,4] For this reason, a
good correspondence between the observed spectra and the calculated ones provides unambiguous absolute configura-tion (AC) assignment. Of course other chiroptical proper-ties may give and will give, as shown later, further confi-dence to the assignment based on VCD.
Results and Discussion
To deepen the results of ref.[1], in this work we will
deter-mine the AC of the two enantiomers of 2a chosen as model
compounds. The first requirement, to carry out this project, was the availability of the pure enantiomers constituting the racemate 2a, by starting from a crude sample. The enanti-oseparation was successfully obtained on a covalently bonded polysaccharide-based stationary phase (Chiralpak IA) with an enantiomeric excess of approximately 97 % for both enantiomers. The first eluted enantiomer, named (–)-2a-I gives [α]D25= –174.76 (c = 0.26 w/v-%, CHCl3) and the
on-line CD-spectrum shows a negative CD band close to 275 nm. The second eluted enantiomer, (+)-2a-II, gives [α]D25= +167.19 (c = 0.26 w/v-%, CHCl3) and a positive CD
band at about 275 nm (Figure 2).
Since enantioselective chromatography and previously determined chiroptical properties ([α]D and on-line ECD)
do not define the AC of enantiomers of 2a, we have deter-mined their ACs by using chiroptical spectroscopy.
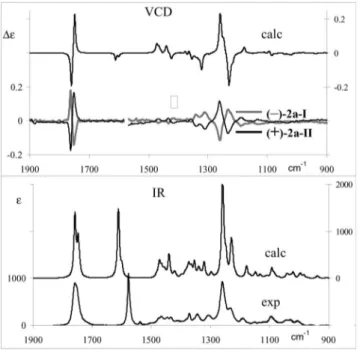
The measured VCD (Figure S1 of the Supporting Infor-mation) and electronic circular dichroism spectra (ECD; Figure S2 of the Supporting Information) for both enantio-mers, (–)-2a-I and (+)-2a-II, have been used to elucidate their AC[5–9]by DFT calculations. Many papers presented,
through the years, very convincing and consistent examples, whereby VCD, in conjunction with DFT calculations, al-lows one to go beyond the ambiguities of ECD spec-troscopy, when the results from the latter technique are ana-lyzed with an insufficient level of theory.[10] Nowadays,
time-dependent DFT (TD-DFT) has, most of the time, al-lowed the correct interpretation of the ECD spectra.[11–12]
However, VCD represents the most powerful technique for the goal of AC elucidation and was even demonstrated to be superior to X-ray diffraction data in a few instances.[13]
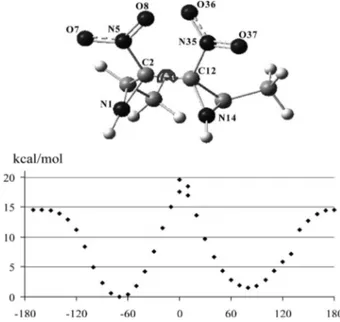
To establish AC, we have adopted the 2R,2⬘S,3R,3⬘R structure for our calculations. We report the schematics and a three-dimensional structure of the (2R,2 ⬘S,3R,3⬘R)-azirid-ine enantiomer in Figure 3, for which we have evidenced the definition of the most important conformational degrees of freedom and have presented the atomic numbering used throughout this paper. Indeed the major task is the confor-mational study of this molecule: VCD and absorption spec-tra are to be calculated on the optimized conformational structures. For conformational analysis, we have to consider all possible allowed values for torsions relative to the dihe-dral angles illustrated in Figure 3.
As a preliminary step, we have focused just on the central part of the molecule (Figure 4) to study the hindered rota-tion defined by the dihedral angle δ (N1–C2–C12–N14). With a scan at the B3LYP/6-31+G** level in Gaussian 03,[14]we obtain just two minima close to what we may call gauche(–) and gauche(+) conformers (corresponding to the
angles δ = –65 and +70°, respectively), the geometry corre-sponding to the trans position, which is associated with a flat maximum, the cis one instead exhibiting a very steep and high barrier.
