UNIVERSITY OF GENOA
School of Medical and Pharmaceutical Sciences
Master’s degree course in Medicine and Surgery
DEGREE THESIS
SURGICAL VENTRICULAR REMODELING IN ISCHEMIC HEART
FAILURE:
THE IMPACT OF OPTIMAL VOLUME REDUCTION ON LONG-TERM
OUTCOME
SUPERVISOR CANDIDATE
Francesco Santini, M.D. Francesca Zanin
CO-SUPERVISORS
Antonio Salsano, M.D.
Serenella Castelvecchio, M.D.
Lorenzo Menicanti, M.D.
Ai miei genitori,
i miei punti cardinali
INDEX
1. Introduction ... 6
2. Heart Failure ... 10
2.1. Definition and classification ... 10
2.2. Epidemiology and Impact on the population ... 15
2.3. Etiology ... 17
2.3.1. Ischemic etiology ... 19
2.4. Pathophysiology ... 22
2.4.1. Left Ventricular Remodeling ... 25
2.5. Diagnosis ... 29
2.5.1. Clinical presentation ... 30
2.5.2. Physical examination ... 31
2.5.3. Investigations ... 33
2.5.3.1. Noninvasive single or combined Imaging techniques ... 36
2.5.3.2. Invasive Imaging techniques ... 38
2.5.4. Algorithm for diagnosis of HF ... 39
2.6. Prognosis ... 40
3. Current therapeutic strategies for chronic ischemic-based HFrEF ... 43
3.1. Medical therapies ... 43
3.1.1. Management of fluid retention ... 45
3.1.2. Prevention of disease progression ... 47
3.2. Device therapy ... 49
3.2.1. Cardiac resynchronization therapy (CRT) ... 50
3.2.2. Implantable cardioverter defibrillator (ICD) ... 51
3.2.3. Left ventricular assist device (LVAD) ... 52
3.3. Surgical strategies ... 53
3.3.1. Cardiac transplantation ... 53
3.3.2. Coronary artery bypass grafting (CABG) ... 54
3.3.3. Mitral valve repair or replacement (MVR) ... 55
3.3.4. Surgical Ventricular Reconstruction (SVR) ... 56
4. Surgical Ventricular Reconstruction (SVR) ... 57
4.1. Rationale to perform SVR ... 57 4.2. Indications ... 58 4.3. History ... 61 4.4. Technique ... 61 4.5. Particular conditions ... 64 4.5.1. Mitral valve (MV) ... 64
4.5.2. Anterior versus Postero-inferior Remodeling ... 67
5. Previous studies ... 71
5.1. Dor and co-authors ... 71
5.2. The Restore Group ... 71
5.3. Menicanti, Castelvecchio et al. ... 72
5.4. The role of LVESVI: White et al., the GUSTO-I trial and Bax et al. ... 73
5.5. The STICH trial ... 74
5.6. Di Donato, Castelvecchio and Menicanti ... 75
5.7. Witkowski et al. ... 76
5.8. Criticisms of the STICH trial ... 77
5.9. Michler et al. ... 79
5.10. ESC/EACTS Guidelines on Myocardial Revascularization ... 80
6. Retrospective analysis at IRCCS Policlinico San Donato ... 82
6.1. Introduction ... 82
6.2. Aim of the study ... 83
6.3. Materials and Methods ... 83
6.3.1. Study design ... 83
6.3.2. Selection of Patients ... 84
6.3.3. End points ... 85
6.3.4. Methods ... 85
6.3.5. Statistical analysis ... 86
7. Results and discussion ... 87
7.1. Results ... 87
7.2. Discussion ... 94
8. Conclusions ... 97
Bibliography ... 98
1. Introduction
Heart failure (HF), to use a term widely popular at present, can be defined as a global pandemic, i.e. “an epidemic occurring worldwide, or over a very wide area, crossing international boundaries and usually affecting a large number of people”1, since it is estimated to involve at least 26 million persons globally, with a 2% overall prevalence in the adult population, and >10% among individuals aged >70 years. Despite medical advances in therapies and implementation of prevention campaigns, its prevalence shows no sign of abating; indeed, it is increasing dramatically as the population ages, weighing heavily on global health expenditures. Moreover, mortality and morbidity of the disease remain high, and the quality of life remains poor. Projections for the coming years in terms of prevalence, hospitalization rates, and healthcare costs are even more alarming, making HF one of the major public health challenges worldwide.
Although it is very difficult to provide a single definition of the complexity of this disease, according to the European Society of Cardiology, HF can be defined as a clinical syndrome, characterized by typical symptoms and signs, caused by a structural and/or functional cardiac abnormality, which results in reduced cardiac output and/or elevated intracardiac pressures at rest or during stress. In short, it is a progressive condition that inevitably leads to frequent hospitalization and decreased life expectancy.
HF may result from a wide range of structural or functional cardiac and non-cardiac disorders. However, in the Western world, ischemic heart disease has become the predominant cause, being accountable for 46% to 68% of cases, with significantly higher mortality than those with non-ischemic etiologies.
To date, the cornerstone of treatment remains guideline-driven medical therapy, with significant improvement in survival and quality of life, followed by use of devices and surgical procedures, such as coronary artery bypass grafting and mitral valve replacement, or ultimately, cardiac transplantation. However, progression of HF, despite optimal pharmacological management, highlights the process of left ventricular (LV) remodeling as the main culprit, due to the mechanical burden produced by the changes that accompany HF. LV remodeling, meant as a set of
changes in LV geometry, architecture, and in the components of the myocardium, eventually leads to a vicious cycle where LV dilation keeps increasing in a wasted effort to maintain normal cardiac output, the ejection fraction (EF) decreases, and hemodynamic overloading worsens, any or all of which are sufficient to continued aggravation of LV dilation, global ventricular function, wall stress, geometric distortion, and, therefore, the progression of HF.
Recent studies have shown that LV remodeling can be reversed, decreasing LV mass and optimizing its shape and therefore improving clinical outcomes in patients with HF and reduced ejection fraction (HFrEF). Indeed, one of the goals of therapy for HF is to prevent and/or reverse LV remodeling, which is the aim of surgery in the present analysis on surgical ventricular remodeling (SVR).
SVR has been introduced as an optional therapeutic strategy to commonly used coronary artery bypass grafting (CABG), and aims to counteract and reverse LV remodeling by reducing the LV to a more physiological volume through the exclusion of scar tissue, and optimizing the shape of the distorted chamber, thereby improving both cardiac function and clinical outcomes of patients with HFrEF. The goal of surgery is also that of decreasing myocardial systolic and diastolic wall stress, resulting in improved wall compliance and reduced filling pressure. Moreover, since wall stress is an important determinant of afterload, it may also enhance LV contractile performance by increasing the extent and velocity of systolic fiber shortening.
This procedure was originally introduced by the French cardiac surgeon Vincent Dor in 1985, based on other prior contributions. From Dor’s group onwards, several studies, mainly observational, have been carried out on HF patients in order to assess SVR true potential. Many reports have confirmed the efficacy of SVR in improving LV systolic function, NYHA functional class, and survival, through reduction in ventricular volume and an increase in the EF, not only in patients with classic dyskinetic aneurysm, but also in those with dilated ischemic cardiomyopathy and severe LV dysfunction, with favorable 5-year outcomes.
However, the surgical procedure was strongly criticized after the publication of the STICH trial in 2007, in which SVR did not meet the expected results. Nowadays,
American surgeons seem reluctant to use the procedure, which has been substantially abandoned. Indeed, several limitations have led to significant uncertainty in making such results generalizable, which relates to the heterogeneous population of patients enrolled in the trial.
