UNIVERSITA’ DEGLI STUDI DI MESSINA
DIPARTIMENTO DI MEDICINA CLINICA E SPERIMENTALE Dottorato di Ricerca in
Biotecnologie Mediche e Chirurgiche XXIX ciclo
INTEGRATION OF HEPATITIS B VIRUS DNA IN CHRONICALLY
INFECTED PATIENTS
PHD STUDENT:
Dott.. TRIPODI GIANLUCA
SUPERVISOR :
Prof. GIOVANNI RAIMONDO
Index
PAGE
1 INTRODUCTION ...3
1.1 The hepatitis B virus ...3
1.1.1 Replication ...4
1.1.2 Viral antigens...7
1.1.3 Genotypes ...8
1.1.5 Natural history of Chronic HBV infection ……….. 10
1.1.6 HBV DNA integration ...13
2 AIMS ...15
3 PATIENTS AND METHODS...16
4 RESULTS...19
5 DISCUSSION...21
6 FIGURES AND TABLES...24
INTRODUCTION
THE HEPATITIS B VIRUS
The hepatitis B virus (HBV) is a DNA virus belonging to the Hepadnaviridae family (Figure 1) (Block, Guo, and Guo 2007). This family is further divided in genera, the, Orthohepadnaviridie, including human and other mammal hepadnaviruses, for example chimp or woodchuck HBV, and the Avihepadnaviridae, such as Duck hepatitis virus (Locarnini et al. 2013). Human HBV only infects humans, chimpanzees and Tupaia Tree shrew as the virus is dependent of attachment to its receptor, the sodium-taurocholate cotransporting polypeptide (NTCP), for infection to occur (Y. Ni et al. 2013).
The HBV genome is relaxed circular partially double stranded DNA, template for five different transcripts. The genome has four open reading frames: the preCore/Core, for the precore and capsid proteins, Pol, for the polymerase, PreS/S, for surface antigens, and X, for the X protein (Figure 2). The longest transcript, precore, is 3.5 kb and translated into the “e” antigen, which is secreted to the blood stream and is thought to have an immune-tolerogenic function. Only 33 bp shorter, the core transcript is the template for capsid proteins but also for the polymerase involved in reverse transcription including the RNase H activity. Two transcripts are translated into the surface proteins. The longest, 2.4 kb, is translated into the L-protein whereas the 2.1 kb transcript encodes the M and S proteins. The X-transcript, 0.8 kb, is template for the X protein, which is suggested to have a transactivating function on many targets as well as involvement in carcinogenesis.
Replication
Viral attachment and entry
The initial stage of HBV infection involves the attachment of virus to the membrane of hepatocyte. A receptor recognition domain has been identified in the preS domain of surface protein (Klingmuller and Schaller, 1993), and the sodium taurocholate cotransporting polypeptide (NTCP) has been recently suggested to be the functional cellular receptor that mediates HBV entry through binding of the preS domain peptide (Ni et al., 2014; Yan et al., 2012). Upon binding to cellular receptors, HBV enters into hepatocyte by endocytosis. Thereafter, the viral capsid is released from endosome into cytoplasm (figure 3)
Transportation of the viral genome into the nucleus
The subsequent stage involves viral disassembly and the transportation of HBV genome into the nucleus, likely involving the modification of viral capsid proteins (Kang et al., 2006). It has been suggested that a nuclear localization signal on viral capsid is exposed following structural changes of capsid, and interacts with nuclear import receptor to mediate the passage of viral genome (Kann et al., 1999).
Conversion of relaxed circular DNA to covalently closed circle DNA
Once the viral genome is released into the nucleus, the relaxed circular DNA (rcDNA), which is incomplete in both strands, is repaired by viral pool protein and circularized to cccDNA. Cellular enzymes are suggested to be involved during conversion of rcDNA to cccDNA (Kock and Schlicht, 1993). The cccDNA serves as the transcriptional template of pgRNA, precore mRNA and all other sub-genomic mRNAs. HBV cccDNA resides in the nucleus of hepatocyte as a viral minichromosome that is organized by histones and non-histone proteins as well as viral proteins, and establishes an intrahepatic HBV reservoir that can produce lifelong persistence in chronically
infected hosts (Newbold et al., 1995). cccDNA is the stable source of progeny viral templates, and is relatively resistant to immune clearance as well as antiviral therapy.
Transcription and translation
Following cccDNA formation, transcription starts with host RNA polymerase II under the regulation of four promoters, two enhancers as well as a GRE. Four groups of unspliced, 5′ capped and 3′ polyadenylated mRNA transcripts are produced: (1) 3.5-kb pgRNA and precore mRNA; (2) 2.4-kb mRNA for LHBs; (3) 2.1-kb mRNA for MHBs and SHBs; and (4) 0.7-kb mRNA for HBx. The pgRNA is bifunctional, serving both as the template for reverse transcriptional synthesis of viral DNA and as messenger RNA for the polymerase and core proteins. The precore mRNA is translated to a precore gene product which is further processed into HBeAg. The rest of the mRNAs serve exclusively as mRNAs for translation of the surface (LHBs, MHBs and SHBs) and HBx proteins.
Synthesis of progeny viral DNA genomes from pregenomic RNA in the cytoplasmic capsids
HBV DNA genome replication begins with encapsidation of viral polymerase and pgRNA inside the nucleocapsids. The encapsidation signal (epsilon), which is a cis-acting element, lies in the pgRNA and folds into a stem-loop structure (Pollack and Ganem, 1993). The epsilon binds to the terminal protein domain of viral polymerase and forms nucleocapsid with capsid proteins. Subsequently, the viral polymerase initiates synthesis of the minus-DNA strand by reverse transcription using the pgRNA as template, and then synthesizes the plus-DNA strand using the minus-DNA strand. Circularization of viral genome is accomplished through DNA strand transfers, and an rcDNA is thus formed. Unlike the situation of the retroviruses, integration of HBV genome into host cellular chromosomes has no role in viral replication. Nevertheless, integration of HBV DNA genome into host chromosomes is frequently observed (Block et al., 2003). It is well-established that HBV DNA integration plays an important role in the oncogenic transformation of infected hepatocytes.
The newly formed nucleocapsids containing the rcDNA are then enveloped and secreted out as mature virions. Alternatively, the nucleocapsids are recycled back to the nucleus and converted into cccDNA to maintain a “pool” of cccDNA. In addition, the subviral particles of SHBs are secreted in excess compared to virions.
Of interest, inhibition of envelope protein secretion and endoplasmic reticulum (ER) retention of LHBs have been implicated when LHBs is overexpressed relative to MHBs and SHBs. This phenomenon may explain the formation of ground-glass hepatocytes (GGH) frequently observed in liver biopsy specimens from chronic HBV carriers and may be associated with the development of LC ( liver cirrhosis) and/or HCC ( Hepatocellular carcinoma).
Viral antigens
The HBV has four different kinds of antigens: precore, core, surface and X antigen. The core antingen (HBcAg) is the main component of the capsid. The precore protein, the precursor of the “ e” antigen (HBeAg), is produced from its own transcript, 33 nt longer than the core transcript. The precore is directed to the ER by means of a leader sequence in its precore part, where it is cleaved before secretion as HBeAg (Milich and Liang 2003). The three different surface proteins, Large, Middle and Small (L, M, S), are translated from two different transcripts. They all contain the S domain but M proteins also contain the preS2 domain and the L protein an additional preS1 domain. The latter domain is also the ligand to the HBV receptor, NTCP. These proteins are present on the surface of virions, but there are also two forms of subviral particles, spherical and filamentous, that lack capsid and hence viral DNA. These are produced in great excess and outnumber the viral particles with an order of 104-105 (Chai et al. 2008).