Considering now the complete molecule (Figure 3), we assume that the two dihedral angles corresponding to the single C–O bonds, O=C–O–Et (C21–O17–C15–O16 and C24–O20–C18–O19), are in a cis position. This is usually the preferred conformation of such groups and, in this
par-Figure 2. Enantioseparation of 2a. UV 220 nm chromatographic traces of crude racemic 2a (A). Purity of first (B) and second (C) collected enantiomers by semi-preparative HPLC. CD 275 nm chromatographic trace of crude racemic 2a (D). On-line ECD spec-tra of both enantiomers (E).
ticular situation, the alternative trans conformation is for-bidden by steric hindrance between the ethyl groups and the adjacent ring. With these considerations in mind, we optimized the structure of the whole molecule at the AM1 level, by starting from initial conditions defined by the sys-tematic combination of the values reported below for the following dihedral angles:
Figure 3. Atomic numbering for heavier atoms mainly involved in the conformational analysis (top). Definition of the relevant dihe-dral angles for the 2R,2⬘S,3R,3⬘R enantiomer of 2a pointing out the most important conformational degrees of freedom (angles: δ,
τ, τ⬘, θ, θ⬘,φ,φ⬘; bottom).
Figure 4. Potential-energy curve (PEC) of the 2R,2⬘S,3R,3⬘R en-antiomer of a model molecule without CO2Et groups for rotation
of the dihedral angle δ (N1–C2–C12–N14) around the central C2– C12 bond. The PEC is obtained on the basis of DFT calculations at the B3LYP/6-31+G** level, the remaining degrees of freedom having been optimized (see text).
1) The dihedral angle δ, at the gauche(+) and gauche(–) positions.
2) The angles τ and τ⬘ (C2–N1–C15–O16 and C12–N14– C18–O19), each one possessing pairs of values differing roughly by 180°.
3) The angles θ and θ⬘ (C31–C21–O17–C15 and C27– C24–O20–C18), each one at the trans, gauche(+) and
gauche(–) positions.
4) Finally the anglesφ and φ⬘ (C15–N1–C2–C12 and C2– C12–N14–C18) for values of about 130° corresponding to an orientation for the CO2Et group relative to the aziridinic
ring plane that we may define as up (u), and of about 0° (cis) corresponding to an orientation for the same group
that we define down (d). These two values ofφ and φ⬘ are related to the configuration of the two aziridinic nitrogen atoms: the up position forφ corresponds to the S configura-tion of nitrogen 1 (Figure 3) and the up posiconfigura-tion ofφ⬘ corre-sponds to the R configuration of nitrogen 14. Since the in-version of nitrogen atoms strongly depends energetically on the other conformational degrees of freedom, at this point, we will consider the anglesφ and φ⬘ in the conformational analysis: the final result will define the absolute configura-tion of the two nitrogen atoms.
All these possibilities together amount to 288 initial geo-metries that need to be tested. However, outside this set, we had also to consider another 36 initial conformations for values of δ larger than 100°, namely beyond the second minimum of Figure 4, forφ values corresponding to the up position, whereas φ⬘ is in the down position, with the two usual possible values for τ and τ⬘ and the three possible values for θ and θ⬘, as above.
The whole 324 initial geometries converged to 286 dis-tinct conformers at the AM1 level, as reported in Table S1 of the Supporting Information, numbered in order of in-creasing energy. The conformers are labeled by evidencing the gross characteristics of the principal dihedral angles, namely the + or – sign for δ, u = up, d = down for both φ andφ⬘, a capital letter ranging from A through P assigned to possible combinations of τ and τ⬘ values, whereas for θ and θ⬘, we do not need to have any particular label, as will become clear further on.