While waiting for further analysis of STICH data, the 2018 Task Force of the European Society of Cardiology/European Association for Cardiothoracic Surgery on Myocardial Revascularization currently recommended SVR at the time of CABG in selected patients undergoing intervention in centers with a high level of surgical expertise.
Despite the disappointing results, when combining SVR with CABG in the STICH trial there was significantly greater reduction in the left ventricular end systolic volume index (LVESVI), and it was hypothesized that a poor reduction in volume may be related to the unsatisfactory outcomes. Later, Witkowski et al. confirmed the importance of a ‘target volume’ to be achieved with SVR, showing that a residual postsurgical LVESVI of at least 60 ml/m2 was independently associated with a favorable outcome. Moreover, a post hoc analysis from the STICH trial showed that a postoperative LVESVI of 70 ml/m2 or lower resulted in improved survival compared with CABG alone.
Several studies have already highlighted the importance of residual LVESVI in patients with ischemic-based HF undergoing SVR, showing the existence of an association between a post-operative LVESVI ≥60 mL/m2 and adverse outcomes. However, its impact on prognosis is still not well-established. Therefore, we aimed to further investigate the role played by residual LVESVI in the definition of the long-term outcomes through retrospective analysis over 14 years focusing on patients with chronic HF with and reduced EF.
This dissertation will begin with a detailed description of HF in its entirety, starting with the definition, classification, and epidemiology, followed by the etiology, pathophysiological mechanisms, diagnostic process, and prognostic factors. In particular, focus will be placed on description of ischemic-based HF, and the effects of left ventricular remodeling and its role as the primary determinant in the progression of disease and as the target of surgical treatment are later explained.
The current medical, device-based, and surgical therapeutic strategies adopted for chronic ischemic-based HF will be then outlined, with particular attention to SVR, the surgical procedure at the core of the present analysis.
After an overview of the relevant literature, the results of the retrospective study will be presented. The analysis was conducted under the direction and expertise of Dr. Serenella Castelvecchio and Dr. Lorenzo Menicanti at IRCCS San Donato Hospital, in Milan, which has the largest worldwide series and represents a reference center for the International surgical community. Since July 2001, the SVR team started to collect data in a prospectively manner, with regular follow-up over time, in order to keep examine changes in LV geometry and their effect on survival. This retrospective study, carried out between January 2002 and December 2016 on 297 patients, assessed the impact of the achievement of optimal left ventricular volume reduction obtained by SVR, expressed as “optimal post-operative LVESVI” < 60 ml/m2 on long-term outcomes.
2. Heart Failure
2.1. Definition and classification
In order to properly understand the relevance of the present analysis, it is essential to overview the basic pathophysiology of ischemic heart failure (HF).
Despite multiple attempts over time to develop an exhaustive definition that encompasses the heterogeneity and complexity of HF, no single conceptual paradigm has been determined. The current American College of Cardiology Foundation (ACCF)/American Heart Association (AHA) guidelines, updated in 2017, define HF as a complex clinical syndrome that results from structural or functional impairment of ventricular filling or ejection of blood. Consequently, there is a failure in delivering oxygen to organs and tissues according to metabolic requirements. The cardinal clinical picture includes symptoms of dyspnea and fatigue, which limit exercise tolerance, and signs of peripheral edema and rales caused by fluid retention, leading to pulmonary and/or splanchnic congestion.2–4
The latest European Society of Cardiology (ESC) guidelines for the diagnosis and treatment of acute and chronic HF define it as a clinical syndrome characterized by typical symptoms (e.g. breathlessness, ankle swelling and fatigue) that may be accompanied by signs (e.g. elevated jugular venous pressure, pulmonary crackles and peripheral oedema) caused by a structural and/or functional cardiac abnormality, resulting in a reduced cardiac output and/ or elevated intracardiac pressures at rest or during stress.5
Basically, both definitions agree that HF is a progressive condition which inevitably leads to frequent hospitalizations, poor quality of life, and decreased life expectancy. HF may result from many disorders at various levels of the cardiovascular system, involving the myocardium, pericardium or endocardium, rhythm and conduction, valves, or large vessels. However, in most patients, the symptoms originate from the Left Ventricular (LV) systolic and/or diastolic dysfunction caused by a myocardial injury.
Although multiple criteria have been proposed to categorize HF over time, the main classification used today in Europe, as illustrated in the 2016 ESC HF Guidelines, is based on measurement of the Left Ventricular Ejection Fraction (LVEF). This value can be measured by several invasive and noninvasive imaging modalities, the most common of which is the echocardiogram. LVEF quantifies ventricular damage by estimating LV systolic function, calculated as the ratio of blood ejected during systole (stroke volume, also calculated as the difference between the end diastolic and the end systolic volume) to blood in the ventricle at the end of the diastole (end-diastolic volume).
HF encompasses a broad spectrum of LV dysfunction, from patients with normal LV size and preserved EF, to those with critical dilatation and/or significantly reduced EF.
Based on LVEF, HF can be divided into three subgroups:
• HF with reduced LVEF (HFrEF, LVEF<40%), previously referred to as systolic HF, It is defined as impaired emptying of the LV and evident as a decreased effective ejection fraction.6 These are patients who are mainly be enrolled in clinical trials on HF.7
• HF with mid-range LVEF (HFmrEF, LVEF=40-49%); studies have shown that patients in this “gray area” present clinical features that are intermediate between HFrEF and HFpEF, but with a clinical profile and outcomes that are largely similar to those with HFpEF.
• HF with preserved LVEF (HFpEF, LVEF>50%), previously termed diastolic HF, “it is defined as a condition in which filling of the LV is sufficient to produce adequate cardiac output but requires elevated pulmonary venous pressure. Thus, diastolic dysfunction is clinically manifested as pulmonary congestion” 6,8. “Patients with HFpEF generally do not have a dilated LV, but instead often have an increase in LV wall thickness and/or increased left atrial (LA) size as a sign of increased filling pressures. Most have additional ‘evidence’ of impaired LV filling or suction capacity, also classified as diastolic dysfunction.” 5 These patients are more likely to be older, female, hypertensive, with atrial fibrillation, and to have a non-ischemic etiology of heart failure.
This classification is relevant because demographics, comorbid conditions, clinical parameters, prognosis, and response to therapies differ significantly among the three categories.
Figure 1- Definition of heart failure with preserved (HFpEF), mid-range (HfmrEF) and reduced
ejection fraction (HfrEF)5
Although the 2016 ESC classification is the most relevant for the present study, there are other ways to define HF.
HF can also be described according to the time course of the disease. The term HF is commonly used to describe a symptomatic syndrome, but a patient can also be rendered asymptomatic by treatment. If a patient has never exhibited the typical clinical picture, but presents HfrEF, is described as having asymptomatic LV systolic
dysfunction. Furthermore, chronic HF describes a patient who has had HF for some
time, and stable refers to a patient with symptoms and signs that have remained generally unchanged for at least 1 month. If the patient’s conditions deteriorate, suddenly or slowly, as the HF is referred to as decompensated, which often leads to hospital admission, negatively impacting prognosis. The acute new onset of HF, termed de novo HF, may be a consequence of Acute Myocardial Infarction (AMI), or may develop subacutely in a patient with a preexisting dilatated cardiomyopathy (DCM). Congestive HF is used to describe acute or chronic HF with evidence of volume overload.