The small X protein has been attributed many potential functions (Feitelson, Bonamassa, and Arzumanyan 2014). It seems to be required for initiation and maintenance of virus replication after infection (Lucifora et al. 2011) and it has been suggested to have transactivating functions on transcription (H. Tang et al. 2005) as well as carcinogenic effects (L. Yang et al. 2008).
Genotypes
Until now, eight HBV genotypes have been identified and are numbered alphabetically from A to H. HBV genotypes differ by at least 8% from each other and several subtypes (at least 24) have been described, except for genotypes E and G. Determination of amino acid variability at amino acids 122 and 160 of the HBV surface protein allows subtyping of HBV. HBV genotypes and subtypes show a distinct geographical distribution, with genotype A being typically isolated in Northern Europe and countries with a strong prevalence of populations of Northern European origin, including the USA, but is occasionally seen also in the Indian subcontinent. Genotype D is highly prevalent in Eastern Europe, Mediterranean countries and Middle East, whereas genotypes B and C are typical of China and Japan. Infections with genotypes E, F, G and H are rare and usually observed in West Africa, Central-South America, Central Europe and Southern USA, and Central America, respectively (Schaefer S.2007 ) ( figure 4).
Infection with more than one genotype is possible and there is evidence that super-infection may be accompanied by acute intensification of the underlying chronic disease, suggesting that adaptive immunity may not always be protective across genotypes.(Chen PJ et al,. 2001).
A relationship between HBV genotypes and clinical outcome of hepatitis B has been reported, but most studies have been limited to compare genotypes B and C or genotypes D and A, because of their geographical distribution, indicating that genotypes A and B are generally associated with a more benign course of infection than genotypes C and D, respectively (Fattovich G et al., 2008, B.Kao JH et al,. 2000)
HBV polymerase lacks a proofreading function, thus the reverse transcription step results in the selection of HBV quasispecies containing several mutations within their viral genome; HBV exhibits a mutation rate more than 10-fold higher than other DNA viruses. Mutations accumulating in individual genomes reflect both the duration of active HBV infection and the strength of immune response. Moreover, apart from viral and host factors, exogenously induce selection pressures, such as immunoprophylaxis and antiviral therapy, may strongly affect substitution rates in HBV. Some of this mutations are detrimental to the virus while others may provide the virus with a survival advantage. Several studies have shown that these HBV
mutations are not distributed randomly but rather tend to cluster in particular portions of the HBV DNA as the Basal Core Promoter/PreCore region and the PreS/S region. Furthermore several studies demonstrated that these specific genetic mutations have been associated with cirrhosis and HCC (Pollicino T, Cacciola I, Saffioti F, Raimondo, 2014 ).
Natural history of chronic HBV infection
Chronic HBV infection is a dynamic process, and its natural history can be schematically divided into five phases, which are not necessarily sequential. Viral factors (HBV replication level), host factors (immune activity against HBV) and interplay between virus and host are highly involved in this dynamic course (Bertoletti et al., 2003; Hadziyannis et al., 2013). (Figure 5) 1) The “immune tolerance” phase
This phase occurs when the infection is frequently acquired in childhood, but is mostly unidentifiable in patients who are infected in adulthood. This phase represents a state of host immune tolerance against HBV, allowing viral replication at very high levels. This phase is characterized by positive HBsAg and HBeAg, high HBV DNA level (usually 107-1011 copies/ml),
normal or slightly abnormal ALT level, no or mild hepatic inflammation, and no or slow progression toward severe chronic hepatitis. (Fattovich, 2003)
2) The “immune reactive HBeAg-positive” phase
After three or four decades of the “immune tolerance” phase (more rapidly in patients infected during adulthood) the liver disease may develop in the second, “immune reactive HBeAg-positive” phase. This phase represents a state of partial breakdown of tolerance and host immunologic arousal for virus clearance. This phase is characterized by positive HBsAg and HBeAg, relatively low HBV DNA level, elevated or fluctuating ALT level, moderate or severe hepatic inflammation, and more rapid progression to fibrosis (Fattovich, 2003; Hadziyannis and Papatheodoridis, 2006; Hoofnagle et al., 2007; McMahon, 2009).
This phase has a variable duration, and may vary from several weeks to several years. The severity of this phase is also variable, and it may lead to unfavorable outcomes such as progression to liver cirrhosis (LC) and even HCC and death. This phase ends in HBeAg seroconversion to anti-HBe, or less frequently in clearance of HBsAg and seroconversion to anti-HBs (Buster et al., 2008).
The HBeAg loss and anti-HBe seroconversion represent a reduced viral replication, and is usually considered an important beneficial milestone in the natural history of CHB infection. However, HBeAg loss alone, which may be due to mutations in the preC gene, cannot be considered as a favorable indication.
3) The “inactive HBV carrier state” phase
Following HBeAg seroconversion to anti-HBe, the chronic infection enters the third phase, which represents an inactive HBV carrier state. This phase is characterized by positive HBsAg and anti-HBe, very low or undetectable HBV DNA level and normal ALT level. The liver histology is nearly normal. However, some inactive HBV carriers have low but persistent HBV DNA level (usually below 2000 IU/ml) with normal ALT levels. These patients may still have elevation of HBV DNA and ALT and may require regular follow-up and careful monitoring.
For some patients, this phase may last for the rest of their life, and they usually have a good long-term outcome with a very low risk of progression to LC or HCC (Chen et al., 2012; de Franchis et al., 1993; Tai et al., 2009). A small portion of them may have HBsAg loss and anti-HBs seroconversion over time (Martinot-Peignoux et al., 2002). On the other hand, reactivation of HBV replication and disease progression may also occur, and these patients have to be included in the fourth phase (Papatheodoridis et al., 2008).
4) The “HBeAg-negative CHB” phase
This is a late phase in the natural history of CHB following the “inactive HBV carrier state” phase, though it may immediately follow HBeAg seroconversion to anti- HBe during the second phase (Liaw et al., 2010; McMahon, 2009). This phase represents a late immune reactive state, and is characterized by positive HBsAg and negative HBeAg, fluctuating HBV DNA level, elevated ALT level and progressive hepatic histological changes (Brunetto et al., 1991; Hadziyannis and Papatheodoridis, 2006). This phase include reactive patients who are HBeAg-negative because of a precore stop codon mutation (G1896A) that abolishes HBeAg secretion (EASL,2012). These patients have a low rate of spontaneous disease remission, and usually have an unfavorable
long-term outcome with a high risk of progression to LC or HCC (Hadziyannis and Papatheodoridis, 2006). Antiviral treatment and careful monitoring should be applied in these subjects.
