Alma Mater Studiorum - Università di Bologna
SCUOLA DI SCIENZE
Dipartimento di Chimica Industriale “Toso Montanari”
Corso di Laurea Magistrale in
Chimica Industriale
Classe LM-71 - Scienze e Tecnologie della Chimica Industriale
Modulazione reversibile dell'attività catalitica
di sistemi tioureidici chirali mediante
interazioni con anioni
Tesi di laurea sperimentale
CANDIDATO
Giacomo Foli
RELATORE
Prof. Luca Bernardi
CORRELATORE
Dott.ssa Mariafrancesca Fochi
Sessione III
___________________________________________________________________________________________________________
Anno Accademico 2013-2014
Abstract
In questo lavoro di tesi viene proposto un approccio semplice e innovativo allo sviluppo di un sistema catalitico modulabile in maniera reversibile, basato su catalizzatori tioureidici chirali. La modulazione dell’attività catalitica avviene attraverso interazioni tra un determinato anione e i due protoni tioureidici di questi catalizzatori. A seconda del carattere coordinante di un anione, l’interazione che si viene a creare con le tiouree può essere forte, nel caso di anioni coordinanti come gli alogenuri, o debole, nel caso di anioni debolmente coordinanti quali borati. Una forte interazione da parte dell’anione con il catalizzatore si traduce in una inibizione dell’attività catalitica, dal momento che il sito tioureidico non è più disponibile per promuovere eventuali reazioni. Al contrario, un anione debolmente coordinante lascia l’attività catalitica fondamentalmente inalterata, anche in termini di stereoselezione. La reversibilità di questa modulazione è ottenuta sfruttando reazioni di metatesi tra due sali che, risultando nella precipitazione di un sale insolubile, riescono a rimuovere un determinato anione dall’ambiente di reazione per sostituirlo con un altro.
This thesis is aimed at the development of a simple and innovative approach for the realization of a switchable catalytic system, based on chiral thioureidic catalysts. The modulation of the catalytic activity is achieved by the formation of an interaction between a coordinating anion and the two thioureidic proton of the catalysts. Depending on the binding ability of the anion, the interaction with the thioureas could be strong, using small coordinating anions such as halides, or feeble, using large, weakly coordinating anions such as borates. A strong interaction between the anion and the catalyst leads to the inhibition of the catalytic activity, due to the unavailability of the thioureidic site to stabilize the transition state of the examined reaction. On the contrary, a weakly coordinating anion leaves unaltered the catalytic activity, even with respect to the stereoselectivity of the process. The reversibility of this modulation is gained through metathesis reactions between two salts, in which the precipitation of an insoluble salt is able to remove a specific anion from the reaction medium.
Sommario
1
Introduzione ... 1
1.1
Sistemi catalitici modulabili ... 1
1.2
Catalizzatori tioureidici ... 6
1.3
Unità tioureidiche nel riconoscimento anionico ... 8
2
Obiettivi ... 10
3
Risultati e discussione ... 12
3.1
Riduzione di immine ... 12
3.2
Riduzione del nitrostirene ... 15
3.3
Riduzione di nitrolefine prochirali ... 23
3.4.1 Riduzione enantioselettiva dell’(E)-(3,3,3-trifluoro-1-nitroprop-1-en-2-il)benzene ... 24
3.4.2 Riduzione enantioselettiva dello (Z)-etil 3-nitro-2-fenilacrilato ... 28
3.4.3 Riduzione enantioselettiva dell’ (E)-(1-nitroprop-1-en-2-il)benzene ... 33
3.4
Reazione di Friedel-Crafts tra indolo e nitrostirene... 40
4
Conclusioni e prospettive future... 46
5
Parte sperimentale... 48
5.1
Metodi generali ... 48
5.2
Materiali ... 48
5.3
Preparazione dei materiali utilizzati... 49
5.3.1 Sintesi del catalizzatore 3a ... 49
5.3.2 Sintesi del catalizzatore 3d26 ... 49
5.3.3 Sintesi dell’immina 1a ... 50
5.3.4 Sintesi dell’immina 1b ... 50
5.3.4 Sintesi degli esteri di Hantzsch 2a e 2b27 ... 51
5.3.5 Sintesi del nitroalchene 6b28 ... 52
5.3.6 Sintesi dell’NaBArF29 ... 52
5.3.7 Preparazione del TBABArF ... 53
5.3.8 Preparazione del Ph4PCl ... 54
5.4
Procedure generali per gli studi cinetici ... 54
5.4.1 Procedura generale per gli studi cinetici sulla riduzione delle immine ... 54
5.4.2 Procedura generale per gli studi cinetici sulla riduzione del nitrostirene ... 55
5.4.3 Procedura generale per gli studi cinetici sulla riduzione enantioselettiva dell’ (E)-(3,3,3-trifluoro-1-nitroprop-1-en-2-il)benzene ... 55
5.4.4 Procedura generale per gli studi cinetici sulla riduzione enantioselettiva dello (Z)-etil 3-nitro-2-fenilacrilato ... 56
5.4.5 Procedura generale per gli sudi cinetici sulla riduzione enantioselettiva dell’(E)-(1-nitroprop-1-en-2-il)benzene ... 56
5.4.5 Procedura generale per gli studi cinetici sulla reazione di Friedel-Crafts tra indolo e nitrostirene ... 57
Introduzione
1
1 Introduzione
1.1
Sistemi catalitici modulabili
La modulazione dell’attività di un catalizzatore è un campo di ricerca che negli ultimi anni ha acquisito sempre maggiore rilevanza. Infatti, la realizzazione di sistemi catalitici controllati da stimoli esterni può avere una serie di potenziali applicazioni. Ad esempio, è possibile programmare processi sintetici multistadio in cui le specie catalitiche vengono attivate/disattivate al momento opportuno. Sempre in campo sintetico, la possibilità di attivare/disattivare un catalizzatore per un processo polimerico porterà ad un maggiore controllo sulla distribuzione dei pesi molecolari nel polimero risultante. Un’altra possibile applicazione della modulazione dell’attività catalitica, è la realizzazione di sistemi di analisi ad elevata sensibilità, dove l’analita in questione, avendo funzione di effettore per una particolare specie catalitica, attiva la formazione di un prodotto catalitico che, essendo presente in quantità maggiori rispetto all’analita, è più facilmente rilevabile (amplificazione del segnale).
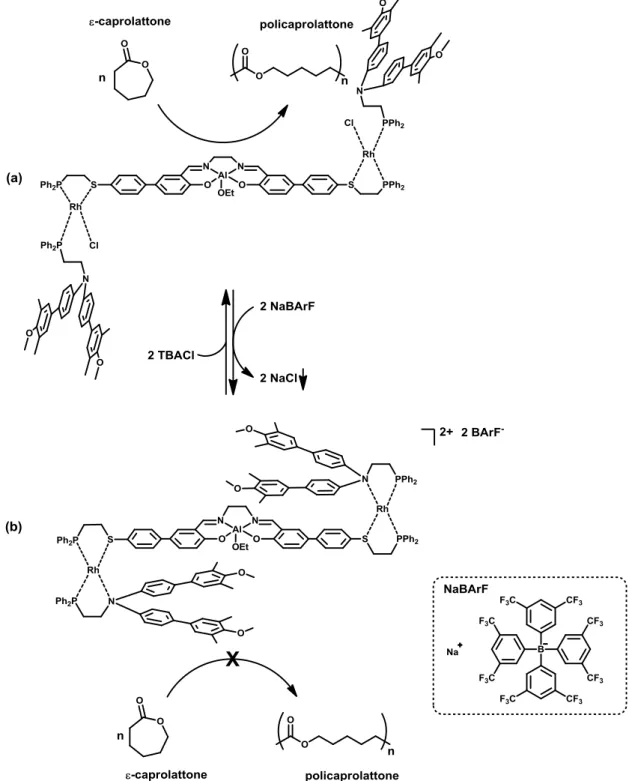
Ispirandosi alla natura, dove l’attività enzimatica viene nella maggior parte dei casi controllata da stimoli chimici, come la variazione del pH o l’interazione degli enzimi stessi con altre molecole, sono stati realizzati, in questi ultimi anni, vari sistemi catalitici che possono essere reversibilmente attivati e disattivati in situ mediante l’aggiunta di stimoli esterni.1 , 2 , 3 Ad esempio, in un lavoro di Mirkin et al. viene riportata la modulazione dell’attività di un sistema catalitico basato su un complesso di Al(III)-Salen (Figura 1).4 Questo sito attivo, che può agire come catalizzatore nella polimerizzazione dell’ε-caprolattone, è collegato, mediante complessi di rodio(I), a due blocchi arilici stericamente ingombranti. Nella struttura aperta (a) del complesso, ciascuno dei due atomi di rodio è coordinato, mediante geometria quadrata planare distorta, alla struttura del catalizzatore tramite due leganti a base di fosforo e uno a base di zolfo. L’ultima posizione di coordinazione è occupata da un cloruro. In questa forma, l’atomo di alluminio centrale acido di Lewis risulta disponibile alla coordinazione di substrati. Il complesso è quindi cataliticamente attivo in questa sua forma aperta (a). L'aggiunta in soluzione di NaBArF (BArF- = tetrakis[3,5-bis(trifluorometil)fenil]borato) comporta la rimozione dal complesso di rodio del cloruro, che precipita come sale di sodio, insolubile
Introduzione
2
nell’ambiente di reazione (diclorometano). Questo anione borato, al contrario del cloruro, non possiede capacità coordinanti, per cui non interagisce con gli atomi di rodio, che completano quindi la loro geometria di coordinazione andando a complessarsi con le aniline presenti nei leganti fosfinici, provocando un cambiamento nella geometria complessiva del complesso, che arriva a mostrare la struttura chiusa (b), nella quale l’accesso al sito catalitico centrale risulta bloccato. Nella sua forma chiusa (b), infatti, il complesso è cataliticamente inattivo. La successiva aggiunta in soluzione dell’anione
Introduzione
3
coordinante cloruro, sotto forma di sale di tetra-n-butilammonio solubile in diclorometano, provoca la riapertura della struttura, dal momento che viene ripristinata l’originaria geometria di coordinazione per gli atomi di rodio: viene quindi ristabilita l’accessibilità al sito catalitico centrale, e quindi l’attività catalitica del complesso. La modulazione dell’attività catalitica di questo sistema è quindi effettivamente realizzabile, in maniera reversibile, mediante l’applicazione di stimoli esterni sotto forma di anioni a differente potere coordinante.