As described later, the present analysis focuses on patients presenting chronic HF with significantly reduced LVEF (HfrEF), and in particular those with an LVEF<35% with concomitant coronary disease.
Another important classification widely used in practice and clinical studies is that of the New York Heart Association (NYHA), which determines the severity of HF in relation to estimation of functional ability and exercise intolerance. It places patients in one of four categories, based on their limitations during physical activities, mainly the amount of exertion needed to provoke the onset of angina and shortness of breath.
NYHA HF classes are as follows: Class Patient Symptoms
I No limitation of physical activity. Ordinary physical activity does not cause undue fatigue, palpitation, dyspnea (shortness of breath).
II Slight limitation of physical activity. Comfortable at rest. Ordinary physical activity results in fatigue, palpitation, dyspnea (shortness of breath).
III Marked limitation of physical activity. Comfortable at rest. Less than ordinary activity causes fatigue, palpitation, or dyspnea.
IV Unable to carry on any physical activity without discomfort. Symptoms of HF at rest. If any physical activity is undertaken, discomfort increases.
Table 1 – New York Heart Association (NYHA) Functional Classification- Patient Symptoms9
Class Objective Assessment
A No objective evidence of cardiovascular disease. No symptoms and no limitation in ordinary physical activity.
B Objective evidence of minimal cardiovascular disease. Mild symptoms and slight limitation during ordinary activity. Comfortable at rest.
C Objective evidence of moderately severe cardiovascular disease. Marked limitation in activity due to symptoms, even during less-than-ordinary activity. Comfortable only at rest.
D Objective evidence of severe cardiovascular disease. Severe limitations. Experiences symptoms even while at rest.
Table 2 – New York Heart Association (NYHA) Functional Classification – Objective Assessment9
In the United States, the most common definition of HF is The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) classification, which describes four stages of HF development, based on structural changes and symptoms.
Figure 2 – American College of Cardiology Foundation/American Heart Association (ACCF/AHA)
Both classifications emphasize the development and progression of disease, focus on exercise capacity and symptoms, and provide useful information about the severity of HF. However, symptom severity correlates poorly with many measures of LV function; although there is a clear relationship between severity of symptoms and survival, patients with mild symptoms may still have an increased risk of hospitalization and death.
2.2. Epidemiology and Impact on the population
“HF has been defined as global pandemic, since it affects around 26 million people worldwide”.11,12
Despite many breakthroughs in cardiovascular medicine, HF still represents a burgeoning problem worldwide, and one of the major public health challenges in Western Countries in terms of the number of patients. HF is one of the leading causes of hospitalizations (>20%) for persons older than 65 years old, and with rates of hospital readmission within 6 months going from 25% to 50%, it appears clear that the burden of economic implications behind HF is more than heavy.13,14 Projections are even more alarming, with total costs that are expected to increase by 127% between 2012 and 2030.12
Although it depends on the definition applied, the overall prevalence of the disease in the adult population is 2%, and increases with age following an exponential pattern, and is >10% among individuals aged >70 years. Among people >65 years of age presenting to primary care with breathlessness on exertion, one in six will have unrecognized HF.5,15
In North America and Europe, the lifetime risk of developing HF is estimated to be 20% for anyone older than 40 years, and at age 55 is 33% for men and 28% for women. Regarding gender distribution, the relative incidence of HF is lower in women than in men, but at least one-half of the patients with HF are women, due to their longer life expectancy.
Figure 3 Prevalence and Incidence of Heart Failure Worldwide12
Moreover, the 5-year survival rate of patients diagnosed with HF is still <50% and might even be underestimated16, 5-year mortality after hospitalization exceeds 40%, and 30-40% of patients die within 1 year after diagnosis.17 The most recent European studies (ESC-HF pilot study) reported that one-year all-cause mortality rates for hospitalized and stable/ambulatory HF patients were 17% and 7%, respectively, with one-year hospitalization rates of 44% and 32%, respectively. Most deaths are attributable to cardiovascular causes, and mortality rates are generally higher in HFrEF than in HFpEF.1819
Furthermore, patients with ischemic-based left ventricular (LV) systolic dysfunction have significantly higher mortality rates than those with non-ischemic etiologies, and HF is associated with ischemic heart disease in 46% to 68% of cases.2012
In the United States, approximately 5.7 million people have HF, leading to 271,000 deaths per year, but the projections are worrisome, since it is expected that by 2030 more than 8 million people will have the condition, accounting for a 46% increase in prevalence.17
While research has been effective in delivering major advances in therapy over the last 30 years, including drugs, devices, and surgery, the prevalence of chronic HF remains high, and prognosis is still poor, in part because improvements in treatments for cardiac diseases have dramatically allowed patients to survive longer
by slowing the progression of HF.2,7,21 This, together with the ageing of the population, can help to explain the increase in prevalence expected in the next few years. 12
2.3. Etiology
HF is a heterogeneous syndrome that may result from a wide range of structural or functional cardiac and non-cardiac disorders. It is not to be considered as a single pathology entity, but rather as a process that may occur in the final stages of most cardiac diseases. Moreover, establishing the etiology of HF is fundamental in order to define the appropriate management for the patient, choose the most suitable investigations and therapeutic strategies, and avoid future episodes of decompensation. Unfortunately, it is often challenging, because while the primary cause may be easy to determine, more than one underlying cause may coexist and contribute to the progression of HF.
Although disorders of the endocardium, pericardium, and large vessels are possible, myocardial disorders are the most common causes of HF, subdivided into those with preserved LVEF and those with reduced LVEF. The etiology of HF in patients with a HFpEF presents slight differences from that of HFrEF, although there is substantial overlap between the etiologies of the two conditions.
Generally, as shown in the table below, any clinical condition that results in alterations of the LV structure or function may predispose to development of HF.
Figure 4 Etiologies of Heart Failure2
In Western countries, coronary artery disease (CAD) has become the predominant cause of HF, being responsible for 46-68% of cases. Hypertension contributes to the development of HF in 75% of patients, most with CAD. CAD and hypertension together augment the risk of HF, as does diabetes mellitus. Other frequent causes include arrhythmias, valvular heart disease, and alcohol. In 20–30% of cases of HFrEF, the exact etiology remains unknown, and patients are referred to as having nonischemic, dilated, or idiopathic cardiomyopathy. Other conditions that may lead to a dilatated cardiomyopathy, and therefore HF, are prior viral infection or toxin exposure (e.g. alcoholic or chemotherapeutic), metabolic conditions, infection, and iatrogenic causes.
Genetic and mitochondrial anomalies have been thought to be less frequent, even if it is becoming increasingly clear that a consistent number of dilated cardiomyopathies previously termed “idiopathic” have a familial link, and are secondary to specific genetic defects, most notably those in the cytoskeleton, some of which inherited in an autosomal dominant fashion. Mutations in genes that encode cytoskeletal proteins (desmin, cardiac myosin, vinculin) and nuclear
19 membrane proteins (laminin) have been identified. Dilated cardiomyopathy can be also associated with Duchenne’s, Becker’s, and limb-girdle muscular dystrophies. Finally, conditions that result in a high cardiac output (e.g. arteriovenous fistula, anemia) are rarely responsible for the development of HF in a normal heart. Nevertheless, in the presence of underlying structural heart disease, these conditions can lead to clinically evident HF.