5) The “HBsAg-negative” phase
In the “HBsAg-negative phase” after HBsAg loss, low-level HBV replication may persist with detectable HBV DNA in the liver. Generally, HBV DNA is not detectable in the serum, while anti-HBc antibody with or without anti-HBs are detectable. HBsAg loss before the onset of cirrhosis is associated with improvement of the outcome with reduced risk of cirrhosis, decompensation and HCC. The clinical relevance of occult HBV infection [detectable HBV DNA in the liver with low-level (<200 IU/ml) or undetectable HBV DNA in blood] is related to possible transmission of the HBV infection, contribution to progression toward cirrhosis as well as to development of hepatocellular carcinoma (HCC) in patients with concomitant other causes of liver disease (namely hepatitis C virus infection, alcoholic hepatitis etc). Furthermore, immunosuppression may lead to HBV reactivation in these patients .
* * * * * * * HBV DNA INTEGRATION
HBV DNA integration into the host genome is a phenomenon well-known since several decades (Marion et al. 1980; Bréchot et al. 1980; Edman et al. 1980). In the early studies, integrated forms of HBV DNA were first detected in a cell line, PLC/PRF/5, derived from a patient with liver cancer. Soon thereafter integrations were also detected in tumour tissue and in liver biopsies from patients with HBeAg-negative chronic hepatitis (Bréchot, Hadchouel, Scotto, Fonck, et al. 1981). It is believed that a linear form of the normally relaxed circular genome of HBV is the precursor for integration (W. Yang and Summers 1999). This form of the viral DNA is imported into the nucleus preferentially during early phases of infection or during periods of extensive cell turnover and reinfection. A preferential site of integration is the viral genomic region between DR1 and DR2 (Z. Jiang et al. 2012; Sung et al. 2012; Takada et al.1990). In an extensive study on HCC from HBV patients Sung et al found that 40% of integrations occurred at a breakpoint within the 1800-bp closer to the DR1.
The accumulating data of integrated HBV DNA sequences suggest that the integrations occur randomly across all human chromosomes (S. Jiang et al.2012). However, by t h e analysis of liver tumours with integrated HBV DNA recurring integrations at level of genes connected to carcinogenesis (i.e. TERT and MLL4) (Sung et al. 2012; Saigo et al. 2008; K.-W. Tang et al.2013; Paterlini-Bréchot et al. 2003; Murakami 2005) have been revealed . Integration sites are quite largely different between tumor and non-tumor tissues, and integration in peculiar sites is considered an event driving cancer development (Sung et al. 2012; Z. Jiang et al.2012).
Integrated HBV DNA was firstly investigated by using restriction enzymes and hybridisation methods (Edman JC et al., 1980). Since then more advanced techniques have been used to identify HBV DNA integrations, such as inverse-PCR{Mason:2016gp}(Tsuei DJ et al., 1994, Huang H-Pet al 2005) (Tu T et al., 2015) Alu-PCR (Minami M et al., 1995, Pollicino T, Vegetti A, Saitta C, Ferrara F, Corradini E, Raffa G, Pietrangelo A, Raimondo G. 2013) or cassette-ligation-mediated PCR (Tamori A et al., 2003) with subsequent Sanger sequencing. Very recently, whole genome sequencing has been
applied for analysing integrated sequences in HCC tissues from patients with a history of chronic HBV infection, but these approaches are still very difficult for several aspects and do not allow examination of large series of samples (Sung W-K et al., 2012 , Toh ST et al., 2013). Integration of HBV DNA has mainly been studied in tumorous and non-tumorous explant liver tissue from patients with HCC undergone liver transplantation (Sung W-K et al., 2012, Hino O et al., 1994) and reviewed in (Bonilla Guerrero R, Roberts LR. 2005). A few studies have shown that integrations are present also in patients with less advanced chronic infections (Huang H-P et al,. 2004, Murakami Y et al., 2005 Fowler MJ et al., 1986, Minami M et al., 2005)
AIM
Aim of this study was to investigate HBV DNA integration in the host’s genome by the analysis of a large series of patients with chronic hepatitis B in different stages of the infection by means of the Alu-PCR method.
Materials and Methods
Patients and samples
From a cross-sectional study of 160 patients with chronic HBV infection focusing on histology and HBV DNA levels (.Lindh M. Et al., 2000), 75 patients, whose liver biopsies were available for analysis by molecular techniques, were included in this study. The patients represented different genotypes and phases of the disease, and 16 were positive, 59 negative for hepatitis B e antigen (HBeAg). None of them were co-infected with hepatitis C or D viruses, or HIV. Serum samples were taken at the time of biopsy. All patients gave written informed consent and the regional ethical review board approved the study.
Serum analyses
Serum HBV DNA was analysed by Cobas Amplicor HBV Monitor (Roche Diagnostic Systems, Branchburg, NJ). HBsAg was quantified using the Architect assay (Abbott, Abbott Park, IL)
Extraction of nucleic acids and DNase treatment
To extract DNA and RNA from liver biopsies, a small piece of liver, approximately 5 mg, was first homogenised in a MagNA Lyser instrument (Roche Applied Science). The homogenate obtained was diluted 1:2 and half of the material was saved for future analyses. The remaining half was inserted in a MagNA Pure robot (Roche Applied Science) and the DNA II tissue kit was used to extract both DNA and RNA. For RNA analysis, DNA was then degraded with DNase (Ambion Inc, TX).
Real-time PCR was performed using specific primers for intrahepatic HBV DNA (Malmström S et al., 2012).The remaining 50 µl of sample was used in the Alu-PCR.
Alu-PCR
Alu-PCR, was used for identification of integrated HBV DNA sequences adjacent to known sequences (Minami M et al., 1995, Murakami Y 2005). First RNA was degraded using RNase (Ambion). Then DNA was digested using HindIII enzyme according to protocol. Next DNA was precipitated using Phenol/Chloroform. The DNA was eluted in 35 µl of H20 and used in PCR-reactions.
First, 10 cycles of tag introducing amplification was performed, followed by destruction of the UTP-containing Alu-primers by Uracil-DNA glycosylase (UDG). This reaction was carried out in a final volume of 50 µl containing, TP buffer 10X + MgCl2, dNTP 10 µM, Alu primer 10 µM, HBV primer 100 µM and Taq High Fidelity enzyme. HBV specific primers for X, Core and S regions were used (table 1). The amplification was carried out with the following cycling conditions: denaturation for 30 sec at 94˚C, annealing for 30 sec at 59˚C and elongation for 3 min at 70˚C with an initial denaturation step of 2 min at 94˚C, and a final step of 10 minutes at 70˚C. During the next amplification a “touchdown” PCR technique was applied. After addition of 10 µM of a new pair of primers (HB2 and Alu-tag primer), 20 cycles of amplification (denaturation for 30 sec at 94˚C, annealing for 30 sec at 65˚C and elongation for 3 min at 70˚C. The annealing temperature was decreased by 1˚C every two cycles, reaching a final temperature of 55˚C. At this step another 20 cycles were performed with a final step of 10 min at 70˚C.
Five µL of PCR product was used in a nested PCR. A new pair of primers (HB3 and Alu-tag primer) was introduced in a mix otherwise similar to the one described above. The cycling conditions were identical with the tag-introducing PCR described above except for an annealing temperature of 62˚C and 40 cycles of amplification. After nested PCR,10 µl product from both first and nested PCR reactions were added to a 1% agarose gel and migrated by electrophoresis. Direct sequencing of PCR products visible on gel was performed to verify viral-host jucntions.