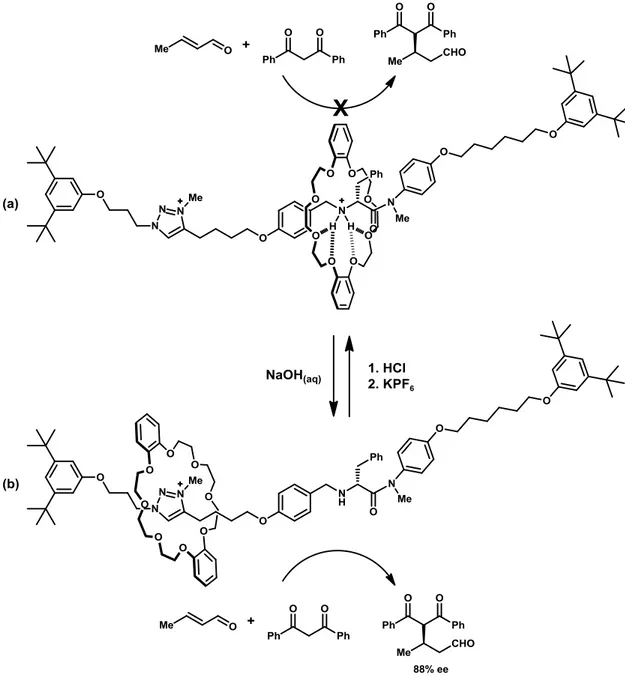
Un altro interessante esempio di sistema catalitico modulabile è stato riportato dal gruppo di Leigh5,6 che ha sviluppato catalizzatori modulabili di natura rotaxanica. I rotaxani sono sistemi molecolari costituiti da un macrociclo all’interno del quale è inserita una catena lineare che termina con due residui stericamente ingombranti che impediscono la “fuga” del macrociclo dall’asse. L’aspetto interessante di questi sistemi risiede nel fatto che la posizione del macrociclo lungo la catena centrale può essere controllata dalle sue interazioni con particolari residui presenti sulla catena stessa. Ad esempio, in un rotaxano costituito da un etere corona macrociclico e un asse centrale contenente sia anelli triazolici che una funzione dibenzilamminica come unità catalitica in grado di promuovere reazioni di addizione di nucleofili ad enali via ione imminio, la posizione del macrociclo dipende dal pH (Figura 2). Infatti, in condizioni acide il macrociclo ha una maggiore affinità per la funzionalità dibenzilamminica in forma protonata rispetto all’anello triazolico. Di conseguenza, l’etere corona andrà a interagire fortemente con il centro catalitico del sistema rendendolo inaccessibile ad eventuali reagenti. Al contrario, a pH basici, il macrociclo mostra una maggiore affinità per l’anello triazolico rispetto alla dibenzilammina in forma non protonata, che risulta quindi libera e disponibile ad interagire con i substrati per promuovere reazioni. In particolare, questo sistema catalitico è stato applicato alla reazione di Michael tra la cinnamaldeide ed un tiolo polifluorurato, dove non solo è stata dimostrata l’efficacia del rotaxano non protonato come catalizzatore, ma anche l’assenza di attività del rotaxano protonato e la sua attivazione in seguito a trattamento basico.
Introduzione
4
Figura 2: Rotaxano protonato (a), catalizzatore disattivo; rotaxano deprotonato (b), catalizzatoe attivo.
Sulla base di questo lavoro preliminare è stato in seguito sviluppato un sistema analogo dove il precedente residuo dibenzilamminico viene sostituito da un gruppo amminico chirale, nel tentativo di realizzare un raro esempio di sistema catalitico asimmetrico modulabile (Figura 3). Infatti, nonostante siano stati sviluppati numerosi sistemi catalitici non chirali, la modulazione di reazioni enantioselettive risulta al momento attuale ancora nelle fasi iniziali dello sviluppo, e quest’ultimo lavoro di Leigh et al. incoraggia fortemente la ricerca in questo campo. I principi su cui si basa questo sistema chirale sono del tutto analoghi a quelli descritti nell’esempio precedente. Anche in questo caso, il macrociclo risulta avere una maggiore affinità per l’ammina in forma protonata (catalizzatore disattivo), mentre l’ammina non protonata risulta interagire debolmente con l’etere corona cha va quindi a posizionarsi sull’anello tiazolico (catalizzatore attivo).
Introduzione
5
Quest’ultimo sistema catalitico è stato applicato all’addizione asimmetrica di Michael del dibenzoilmetano all’(E)-crotonaldeide. Anche in questo secondo caso si è dimostrato come il rotaxano sia attivo come catalizzatore in condizioni basiche, mentre in presenza di acidi la reazione non procede. Anche in questo caso l’attivazione in situ di quest’ultimo rotaxano mediante trattamento basico è stata riportata con successo. Inoltre, occorre sottolineare come il catalizzatore riattivato in situ mediante trattamento con una base abbia portato allo stesso valore di eccesso enantiomerico relativo al prodotto di reazione (88% ee) rispetto al catalizzatore preparato in forma attiva.
Introduzione
6
1.2
Catalizzatori tioureidici
Figura 4
Le tiouree (Figura 4) sono una classe di molecole che recentemente hanno trovato largo impiego nel campo dell’organocatalisi. La loro attività è dovuta alla presenza dei due protoni tioureidici che, avendo una certa acidità (pKa = 21.1 in dimetilsolfossido),7
rendono le tiouree dei buoni donatori di legame a idrogeno. Inoltre il gruppo tiocarbonilico, essendo lo zolfo uno scarso accettore di legame a idrogeno, minimizza possibili aggregazioni fra due unità tioureidiche in soluzione, aggregazioni che invece risultano essere più pronunciate quando è presente il semplice gruppo carbonilico, ovvero nel caso delle uree che d’altra parte sono anche meno acide.8
I sistemi catalitici tioureidici vengono spesso paragonati alle buche ossoanioniche degli enzimi. In un grande numero di reazioni catalizzate da enzimi, intermedi o stati di transizione anionici, derivanti ad esempio dall’addizione di una specie nucleofila ad un carbonile, vengono efficacemente stabilizzati da una rete di legami a idrogeno. Allo stesso modo, la donazione di due legami a idrogeno da parte dei sistemi tioureidici è in grado di promuovere reazioni che coinvolgono intermedi o stati di transizione con forte carattere negativo. E’ interessante notare come queste similitudini abbiano portato a verificare se fosse possibile realizzare sistemi organocatalitici tioureidici che mimassero sistemi biologici.9 Ad esempio, ispirandosi alla riduzione di nitroalcheni mediata dall’Old Yellow Enzyme (OYE),10
il gruppo di Schreiner ha realizzato la riduzione biomimetica di nitroolefine utilizzando esteri di Hantzsch, donatori di idrogeno analoghi al NADH, mediata dalla tiourea di Schreiner come sostituto dell’OYE (Figura 5).11
Introduzione
7
Figura 5
Il meccanismo proposto evidenzia come la tiourea, interagendo con il gruppo nitro via donazione di legami a idrogeno, riesca a stabilizzare lo stato di transizione che presenta una forte carica negativa sui due ossigeni del gruppo nitro, e quindi facilitare la riduzione.
La stabilizzazione dello stato di transizione mostrata dalle tiouree sembra giocare un ruolo fondamentale anche nell’indirizzare la stereochimica di reazioni catalitiche asimmetriche, che prevedono l’utilizzo di tiouree chirali come organocatalizzatori.12 Per esempio, nel nostro laboratorio è stata sviluppata recentemente13 la riduzione di una nitroolefina prochirale β-trifluorometil sostituita, sempre con esteri di Hantzsch (Figura
6). Il meccanismo proposto evidenzia come la stereoselettività del processo non sia
dovuta a repulsioni steriche tra il catalizzatore e i substrati, ovvero allo schermaggio di una delle due facce prochirali del nitroalchene, ma piuttosto ad una buona sovrapposizione geometrica tra le funzionalità del catalizzatore tioureidico e lo stato di transizione che porta all’enantiomero maggioritario del prodotto.