Figure 5 Causes of heart failure in population-based studies: “The variation in frequencies of causes
of heart failure reported in different studies can be explained by differences in study population, from the highly selected group of participants in clinical trials to relatively unselected participants in population-based studies, differences in definitions, and time differences”15
2.3.1. Ischemic etiology
HF is correlated with ischemic heart disease (IHD) in 46% to 68% of cases. As discussed later, the present analysis involves only patients suffering from ischemic-based HF, and thus it is necessary to place focus on this specific etiology.
IHD is responsible for more deaths and disabilities and greater economic costs than any other disease in the western world. According to the ISTAT 2017 Report on the 25 leading causes of death in Italy in 2003 and 2014, IHD is the leading cause, with 69,653 deaths in 2014, followed by cerebrovascular diseases (57,230), and other cardiac disorders (49,554). Together, these three pathologies are responsible for 29.5% of deaths in Italy each year, even though from 2003 to 2014 there has been a significant decrease in mortality rates.22
Trends in heart failure incidence
Bonneux et al predicted a steady increase in the number of patients with heart failure: the ageing of the population, improvements in the treatment of acute coronary syndromes, and a longer survival of heart failure patients all contribute to a larger pool of (potential) heart failure patients.14w42The ageing
of the population is undisputed as is the improvement of prognosis in heart failure patients.
Few studies have addressed trends in the incidence of heart failure post-myocardial infarction. Despite a decrease in coronary heart disease and all cause mortality in 546 Framingham heart study participants who suffered a non-Q wave myocardial infarction between 1950 and 1989, the percentage of them developing heart failure remained stable.w43 In a group of 1537 patients who suffered a myocardial infarction (not excluding non-Q wave myocardial infarctions) between 1979 and 1994 in Olmsted County, Minnesota, a 28% reduction in the occurrence of post-myocardial infarction heart failure was documented.18 The
Worcester heart attack study reported a decline in heart failure during hospitalisation for myocardial infarction between 1975 and 1995.w44
The reduction of post-myocardial infarction heart failure is consistent with the declining severity of myocardial infarction following the introduction of reperfusion treatment—a decline that may well continue given the increasingly aggressive (primary percutaneous interventions) and timely interventions in patients with acute coronary syndromes.w45 These data
suggest that improved survival following myocardial infarction is not a major contributor to the occurrence of heart failure.
The incidence of heart failure in men participating in the Framingham heart study did not change over the last 50 years (1950–1999), whereas the incidence in women declined 30– 40%.w46 A larger population-based study in Olmsted County, Minnesota (4537 heart failure patients, 42% of whom were diagnosed as outpatients) reported no change in heart failure incidence between 1979 and 2000.23
Taken together, the data indicate that the incidence of heart failure has not declined over the last two decades and that the ageing of the population in combination with improved prognosis fuel the heart failure epidemic. It follows that prevention of the occurrence of heart failure is needed to stem the epidemic.w42
PROGNOSIS OF HEART FAILURE
‘‘A poor prognosis’’ and prognostication in daily practice
There is no doubt that the prognosis of heart failure patients remains poor, even in the realm of the development of a myriad of effective pharmacological and non-pharmacological inter-ventions. This is illustrated by the title of a paper on the prognosis of the syndrome: ‘‘More malignant than cancer’’.w47 Any doctor treating heart failure patients will confirm that life expectancy in heart failure patients is ‘‘reduced’’ and that sudden cardiac death is a ‘‘major’’ cause of death, that (acute) worsening of CHF occurs ‘‘quite often’’, leading to ‘‘frequent’’ hospitalisations, and that quality of life in these patients is ‘‘impaired considerably’’. We included the quotation marks in the latter sentence to indicate the implicit nature of prognos-tication in clinical practice.
Although information on the natural history of a disease is relevant to illustrate its burden for health care and the society at large, prognostication in individual patients plays a crucial role in daily clinical practice. After the diagnosis (and possible aetiology) of heart failure has been established, a doctor will estimate an individual patient’s probability of developing clinically relevant prognostic outcomes—for example, a 5 year survival probability. Such estimates are typically based on patients’ characteristics, including age, comorbidity, severity and cause of heart failure that are known to influence prognosis. This information, together with the anticipated, preferably evidence-based, effect of possible therapeutic inter-ventions and patient preferences, is instrumental in the Table 5 Causes of heart failure in population based studies
Cause Framingham heart study15 Hillingdon heart failure studyw24 Bromley heart failure study16 Men Women Ischaemic 59 48 36 52 Non-ischaemic: Hypertension 70 78 14 4
Valvular heart disease 22 31 7 10
Atrial fibrillation 5 3
Alcohol 4
Other 7 7 4 5
Unknown 34 23
Because of rounding, the percentages do not always add up to 100. Framingham heart study: ischaemic heart disease and hypertension could be co-named as causing heart failure.
Table 6 Risk factors for the occurrence of heart failure in three population based studies15w13 w31
Risk factor
Framingham heart study
Cardiovascular health study
Rotterdam study
Men Women Men Women
RR (95% CI) PAR RR (95% CI) PAR RR PAR RR (95% CI) RR (95% CI)
Hypertension 2.1 (1.3 to 3.2) 39 3.4 (1.7 to 6.7) 59 1.4 13 1.0 (0.5 to 1.9) 2.6 (1.6 to 4.2) MI 6.3 (4.6 to 8.7) 34 6.0 (4.4 to 8.3) 13 – – 1.9 (1.1 to 3.6) 1.8 (0.9 to 3.5) Angina pectoris 1.4 (1.0 to 2.0) 5 1.7 (1.2 to 2.3) 5 – – 1.3 (0.6 to 2.8) 1.3 (0.7 to 2.6) Diabetes mellitus 1.8 (1.3 to 2.6) 6 3.7 (2.7 to 5.2) 12 1.8 8 2.1 (1.0 to 4.4) 1.6 (0.8 to 3.2) LVH 2.2 (1.5 to 3.2) 4 2.9 (2.0 to 4.1) 5 2.3 6 1.6 (0.4 to 6.7) 0.8 (0.1 to 5.5) Valvular disease 2.5 (1.7 to 3.6) 7 2.1 (1.5 to 2.9) 8 – – – – Atrial fibrillation 2.1 2 1.5 (0.5 to 5.1) 0.6 (0.1 to 4.6) COPD 1.4 6 0.8 (0.3 to 2.6) 3.2 (1.7 to 7.4)
CI, confidence interval; COPD, chronic obstructive pulmonary disease; LVH, left ventricular hypertrophy; MI, myocardial infarction; PAR, population attributable risk (%); RR, relative risk.
1142
EDUCATION IN HEART
Protected by copyright.
on March 25, 2020 at Sistema Bibliotecario - Università degli Studi di Genova.
http://heart.bmj.com/
IHD is the most common and fatal illness in the United States as well, where it affects over than 15.5 million persons ≥20 years of age, as reported in the 2016 Heart Disease and Stroke Statistics update of the American Heart Association (AHA)23,24, and it kills approximately one person every minute. Moreover, according to the WHO 2000-2016 Report, IHD is the leading cause of death not only in Italy or the U.S., but also worldwide, accounting for more than 9 million deaths in 2016.25 IHD, also known as Coronary Heart Disease (CHD) or Coronary Artery Disease (CAD), is the main cause of HF in the western world16; CAD remarkably increases the chance of developing HF.15 Furthermore, it is estimated that for 7–8 years after AMI, up to 36% of patients will experience HF, especially those with LV systolic dysfunction documented during admission15.