Direct sequencing
Sequences of the viral-host junctions were determined using the primers used for PCR reactions, either on purified PCR products (PureLinkPCR Purification kit, Invitrogen, Carlsbad, CA) or bands extracted from gel (Wizard SV Gel and PCR Clean-Up System, Madison, WI). The BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied biosystems) was used according to the manufacturer’s instructions followed by clean-up of the dye (USB® PrepEase® Sequencing Dye Clean-Up Kit). A DNA sequencer (ABI PRISM 310 Genetic Analyzer; Applied or ABI 3130x Genetic Analyzer) was used to resolve the sequencing products.
Bioinformatics and Statistics
The BLAST tool from NCBI was used to find the flanking cellular sequence and to verify the detection of HBV DNA.
Results
Patient profiles
Clinical data on patients are summarised in table 2. Patients where categorised as high replication low inflammation (HRLI, HBeAg positive, HBV DNA >6 log10 copies/ml, normal ALT and inflammation score ≤3), HBeAg-positive hepatitis (EPH, HBeAg-positive, inflammation score ≥4 and/or ALT>ULN); HBeAg-negative hepatitis (ENH, HBeAg-negative with HBV DNA >4 log10 copies/ml and/or ALT >ULN); low replication low inflammation(LRLI, HBeAg negative, HBV DNA <4 log10 copies/ml, inflammation score ≤3 and normal ALT). There was a significantly lower proportion of women with detectable integrations as compared to men. Integrations were found in all patients in the immune tolerant stage.
Sequence analysis of integrated HBV DNA in liver biopsies
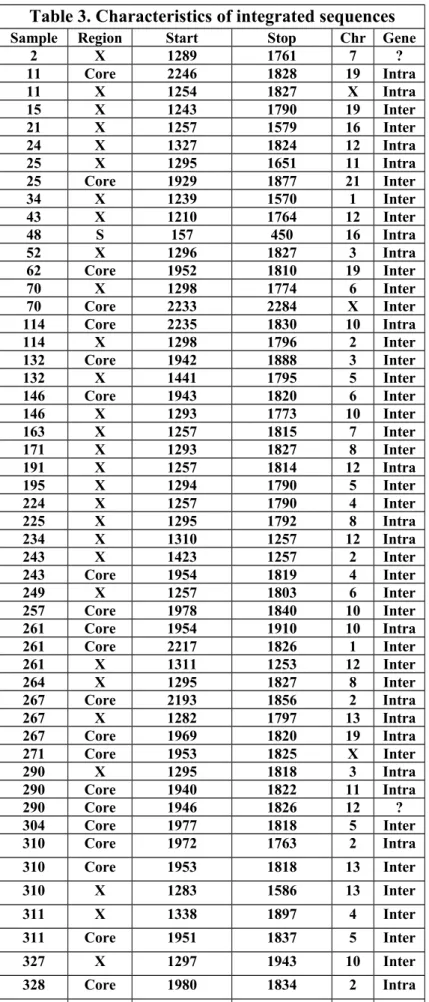
After Alu-PCR, gel electrophoresis indicated presence of integration in the samples by presence of one or several bands (Figure 6). Table 3 summarises results of direct sequencing that was performed on 44 positive samples (3/3 HRLI, 7/13 EPH, 24/38 ENH, 10/21 LRLI). Most integrated HBV sequences terminated in the vicinity of DR1 (nt 1833 to 1844), a proposed integration junction corresponding with the end of double stranded linear HBV DNA (Figure 7). Only two samples contained sequences spanning this region.
Two samples contained integrated HBV sequences that seemed to have undergone a rearrangement. In particular from patient 381, the integrated sequence contained DNA representing nt 1295-1683 in the HBV genome, followed by nt 2587-2471 in reverse direction, whereas from patient 392, DNA from one of the two chimeric sequences detected ccontained HBV DNA in reverse (nt 1934-1815) followed by nt 2734-2815 in forward direction (Figure 8).
Southern blot hybridization analysis
On 18 samples hybridisation with Southern blot was performed to test the HBV specificity of the assay. It distinguished all bands visible by agarose gel electrophoresis as containing HBV DNA as well as an additional ten cases (Figure 9, and data not shown). Southern blot was not performed on additional samples, on which direct sequencing was used to confirm the viral integrations in samples with visible bands on gel.
Chromosomal distribution of viral-host junctions
When submitted to BLAST, the samples mapped to 17 different human chromosomes (all but Chr# 14, 15, 17, 18, 20 and Y), as indicated in Table 3 and Figure 10. Of the integrated sequences, 20 were intragenic, whereas 36 were intergenic and two were undetermined. The latter two mapped to BAC clones of Chr 7 and 12 respectively when submitted to BLAST. In 33 cases with a visible band on gel, sequencing revealed only HBV DNA (data not shown).
Multiple integrations in the same patient
In 15 patients two or more unique integrations were found. In some cases we found integrations using different primers (X, Core or S) whereas in others multiple bands from the same PCR reaction were extracted from agarose gel and sequenced.
Discussion
Using the Alu-PCR method followed by Sanger sequencing we obtained 58 integrated sequences from a total of 44 samples. To our knowledge this is the first study where HBV DNA integration was systematically investigated in patients belonging to different stages of disease and representing different genotypes and where intrahepatic levels of HBV DNA were also available.
There have been several studies on HBV DNA integration in HCC tumours (Sung et al. 2012., Toh ST et al., 2013) and review ( Bonilla Guerrero R, Roberts LR. 2005) , whereas less has been reported from CHB patients without HCC (Huang H-Pet al 2005., Murakami Y et al., 2004., Minami M et al., 2005, Fowler MJ et al., 1986). Integrations found in tumours are probably the result of clonal expansion of cells containing certain patterns of HBV DNA integration. In non-tumour cells, fewer events of integration have been observed by whole genome sequencing, probably due to fewer number of cells containing the same integration. We found at least one integration in 59 % of samples analysed, and the integrated sequences were evenly distributed on different chromosomes. Interestingly, all three patients in the HRLI stage had detectable integrations compared with less than half of the patients in the LRLI stage. This suggests that in the HRLI stage, when essentially all hepatocytes are infected and may produce large amounts of ds linear DNA, the presumed substrate for integration, integrations may be relatively frequent despite the low cell turnover, in agreement with a recent study by Mason et al. {Mason:2016gp}. A lower frequency of integrations in the LRLI stage might be a result of hepatocytes carrying integrations being replaced with uninfected hepatocytes without integrations during the course of chronic infection.
Our data also support the notion of a random selection of integration site, at least with regards to chromosome. However, almost 40% of integrations were intragenic, the apparent preference is for these regions could be due to the fact that coding regions of the human genome account for only 1-5% of the total nuclear DNA. Moreover to some extent this could be explained by the fact that Alu repeats are more frequent in intragenic regions as compared with intergenic regions.
In line with previous studies, 88% of the viral-host junctions were located to the region between DR1 and DR2, which constitutes the cohesive overlap region in the incomplete circular genome that is contained in the viral particle. Sung et al found that 40% of integration breakpoints were
restricted to genomic regions adjacent to position 1800. Several groups have suggested mechanisms for integration where this region participates in the integration process, and a linear precursor ending at either DR1 or DR2 seems to be the preferred substrate for the integration event. However, we found two integrations spanning this region, one of which was located in an intergenic region of chromosome 10 and one on chromosome 4, suggesting other forms of HBVDNA might be possible candidates for integration.