Introduzione
8
1.3
Unità tioureidiche nel riconoscimento anionico
La realizzazione di sistemi che riescano ad interagire con anioni (recettori anionici) ha stimolato, negli ultimi anni, una intensa attività di ricerca.14,15,16 I recettori anionici che si sono sviluppati possono essere divisi in due categorie: recettori carichi positivamente e recettori neutri.17 Se i primi sono caratterizzati dalla presenza di funzionalità ammonio o dalla incorporazione di ioni metallici che riescono a legare gli anioni, per quanto riguarda i secondi il riconoscimento anionico avviene prevalentemente mediante una interazione di tipo legame a idrogeno tra gli anioni e gruppi funzionali presenti sul recettore. Il frammento N-H risulta essere adeguato per questo scopo e nella maggior parte dei recettori neutri è presente questo gruppo funzionale. È a questo punto evidente come la funzionalità tioureidica possa tornare utile nella realizzazione di recettori anionici neutri ed infatti sono stati sintetizzati diversi composti che coordinano anioni mediante interazioni con protoni tioureidici.18
In generale, affinché si realizzi una buona interazione tra l’accettore di legame a idrogeno (l’anione) e il donatore (il recettore), la topologia del sito di coordinazione sembra rivestire una certa importanza. Nel caso specifico delle tiouree, la disposizione dei due gruppi NH sembra prestarsi a una buona interazione con ossoanioni bidentati (come per esempio l’acetato). Ad esempio, Amendola et al.17
hanno studiato le costanti di associazione (K) tra una particolare tiourea (Figura 7) e vari anioni eseguendo esperimenti in dimetilsolfossido in presenza di diversi sali di tetra-n-butilammonio a temperatura ambiente. In accordo con l’ipotesi espressa precedentemente, è emerso come sia l’acetato che il benzoato riescano ad interagire fortemente con la tiourea: per l’acetato log K = 6.02 mentre per il benzoato log K = 5.77. Esperimenti condotti in presenza di diidrogenofosfato hanno confermato ulteriormente l’affinità mostrata dalle tiouree nei confronti di questo tipo di ossoanioni. Anche in questo caso, è stata registrata una costante di associazione piuttosto elevata (log K = 5.44). Queste interazioni vengono giustificate dalla direzionalità dei legami a idrogeno che riescono a coordinare in maniera bidentata gli ossigeni sui quali viene delocalizzata la carica negativa di questa tipologia di anioni. Inoltre è stata osservata una correlazione tra la forza dell’associazione e la basicità dell’anione. L’acetato, che mostra il valore di costante di associazione più elevato, è l’anione con maggiore carattere basico; al contrario il diidrogenofosfato, il meno basico dei tre ossoanioni esaminati, presenta la costante di associazione minore.
Introduzione
9
Figura 7
Queste correlazioni tra basicità dell’anione e forza dell’associazione, hanno portato gli autori del lavoro a ipotizzare che l’interazione tra la tiourea e l’anione altro non sia che un trasferimento protonico incipiente e “congelato” dal donatore all’accettore.
Andando a studiare il comportamento degli alogenuri, anioni a cui si può attribuire una geometria sferica, la medesima tiourea ha mostrato buoni valori di associazione interagendo sia con il fluoruro (log K = 5.70) che con il cloruro (log K = 4.88). È interessante osservare come anche in questo caso l’anione più basico interagisce meglio con i protoni tioureidici, seppure la correlazione non risulti così stringente.
In generale, è possibile affermare che recettori anionici di questo tipo presentano interazioni molto forti con anioni piccoli e poco polarizzabili (alogenuri, carbossilati, idrogenofosfato, nitrato, ecc.), interazioni che dipendono anche dalla topologia (geometria) dell’anione, e dalle sue dimensioni che devono essere compatibili con quelle del recettore. Al contrario, anioni molto grandi e con carica diffusa, quali l’esafluorofosfato o il perclorato, ovvero anioni generalmente definiti come “debolmente coordinanti”,19
Obiettivi
10
2 Obiettivi
È stata precedentemente riportata l’affinità che i sistemi tioureidici mostrano nei confronti di determinati anioni. Inoltre si è descritto come l’azione organocatalitica di queste molecole si esplichi nella stabilizzazione dello stato di transizione carico negativamente delle reazioni, mentre interazioni con i substrati neutri sono deboli e geometricamente non definite (condizioni di Curtin-Hammett). Sulla base di queste due proprietà, ci si pone l’obiettivo, in questo lavoro di tesi, di verificare se sia possibile realizzare una modulazione reversibile dell’attività catalitica di catalizzatori chirali a base tioureidica attraverso interazioni con diversi tipi di anioni. In particolare, ci si è proposti di sfruttare la diversa capacità coordinante di anioni a diversa dimensione e polarizzabilità, supponendo che anioni fortemente coordinanti (es. alogenuri) fossero in grado di inibire fortemente il sistema catalitico, mentre anioni poco coordinanti (es. arilborati) non perturbassero il sistema catalitico, né per quanto riguarda l’attività né per quanto riguarda la stereoselettività (Figura 8). Inoltre, la possibilità di eseguire operazioni di metatesi anionica in condizioni semplici, come mostrato nella prima parte del capitolo introduttivo (metatesi tra TBACl e NaBArF nel lavoro di Mirkin e al.4), dovrebbe garantire la reversibilità di questo controllo.
Figura 8
Questo tipo di approccio alla realizzazione di un sistema catalitico modulabile presenta infatti un vantaggio rispetto a quelli illustrati brevemente nell’introduzione e, più in generale, rispetto a quelli realizzati fino ad ora, nonostante risulti chiaramente meno elegante. Infatti, tutti i sistemi catalitici modulabili fino ad ora riportati sono stati realizzati da zero, ovvero per andare a modulare un determinato processo sono stati
Obiettivi
11
progettati nuovi catalizzatori, presentanti generalmente strutture ad elevata complessità, con l’unico scopo di poter appunto realizzare un sistema modulabile. Appare evidente come questa procedura richieda un dispendio sia di tempo che di energie. Inoltre, la sviluppo di un nuovo sistema catalitico chirale in grado di indurre elevate enantioselezioni non è sicuramente un progetto di facile realizzazione. Al contrario, ci sono numerosissimi esempi di organocatalizzatori semplici basati su funzionalità tioureidiche, studiati da tempo e con attività catalitica nota e di grande efficacia, anche in termini di enantioselezione, utilizzati in numerose reazioni estremamente diverse tra loro.12
Più in dettaglio, in questo lavoro di tesi si è inizialmente verificata la correttezza delle ipotesi proposte studiando un sistema catalitico il più semplice possibile, quale la tiourea di Schreiner, non chirale, per poi portarsi verso l’utilizzo di catalizzatori tioureidici chirali (Figura 9). Questi catalizzatori sono stati impiegati in reazioni standard, in cui è nota la loro attività catalitica.
Risultati e discussione
12
3 Risultati e discussione
3.1
Riduzione di immine
Per cercare di valutare l’effetto di inibizione o di non inibizione che alcuni determinati ioni possono avere sull’attività catalitica della tiourea di Schreiner 3a, è stata presa in esame la reazione di riduzione di N-aril immine 1 con esteri di Hantzsch 2 (Figura 10).20
Figura 10
Dal momento che, nel lavoro originario,20 la riduzione catalitica dell’immina 1a con l’estere 2a, effettuata, con un carico catalitico dell’1% molare, in diclorometano 0.2 M a temperatura ambiente mostra a 15 h una conversione pari all’89%, si è in un primo momento deciso di utilizzare lo stesso carico catalitico, ma lavorando in CDCl3 1 M a 0
°C, per cercare di allungare i tempi di reazione e rendere più semplici gli studi cinetici. In queste condizioni, uno studio cinetico della reazione effettuato tramite spettroscopia 1H NMR ha permesso di costruire la curva (■) ripotata nel Grafico 1, che mostra una conversione pari al 74% dopo 23 h.
Risultati e discussione
13
Grafico 1: Andamento cinetico della riduzione catalitica di 1a con 2a in CDCl3 1M (■); comportamento
cinetico della reazione catalitica in presenza di 1.5% molare di TBANO2 e attivazione inaspettatamente
veloce a 28 h in seguito ad aggiunta di 1.5 mol% di NaBArF (▲)
Si è quindi studiato l’effetto sulla reazione catalitica di due sali di ammonio, TBAOAc e TBANO2, in queste condizioni di reazione, caricati in rapporto 1.5 rispetto al
catalizzatore 3a. In accordo con le aspettative iniziali, in entrambi i casi l’anione ha fortemente inibito la riduzione: a 28 h la conversione era ancora trascurabile per entrambe le reazioni. La riattivazione dell’attività catalitica in queste reazioni mediante l’aggiunta di NaBArF equimolare con il sale ha però mostrato un comportamento anomalo: la reazione è andata ad elevate conversioni in breve tempo, mostrando quindi una cinetica decisamente più rapida rispetto alla reazione catalitica standard. Nel caso della riattivazione da TBANO2, ad esempio, la reazione in seguito a trattamento con
NaBArF ha portato all’83% di conversione nel giro di 2 ore circa (Grafico 1, linea rossa,
▲). Si è ipotizzato che la motivazione di ciò fosse dovuta alla scarsa omogeneità della reazione (l’estere 2a è molto poco solubile). Si è quindi deciso di utilizzare l’estere 2b (R = t-Bu), relativamente più solubile rispetto al 2a. Sono quindi state individuate nuove condizioni di reazione mantenendo sempre lo stesso carico catalitico e lavorando a 0 °C, ma in CDCl3 0.1 M, per rallentare la reazione che risultava molto più veloce con questo
estere 2b più reattivo. Tuttavia sono da subito emersi problemi di riproducibilità, per esempio in un primo caso la reazione catalitica ha mostrato a 18 h una conversione pari al 90% mentre in una prova successiva la conversione a 24 h era ancora inferiore al 75%. Sono state effettuate numerose altre prove su questa reazione a differenti concentrazioni, con diversi carichi catalitici, in vari solventi (toluene, diclorometano, MTBE) e provando ad utilizzare immine meno reattive, come la 2b, ma in tutti i casi si sono sempre
0 10 20 30 40 50 60 70 80 90 100 0 10 20 30 40 Conv e rsi on e [% ] tempo[h]
Risultati e discussione
14
presentati grossi problemi di riproducibilità. Probabilmente la riduzione è fortemente sensibile a piccole variazioni dei parametri di reazione, come per esempio tracce di acqua o di ossigeno presenti nei solventi, fenomeni di aggregazione molecolare, e ciò la rende difficilmente controllabile. Si è quindi giunti alla conclusione che questa riduzione non fosse utile ai nostri scopi.