The term IHD refers to all cardiovascular disorders caused by narrowed coronary arteries. At the basis of the disease there is an inadequate supply of blood and oxygen to a portion of the myocardium, which typically happens when there is an inequality between supply and demand of myocardial oxygen. This is due to blockage of one or more blood vessels, as a result of a blood clot or constriction of the vessel, or, most commonly, of an atherosclerotic plaque. It is a chronic and progressive condition defined by the ESC as “a pathological process characterized by atherosclerotic plaque accumulation in the epicardial arteries, whether obstructive or non-obstructive”26, but still sufficient to provoke reduction of myocardial blood flow and hence poor perfusion in the region supplied by that artery. IHD may present with several different clinical pictures, generally stable, but with a chance of becoming unstable at any time, due to its dynamic process: AMI, chronic ischemia, stable or unstable angina pectoris and/or dyspnea, acute, chronic or decompensated HF, arrhythmia, and also sudden cardiac death (SCD)1 or clinically asymptomatic reduced LV function.
1 Sudden cardiac death describes the unexpected natural and non-traumatic death from a cardiac cause
occurring within a short time period, generally ≤1 hour from the onset of symptoms, in an apparently healthy subject. The term SCD is used when a congenital or acquired potentially fatal cardiac condition was known to be present during life, or Autopsy has identified a cardiac or vascular anomaly as the probable cause of the event, or No obvious extra-cardiac causes have been identified by post-mortem examination and therefore an
If the narrowing involves less than 50% of the vessel, symptoms and discomfort may only occur when the demand for oxygen increases, such as during physical exertion, which is referred to as stable angina pectoris. This stage is usually characterized by chest and arm discomfort and is relieved promptly with rest or nitroglycerin. The Canadian Cardiovascular Society (CCS) provides one of the main classifications of angina:
Figure 6 Canadian Cardiovascular Society grading of angina pectoris28,29
As the narrowing increases, symptoms may worsen and also be present at rest, with the clinical picture becoming overt. If the blood flow to a portion of the myocardium is completely blocked, myocytes die, leading to AMI. Unfortunately, patients may have silent ischemia, ischemia without pain, and even AMI without prior warning.30 HF in the setting of CHD may be the result of multiple factors that can possibly contribute to the development of HF and LV systolic dysfunction.
Firstly, as detailed in the next chapter, previous AMI leads to the loss of myocytes, development of myocardial fibrosis, and consequent LV remodeling, resulting in
dilation of the chamber and neurohormonal activation. LV remodeling represents the anatomical substrate of ischemic-based HF. This leads to progressive deterioration of the remaining functioning myocardium.
Second, patients with a history of CAD or AMI commonly have relevant atherosclerotic disease in other coronary arteries, in addition to the infarcted artery. Therefore, in addition already irreversibly damaged tissue, there is often a significant portion of jeopardized myocardium, only partially perfused by a stenotic artery, which may deteriorate in myocardial ischemia/hibernation. As a consequence, there is the risk of recurrent MI, which may provoke, as in a loop, further deterioration in LV function and even SCD.
Lastly, endothelial dysfunction, a characteristic feature of atherosclerosis, may independently contribute to progression of the dysfunction.31
In the next chapter, the pathophysiology of ischemic HF will be detailed, as well its consequences on the myocardium.
2.4. Pathophysiology
The classical definition of HF as the “inability of the heart to provide sufficient oxygen to the metabolizing tissues despite an adequate filling pressure”32 is essentially a pathophysiological description. However, despite multiple attempts to define a single pathophysiological process that can exhaustively explain every aspect of this complex syndrome, a consensus has not been reached yet.
In describing the pathophysiology of HF, focus will be placed on the molecular and cellular changes that underlie HFrEF, placing further emphasis on the role of LV remodeling as a primary determinant in the progression of HF and as the target of surgical treatment.
HF is a progressive disorder that originates after an index event that either damages the heart muscle, resulting in a loss of function of cardiac myocytes or interferes with the ability of the myocardium to generate force, thereby keeping the heart from contracting properly. The index event may have an acute presentation, such as in the case of AMI, a more gradual onset, as for example in hemodynamic pressure or
volume overloading, or it can also be hereditary as in several genetic cardiomyopathies.
Aside from its mechanism of presentation, each of these index events affects and worsens the heart’s ability to pump blood and therefore LV function.
However, most patients remain asymptomatic or minimally symptomatic for long periods after the initial episode, thanks to the introduction of several compensatory mechanisms activated by the decrease of the cardiac output, which allow the patient to maintain LV function that is only minimally depressed, modulated within a homeostatic range varying from months to years. These mechanisms consist in the activation of:
• the renin-angiotensin-aldosterone system (RAAS), triggered by increased sympathetic stimulation of the kidney and renal hypoperfusion due to impaired cardiac output. The RAAS is responsible for maintaining cardiac output by increased reabsorption of sodium and water in exchange for potassium, promoting release of arginine vasopressin and catecholamines, vasoconstriction, and cell growth in vasculature through AT1 receptors, whereas activation of AT2, predominant in the myocardium, leads to vasodilatation, inhibition of cell growth, natriuresis, and release of bradykinin; • the adrenergic nervous system: the increased activation of β1-adrenergic receptors enhances myocardial contractility and increases heart rate, with a resultant improvement in cardiac output. Moreover, the stimulation of myocardial α1 -adrenergic receptors is responsible for peripheral arterial vasoconstriction and a modest inotropic effect; it is accompanied by a contemporaneous withdrawal of parasympathetic tone.
• a family of countervailing vasodilatory molecules and inflammatory
mediators, including atrial and brain natriuretic peptides (ANP and BNP),
bradykinin, prostaglandins (PGE2 and PGI2), and nitric oxide (NO), which counterbalance the excessive peripheral vascular vasoconstriction, and unload the heart by counteracting RAAS and the salt and water homeostatic dysregulation. These mechanisms are responsible for cardiac repair and remodeling.
24 Although patients may remain minimally symptomatic or even asymptomatic for months, at some point they become frankly symptomatic. The progression to overt HF is the result of the overexpression of the neurohormonal, adrenergic, and cytokine systems previously mentioned, sustained by the loss of inhibitory input from baroreceptors in the left ventricle, aortic arch, carotid sinus, and renal afferent arterioles. While in the short term they succeed in preserving the patient’s functional capacity, in the long term they have a deleterious impact on the heart and circulation, ultimately leading to a series of unfavorable adaptive changes within the myocardium, termed LV remodeling. Some of these negative effects include augmented myocardial energy requirements (and therefore possible ischemia if oxygen delivery is restricted), triggering of ventricular tachycardia and SCD, decreased NO levels, excessive peripheral arterial vasoconstriction, increased inflammation and oxidative stress, high norepinephrine levels, and fibrosis and hypertrophy of the heart, vessels, kidneys, and other organs.
Figure 7 Pathogenesis of HF32
Unlike HFrEF, understanding of the pathophysiological mechanisms of HFpEF is still in progress. “That is, although diastolic dysfunction was thought to be the only
Pathophysiology of Heart Failur
e
22 455
elicits a modest positive inotropic effect, as well as peripheral arterial vasoconstriction (Fig. 22-2). Although NE enhances both contrac-tion and relaxacontrac-tion and maintains blood pressure, myocardial energy requirements are augmented, which can intensify ischemia when myocardial O2 delivery is restricted. The augmented adrenergic outflow from the central nervous system also may trigger ventricular tachycardia or even sudden cardiac death, particularly in the pres-ence of myocardial ischemia. Thus activation of the sympathetic nervous system provides short-term support that has the potential to become maladaptive over the long term (see Fig. 22-2). Moreover, increasing evidence suggests that apart from the deleterious effects of sympathetic activation, parasympathetic withdrawal also may con-tribute to the pathogenesis of heart failure. Withdrawal of parasym-pathetic nerve stimulation has been associated with decreased nitric oxide (NO) levels, increased inflammation, increased sympathetic activity and worsening LV remodeling. Two ongoing clinical trials, INOVATE-HF (Increase of Vagal Tone in CHF) (NCT01303718) and NECTAR-HF (Neural Cardiac Therapy for Heart Failure Study) (NCT01385176), are examining the effects of vagal nerve stimulation on LV structure and clinical outcomes in patients with New York Heart Association (NYHA) class III heart failure.