Two sequences contained what seems to be rearrangement events, and this has also been observed by others (Takada et al.1990). Takada et al. showed a one- to three-nucleotide identity between the two viral sequences as well as adjoining sequences weakly homologous to each other. This was the case in patients 381 and 392, where homologous regions between the + and – strands of HBV were found. According to Takada et al. these rearrangement events occur before integration and could be another form of HBV DNA used as substrate for integration. That other forms of HBV DNA than double stranded linear form can be substrate for integration fits with our observation that not all breakpoints in the viral genome are found at nt 1830.
More than one third of the patients had more than one integrated HBV DNA sequence detected by Alu-PCR and characterised by direct sequencing. Additional samples had multiple bands on gel electrophoresis or Southern blot. Taking into account that the analyses were performed on only 5 mg of liver tissue (containing ≈ 500,000 hepatocytes as compared with the total number of hepatocytes in the liver being≈1011it is likely that a million or more different integrations can be
present at a given time point in one patient). Such a vast number of different integrations would impose a significant risk that some might have oncogenic effects, in particular if combined with a high rate of cell regeneration (Pollicino T, Vegetti A, Saitta C, Ferrara F, Corradini E, Raffa G, Pietrangelo A, Raimondo G. 2013). However, there is currently no evidence of a preferential site of integration, as is the case for woodchuck hepatitis virus, that frequently integrates in N-myc genes. Most CHB patients do however not develop cancer. Our findings underline the possibility that integrated HBV DNA in HCC may often be a coincidence and might not always have an implication in the carcinogenesis. Curiously, integrations were less frequent among female patients. Whether this is of relevance for the lower frequency of HCC among women is not known.
When comparing the samples where integrations were found with those without detectable integration by Alu-PCR, no statistically significant differences where found as regards age, serum
levels of HBV DNA or HBsAg, ALT levels or total intrahepatic HBV DNA. Given the low sensitivity of the Alu-PCR one cannot rule out that integrations are present in all the biopsies analysed in this study. As integration of HBV DNA seems to occur already early during chronic infection, when viral levels are high, it is possible that early intervention with antiviral treatment might reduce future integrations and the risk for cancer.
There are some limitations with our study. First, the Alu-PCRis less likely to identify integrations located distant to (>1500 base pairs) an Alu-repeat. Considering that the distance between Alu repeats is often >3000 bp this might explain why we did not find any integration in one third of the samples. In a recent study using whole-genome sequencing, integration breakpoints were detected in 92.6% of HCCs from HBsAg-positive individuals (Sung et al. 2012). They found that 7.8% of the detected breakpoints were located more than 10 kb from the nearest Alu-repeat and 28% were located more than 3 kb from an Alu-repeat. In addition, the assay requires a minimum of about 100 copies of the integration for detection (Murakami Y et al., 2004.,). Thus a hepatocyte with an integrated HBV DNA sequence needs to divide at least seven times to produce a sufficient number of copies. Recently integrated sequences or those hampering cell division would thus not be detected. The high rate of integration in our study, despite these limitations of the Alu-PCR method suggest that integrated HBV DNA might be present in virtually all chronically infected livers. To confirm this, studies with alternative and more sensitive techniques, such as deep sequencing by next generation sequencing would be required.
Another limitation with Alu-PCR is that only one of the two viral-host junctions is identified. Thus, the full size of the integrated sequence is not obtained and it cannot be deduced if the integrated HBV DNA is functional, i.e. if viral proteins are expressed. To study this, whole genome sequencing (Sung et al. 2012) would be the preferred method, but the complete integrated HBV sequences were also obtained in earlier studies on hepatoma cell lines and HCC specimens that used cloning methods by phage library construction.
In summary, this study is the first to characterise integrated HBV DNA sequences in CHB patients in different stages of disease. The results indicate that integration of viral DNA is a very common and early event during chronic infection, occurring in the majority of, probably all, patients.
Figures and tables
Figure 1: Virology HBV. (Figure adapted from Pollicino and Raimondo, 2014 with slight modification
Figure 2. The partially double stranded HBV-genome with the four open reading frames indicated. Numerotation of nucleotides start at the EcoR1-site (the genome is ~3.2 kb).
Figure 3 Schematic representation of HBV infection cycle. The 6 stages of HBV infection cycle: 1) viral attachment and entry; 2) transportation into the nucleus; 3) conversion of rcDNA to cccDNA; 4) transcription and translation; 5) synthesis of progeny viral DNA genomes; and 6) assembly of viral particles and secretion (Figure adapted from Pollicino and Raimondo,2014 With slight modification).
Figure 4: HBV Genotypes (Figure adapted from Pollicino and Raimondo, with slight modification
Figure 5: Natural history of HBV infection and disease progression.
Figure 6: Left: Four samples positive on gel (lane 2, 21, 52, 57) and one possibly positive (lane 9); Right:Multiple integrations on gel visible as several bands (25, 43, 90) or smear (34, 37).
Figure 3. Nucleo de sequences of virus-virus junc ons. Shading shows region of par al homology between HBV strands. Top sequence: Pa ent 381. Bo om sequence: Pa ent 392.
GGCAGAGGTGAAAAAGTTGCATGGTGCTGGTGCGCAG
ATCTAGTTAATCATTACTTCCAAACCAGACATTATTT
1810 2720 2740AGGCAGAGGTGAAAAAGTTCCAAACCAGACATTATTTA
HBV(+)
392
HBV(–)
1820 1800. . . . .
. . . . . . . .
2730GACTCTTGGACTCTCAGCAATGTCAACGACCGACCTTGAGG
TGTAAGAGGGCCCACATATTGTTGACATCTATTAATAATGT
1680 2590 2580CTCTTGGACTCTCAGCAATGTTGACATCTATTATTAATGTC
HBV(–)
381
HBV(+)
1670 1690. . . . . . . . .
. . . . . . .