Risultati e discussione
15
3.2
Riduzione del nitrostirene
Visti i problemi di riproducibilità sulla reazione di riduzione delle immine 1, è stata presa in esame una reazione simile, la reazione di riduzione di substrati nitroolefinici, in quanto dati di letteratura sembravano mostrare come questa trasformazione necessitasse di un’attivazione maggiore. Abbiamo quindi supposto che piccole variazioni nelle condizioni di reazione potessero essere tollerate da questa reazione, portando a risultati riproducibili ed utili ai nostri scopi.
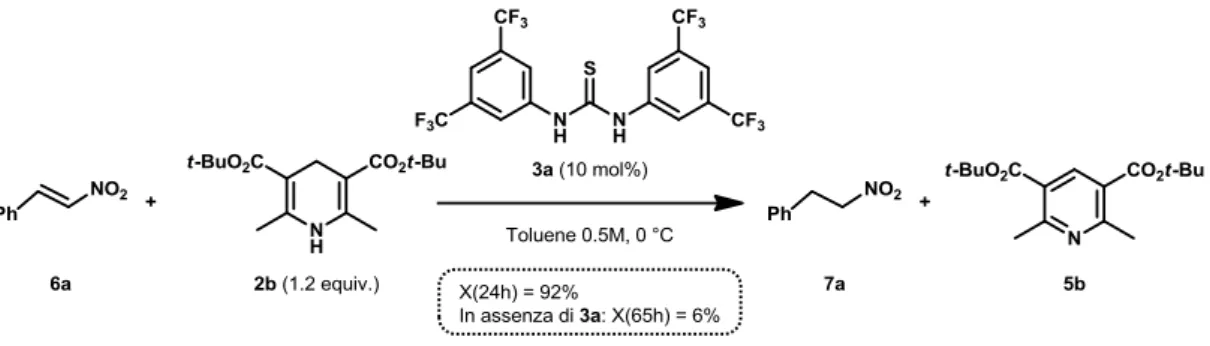
In particolare, ci si è quindi concentrati sulla riduzione del nirostirene 6a con esteri di Hantzsch 2 catalizzata dalla tiourea 3a (Figura 11).
Figura 11
In letteratura,11 viene riportato che la riduzione di 6a con l’estere etilico 2a in diclorometano 0.2 M a riflusso in presenza di 10 mol% di 3a mostra a 24 h una conversione dell’88%. Da subito si è deciso di lavorare con l’estere t-butilico 2b, che seppur più reattivo del 2a, come detto prima, risulta avere meno problemi di solubilità. La reazione si è dimostrata decisamente più riproducibile rispetto alla riduzione delle immine e sono stati quindi effettuati studi preliminari sulla cinetica di reazione in diclorometano e toluene a differenti concentrazioni e temperature, mantenendo sempre il carico catalitico di 10% molare. Da questi studi è stato possibile individuare le condizioni di reazione accettabili per i nostri scopi, che consistono nel condurre la reazione in toluene, con una concentrazione 0.5 M rispetto al nitrostirene 6a, a 0 °C (Figura 12). In queste condizioni, la reazione a 48 h mostra una conversione pressoché completa mentre la reazione in assenza di catalizzatore praticamente non procede (conversione pari al 6% dopo 65 h). L’andamento della reazione catalitica standard è riportato nel Grafico 2.
Risultati e discussione
16
Figura 12: Condizioni di reazione individuate per la reazione di riduzione catalitica di 6a.
Grafico 2: Andamento cinetico della reazione di riduzione catalitica standard di 6a.
Avendo determinato condizioni di reazione considerate idonee allo scopo del lavoro, si è passati a valutare l’effetto di diversi sali di tetra-n-butilammonio (TBA) sull’attività catalitica, sempre aggiunti in rapporto 1.5:1 rispetto al catalizzatore. Sono state quindi valutate le conversioni a 24 h della reazione catalitica in presenza di 15% molare di diversi sali (Grafico 3).
0 10 20 30 40 50 60 70 80 90 100 0 5 10 15 20 25 30 35 40 45 50 Conv e rsi on e [% ] tempo[h]
Risultati e discussione
17
Grafico 3: Conversioni registrate a 24 h per la reazione di riduzione catalitica standard di (3a 10mol%) e
per la stessa reazione in presenza di sali di ammonio (3a 10 mol% + TBAX 15 mol%).
Come si può osservare dal Grafico 3, gli alogenuri di TBA mostrano una chiara inibizione sull’attività catalitica. Questo comportamento è in linea con le aspettative iniziali, dal momento che, per quanto riguarda anioni di piccole dimensioni, come appunto gli alogeni, ci si aspetta una buona interazione con le tiouree, interazione che nel nostro caso si traduce in una riduzione dell’attività catalitica o, nella migliore delle ipotesi, in una sua totale disattivazione. Il dato che però incuriosisce è quello relativo allo ioduro. Questo è infatti il più grande tra gli alogeni, ma appare evidente come sia questo l’anione che provoca una maggiore inibizione dell’attività catalitica di 3a. Per quanto riguarda invece anioni di grandi dimensioni, come il BArF- o il PF6-, ci si aspetta, invece,
un comportamento opposto: l’interazione di questi con le tiouree dovrebbe essere praticamente nulla, a causa appunto delle loro dimensioni, e, per questo motivo, non ci si attende una particolare discordanza tra i valori delle conversioni registrati per la reazione catalitica standard e quelli registrati per la reazione catalitica in presenza di questi anioni. Contrariamente a ciò, sia il TBABArF che il TBAPF6 hanno però un piccolo effetto
negativo sulla cinetica di reazione, rallentandola solo leggermente. Tuttavia il loro comportamento, in particolare nel caso del TBABArF, è stato ritenuto accettabile per proseguire i nostri studi. Da questi dati si è quindi giunti alla conclusione che la reazione viene fortemente rallentata dalla presenza di TBAI, mentre il TBABArF non provoca un’eccesiva variazione dell’attività catalitica.
L’individuazione di questa coppia di anioni è di fondamentale importanza per gli scopi di questo lavoro, ovvero la disattivazione in situ dell’attività catalitica, mediante aggiunta in reazione di un anione inibente, in questo caso lo ioduro, e la successiva riattivazione di
0 10 20 30 40 50 60 70 80 90 100 3a 3a + TBACl 3a + TBABr 3a + TBAI 3a + TBABArF 3a + TBAPF6 Conv e rsi on e [% ]
Risultati e discussione
18
questa attraverso l’aggiunta di un anione non inibente, nello specifico il BArF
-, con contemporanea rimozione dalla soluzione dell’anione inibente. Come già descritto nell’introduzione nel caso del TBACl/NaBArF usato da Mirkin,4
la concretizzazione di questa operazione consiste prima nell’aggiunta in reazione di un sale di iodio solubile nel solvente di reazione, ad esempio il TBAI, che provoca la disattivazione di 3a. In un secondo momento, viene effettuata l’aggiunta di un sale di BArF- solubile nel solvente di reazione il cui controione riesca a rimuovere lo ioduro dalla soluzione, riattivando quindi il catalizzatore. Un composto che presenta queste caratteristiche è il NaBArF. Una volta disciolto ci si aspetta un rapido scambio ionico tra i due sali (TBAI e NaBArF) che ha come risultato la precipitazione di NaI (insolubile in toluene) e la permanenza in soluzione del solo TBABArF, che non avendo particolari effetti sulla tiourea, dovrebbe provocare la riattivazione dell’attività catalitica.