Activation of the Renin-Angiotensin System
In contrast with the sympathetic nervous system, the components of the RAS are activated comparatively later in heart failure. The pre-sumptive mechanisms for RAS activation in heart failure include renal hypoperfusion, decreased filtered sodium reaching the macula densa in the distal tubule, and increased sympathetic stimulation of the kidney, leading to increased renin release from jutaglomerular apparatus (Fig. 22-3). As shown in Figure 22-4, renin cleaves four amino acids from circulating angiotensinogen, which is synthesized in the liver, to form the biologically inactive decapeptide angiotensin I. Angiotensin-converting enzyme (ACE) cleaves two amino acids from angiotensin I to form the biologically active octapeptide(1-8) angiotensin II. Most ACE activity (approaching 90%) in the body is found in tissues; the remaining 10% is found in a soluble (non– membrane-bound) form in the interstitium of the heart and vessel wall. The importance of tissue ACE activity in heart failure is suggested by the observation that ACE messenger RNA (mRNA) and ACE-binding sites and ACE activity are increased in explanted human hearts.3 Angiotensin II also can be synthesized using renin-independent pathways through the enzymatic conversion of angio-tensinogen to angiotensin I by kallikrein and cathepsin G (see Fig. 22-4). The tissue production of angiotensin II also may occur along ACE-independent pathways, through the activation of chymase. This latter pathway may be of major importance in the myocardium, par-ticularly when the levels of renin and angiotensin I are increased by the use of ACE inhibitors. Angiotensin II itself can undergo further proteolysis to generate three biologically active fragments: angioten-sin III2-8 and angiotensin IV,3-8 which promote vasconstriction,4 and angiotensin,1-7 which may act to counteract the deleterious effects of angiotensin II on endothelial function.
Angiotensin II exerts its effects by binding to two G protein–coupled receptors, the angiotensin type 1 (AT1) and angiotensin type 2 (AT2) receptors. The predominant angiotensin receptor in the vasculature is the AT1 receptor. Although both the AT1 and AT2 receptor subtypes are present in human myocardium, the AT2 receptor predominates in a 2:1 molar ratio. Cellular localization of the AT1 receptor in the heart is most abundant in nerves distributed in the myocardium, whereas the AT2 receptor is localized more specifically in fibroblasts and the interstitium. Activation of the AT1 receptor leads to vasoconstriction, cell growth, aldosterone secretion, and catecholamine release, whereas activation of the AT2 receptor leads to vasodilation, inhibi-tion of cell growth, natriuresis, and bradykinin release. Studies have shown that the AT1 receptor and mRNA levels are downregulated in failing human hearts, whereas AT2 receptor density is increased or unchanged, so that the ratio of AT1 to AT2 receptors decreases.4 As a result of the increase in sympathetic tone, there is an increase
in circulating levels of NE, a potent adrenergic neurotransmitter. The elevated levels of circulating NE result from a combination of increased release of NE from adrenergic nerve endings and its con-sequent “spillover” into the plasma, as well as reduced uptake of NE by adrenergic nerve endings. In patients with advanced heart failure, the circulating levels of NE in resting patients are two to three times those found in normal subjects. Indeed, plasma levels of NE predict mortality in patients with heart failure. Whereas the normal heart usually extracts NE from the arterial blood, in patients with moderate heart failure the coronary sinus NE concentration exceeds the arterial concentration, indicating increased adrenergic stimulation of the heart. However, as heart failure progresses there is a significant decrease in the myocardial concentration of NE. The mechanism responsible for cardiac NE depletion in severe heart failure is not clear and may relate to an “exhaustion” phenomenon resulting from the prolonged adrenergic activation of the cardiac adrenergic nerves in heart failure. In addition, there is decreased activity of myocardial tyrosine hydroxylase, which is the rate-limiting enzyme in the synthesis of NE. In patients with cardiomyopathy, iodine-131– labeled metaiodobenzylguanidine (MIBG), a radiopharmaceutical that is taken up by adrenergic nerve endings, is not taken up normally, suggesting that NE reuptake also islso impaired in heart failure.
Increased sympathetic activation of the beta1-adrenergic receptor results in increased heart rate and force of myocardial contraction, FIGURE 22-1 Pathogenesis of heart failure. A, Heart failure begins after a so-called index event produces an initial decline in pumping capacity of the heart. B, After this initial decline in pumping capacity of the heart, a variety of compensa-tory mechanisms are activated, including the adrenergic nervous system, the RAS, and the cytokine systems. In the short term, these systems are able to restore cardiovascular function to a normal homeostatic range, with the result that the patient remains asymptomatic. With time, however, the sustained activation of these systems can lead to secondary end-organ damage within the ventricle, with worsening LV remodeling and subsequent cardiac decompensation. As a result of these changes, patients undergo the transition from asymptomatic to symptomatic heart failure. ANP/BNP = atrial natriuretic peptide/brain type natriuretic peptide; NOS/ROS = nitric oxide synthase/reactive oxygen species; SNS = sympathetic nervous system. (From Mann DL: Mechanisms and models in HF: a combinatorial approach. Circulation 100:99, 1999; and Kaye DM, Krum H: Drug discovery for heart failure: A new era or the end of the pipeline? Nat Rev Drug Discov 6:127, 2007.)
A B 20 60 EJECTION FRACTION (%) TIME (yr) Secondary damage Compensatory mechanisms Index event ASYMPTOMATIC SYMPTOMATIC Secondary damage • LV remodeling • Contractility↓ • Hypertrophy • Apoptosis • Fibrosis • NOS/ROS • Electrophysiology Neurohormones • ↑SNS activity • ↑RAS • ↑Endothelin • ↑ANP/BNP • ↑Cytokines Index event Endothelium • Vasoconstriction • NOS/ROS • Structural change • Cytokines
mechanism responsible, community-based studies suggest that additional extracardiac mechanisms may be important, such as increased vascular stiffness and impaired renal function.” 21 However, as already said, this type of HF will not be discussed in this thesis.
2.4.1. Left Ventricular Remodeling
Despite the pharmacological management of the neurohormonal compensatory mechanisms previously described, in the vast majority of patients, HF is progressive. This points to a role for LV remodeling as one of the main culprits in contributing to the progression of HF.32,33
LV remodeling refers to changes in LV mass, volume, shape, and composition at the cellular and molecular levels that the heart develops in reaction to cardiac injury, for example after a myocardial infarction, and dysfunctional hemodynamic loading conditions, like in HF. LV remodeling plays an important part in the progression of HF due to the mechanical burden produced by changes in LV geometry and architecture and in the components of the myocardium.
These changes comprise32:
• Alterations in the biology of cardiac myocytes:
- Eccentric hypertrophy à this typical hypertrophy pattern develops in response to chronic volume overload: increased diastolic wall stress results in an extension in the length of myocytes with the addition of sarcomeres in series, thus worsening LV dilation. It may also be accompanied by wall thinning, due to the addition of sarcomeres in parallel as a consequence of pressure overload.