From the calculations carried out at the AM1 level, the most probable conformations appear to be the ones with φ and φ⬘ up. We optimized the lower-energy conformers’ geometries at B3LYP/6-31+G** level and we calculated VCD and absorption spectra within the harmonic approxi-mation.[15,16]For a few cases, we checked that a change in
just θ and θ’ has little influence on the shape of the VCD spectrum. A comparison is shown in Figure S3 of the Sup-porting Information. In Figure S4 of the SupSup-porting Infor-mation we show the VCD spectra for a first set of eight conformers with φ and φ⬘ in the up position, with the two possible + and – values for δ, and for the four combinations A–D of two possible values for each angle τ and τ⬘: the eight conformations are indicated by the four-symbol nota-tion introduced above, namely –uuA, –uuB, –uuC, –uuD, +uuA, +uuB, +uuC, and +uuD.
The results obtained by considering the conformers with a low AM1 energy are unsatisfactory, especially with regard to the simultaneous match of the sign of the two VCD cou-plets centered at about 1765 and 1250 cm–1; for this reason,
we were forced to consider other conformations besides the ones discussed above, even if they exhibited high energy val-ues in the semi-empirical analysis. In this case, AM1 calcu-lations do not adequately represent the relative energies, es-pecially for the conformers differing in theφ and φ⬘ angles. Following this hypothesis, we have examined the other cases:φ = down and φ⬘ = up, φ = up and φ⬘ = down, and finallyφ = down and φ⬘ = down, obviously still considering the different possibilities for δ, τ, and τ⬘. To conclude, we took into account the lowest AM1 energy conformers in
each homologous δ, φ, φ⬘, τ, τ⬘ subset and optimized them at the B3LYP/6-31+G** level. To obtain the correct pop-ulation values, for the most probable cases thus obtained, we had to consider also the different possible values for θ and θ⬘ at the same level. All these tested examples are re-ported in Table S2 of the Supporting Information with the numbering adopted from AM1 results given in the first col-umn. It is evident that the preferred conformations have φ = up andφ⬘ = down.
Despite the large number of possible conformations, just a few conformers are populated (if one disregards ethyl ro-tations, i.e. angles θ and θ⬘); all of them have a δ value close to –60°, withφ always up. The different conformational be-havior with respect to angleφ and φ⬘ is due to the different chirality of the asymmetric carbon atoms C3 and C13: φ⬘
down lowers the steric hindrance between the ring methyl
and the CO2Et group. In Figure 5, the three most populated
Figure 5. Representation of the three most populated conformers: –udH (63 %), –uuC (17 %), and –udG (13 %) as obtained on the basis of DFT calculations at the B3LYP/6-31+G** level. The pop-ulation factors are summed over all possible conformations of the CO2Et groups (H atoms are not reported).
Figure 6. Comparison of the (–)-2a-I and (+)-2a-II experimental data (lower traces) with the Boltzmann weighted average calculated spectra of the 2R,2⬘S,3R,3⬘R enantiomer (calculated frequencies are scaled by a factor of 0.975).
conformers are reported with their population factors ob-tained by summing up over all the contributions due to dif-ferent θ and θ⬘ values. For the most probable conformers of Table S2 of the Supporting Information, we observe that the two carboxylic groups avoid being mutually parallel and the four possible combinations of values for τ and τ⬘, once fixed δ,φ, and φ⬘, correspond to exchanging the position of the two oxygen atoms of the carboxylic group (the confor-mation of the two carboxylic groups is indicated with the capital letter introduced above).