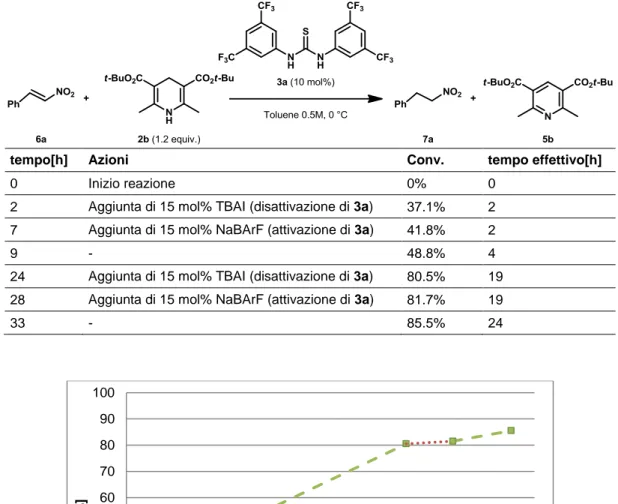
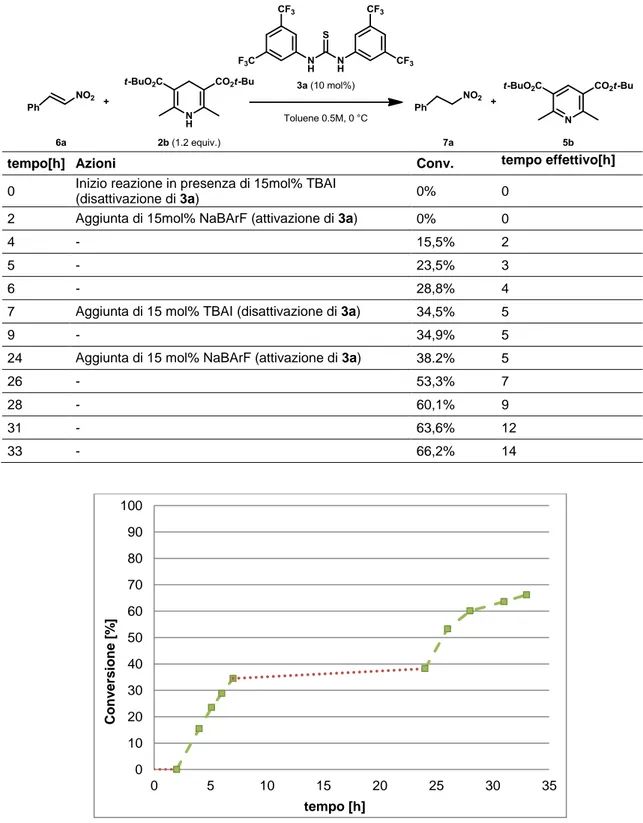
Si è quindi deciso di organizzare un primo esperimento di disattivazione/riattivazione in situ (esperimento ON/OFF) sulla reazione di riduzione di 6a utilizzando la coppia di sali TBAI/NaBArF. Le azioni eseguite durante l’esperimento e le conversioni registrate sono riportate nella Tabella 1. Tutti i prelievi per determinare la conversione sono stati effettuati subito dopo l’esecuzione di ciascuna azione, tranne per il tempo 0 h dove non è stato eseguito alcun prelievo. Dove non è stata riportata alcuna azione, si è provveduto solo ad eseguire il prelievo per la valutazione della conversione. Nella tabella vengono inoltre riportati i tempi effettivi di reazione, ovvero i tempi che si ottengono escludendo dalla scala temporale gli intervalli di tempo in cui il catalizzatore è disattivato. L’andamento della reazione è riportato nel Grafico 4.
Risultati e discussione
19
Tabella 1: Primo esperimento ON/OFF sulla riduzione catalitica di 6a.
tempo[h] Azioni Conv. tempo effettivo[h] 0 Inizio reazione 0% 0
2 Aggiunta di 15 mol% TBAI (disattivazione di 3a) 37.1% 2 7 Aggiunta di 15 mol% NaBArF (attivazione di 3a) 41.8% 2
9 - 48.8% 4
24 Aggiunta di 15 mol% TBAI (disattivazione di 3a) 80.5% 19 28 Aggiunta di 15 mol% NaBArF (attivazione di 3a) 81.7% 19
33 - 85.5% 24
Grafico 4: Andamento cinetico della prima riduzione catalitica ON/OFF di 6a (catalizzatore ON, linea a
tratti; catalizzatore OFF, linea a punti).
Nel Grafico 4 si riesce ad osservare facilmente come l’aggiunta di TBAI diminuisca effettivamente l’attività del catalizzatore e come questa disattivazione sia grosso modo reversibile, dal momento che la successiva aggiunta di NaBArF riesce a ripristinare l’attività catalitica.
Visti i risultati accettabili ottenuti da questo primo esperimento di attivazione/disattivazione, si è deciso di eseguire un secondo esperimento di questo tipo sulla medesima reazione, aggiungendo il TBAI a 0 h per poi attivare il catalizzatore con NaBArF in un secondo momento (dopo 2 h). Le azioni eseguite durante l’esperimento e
0 10 20 30 40 50 60 70 80 90 100 0 5 10 15 20 25 30 35 Conv e rsi on e [% ] tempo[h]
Risultati e discussione
20
le conversioni registrate sono riportate nella Tabella 2 e l’andamento della reazione nel
Grafico 5.
Tabella 2: Secondo esperimento ON/OFF sulla riduzione catalitica di 6a.
tempo[h] Azioni Conv. tempo effettivo[h]
0 Inizio reazione in presenza di 15mol% TBAI
(disattivazione di 3a) 0% 0 2 Aggiunta di 15mol% NaBArF (attivazione di 3a) 0% 0
4 - 15,5% 2
5 - 23,5% 3
6 - 28,8% 4
7 Aggiunta di 15 mol% TBAI (disattivazione di 3a) 34,5% 5
9 - 34,9% 5
24 Aggiunta di 15 mol% NaBArF (attivazione di 3a) 38.2% 5
26 - 53,3% 7
28 - 60,1% 9
31 - 63,6% 12
33 - 66,2% 14
Grafico 5: Andamento cinetico della seconda riduzione catalitica ON/OFF di 6a (catalizzatore ON, linea
a tratti; catalizzatore OFF, linea a punti) 0 10 20 30 40 50 60 70 80 90 100 0 5 10 15 20 25 30 35 Conv e rsi on e [% ] tempo [h]
Risultati e discussione
21
Per cercare di valutare se effettivamente il comportamento della reazione catalitica attivata e disattivata in situ possa essere confrontabile con la reazione catalitica standard, si è provato a riportare in un unico grafico l’andamento della reazione catalitica standard e l’andamento delle reazioni ON/OFF, andando a sostituire, per queste ultime due, i tempi reali con i tempi effettivi di reazione. Il risultato di questa manipolazione grafica è riportato nel Grafico 6, in cui la curva (■) rappresenta i risultati della reazione standard, la curva (▲) i risultati del primo esperimento ON/OFF (disattivazione e attivazione a 2 e 19 h di tempo effettivo, vedi Tabella 1, Grafico 4), e la curva (▬) i risultati del secondo esperimento ON/OFF (attivazione a 0 h di tempo effettivo, disattivazione e attivazione a 5 h di tempo effettivo, vedi Tabella 2, Grafico 5)
Grafico 6: Confronto tra la cinetica della reazione catalitica standard (■), della prima riduzione ON/OFF di 6a (▲) e della seconda riduzione ON/OFF (▬)utilizzando i tempi di reazione effettivi.
Per quanto riguarda il primo esperimento ON/OFF, si può notare che la cinetica ricalcolata con i tempi effettivi non si discosta eccessivamente da quella registrata per la catalitica standard, seppure siano presenti delle discrepanze notevoli. Infatti, a partire dalla prima riattivazione dell’attività catalitica (a 2 h tempo effettivo) i valori mostrano una certa deviazione da quelli registrati per la catalitica standard: si veda per esempio il dato delle 4 h tempo effettivo. Per quanto riguarda il secondo esperimento ON/OFF, si può osservare come la cinetica della reazione nei primi tempi mostri un certo discostamento da quello che è l’andamento delle reazione standard.
Questi risultati sono stati interpretati considerando innanzitutto che la solubilità di NaBArF in toluene a 0 °C non è ottimale. Ciò ha portato a uno scambio ionico in
0 10 20 30 40 50 60 70 80 90 100 0 5 10 15 20 25 30 C onv ersio ne [% ] tempo effettivo[h]
Risultati e discussione
22
soluzione lento, o perlomeno non immediato, che ha comportato la permanenza in soluzione di una certa quantità di ioduro, se non tutto, nei primi momenti successivi all’aggiunta di NaBArF. L’attività catalitica non si è quindi ripristinata in breve tempo e ciò potrebbe spiegare il valore di conversione registrato a 4 h di tempo effettivo nel primo esperimento ON/OFF. D’altra parte, le conversioni generalmente minori rispetto alla reazione standard osservate in entrambi gli esperimenti ON/OFF, sintomi di cinetiche di reazione più lente, possono venire interpretate considerando che il catalizzatore 3a è nel suo stato attivo in presenza di TBABArF in soluzione. Come osservato in precedenza (Grafico 3), TBABArF ha un leggero, ma non trascurabile, effetto di inibizione sull’attività catalitica.
Nonostante i risultati ottenuti non fossero ottimali, è stato possibile concludere dai questi risultati che l’attività catalitica della tiourea 3a può effettivamente essere controllata in maniera reversibile mediante interazione con anioni, ovvero l’ipotesi iniziale si è dimostrata fondata.
Si è passati quindi al vero obiettivo del lavoro di tesi, ovvero lo studio dell’interazione con anioni di sistemi tioureidici chirali che catalizzino reazioni enantioselettive, iniziando logicamente a valutare reazioni analoghe alla riduzione di 6a, ma che operino su substrati nitroolefinici prochirali.
Risultati e discussione
23
3.3
Riduzione di nitrolefine prochirali
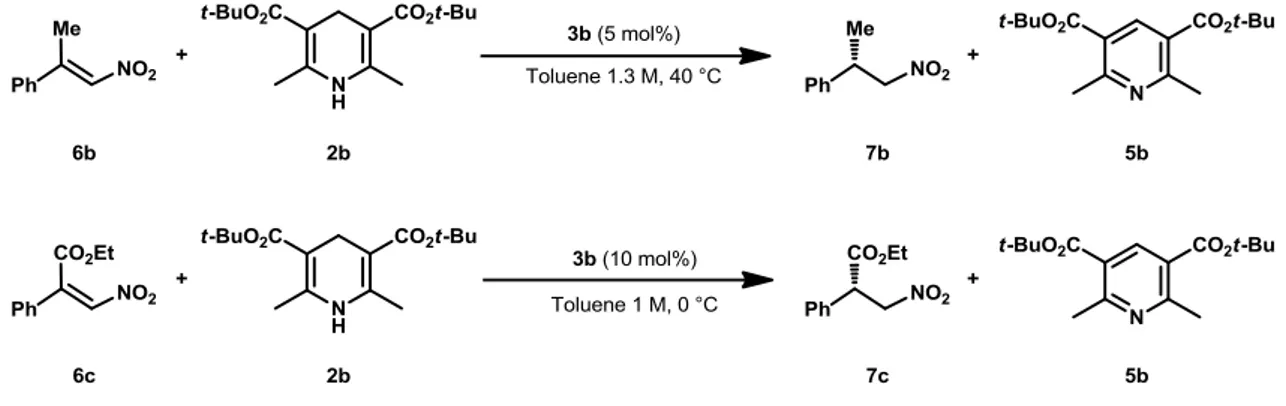
A questo punto del lavoro, ci si è quindi concentrati sulla modulazione di un sistema tioureidico chirale che catalizzasse reazioni che avessero i requisiti espressi prima, ovvero riduzione enantioselettiva di substrati prochirali di natura nitroolefinica. Reazioni di questo genere sono state investigate negli anni passati21,22 e si è osservato come la riduzione di nitroolefine trisostituite con l’estere di Hantzsch 2b catalizzata dalla tiourea
3b (Grafico 13) portasse ad alte rese e elevati valori di enantioselezione.