- Disruptions in cellular organization à there is upregulation of the cytoskeletal protein desmin and membrane-associated proteins such as vinculin and dystrophin, an increase in the number of myofibrils and mitochondria, an expansion of nuclei and mitochondria, myofibril displacement, alterations in contractile elements (myofibrillar ATPase,
actomyosin, myosin, troponin, tropomyosin, titin activity and/or expression reduced) and regulatory proteins.
- β-adrenergic signaling desensitization à downregulation of beta adrenergic receptor density takes place probably due to the increased number of NE nearby, and reduced contractile response to beta adrenergic agonists; as a consequence, there is a reduction in LV contractility.
- increased expression of β myosin and decreased expression of a myosin heavy chain gene à reactivation of fetal genes generally not expressed postnatally, and silencing of others normally expressed, may play a relevant role in contractile dysfunction. This genetic reprogramming is triggered by local and systemic mechanisms: the mechanical stretching of the myocyte, neurohormones, such as norepinephrine or angiotensin II, inflammatory cytokines like TNF or IL-6, other peptides and growth factors, and reactive oxygen species.
- Myocytolysis à in the end stage of hypertrophy, myocytolysis occurs, with disruption of Z-bands and the normal parallel arrangement of sarcomeres, and tortuosity of T tubules.
- Alterations in excitation-contraction coupling à failure in the mechanism that, starting from the cardiac action potential, under normal circumstances leads to myocyte contraction and relaxation, most evident at high heart rates, with resultant impaired force-frequency coupling. This is due to the decline of the systems usually responsible for the adjustment of cardiac performance to a higher contraction frequency. Contrary to what normally occurs, in HF there is a reduction in the amount of intracellular Ca2+, a protracted deterioration of transient Ca2+, and an increased quantity of diastolic Ca2+. At the basis of the reduced transient intracellular Ca2+ there are three critical defects in Ca2+cycling, resulting in depletion of Ca2+ from the sarcoplasmic reticulum (SR): (1) increased Ca2+leakage through the ryanodine receptor, (2) impaired calcium uptake by sarcoplasmic reticulum due to reduced levels and function of SERCA2a (a sarcoplasmic reticulum Ca2+pump) , and (3) increased expression and function of the sarcolemmal Na+/Ca2+ exchanger (NCX). - abnormal energetics and metabolism;
• Myocardial Changes:
- progressive loss of myocytes à this is due to a continuum of cell death responses: necrosis, apoptosis, and autophagic cell death pathways; - reorganization of the extracellular matrix à the organized structural type
I and III collagen weave that usually ensures the integrity of myocytes and the alignment of myofibrils is degraded and replaced by an interstitial collagen matrix that does not provide structural support, but ultimately leads to fibrosis.2,21
• Alterations in Left Ventricular Chamber Geometry:
As a consequence of the changes in myocyte biology and myocardial structure, the LV undergoes several alterations in its geometry that contribute to worsening HF.
- LV dilation à leads to an increase in LV end-diastolic volume and afterload, worsened by delayed LV filling due to reduced LV compliance from hypertrophy or fibrosis.
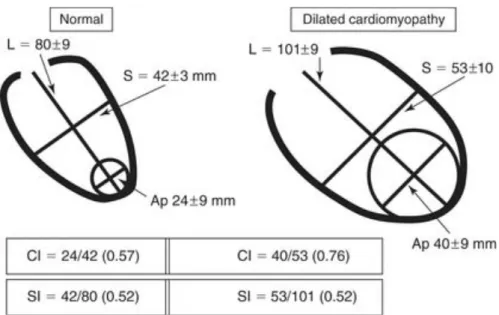
- Increased LV sphericity index à there is a shift from an elliptical to a spherical shape that has the effect of increasing wall stress, maintaining HF. The sphericity index (SI) is an effort to quantify the abnormal geometric changes that accompany HF and assess the shape by measuring the ratio of either the long axis to short axis or end diastolic volume to the volume of the sphere having the measured long axis diameter. An inferior MI induces a regional remodeling of the basal and mid segments of the inferopostero-lateral wall, with a significant increase in LV transverse diameters and consequently in the SI. However, as the SI evaluates the entire LV chamber, in some cases the it fails to detect regional abnormalities, like those at the apical level. For that reason, the apical conicity index (ACI) was introduced, described as the ratio of the apex to the short axis. This ACI, easily measured by echocardiography, is significantly greater in patients with an anterior MI, where the apex is primarily involved, quantifying the changes in enlarged, less conical apex.34 (Figure 8 and 9)
- LV wall thinning à along with LV dilation, contributes to the “afterload mismatch” that worsens cardiac output. It also causes the constant expression or activation of stretch-activated genes and hypertrophic signaling pathways. Furthermore, it may provoke episodes of hypoperfusion of the subendocardium and increase oxidative stress. - Mitral valve incompetence à distancing of papillary muscles leads to
functional mitral regurgitation and further hemodynamic volume overloading of the ventricle.
Taken together, LV remodeling lead to a vicious cycle where LV dilation keeps increasing in a wasted effort to maintain a normal cardiac output, the EF decreases, and hemodynamic overloading worsens, any or all of which are sufficient to continued aggravation of LV function, wall stress, geometric distortion and therefore the progression of HF.32,35–40
Furthermore, mitral regurgitation may occur as a result of the LV dilation, due to the displacement of the papillary muscles and annular enlargement, worsening the prognosis.41
Recent studies have shown that LV remodeling can be reversed, decreasing LV mass and optimizing its shape. Reverse LV remodeling is also associated with improved clinical outcomes in patients with HFrEF. Indeed, one of the goals of therapy for HF is to prevent and/or reverse LV remodeling, which is the aim of surgery in the present analysis.20,42
Figure 8 Left panel: the LV apex is primarily involved after an anterior MI. As a consequence, the conicity index (CI, obtained from the apical – c – to short axis ratio – b)
is significantly greater in the anterior remodeling compared to posterior remodeling group. Right panel: a previous inferior MI induces a regional remodeling of the basal and mid segments of the inferopostero-lateral wall, with a sig- nificant increase in LV transverse diameters and conse- quently in the sphericity index (SI, obtained from the short – b -to
long axis ratio – a)43
Figure 9 Geometric measures in normal and dilated cardiomyopathy. Sphericity index (SI) is calculated as the short- to long-axis ratio (S/L), and conicity index (CI) as the apical to short-axis ratio (Ap/S). Apical diameter is determined by using the diameter of the sphere that best fits the apex. Note that the SI has the same value in normal subjects and in patients with dilated cardiomyopathy, because the elongation of the ventricle is proportional to the increase in width, so the ratio remains stable, whereas the CI is markedly abnormal in the
patients.44
2.5. Diagnosis
Although HF is a complex clinical syndrome that may present with several different clinical pictures, the vast majority of patients show a classical constellation of symptoms related to impairment of myocardial performance, with findings that may range from normal ventricular size and function to severe dilation and reduced ejection fraction. If HF is suspected, the goals of the clinician are to confirm diagnosis, outline the underlying cause(s), estimate the severity and prognosis, and choose the most suitable therapy.
Diagnosis of HF can be relatively straightforward when the patient presents with the classic manifestations. Nevertheless, as the signs and symptoms of HF and the diagnostic physical findings are neither specific nor sensitive, the clinical picture
alone is not sufficient to establish a diagnosis, and additional laboratory testing, cardiac imaging, and functional studies should be performed.