2580Figure 8 : Nucleotide sequenze of virus-virus junctions. Shading shows region of partial homology between HBV strands. Top sequences : Patient 381. Bottom sequences: Patient 392
Figure 9 : Left : Gel electrophoresis with three positive samples (and one borderline). Right: Southern blot showing the specificity of the amplification. P.C: positive control
Table 1
. Sequence of primers used in Alu-PCR
(from Murakami et al, Gut, 2005)Code sequence (5'-3') Position (HBV) Description
UP5 CAGUGCCAAGUGUUUGCUGACGCCAAAGUGCUGGGAUUA Alu-sense
T3-515 AUUAACCCUCACUAAAGCCUCGAUAGAUYRYRCCAYUGCAC Alu-antisense
UP6 CAAGTGTTTGCTGACGCCAAAG Alu-sense (tag)
midT3 ATTAACCCTCACTAAAGCCTCG Alu-antisense (tag)
pUTP ACAUGAACCUUUACCCCGUUGC 1131-1152 HB1 (HBV X) MM37 TGCCAAGTGTTTGCTGACGC 1174-1193 HB2 (HBV X) MM60 CTGCCGATCCATACTGCGGAAC 1258-1279 HB3 (HBV X) uPre 31 GAGUUCUUCUUCUAGGGGACCUG 2350-2328 HB1 (HBV Core) MM31 AGTGCGAATCCACACTC 2288-2269 HB2 (HBV Core) MM25 GGAAGGAAAGAAGTCAGAAGG 1978-1960 HB3 (HBV Core) uPreS2 ACACGGCGGUAUUUUGGGGTGGAG 3042-3065 HB1 (HBV S) MM2R CAGGCTCAGGGCATATTGACAA 3070-3091 HB2 (HBV S) MD71 YCCTGCTGGTGGCTCCAGTTC 55-75 HB3 (HBV S) U = dUTP, Y =C or T, R = A or G
Table 2
.Patient profiles
Integration No integration p Agea 33.5 (16-60) 33 (18-50) >0.05 Gender (F/M)b 11/33 18/13 0.008 Serum HBV DNAa 4.63 (2.78-10.06) 4.1 (2.2-9.71) >0.05 Serum HBsAga 3.66 (0.92-5.13) 3.68 (1.08-5.29) >0.05 ALT/ULNa 0.77 (0.24-9) 0.8 (0.3-8.63) >0.05 Genotype (A/B/C/D) 10/7/2/25 7/2/3/19 Clinical stage (IT/EPH/ENH/IC) 3/7/24/10 0/6/14/11
total ihHBV DNAa 2.7 (0.83-4.73) 2.63 (0.76-4.81) >0.05
Table 3. Characteristics of integrated sequences
Sample Region Start Stop Chr Gene
2 X 1289 1761 7 ? 11 Core 2246 1828 19 Intra 11 X 1254 1827 X Intra 15 X 1243 1790 19 Inter 21 X 1257 1579 16 Inter 24 X 1327 1824 12 Intra 25 X 1295 1651 11 Intra 25 Core 1929 1877 21 Inter 34 X 1239 1570 1 Inter 43 X 1210 1764 12 Inter 48 S 157 450 16 Intra 52 X 1296 1827 3 Intra 62 Core 1952 1810 19 Inter 70 X 1298 1774 6 Inter 70 Core 2233 2284 X Inter 114 Core 2235 1830 10 Intra 114 X 1298 1796 2 Inter 132 Core 1942 1888 3 Inter 132 X 1441 1795 5 Inter 146 Core 1943 1820 6 Inter 146 X 1293 1773 10 Inter 163 X 1257 1815 7 Inter 171 X 1293 1827 8 Inter 191 X 1257 1814 12 Intra 195 X 1294 1790 5 Inter 224 X 1257 1790 4 Inter 225 X 1295 1792 8 Intra 234 X 1310 1257 12 Intra 243 X 1423 1257 2 Inter 243 Core 1954 1819 4 Inter 249 X 1257 1803 6 Inter 257 Core 1978 1840 10 Inter 261 Core 1954 1910 10 Intra 261 Core 2217 1826 1 Inter 261 X 1311 1253 12 Inter 264 X 1295 1827 8 Inter 267 Core 2193 1856 2 Intra 267 X 1282 1797 13 Intra 267 Core 1969 1820 19 Intra 271 Core 1953 1825 X Inter 290 X 1295 1818 3 Intra 290 Core 1940 1822 11 Intra 290 Core 1946 1826 12 ? 304 Core 1977 1818 5 Inter 310 Core 1972 1763 2 Intra 310 Core 1953 1818 13 Inter 310 X 1283 1586 13 Inter 311 X 1338 1897 4 Inter 311 Core 1951 1837 5 Inter 327 X 1297 1943 10 Inter 328 Core 1980 1834 2 Intra 328 Core 2276 1902 7 Intra 381 X 1295/2587 1683/2471 22 Inter 388 Core 1978 1821 1 inter
392 Core 1934/2734 1815/2815 9 intra 392 Core 1926 1822 9 inter 404 Core 2200 1850 2 inter 404 X 1226 1813 11 inter * BAC clones
References
1. Block, Timothy M, Haitao Guo, and Ju-Tao Guo. 2007. “Molecular Virology of Hepatitis B Virus for Clinicians..” Clinics in Liver Disease 11 (4):
685–706–vii. doi:10.1016/j.cld.2007.08.002.
2. Locarnini, Stephen, Margaret Littlejohn, Muhammad Nazri Aziz, and Lilly Yuen. 2013. “Possible Origins and Evolution of the Hepatitis B Virus (HBV).” Seminars in Cancer Biology. Elsevier Ltd: 1–15. doi:10.1016/j.semcancer.2013.08.006.
3. Ni, Yi, Florian A Lempp, Stefan Mehrle, Shirin Nkongolo, Christina Kaufman, Maria Fälth, Jan Stindt, et al. 2013. “Hepatitis B and D Viruses Exploit Sodium Taurocholate
Co-Transporting Polypeptide for Species-Specific Entry Nto Hepatocytes..” Gastroenterology. doi:10.1053/j.gastro.2013.12.024.
4. Klingmuller, U., and Schaller, H. (1993). Hepadnavirus infection requires interaction between the viral pre-S domain and a specific hepatocellular receptor. J Virol, 67(12),7414-7422.
5. Ni, Y., Lempp, F. A., Mehrle, S., Nkongolo, S., Kaufman, C., Falth, M., Stindt, J., Koniger, C., Nassal, M., Kubitz, R., Sultmann, H., and Urban, S. (2014). Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology, 146(4), 1070-1083.
6. Yan, H., Zhong, G., Xu, G., He, W., Jing, Z., Gao, Z., Huang, Y., Qi, Y., Peng, B., Wang, H., Fu, L., Song, M., Chen, P., Gao, W., Ren, B., Sun, Y., Cai, T., Feng, X., Sui, J., and Li, W. (2012). Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife, 1, e49.
7. Kang, H. Y., Lee, S., Park, S. G., Yu, J., Kim, Y., and Jung, G. (2006). Phosphorylation of Hepatitis B virus Cp at Sert 87 facilitates core assembly. Biochem J, 398 (2), 311-317
8. Kann, M., Sodeik, B., Vlachou, A., Gerlich, W. H., and Helenius, A. (1999). Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol, 145(1), 45-55.
9. Kock, J., and Schlicht, H. J. (1993). Analysis of the earliest steps of hepadnavirus replication: genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J Virol, 67(8), 4867-4874.
10. Newbold, J. E., Xin, H., Tencza, M., Sherman, G., Dean, J., Bowden, S., and Locarnini, S. (1995). The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol, 69(6),
11. Pollack, J. R., and Ganem, D. (1993). An RNA stem-loop structure directs hepatitis B 3350-3357.
12. Block, T. M., Mehta, A. S., Fimmel, C. J., and Jordan, R. (2003). Molecular viral oncology
13 .Milich, David, and T Jake Liang. 2003. “Exploring the Biological Basis of Hepatitis B E Antigen in Hepatitis B Virus Infection..” Hepatology 38 (5): 1075–86. doi:10.1053/jhep.2003.50453.
14 .Chai, Ning, Ho Eun Chang, Emmanuelle Nicolas, Ziying Han, Michal Jarnik, and John Taylor. 2008. “Properties of Subviral Particles of Hepatitis B Virus..” Journal of Virology 82 (16): 7812–17.
doi:10.1128/JVI.00561-15 .Feitelson, Mark A, Barbara Bonamassa, and Alla Arzumanyan. 2014. “The Roles of Hepatitis B Virus-Encoded X Protein in Virus Replication and the Pathogenesis of Chronic Liver Disease.” Expert Opinion on Therapeutic Targets: 1–14. doi:10.1517/14728222.2014.867947.
16. Lucifora, Julie, Silke Arzberger, David Durantel, Laura Belloni, Michel
Strubin, Massimo Levrero, Fabien Zoulim, Olivier Hantz, and Ulrike Protzer. 2011. “Hepatitis B Virus X Protein Is Essential to Initiate and Maintain Virus Replication After Infection..” Journal of Hepatology 55 (5): 996–1003. doi:10.1016/j.jhep.2011.02.015.08.