Figura 13: Catalizzatore 3b
In particolare, è stata riportata la riduzione di nitroolefine β-sostituite sia da gruppi alifatici,21 come per esempio il 6b, che da gruppi esterei,22 come il 6c (Figura 14).
Figura 14: Riduzioni di substrati nitroolefinici prochirali catalizzate da 3b.
In tempi successivi, è stata sviluppata una nuova tipologia di sistemi tioureidici23 aventi una struttura semplificata (Figura 15) ed è stato dimostrato come questi possano spesso andare efficientemente a sostituire catalizzatori tioureidici di vecchia generazione e dalla struttura più complessa,24 simili al 3b.
Risultati e discussione
24
Figura 15: Catalizzatori tioureidici di nuova generazione
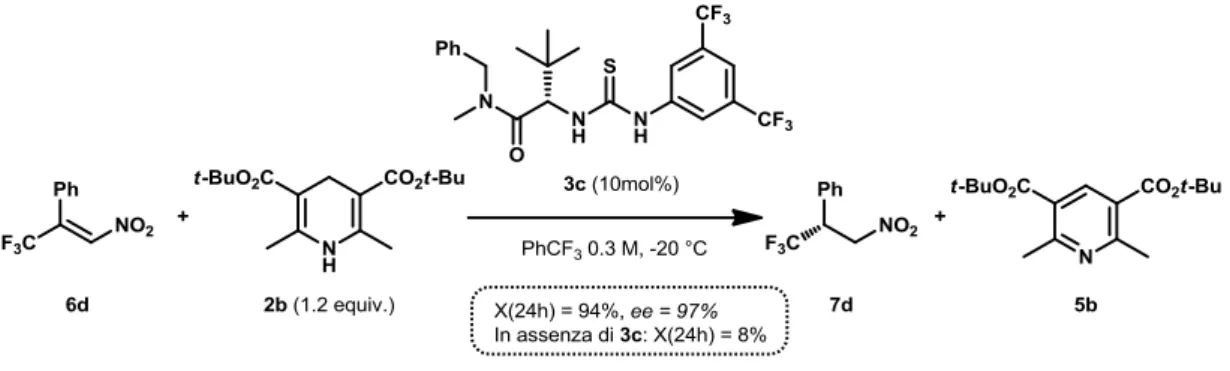
Recentemente,13 il nostro laboratorio ha riportato come la tiourea 3c (Figura 16) catalizzi la riduzione dell’(E)-(3,3,3-trifluoro-1-nitroprop-1-en-2-il)benzene (6d) (Figura
17) con alti valori di resa ed enantioselezione.
Figura 16: Catalizzatore 3c
Figura 17: Riduzione di 6d catalizzata da 3c.
Catalizzatori come la tiourea 3c hanno una struttura relativamente più semplice rispetto a quelli di vecchia generazione e ciò li rende sistemi catalitici di più facile applicabilità. Si è quindi deciso di studiare in un primo momento la reazione di riduzione di 6d, per poi prendere in esame anche le riduzioni di 6b e 6c utilizzando però la tiourea semplificata
3c.
3.4.1 Riduzione enantioselettiva dell’(E)-(3,3,3-trifluoro-1-nitroprop-1-en-2-il)benzene
Nel lavoro precedente,13 la riduzione di 6d con 2b (Figura 18) viene condotta in trifluorotoluene a -20 °C, con una concentrazione 0.3 M rispetto al substrato 6d. In presenza del catalizzatore 3c (10% molare), la reazione mostra una conversione del 94% dopo 24 h. Per la reazione in assenza di catalizzatore, invece, dopo 24 h la conversione è trascurabile.
Risultati e discussione
25
Figura 18: Condizioni di reazione per la reazione di riduzione catalitica di 6d riportate in letteratura.
Sulla base di questi risultati, sono stati effettuati degli studi preliminari su questa reazione, usando però come solvente diclorometano, visti i problemi riscontrati con un solvente aromatico come il toluene (bassa solubilità dei sali e scarsa efficienza negli scambi ionici) nella reazione di riduzione del semplice nitrostirene 6a. Lavorando quindi in condizioni analoghe a quelle del lavoro precedente, ma conducendo la reazione in diclorometano 0.25 M, è stata osservata una notevole accelerazione della cinetica di reazione rispetto alle condizioni originarie, con una conversione pressoché completa a 4 h (maggiore del 90%). Ritenendo una cinetica di questo tipo difficilmente gestibile, si è provato ad abbassare la concentrazione della reazione a 0.125 M, ma, anche in queste condizioni, già a 2 h la reazione catalitica ha mostrato una conversione piuttosto elevata (circa del 60%). Tuttavia, è stato deciso di provare comunque preliminarmente l’effetto di alcuni sali di ammonio sul catalizzatore 3c in queste ultime condizioni. Utilizzando un rapporto 1:1.5 fra catalizzatore 3c e sali di ammonio, sono stati provati sia anioni dai quali ci si aspettava una qualche inibizione sull’attività catalitica (ioduro e acetato) sia BArF- che al contrario avrebbe dovuto lasciare inalterata la cinetica di reazione. È emerso che effettivamente il TBABArF non presenta alcun effetto sulla cinetica di reazione, ma un comportamento analogo è stato osservato anche nel caso del TBAI: a 2 h entrambe le reazioni hanno mostrato una conversione paragonabile a quella della catalitica standard. Per quanto riguarda il TBAOAc, a 2 h è stata registrata una conversione inferiore rispetto alla catalitica standard, ma non così bassa da poter essere considerata accettabile (45% vs. 60%). A questo punto, si è ipotizzato che la mancata inibizione dell’attività catalitica in questa reazione fosse dovuta in qualche modo alla elevata reattività del processo e non alla impossibilità di interazione tra la tiourea 3c e gli anioni. Partendo dal presupposto che una cinetica più lenta sia in qualche modo correlabile a un sistema meno reattivo e che un sistema meno reattivo possa quindi essere
Risultati e discussione
26
più facilmente modulabile attraverso interazioni anioniche con il catalizzatore tioureidico, si è deciso di utilizzare le medesime condizioni del lavoro precedente (trifluorotoluene 0.3 M, -20 °C), dal momento che in queste condizioni la riduzione di 6d mostra una cinetica più lenta rispetto a quelle registrate in diclorometano (ca 90% di conversione dopo 24 h).
Inaspettatamente, si è osservato che, in queste ultime condizioni, la reazione catalitica in presenza di TBABArF procede con una cinetica più veloce rispetto a quella riportata per la catalitica standard. Mentre quest’ultima a 4 h mostra una conversione pari al 60%, la catalitica in presenza di TBABArF a 3 h mostra già una conversione prossima al 70%. Questo fenomeno di accelerazione della cinetica è stato osservato anche nella reazione catalitica in presenza di TBACl (Grafico 7) dove l’incremento nei valori della conversione è risultato addirittura più marcato rispetto a quello osservato per la catalitica in presenza di TBABArF.
Grafico 7: Andamento cinetico della reazione di riduzione catalitica standard di 6d in trifluorotoluene 0.3
M a -20 °C (■) e andamento cinetico della riduzione catalitica in presenza di TBACl nelle medesime condizioni (▲)
Questi comportamenti anomali ci hanno spinto all’abbandono del trifluorotoluene come solvente, dal momento che è stato ipotizzato essere questo il responsabile di tali fenomeni. Si è quindi deciso di ritornare al diclorometano come solvente di reazione dal momento che i precedenti studi non avevano mai mostrato accelerazioni cinetiche correlate alla presenza di sali in soluzione, evidenza che invece è emersa dagli studi in trifluorotoluene. Ritenendo sempre che la fallita inibizione della reazione in
0 10 20 30 40 50 60 70 80 90 100 0 1 2 3 4 5 Conv e rsi on e [% ] tempo[h]
Risultati e discussione
27
diclorometano precedentemente osservata fosse dovuta in qualche modo alla reattività elevata del processo e non alla impossibilità di interazione tra tiourea e anioni, si è deciso di lavorare a temperature più basse, eseguendo la reazione in diclorometano 1 M a -50 °C. In queste condizioni, la catalitica standard mostra una conversione pari al 90% a 48 h (la reazione effettuata in assenza di catalizzatore presenta una conversione pari al 14%). In queste condizioni è stato quindi preliminarmente valutato l’effetto di alcuni sali di ammonio con anioni a potenziale attività inibitrice (cloruro, bromuro, nitrito). Purtroppo, non è stato osservato alcun effetto sulla velocità di reazione. In Grafico 8 sono riportate le conversioni di queste prove registrate a 5 h di reazione.