The assessment of patients with HF should always begin with complete medical history, in order to explore possible causes of the disease and exacerbating factors, as well as obtain crucial data for proper management.
2.5.1. Clinical presentation
As mentioned above, HF may present with a wide range of possible clinical pictures, none of which are specific to HF, and much less to HFpEF versus HFrEF. However, the classic set of symptoms usually includes fatigue, due to low cardiac output, and shortness of breath, as a result of pulmonary congestion, restricted cardiac output, and increased filling pressure. Other factors may also contribute to worsening of fatigue, such as reduction in pulmonary compliance, increased airway resistance, skeletal muscle (respiratory muscles and diaphragm) abnormalities, and other noncardiac comorbidities (e.g., anemia, renal dysfunction, endocrinologic abnormalities)45.
In the early stages of HF, dyspnea develops only during exercise. As the disease degenerates, it can occur even with less demanding activity, until it eventually becomes present at rest. Moreover, 40% of patients with advanced HF shows Cheyne-Stokes respiration, also known as periodic respiration, characterized by cycles of apneic phases followed by hyperventilation and hypocapnia. It is attributable to the increased sensitivity of the respiratory center to arterial pCO2 and a lengthy circulatory time.
Although dyspnea is considered a cardinal symptom of HF, its initial absence does not allow to exclude a diagnosis, just like its presence is not sufficient to establish a diagnosis, since it is a possible red flag of many other medical conditions.
With progression of the disease, the so-called ‘air hunger’ starts to develop in recumbency (orthopnea), especially on the left side (trepopnea), due to the redistribution of fluids from the splanchnic to the central circulation and the consequent increase in pulmonary capillary pressure. Therefore, it is most evident during the night (paroxysmal nocturnal dyspnea), with acute episodes of severe
shortness of breath frequently accompanied by coughing or wheezing, awakening the patient and forcing them to sleep with the head elevated in order to relieve dyspnea. Orthopnea and paroxysmal nocturnal dyspnea are high reliable indicators of HF.
Symptoms related to right heart congestion may also be present, such as weight gain, increasing abdominal girth, early satiety, upper quadrant pain related to liver congestion and the onset of edema in dependent organs.
Other possible manifestations include gastrointestinal symptoms, like anorexia, nausea, cachexia, cerebral symptoms such as confusion and disorientation, especially in elderly patients, sleep and mood disturbances, and nocturia caused by the reabsorption of fluid during sleep.
2.5.2. Physical examination
Scrupulous physical examination completes the information collected through the medical history; therefore, it is the basis in the evaluation of patients with HF, in order to determine the aetiology, assess severity, and choose best treatment. The first aspect that should be considered is the patient’s general appearance, immediately followed by measurement of vital signs in seated and standing positions, evaluation of heart and pulse, and assessment of other organs, in search of signs of congestion or hypoperfusion or indications of comorbidities.
The clinician should inspect the patient’s habitus and state of alertness, with particular attention to the possible presence of discomfort, shortness of breath, coughing, pain, or Cheyne-Stokes respiration. In the initial stages of HF, the patient usually complains of moderate distress only when recumbent, but when HF worsens, the shortness of breath becomes disabling, preventing them from carrying out daily activities, even speaking, and forcing them to sit in the upright position in order to breath.
Pulse and systolic blood pressure are generally reduced in severe HF, due to LV dysfunction, but in the initial stages may be normal or even high.
The color of the skin, particularly of the extremities, may suggest underperfusion due to peripheral vasoconstriction if pallid or cyanotic, but it can also show signs of
alcohol abuse, such as spider angioma or palmar erythema, or even underlying diseases like bronzing due to hemochromatosis, or easy bruising from amyloidosis. Despite its importance, cardiac examination may not be always helpful to assess the severity of the disease. Heart size and the quality of the point of maximal impulse can be estimated through inspection and palpation. The apical impulse may be palpable and displaced over two interspaces below the fifth intercostal space and/or lateral to the midclavicular line, if cardiomegaly is present. In advanced HF, a palpable third sound may be present.
Instead, auscultation plays a key role in HF evaluation. A third heart sound (S3), defined as protodiastolic gallop, is generally audible, and is a highly specific red flag of ventricular volume overload. A fourth sound may be also present in the case of diastolic dysfunction. In severe HF, these two sounds may overlap, resulting in a summation gallop.
Moreover, a typical holosystolic murmur of mitral regurgitation and a left sternal border murmur of tricuspid insufficiency are common findings in many patients with severe HF.
Recognizing reduced cardiac output is also of great importance: poor mentation, reduced urine output, mottled skin, and cool extremities may suggest a condition of systemic hypoperfusion.
One of the aims of the clinician in assessment of HF is to quantify the presence of volume retention, with or without pulmonary and/or systemic congestion. The measurement of jugular venous pressure (JVP) on the recumbent patient, with the head tilted at 45°, provides an estimation of right atrial pressure and therefore left-sided filling pressure, being an indirect way to evaluate the volume status of the patient. JVP is measured by calculating the height of the venous column of blood above the sternal angle and then adding 5 cm. It is expressed in cm of water (normal ≤8 cm). Especially in the early stages, where JVP may be normal at rest, it can be accentuated by applying pressure on the right upper quadrant of the abdomen while determining venous pulsations in the neck (hepatojugular reflux). If right failure or tricuspid regurgitation are present, JVP may not be reliable, as well as in patients with pulmonary arterial hypertension.
Pulmonary congestion is another typical sign of HF, with variable clinical manifestations. Transudation of fluid from pulmonary capillaries into alveoli results in fine pulmonary crackles rising from the base upwards over both lung fields, namely rales and crepitations or rhonchi, accompanied by expiratory wheezing (cardiac asthma) if reactive bronchoconstriction is also present. However, these sounds are frequently absent in patients with severe chronic HF, thanks to the compensatory increase in local lymphatic drainage. Leakage of fluids may also occur into the pleural cavities, resulting in pleural effusions, which is clinically evident in dullness to percussion and diminished breath sounds. Bilateral effusions are more frequent, but if unilateral, is usually right sided.
Other common but late findings in volume-overloaded patients with HF include hepatomegaly, ascites as a result of increased pressure in the hepatic veins, and jaundice secondary to the hepatic disfunction. Furthermore, in these patients lower-extremity edema is commonly found, but it needs to be related with JVP measurement, as it is non-specific and may be the result of venous insufficiency, or as a side effect of medications such as calcium channel blockers. Peripheral edema is usually symmetric and bilateral, mainly affecting the ankles and pretibial region, or the sacral area and the scrotum in bedridden patients.
2.5.3. Investigations
Routine Laboratory Testing Patients with new-onset HF as well as those with
chronic HF should undergo a complete laboratory panel including electrolytes, blood urea nitrogen, serum creatinine, hepatic enzymes, fasting lipid profile, thyroid-stimulating hormone, transferrin saturation, uric acid, complete blood count, and urinalysis. If comorbidities are suspected, patients should be tested for diabetes mellitus (fasting serum glucose or oral glucose tolerance test), HIV infection, hemochromatosis, rheumatologic diseases, amyloidosis, or pheochromocytoma. Frequent findings in patients with HF include abnormalities of sodium (hyponatremia is the most common) and potassium (mostly hypokalemia, common in patients treated with diuretics), abnormalities in renal function, urea nitrogen and serum creatinine (suggesting a cardiorenal syndrome), hyperglycemia, elevated serum uric