17. Tang, Hong, Luvsanjav Delgermaa, Feijun Huang, Naoki Oishi, Li Liu, Fang He, Liansan Zhao, and Seishi Murakami. 2005. “The Transcriptional Transactivation Function of HBx Protein Is Important for Its Augmentation Role in Hepatitis B Virus Replication..” Journal of Virology 79 (9): 5548–56. doi:10.1128/JVI.79.9.5548-5556.2005.
X Protein Upregulates Expression of SMYD3 and C-MYC in HepG2 Cells.” Medical Oncology (Northwood, London, England) 26 (4): 445–51. doi:10.1007/s12032-008-9144-1.
19. Schaefer S.World J Gastroenterol. 2007 Jan 7;13(1):14-21. Review. Hepatitis B virus taxonomy and hepatitis B virus genotypes.
20. Kao JH, Chen PJ, Lai MY, Chen DS. Hepatology. 2001 Oct;34(4 Pt
1):817-23 Acute exacerbations of chronic hepatitis B are rarely associated with superinfection of hepatitis B virus.
21. Fattovich G, Bortolotti F, Donato F.J Hepatol. 2008 Feb;48(2):335-52. Epub 2007 Dec 4 Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors.
22. Kao JH, Chen PJ, Lai MY, Chen DS. Gastroenterology. 2000 Mar;118(3):554-9 Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B.
23. Pollicino T, Cacciola I, Saffioti F, Raimondo G J Hepatol. 2014 Aug;61(2):408- 17 Hepatitis B virus PreS/S gene variants: pathobiology and clinical implications.
24. Bertoletti, A., Maini, M., and Williams, R. (2003). Role of hepatitis B virus specific cytotoxic T cells in liver damage and viral control. Antiviral Res, 60(2), 61-66.
25. Hadziyannis, S. J., Vassilopoulos, D., and Hadziyannis, E. (2013). The natural course of chronic hepatitis B virus infection and its management. Adv Pharmacol, 67,247-291.
26. Fattovich, G. (2003). Natural history and prognosis of hepatitis B. Semin Liver Dis,23(1),47-58.
27. Hadziyannis, S. J., and Papatheodoridis, G. V. (2006). Hepatitis B e antigen-negative chronic hepatitis B: natural history and treatment. Semin Liver Dis, 26(2), 130-141.
28. Hoofnagle, J. H., Doo, E., Liang, T. J., Fleischer, R., and Lok, A. S. (2007). Management of hepatitis B: summary of a clinical research workshop. Hepatology,45(4), 1056-1075.
29.McMahon,B.J. (2009). The natural history of chronic hepatitis B virus infection. Hepatology,49( 5 suppl), S45-S55.30.
30. Buster, E. H., Flink, H. J., Cakaloglu, Y., Simon, K., Trojan, J., Tabak, F., So, T. M., Feinman, S. V., Mach, T., Akarca, U. S., Schutten, M., Tielemans, W., van Vuuren, A. J.,Hansen, B. E., and Ja Shafritz, D. A., Shouval, D., Sherman, H. I., Hadziyannis, S. J., and Kew, M. C. (1981). Integration of hepatitis B virus DNA into the genome of liver cells in chronic liver disease and hepatocellular carcinoma. Studies in percutaneous liver biopsies and post-mortem tissue specimens, N Engl J Med, 305 (18), 1067-1073.
31.Chen, Y. C., Huang, S. F., Chu, C. M., and Liaw, Y. F. (2012). Serial HBV DNA levels in patients with persistently normal transaminase over 10 years following spontaneous HBeAg seroconversion. J Viral Hepat, 19(2), 138-146.
32. de Franchis, R., Meucci, G., Vecchi, M., Tatarella, M., Colombo, M., Del, N. E., Rumi, M. G., Donato, M. F., and Ronchi, G. (1993). The natural history of asymptomatic hepatitis B surface antigen carriers. Ann Intern Med, 118(3), 191-194.
33. Tai, D. I., Lin, S. M., Sheen, I. S., Chu, C. M., Lin, D. Y., and Liaw, Y. F. (2009). Long-term outcome of hepatitis B e antigen-negative hepatitis B surface antigen carriers in relation to changes of alanine aminotransferase levels over time. Hepatology, 49(6), 1859-1867.
34. Martinot-Peignoux, M., Boyer, N., Colombat, M., Akremi, R., Pham, B. N., Ollivier, S.,Castelnau, C., Valla, D., Degott, C., and Marcellin, P. (2002). Serum hepatitis B virus DNA levels and liver histology in inactive HBsAg carriers. J Hepatol, 36(4),543-546.
35. Papatheodoridis, G. V., Chrysanthos, N., Hadziyannis, E., Cholongitas, E., and Manesis, E. K. (2008). Longitudinal changes in serum HBV DNA levels and predictors of progression during the natural course of HBeAg-negative chronic hepatitis B virus infection. J Viral Hepat, 15(6), 434-441.
36. Liaw, Y. F., Brunetto, M. R., and Hadziyannis, S. (2010). The natural history of chronic HBV infection and geographical differences. Antivir Ther, 15 Suppl 3, 25-33.
37. Brunetto, M. R., Giarin, M., Oliveri, F., Saracco, G., Barbera, C., Parrella, T., Abate, M. L., Chiaberge, E., Calvo, P. L., Manzini, P., and Et, A. (1991). 'e' antigen defective hepatitis B virus and course of chronic infection. J Hepatol, 13 Suppl 4, S82-S86.
38.EASL. (2012). EASL clinical practice guidelines: Management of chronic hepatitis virus infection. J Hepatol, 57(1), 167-185.
39.Marion, P L, F H Salazar, J J Alexander, and W S Robinson. 1980. “State of Hepatitis B Viral DNA in a Human Hepatoma Cell Line..” Journal of
Virology 33 (2): 795–806.
40.Bréchot, C, C Pourcel, A Louise, B Rain, and P Tiollais. 1980. “Presence of Integrated Hepatitis B Virus DNA Sequences in Cellular DNA of Human Hepatocellular Carcinoma..” Nature 286 (5772): 533–35.
41.Edman, J C, P Gray, P Valenzuela, L B Rall, and W J Rutter. 1980. “Integration of Hepatitis B Virus Sequences and Their Expression in a Human Hepatoma Cell..” Nature 286 (5772):535-38
42.Bréchot, C, M Hadchouel, J Scotto, M Fonck, F Potet, G N Vyas, and P Tiollais. 1981. “State of Hepatitis B Virus DNA in Hepatocytes of Patients with Hepatitis B Surface Antigen-Positive and -Negative Liver Diseases..” Proceedings of the National Academy of Sciences of the United States of America 78 (6): 3906–10.
43.Yang, W, and J Summers. 1999. “Integration of Hepadnavirus DNA in Infected Liver: Evidence for a Linear Precursor..” Journal of Virology 73 (12): 9710–17.
44.Jiang, Zhaoshi, Suchit Jhunjhunwala, Jinfeng Liu, Peter M Haverty, Michael I Kennemer, Yinghui Guan, William Lee, et al. 2012. “The Effects of Hepatitis B Virus Integration Into the Genomes of Hepatocellular Carcinoma Patients..” Genome Research 22 (4): 593–601. doi:10.1101/gr.133926.111.