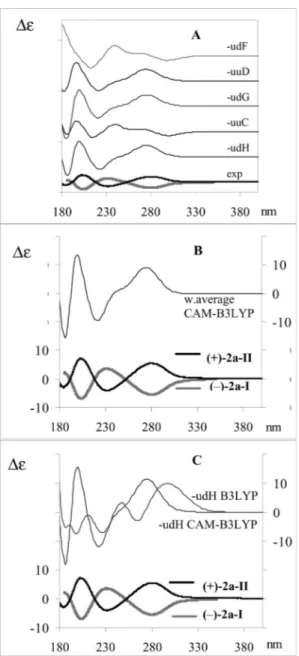
Figure 7. A) Comparison of experimental (lowest traces) with cal-culated (upper traces) ECD spectra of (–)-2a-I and (+)-2a-II for the five most populated conformers of the 2R,2⬘S,3R,3⬘R enantiomer, based on DFT calculations at the CAMB3LYP/6-31+G** level. B) Comparison of experimental (lower traces) with CAMB3LYP/6-31+G** level calculated (upper traces) ECD spectra for the confor-mational average. C) Comparison of experimental (lower traces) with calculated (upper traces) ECD spectra for the most populated conformer (–udH) obtained on the basis of DFT calculations at the B3LYP/6-31+G** level and at the CAMB3LYP/6-31+G** level.
We report in Figure S5 of the Supporting Information the VCD and absorption spectra of the most populated conformers (corresponding to a 93 % population within the subset of 70 cases reported in Table S2 of the Supporting Information). In Figure 6, finally we compare the experi-mental data with the Boltzmann-weighted average calcu-lated spectra, after scaling the calcucalcu-lated frequencies by a 0.975 factor for ease of comparison. A quite good corre-spondence is obtained: this allows us to establish unequivo-cally that the second eluted compound (+)-2a-II has a 2R,2⬘S,3R,3⬘R configuration. The fact that we obtain a good match of experimental and calculated spectra means also that the conformational analysis conducted here gives a good representation of the molecular system in CCl4
solu-tion, that is, the obtained population factors are reliable; in particular, the conformation here called –udH (allowing for complete mobility of end-ethyls) is populated at 63 %, and shows already almost the correct VCD spectrum. Two other probable conformers are –uuC and –udG, which are popu-lated at 17 and 13 %, respectively.
The assignment is also confirmed by the calculations of ECD spectra based on the TD-DFT method,[11,12] which
are presented in Figure 7. Also in this case, the conforma-tion of the terminal ethyl groups does not influence the shape of the spectra at the wavelengths of interest. We no-tice that the B3LYP functional does not perfectly account for the observed data, even though the assignment is still correct. It is known that the Coulomb-attenuated CAM-B3LYP functional[17,18]gives better results, especially for
N-containing molecules.[19]A comparison of the performance
of the two functionals on the most populated conformer is given in part C of Figure 7. The calculated average spec-trum at the CAM-B3LYP level is in good correspondence with experimental observations.
Conclusions
We reiterate that this work is strictly linked to our pre-vious work:[1] by allowing the complete definition of the
major pairs of enantiomers among the possible ones re-sulting from the reaction of Scheme 1. This work may pro-vide information concerning future work on catalysts for asymmetric syntheses. In addition, by definitively confirm-ing the structure of the isolated racemate, this work can also help the elucidation of the reaction mechanism, with respect to the generation and control of the four “stable” chiral centers involved in the bisaziridination process, which in turn may give rise to multiple conformations that need to be examined.
DFT calculations in vacuo are usually accepted as well suited to represent molecules in apolar solvents, such as CCl4. For this reason, after careful investigation of the
pos-sible values of important dihedral angles, one can adopt the population factors obtained on the basis of free-energy calculations in vacuo. This analysis shows that just a few conformers are important, and due to the fact that the quite large signals recorded in VCD spectra are nearly
unper-turbed by conformations of end-ethyls, we can state with confidence that the absolute configuration of the (+)-2a-II eluted biaziridine previously described by us[1] is
2R,2⬘S,3R,3⬘R. The study of this molecule has once more evidenced the inadequacy of semi-empirical calculations for even preliminary conformational screening, if adopted without a careful check. The number of possible conform-ers being high, one should consider the conformational analysis, even conducted at the DFT level, to be reliable only if supported by experimental data. For this reason, we think that it is quite important to be able to reproduce the VCD spectrum, which possesses many independent data, as well as the ECD spectrum, as conducted in the present work.