Grafico 8: Conversioni registrate in diclorometano 1 M a -50 °C a 5 h per la reazione di riduzione
catalitica standard di 6d (3c 10 mol%) e per la stessa reazione in presenza di sali di ammonio che non inibiscono l’attività catalitica (3c 10 mol% + TBAX 15 mol%).
Visti questi ultimi risultati, si è giunti alla conclusione che le interazioni fra il substrato
6d e il catalizzatore 3c fossero troppo forti, presumibilmente per la presenza del gruppo
trifluorometile, per provare a realizzare una qualche inibizione anionica sull’attività catalitica. La reazione non risultava quindi utile per gli obbiettivi di questo lavoro, e ci si è spostati sulla riduzione degli altri due nitroalcheni 6b e 6c.
0 10 20 30 40 50 60 70 80 90 100 3b 3b + TBACl 3b + TBABr 3b + TBANO2 Conv e rsi on e [% ]
Risultati e discussione
28
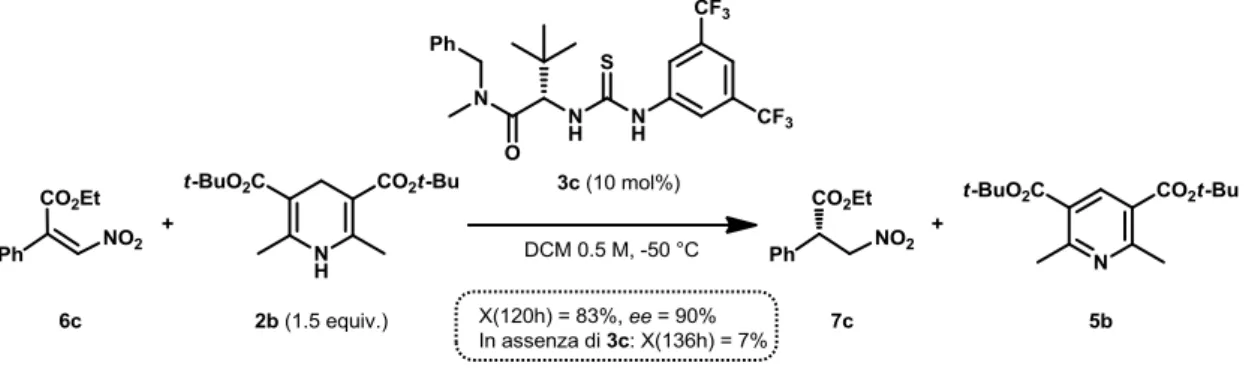
3.4.2 Riduzione enantioselettiva dello (Z)-etil 3-nitro-2-fenilacrilato
Vista l’impossibilità di modulazione anionica dell’attività catalitica di 3c per quanto riguarda la riduzione di 6d, ci si è concentrati quindi su un substrato meno reattivo, lo (Z)-etil 3-nitro-2-fenilacrilato (6c). Rispetto alla procedura di letteratura, le reazioni sono state effettuate sempre utilizzando il catalizzatore semplificato 3c.
Procedendo nella consueta maniera, sono state individuate le condizioni di reazioni più adatte alle esigenze del lavoro e ci si è accertati che in queste la reazione in assenza di catalizzatore mostrasse conversioni trascurabili. Dopo una serie di esperimenti, è stato trovato che la reazione di riduzione in diclorometano 0.5 M a -50 °C con un carico catalitico del 10% molare di 3c mostri a 120 h una conversione pari a circa l’83%, mentre la reazione in assenza di 3c a 136 h porti a una conversione relativamente trascurabile (pari al 7%). Per la reazione di riduzione catalitica standard (Figura 19), la cui cinetica è riportata nel Grafico 9, è stato inoltre valutato l’eccesso enantiomerico che è risultato essere del 90%, confermando quindi la possibilità di utilizzare il catalizzatore semplificato 3c anche per la riduzione di questo nitroalchene 6c.
Risultati e discussione
29
Grafico 9: Andamento cinetico della reazione di riduzione catalitica standard di 6c.
È stato quindi determinato l’effetto di vari sali sulla reazione catalitica, valutando le conversioni a 60 h in presenza di questi sali. I risultati sono riportati nel Grafico 10. Inoltre si è provveduto a valutare gli eccessi enantiomerici delle reazioni catalitiche in presenza di TBABArF e TBAPF6, ovvero anioni poco coordinanti che dovrebbero dare
interazioni minime con il catalizzatore. Gli eccessi enantiomerici sono risultati essere pari all’88% sia nel caso della riduzione catalitica in presenza di TBABArF che nel caso della riduzione catalitica in presenza di TBAPF6, ovvero confrontabili con quello della
reazione catalitica standard, confermando quindi in tutto e per tutto l’ipotesi di lavoro.
Grafico 10: Conversioni registrate a 60 h per la reazione di riduzione catalitica standard di 6c (3c 10
mol%) e per la stessa reazione in presenza di diversi sali di ammonio (3c 10 mol% + TBAX 15 mol%). 0 10 20 30 40 50 60 70 80 90 100 0 50 100 150 Conv e rsi on e [% ] tempo[h] 0 10 20 30 40 50 60 70 80 90 100 3c 3c + TBABr 3c + TBANO2 3c + TBANO3 3c + TBAOAc 3c + TBABArF 3c + TBAPF6 Conv e rsi on e [% ] ee = 90% ee = 88% ee = 88%
Risultati e discussione
30
Seppure la reazione in presenza di TBABArF non avesse una cinetica del tutto paragonabile alla reazione standard, e allo stesso tempo l’inibizione ad opera del bromuro non potesse essere considerata totale, si è deciso di verificare comunque la potenziale reversibilità dell’inibizione/attivazione del catalizzatore 3c in questa reazione. E’ stato quindi organizzato un esperimento di attivazione/disattivazione utilizzando TBABr per disattivare il catalizzatore e NaBArF per riattivarlo. Più in dettaglio (Tabella
3), alla reazione catalitica standard è stato aggiunto a 24 h il TBABr (disattivazione del
catalizzatore) e a 48 h NaBArF (riattivazione del catalizzatore). Le procedure di disattivazione e attivazione vengono ripetute rispettivamente a 72 e 96 h. Le conversioni valutate sono riportate nella Tabella 3 e l’andamento della reazione è riportato nel
Grafico 11.
Tabella 3: Esperimento ON/OFF sulla riduzione catalitica di 6c.
tempo[h] Azioni Conv. tempo effettivo[h]
0 Inizio reazione 0% 0
17 - 20.7% 17
24 Aggiunta di 15 mol% TBABr (disattivazione di 3c) 28.5% 24 48 Aggiunta di 15 mol% di NaBArF (attivazione di 3c) 29.1% 24
65 - 49.8% 41
72 Aggiunta di 15 mol% TBABr (disattivazione di 3c) 56.5% 48 96 Aggiunta di 15 mol% di NaBArF (attivazione di 3c) 57.0% 48
160 - 77.9% 112
168 - 80.3% 120
Risultati e discussione
31
Grafico 11: Andamento cinetico della riduzione catalitica ON/OFF di 6c (catalizzatore ON, linea a tratti;
catalizzatore OFF, linea a punti).
Dal Grafico 11 si osserva facilmente come l’aggiunta di TBABr riesca effettivamente a disattivare l’attivare catalitica di 3c, mentre la successiva aggiunta di NaBArF la ripristini. Riportando su un unico grafico l’andamento della riduzione catalitica standard e l’andamento della riduzione ON/OFF andando a sostituire i tempi reali con quelli che sono i tempi effettivi di reazione si ottiene il Grafico 12.
Grafico 12: Confronto tra la cinetica della riduzione catalitica standard (■) e della riduzione ON/OFF di
6c (▲) utilizzando i tempi di reazione effettivi. 0 10 20 30 40 50 60 70 80 90 100 0 50 100 150 200 Conv e rsi on e [% ] tempo[h] 0 10 20 30 40 50 60 70 80 90 100 0 20 40 60 80 100 120 140 Conv e rsi on e [% ] tempo effettivo[h]
Risultati e discussione
32
Come si può osservare dal grafico, gli andamenti cinetici delle due reazioni sono praticamente coincidenti, dimostrando quindi la perfetta reversibilità dei processi di attivazione e disattivazione del catalizzatore 3c in questa reazione. Al termine della riduzione ON/OFF è stato valutato l’eccesso enantiomerico del prodotto 7c ottenuto, che è risultato essere pari all’80%, ovvero leggermente inferiore a quello registrato per la reazione catalitica standard, e anche a quello ottenuto in presenza di TBABArF. Questa lieve diminuzione di enantioselezione è stata attribuita alla presenza di una quantità considerevole di sali di ammonio in soluzione, in grado quindi di alterare la ionicità del mezzo di reazione.
Risultati e discussione
33
3.4.3 Riduzione enantioselettiva dell’ (E)-(1-nitroprop-1-en-2-il)benzene
Avendo ottenuto risultati più che accettabili per quanto riguarda la riduzione di 6c catalizzata dalla tiourea 3c, ci si è spostati a valutare la riduzione dell’(E)-(1-nitroprop-1-en-2-il)benzene (6b), catalizzata sempre dal sistema tioureidico semplificato 3c. Cercando sempre di lavorare in diclorometano, per i già citati motivi di solubilità dei sali di ammonio e del NaBArF, è stato come nelle precedenti reazioni inizialmente eseguito un lavoro preliminare di screening di condizioni di reazione, al fine di individuare quelle più adeguate per i nostri scopi. Dopo una serie di esperimenti a diverse concentrazioni e temperature, è stato individuato che conducendo la reazione a 0 °C in diclorometano 0.5 M rispetto a 6b con un carico catalitico del 10% molare di 3c la conversione nel prodotto
7b raggiungeva il valore di 90% dopo 46 h (Figura 20), e allo stesso tempo la reazione
non catalizzata essenzialmente non procedeva. In queste condizioni, l’eccesso enantiomerico valutato sul prodotto 7b è risultato essere del 90%, confermando quindi come il catalizzatore semplificato 3c fosse in grado di promuovere anche questa reazione in maniera altamente stereoselettiva. La cinetica della reazione catalitica standard è riportata nel Grafico 13.