45.Sung, Wing-Kin, Hancheng Zheng, Shuyu Li, Ronghua Chen, Xiao Liu, Yingrui Li, Nikki P Lee, et al. 2012. “Genome-Wide Survey of Recurrent HBV Integration in Hepatocellular Carcinoma..” Nature Genetics. doi:10.1038/ng.2295.
46.Takada, S, Y Gotoh, S Hayashi, M Yoshida, and K. Koike. 1990. “Structural Rearrangement of Integrated Hepatitis B Virus DNA as Well as Cellular Flanking DNA Is Present in Chronically Infected Hepatic Tissues..” Journal of Virology 64 (2): 822– 28.
47.Tsuei, D J, P J Chen, M. Y. Lai, D S Chen, C S Yang, J Y Chen, and T Y Hsu. 1994. “Inverse Polymerase Chain Reaction for Cloning Cellular Sequences Adjacent to Integrated Hepatitis B Virus DNA in Hepatocellular Carcinomas..” Journal of Virological Methods 49 (3):
48 .Minami, M, K Poussin, C Bréchot, and P Paterlini. 1995. “A Novel PCR Technique Using Alu-Specific Primers to Identify Unknown Flanking Sequences From the Human Genome..” Genomics 29 (2): 403–8. doi:10.1006/geno.1995.9004.
49. Tamori, Akihiro, Shuhei Nishiguchi, Shoji Kubo, Masaru Enomoto, Noritoshi Koh, Tadashi Takeda, Susumu Shiomi, Kazuhiro Hirohashi, Hiroaki Kinoshita, and Shuzo Otani. 2003. “Sequencing of Human-Viral DNA Junctions in Hepatocellular Carcinoma From Patients with HCV and Occult HBV Infection..” Journal of Medical Virology 69 (4): 475–81. doi:10.1002/jmv.10334.
50.Jiang, Suzhen, Ziwei Yang, Weijie Li, Xiaojun Li, Yongfeng Wang, Jiangbo
Zhang, Chunhui Xu, et al. 2012. “Re-Evaluation of the Carcinogenic Significance of Hepatitis B Virus Integration in Hepatocarcinogenesis..” PLoS ONE 7 (9): e40363. doi:10.1371/journal.pone.0040363.
51. Saigo, Kenichi, Kenichi Yoshida, Ryuji Ikeda, Yoshiko Sakamoto, Yoshiki
Murakami, Tetsuro Urashima, Takehide Asano, Takashi Kenmochi, and Ituro Inoue. 2008. “Integration of Hepatitis B Virus DNA Into the Myeloid/Lymphoid or Mixed-Lineage Leukemia (MLL4) Gene and Rearrangements of MLL4 in Human Hepatocellular Carcinoma..” Human Mutation 29 (5): 703–8. doi:10.1002/humu.20701. 52. Tang, Ka-Wei, Babak Alaei-Mahabadi, Tore Samuelsson, Magnus Lindh, and Erik Larsson. 2013. “The Landscape of Viral Expression and Host Gene Fusion and Adaptation in Human Cancer..” Nature Communications 4: 2513. doi:10.1038/ncomms3513.
53. Paterlini-Bréchot, Patrizia, Kenichi Saigo, Yoshiki Murakami, Mounia Chami, Devrim Gozuacik, Claude Mugnier, David Lagorce, and Christian Bréchot. 2003. “Hepatitis B Virus-Related Insertional Mutagenesis Occurs Frequently in Human Liver Cancers and Recurrently Targets Human Telomerase Gene..” Oncogene 22 (25):
54. Murakami, Y. 2005. “Large Scaled Analysis of Hepatitis B Virus (HBV) DNA Integration in HBV Related Hepatocellular Carcinomas.” Gut 54 (8): 1162–68. doi:10.1136/gut.2004.054452.
55. Huang P, Tsuei DJ, Wang K-J, Chen Y-L, Ni Y-H, Jeng Y-M, Chen L, Hsu Y, Chang M-H. 2005. Differential integration rates ofhepatitis B virus DNA in the liver ofchildrenwithchronichepatitis B virus infection and hepatocellularcarcinoma. Journal of Gastroenterology and Hepatology20:1206–1214.
56. Tu T, Mason WS, Clouston AD, Shackel NA, McCaughan GW, Yeh MM, Schiff ER, Ruszkiewicz AR, Chen JW, Harley HAJ, Stroeher UH, Jilbert AR. 2015. Clonal expansion of hepatocytes with a selective advantage occurs during all stages ofchronichepatitis B virus infection. J Viral Hepat22:737–753.
57. Pollicino T, Vegetti A, Saitta C, Ferrara F, Corradini E, Raffa G, Pietrangelo A, Raimondo G. 2013. Hepatitis B virus DNA integration in tumourtissueof a non-cirrhoticHFE-haemochromatosis patient withhepatocellularcarcinoma. J Hepatol58:190–193.
58. Sung W-K, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, MulawadiFH, Wong KF, Liu AM, Poon RT, Fan ST, Chan KL, Gong Z, Hu Y, Lin Z, Wang G, Zhang Q, Barber TD, Chou W-C, Aggarwal A, Hao K, Zhou W, Zhang C, Hardwick J, Buser C, Xu J, Kan Z, Dai H, Mao M, Reinhard C, Wang J, Luk JM. 2012. Genome-wide survey of recurrent HBV integration in hepatocellula rcarcinoma. Nat Genet.
59. Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK-H, Chung AY-F, Ooi LL-P-J, Lee CG-L. 2013. Deep sequencing of the hepatitis B virus in hepatocellularcarcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis34:787–798.
60. Hino O, Kitagawa T, Koike K, Kobayashi M, Hara M, Mori W, Nakashima T, Hattori N, Sugano H. 1984. Detection of hepatitis B virus DNA in hepatocellula rcarcinomas in Japan. Hepatology4:90–95.
61 . Bonilla Guerrero R, Roberts LR. 2005. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J Hepatol42:760–777.
62. Murakami Y, Minami M, Daimon Y, Okanoue T. 2004. Hepatitis B virus DNA in liver, serum, and peripheral blood mononuclear cells after the clearance of serum hepatitis B virus surface antigen. J Med Virol72:203–214.
63. Fowler MJ, Thomas HC, Monjardino J. 1986. Cloning and analysis of integrated hepatitis B virus DNA of the adrsubtype derived from a human primary liver cell carcinoma. Journal of General Virology67 ( Pt 4):771–775.
64. Minami M, Daimon Y, Mori K, Takashima H, Nakajima T, Itoh Y, Okanoue T. 2005. Hepatitis B virus-relatedinsertional mutagenesis in chronichepatitis B patients as an earlydrasticgeneticchange leading tohepatocarcinogenesis. Oncogene24:4340–4348. 65. Lindh M, Horal P, Dhillon AP, Norkrans G. 2000. Hepatitis B virus DNA levels, precore mutations, genotypes and histologicalactivity in chronichepatitis B. J Viral Hepat7:258–267.
66. Malmström S, Larsson SB, Hannoun C, Lindh M. 2012. Hepatitis B Viral DNA Decline at Loss ofHBeAg Is MainlyExplained by ReducedcccDNALoad – Down-RegulatedTranscriptionofPgRNA Has LimitedImpact. PLoSONE7:e36349