Coming finally to comment on the predominant confor-mation for the assigned configuration, namely the one we called –udH, we first observe that the negative central dihe-dral angle δ is predicted even at the semi-empirical level (see Table S1 of the Supporting Information). The succession ud attributed toφ and φ⬘, respectively, is also quite important: as previously noted, we observe that the conformational preference ud assigns the same S configuration to the N atoms of the two aziridine rings, if the lone pair is included in the definition of the AC of the N atom. Earlier in the literature,[20]VCD already succeeded to define the AC of N
atoms or, as defined in ref.[20], to choose between
“in-vertomers”. The conformational preference denoted as H is related to the dihedral angles τ and τ⬘, the first one with values at approximately –60° and the second one at approxi-mately +120°. The ensemble of the three conformational characteristics udH is seen (see Figure S5 in the Supporting Information) to be related to the correct prediction of the VCD doublet at approximately 1750 cm–1; indeed, they
en-sure the correct relative position of the C=O bonds of the two carboxylic groups. Regarding the VCD doublet at ap-proximately 1250 cm–1, almost all calculated VCD spectra
of Figure S5 of the Supporting Information are correct in that respect; the wider bandwidth observed for this doublet is due to the differences in calculated frequencies of the doublet depending on conformation. Finally, the relative position of the two nitro groups is mostly determined by the negative δ angle, the stretching vibrations of these groups are responsible of the sharp intense absorption band at 1590 cm–1.
Experimental Section
General: Full details concerning materials (2a, 3a, CH2Cl2, CHCl3,
CCl4, and MeCN), separation of 2a by chiral chromatography,
de-termination of polarimetric, mid-IR VCD, and ECD measure-ments, as well as DFT and TD-DFT calculations for the 2R,2⬘S,3R,3⬘R enantiomer of 2a are reported in the Supporting Information.
Superimposed experimental VCD spectra of (–)-2a-I and (+)-2a-II and superimposed experimental ECD spectra of (–)-2a-I and
(+)-2a-II are reported in Figures S1 and S2, respectively (Supporting
Information).
Energy values and geometrical characteristics of the conformers obtained with AM1 optimization are reported in Table S1 (Sup-porting Information). Energy values and geometrical characteris-tics of the conformers obtained with B3LYP/6-31+G** optimiza-tion are reported in Table S2 (Supporting Informaoptimiza-tion). Compari-son of VCD spectra on two groups of conformers discussed in the text to analyze the dependence of VCD spectra on the different dihedral angles are reported in Figures S3 and S4 (Supporting In-formation). In Figure S5 (Supporting Information) full comparison of calculated IR and VCD spectra for the first most populated 20 conformers of (2R,2⬘S,3R,3⬘R)-biaziridine with the superimposed experimental spectra of (–)-2a-I and (+)-2a-II is given. This mate-rial is available free of charge via Internet at http://www.chemeur-j.org.
Supporting Information (see also the footnote on the first page of
this article): In this section the complete description of experimen-tal procedures and DFT calculations with references are listed.
Acknowledgments
The Italian Ministero dell’Università e della Ricerca (MIUR) and Università degli Studi di Roma “La Sapienza” are gratefully ac-knowledged for financial support (PRIN 2007FJC4SF_005, PRIN 20078J9L2A_005, PRIN, 2008LYSEBR_003). The Consorzio In-teruniversitario Lombardo per l’Elaborazione Automatica (CILEA), via R. Sanzio 4, 20090 Segrate (MI), Italy, is acknowl-edged for kindly allowing the use of computational facilities.