Risultati e discussione
34
Grafico 13: Andamento cinetico della reazione di riduzione catalitica standard di 6b.
Si è quindi studiato l’effetto di alcuni sali (caricati sempre in rapporto 1.5 rispetto al catalizzatore) sull’attività catalitica, confrontando le conversioni registrate a 46 h. Nel
Grafico 14 viene confrontata la conversione della reazione catalitica standard con le
conversioni registrate per le reazioni catalitiche in presenza di sali con anioni coordinanti, mentre nel Grafico 15 la conversione per la catalitica standard viene confrontata con quelle registrate per le reazioni catalitiche in presenza di anioni poco coordinanti e quindi potenzialmente innocui rispetto all’attività di 3c. Nello stesso grafico sono riportati anche gli eccessi enantiomerici relativi al prodotto 7b per ciascuna reazione.
Grafico 14: Conversioni registrate a 46 h per la reazione di riduzione catalitica standard di 6b (3c 10
mol%) e per la stessa reazione in presenza di sali che inibiscono l’attività catalitica (3c 10 mol% + sale 15 mol%). 0 10 20 30 40 50 60 70 80 90 100 0 10 20 30 40 50 60 Conv e rsi on e [% ] tempo[h] 0 10 20 30 40 50 60 70 80 90 100 3c 3c + TBACl 3c + TBABr 3c + TBAI 3c + TBANO2 3c + TBANO3 3c + TBAOAc 3c + Ph4PCl C onv ersio ne [% ]
Risultati e discussione
35
Dal Grafico 14 si può facilmente osservare come, in questo caso, sia il cloruro l’anione che riesce a inibire meglio l’attività del catalizzatore. Inoltre, guardando il comportamento degli alogenuri (TBACl, TBABr e TBAI) si può osservare come esista una dipendenza tra le dimensioni dell’anione (e quindi la forza di coordinazione) e la capacità di inibizione: il cloruro, l’anione più piccolo tra questi tre, è quello che inibisce meglio il catalizzatore, mentre lo ioduro, il più grande, è quello che inibisce peggio. È inoltre interessante notare come anche la natura del controione giochi un ruolo fondamentale nell’inibizione. Infatti, si è deciso di verificare se un anione coordinante che derivasse da un sale diverso dal tetra-n-butilammonio potesse avere un’interazione differente con il catalizzatore. È stata quindi condotta una reazione catalitica in presenza di tetrafenilfosfonio cloruro (Ph4PCl) e osservando il Grafico 14 si nota facilmente che
se il cloruro del TBACl è l’anione con migliori capacità di inibizione sul catalizzatore 3c in questa reazione rispetto agli altri sali di ammonio, il cloruro del tetrafenilfosfonio ha, per quanto riguarda l’inibizione, un comportamento anche migliore, portando ad un risultato più che soddisfacente (inibizione quasi completa). Questo comportamento, del tutto in linea con le attese, è interpretabile considerando la minore affinità per il cloruro del catione tetrafenilfosfonio rispetto al tetra-n-butilammonio,25 che rende quindi l’anione cloruro maggiormente disponibile per la coordinazione alla tiourea 3c. Il Ph4PCl
è stato quindi individuato come sale ottimale per disattivare il catalizzatore 3c in questa reazione.
Infine, occorre riportare che per quanto riguarda le reazioni catalitiche condotte in presenza di TBANO2, TBANO3 e TBAOAc è stata osservata la comparsa in reazione di
un sottoprodotto che è stato identificato come l’isomero del reagente 6b (6e, Figura 21). Evidentemente, questi anioni promuovono l’isomerizzazione di 6b a 6e.
Risultati e discussione
36
Grafico 15: Conversioni registrate a 46 h per la reazione di riduzione catalitica standard di 6b (3c 10
mol%) e per la stessa reazione in presenza di sali che non inibiscono l’attività catalitica (3c 10 mol% +
sale 15 mol%).
Per quanto riguarda gli anioni non-inibenti, in questa reazione visti i risultati ottimali ottenuti con il cloruro di fosfonio, non solo sono stati testati gli usuali TBAPF6 e
TBABArF, ma anche il corrispondente sale di fosfonio di quest’ultimo anione, ovvero il Ph4PBArF. Tutte e tre le reazioni hanno mostrato sia conversioni paragonabili tra loro e
confrontabili con quella della catalitica standard sia eccessi enantiomerici essenzialmente identici, confermando quindi l’adeguatezza di questa reazione per i nostri scopi. Anche se il PF6- sembrerebbe avere un effetto persino migliore del BArF-, questo anione risulta
essere difficile da utilizzare durante l’esperimento di attivazione/disattivazione in situ. Infatti, utilizzando la procedura precedentemente collaudata per l’attivazione/disattivazione, il PF6- dovrebbe essere introdotto in reazione come NaPF6,
ma questo sale non è però solubile in diclorometano. Allo stesso modo, test di solubilità effettuati con LiPF6 e KPF6 hanno indicato come anche questi sali, con un diverso
metallo alcalino, fossero essenzialmente insolubili nello stesso solvente. La mancata solubilizzazione comporta l’impossibilità dello scambio anionico tra il sale inibente e il sale non inibente. Questo mancato scambio si traduce, in ultima analisi, nella mancata riattivazione del catalizzatore. L’utilizzo del PF6- sarebbe invece forse possibile
utilizzando un sale d’argento (AgPF6). Tuttavia questo sale è molto igroscopico e
difficilmente maneggiabile. Anche in questo caso, quindi, si è deciso di utilizzare l’NaBArF come sale per ripristinare l’attività catalitica.
0 10 20 30 40 50 60 70 80 90 100 3c 3c + TBABArF 3c + Ph4PBArF 3c + TBAPF6 Conv e rsi on e [% ] ee = 90% ee = 89 % ee = 90% ee = 89%
Risultati e discussione
37
È stato quindi organizzato un esperimento di disattivazione/riattivazione in situ dell’attività catalitica. Si è deciso di aggiungere alla reazione catalitica il Ph4PCl a 6 h di
reazione (disattivazione del catalizzatore) seguita dall’aggiunta, a 22.5 h di reazione, di NaBArF (attivazione del catalizzatore). Le procedure di disattivazione e attivazione vengono ripetute rispettivamente a 29.5 e 47 h. Le conversioni valutate sono riportate nella Tabella 4 e l’andamento della reazione nel Grafico 14.
Tabella 4: Esperimento ON/OFF sulla riduzione catalitica di 6b.
tempo[h] Azioni Conv. tempo effettivo[h]
0 Inizio reazione 0% 0
3 - 20.2% 3
6 Aggiunta di 15 mol% Ph4PCl (disattivazione di 3c) 34.5% 6 22.5 Aggiunta di 15 mol% di NaBArF (attivazione di 3c) 37.7% 6
25.5 - 46.2% 9
29.5 Aggiunta di 15 mol% Ph4PCl (disattivazione di 3c) 54.3% 13 47 Aggiunta di 15 mol% di NaBArF (attivazione di 3c) 54.9% 13
50 - 60.0% 16
54 - 65.3% 20
Risultati e discussione
38
Grafico 16: Andamento cinetico della riduzione catalitica ON/OFF di 6b (catalizzatore ON, linea a tratti;
catalizzatore OFF, linea a punti).
Riportando sullo stesso grafico l’andamento della reazione catalitica standard e l’andamento della reazione ON/OFF andando a sostituire i tempi reali con quelli che sono i tempi effettivi di reazione si ottiene il Grafico 17.
Grafico 17: Confronto tra l’andamento cinetico della riduzione catalitica standard (■) e l’andamento cinetico della riduzione ON/OFF di 6b (▲) utilizzando i tempi di reazione effettivi.
Dal grafico, si può osservare come a tempi brevi le due cinetiche siano paragonabili. A partire dalle 16 h di tempo effettivo si osserva invece un rallentamento per quanto riguarda la reazione di riduzione ON/OFF. Andando ad osservare nel dettaglio gli spettri NMR relativi ai prelievi effettuati dalle 16 h di tempo effettivo in poi, si osserva una
0 10 20 30 40 50 60 70 80 90 100 0 20 40 60 80 Conv e rsi on e [% ] tempo[h] 0 10 20 30 40 50 60 70 80 90 100 0 10 20 30 40 Conv e rsi on e [% ] tempo effettivo[h]
Risultati e discussione
39
graduale scomparsa dei segnali relativi all’estere 2b, che risulta praticamente assente nei prelievi finali. Probabilmente i continui prelievi effettuati sulla reazione e il prolungamento di questa dovuto agli spegnimenti ha portato alla completa ossidazione dell’estere 2b da parte dell’ossigeno atmosferico. Non essendo più presente il riducente, la reazione non riesce ad arrivare a completezza. Utilizzare un eccesso più importante di
2b (2 equivalenti), probabilmente eliminerebbe questo problema. Nonostante questo,
l’aspetto più importante emerso da questa prova di disattivazione/attivazione in situ è l’eccesso enantiomerico registrato per il prodotto 7b, che è risultato essere identico a quello registrato al termine della reazione catalitica standard (90% ee).