[1] A. Ciogli, S. Fioravanti, F. Gasparrini, L. Pellacani, E. Rizzato, D. Spinelli, P. A. Tardella, J. Org. Chem. 2009, 74, 9314–9318. [2] Several 1,2,4-oxadiazole derivatives, which can be considered hydrolysis-resistant bioisosters of esters or amides, have been suggested and/or used as antitussive agents, coronary vasodila-tors, local anaesthetic, antispasmodic, antiinflammatory and analgesic agents, muscarinic agonists and antagonists, depress-ant of CNS, etc. (A. R. Katritzky, C. A. Ramsdem, E. F. V. Scriven, R. J. K. Tylot (Eds.), Comprehensive Heterocyclic
Chemistry III, vol. 5, Elsevier, London, UK). Very recently,
some derivatives have shown interesting properties as inhibitors of MDR1 activity (M. Viale, C. Cordazzo, B. Cosimelli, D. de Totero, P. Castagnola, C. Aiello, E. Severi, G. Petrillo, M. Cianfriglia, D. Spinelli, J. Med. Chem. 2009, 52, 259–266). [3] P. J. Stephens, F. J. Devlin, F. Gasparrini, A. Ciogli, D. Spinelli,
B. Cosimelli, J. Org. Chem. 2007, 72, 4707–4715.
[4] P. M. Wood, L. W. L. Woo, J. R. Labrosse, M. N. Trusselle, S. Abbate, G. Longhi, E. Castiglioni, F. Lebon, A. Purohit, M. J. Reed, B. V. L. Potter, J. Med. Chem. 2008, 51, 4226–4238. [5] P. J. Stephens, F. J. Devlin, J. J. Pan, Chirality 2008, 20, 643–
663.
[6] L. A. Nafie, T. B. Freedman, Vibrational Optical Activity
Theory, in: Circular Dichroism: Principles and Applications, 2nd
ed. (Eds.: N. Berova, K. Nakanishi, R. W. Woody), Wiley-VCH, New York, 2000, chapter 4, pp. 97–132.
[7] P. L. Polavarapu, J. He, Anal. Chem. A 2004, 76, 61A–67A. [8] T. Kuppens, P. Bultinck, A. Langeneker, Drug Discovery
To-day:. Technologies 2004, 269–275.
[9] G. Longhi, S. Abbate, R. Gangemi, E. Giorgio, C. Rosini, J.
Phys. Chem. A 2006, 110, 4958–4968.
[10] A. Aamouche, F. J. Devlin, P. J. Stephens, J. Am. Chem. Soc.
2000, 122, 2346–2354.
[11] C. Diedrich, S. Grimme, J. Phys. Chem. A 2003, 107, 2524– 2539.
[12] A. E. Hansen, T. D. Bouman, Adv. Chem. Phys. 1980, 44, 454– 644.
[13] P. J. Stephens, F. J. Devlin, P. Besse, Tetrahedron: Asymmetry
[14] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morok-uma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzew-ski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian 03, rev. B.05, Gaussian, Inc., Pittsburgh, PA, 2003.
[15] P. J. Stephens, J. Phys. Chem. 1985, 89, 748–752.
[16] a) P. J. Stephens, M. A. Lowe, Ann. Rev. Phys. Chem. 1985,
36, 213–241; b) P. J. Stephens, C. S. Ashvar, F. J. Devlin, J. R.
Cheeseman, M. J. Frisch, Mol. Phys. 1996, 89, 579–594. [17] T. Yanai, D. P. Tew, N. C. Handy, Chem. Phys. Lett. 2004, 393,
51–57.
[18] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morok-uma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzew-ski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian 09, Gaussian, Inc., Pittsburgh, PA, 2009.
[19] M. J. G. Peach, C. R. Le Sueur, K. Ruud, M. Guillaume, D. J. Tozer, Phys. Chem. Chem. Phys. 2009, 11, 4465–4470. [20] A. Rauk, T. Eggimann, H. Wieser, G. V. Shustov, D. Y. Yang,
Can. J. Chem. 1994, 72, 506–513.
Received: May 27, 2010 Published Online: September 23, 2010