1
UNIVERSITA’ DI PISA
Facoltà di Scienze Matematiche, Fisiche e Naturali
Corso di LAUREA MAGISTRALE in CHIMICA INDUSTRIALE
Curriculum: Industriale
Classe: LM 71 (Scienze e Tecnologie Chimiche)
“Il contenuto di questa relazione è strettamente riservato, essendo presenti
argomenti tutelati dalla legge come segreti. Pertanto tutti coloro che ne
prendono conoscenza sono soggetti all’obbligo, sanzionato anche penalmente
dagli articoli 325 e 623 del codice penale, di non divulgare e di non utilizzare le
informazioni acquisite.”
“Nuovi concianti furanici ottenuti per conversione
catalitica di biomasse”
Candidato:
Marco CAMPANI
Relatore Interno:
Prof.ssa Anna Maria RASPOLLI GALLETTI
Relatore Esterno:
Dott. Domenico CASTIELLO
Controrelatore:
Prof. Valter CASTELVETRO
2
Le seguenti persone dichiarano di prendere visione della tesi di Laurea in Chimica
Industriale sotto secratazione dal titolo: “Nuovi concianti furanici ottenuti per
conversione catalitica di biomasse”.
Nome Cognome Qualifica Ruolo nella commissione Firma
Roger Fuoco Prof. Ordinario Presidente Anna M. Raspolli Galletti Prof. Associato Relatore Domenico Castiello Docente Esterno Relatore Valter Castelvetro Prof. Associato Controrelatore
Benedetta Mennucci Prof. Ordinario Membro Pres. Supplente Gennaro Pescitelli Ricercatore Membro
Corrado Malanga Ricercatore Supplente Chiara Cappelli Ricercatore Supplente Maurizio Persico Prof. Ordinario Supplente Giacomo Ruggeri Prof. Associato Supplente Giorgio Valentini Prof. Associato Supplente
3
Sommario
INTRODUZIONE E SCOPO DELLA TESI ... 8
1. Le biomasse: stato dell’arte ... 10
1.1. Tipologie di biomasse ... 11
1.2. Struttura e componenti della biomassa lignocellulosica ... 12
1.2.1. Cellulosa ... 12
1.2.2. Emicellulosa ... 14
1.2.3. Pectine ... 19
1.2.4. Lignina ... 20
1.2.5. Componenti non-strutturali del legno ... 24
1.3. Riferimenti bibliografici ... 27
2. Furaldeide ottenuto da materie prime rinnovabili ... 29
2.1. L’idrolisi acida dei costituenti C5 della biomassa lignocellulosica a furfurale ... 29
2.2. Potenziali applicazioni del furfurale e dei suoi derivati ... 36
2.2.1. Impiego del furfurale come tale ... 37
2.2.2. Impiego dei derivati del furfurale ... 39
2.3. Impiego del furfurale in ambito conciario ... 42
2.4. Riferimenti bibliografici ... 43
3. Produzione del HMF e del BHMF da materie prime rinnovabili ... 45
3.1. Riferimenti bibliografici ... 54
4. La pelle e le proprietà del collagene ... 56
4.1. Istologia della pelle ... 56
4.1.1. L’epidermide ... 57
4.1.2. Il pelo ... 58
4.1.3. Derma ... 58
4
4.2. La chimica dei componenti della pelle... 59
4.2.1. Componenti proteici ... 59 4.2.2. Collagene ... 60 4.2.3. Cheratine ... 61 4.2.4. Elastina ... 62 4.2.5. Albumine e globuline ... 62 4.2.6. Reticolina ... 62
4.3. Chimica delle proteine ... 62
4.3.1. Struttura degli amminoacidi ... 63
4.3.2. Amminoacidi come ioni dipolari ... 64
4.3.3. Classificazione, funzione e struttura delle proteine ... 65
4.3.4. Legami tra catene polipeptidiche... 67
4.4. Proprietà del collagene ... 68
4.4.1. Punto isoelettrico del collagene ... 69
4.4.2. Gonfiamento del collagene ... 69
4.4.3. Temperatura idrotermica di contrazione del collagene ... 70
4.5. Riferimenti bibliografici... 72 5. Il processo conciario ... 73 5.1. Operazioni preliminari ... 75 5.2. Lavori di riviera ... 76 5.2.1. Rinverdimento ... 76 5.2.2. Depilazione e calcinazione ... 77 5.2.3. Decalcinazione e macerazione ... 80 5.3. La concia ... 81 5.3.1. Il Pickel ... 81 5.3.2. La Concia ... 82 5.3.3. Pressatura e rasatura ... 82
5 5.4. Caratterizzazione ... 82 5.4.1. Riconcia ... 83 5.4.2. Tintura ... 84 5.4.3. Ingrasso ... 85 5.5. Asciugatura ... 85 5.5.1. Sosta a cavalletto ... 86 5.5.2. Messa a vento ... 86 5.5.3. Rullo caldo ... 86 5.5.4. Essiccazione sottovuoto ... 87
5.5.5. Essiccazione alla termoplacca ... 87
5.5.6. Sosta a catena aerea e tunnel condizionato ... 88
5.6. Rifinizione ... 88
5.7. Riferimenti Bibliografici ... 90
6. Il processo di concia ... 91
6.1. Proprietà delle pelli conciate ... 94
6.2. La concia minerale ... 94
6.3. La concia con tannini, vegetali e sintetici ... 95
6.4. La concia all’aldeide ... 97
6.5. Riferimenti Bibliografici ... 101
7. Risultati ... 102
7.1. Introduzione ... 102
7.1.1. Vantaggi dell’impiego delle Biomasse ... 102
7.2. Determinazione della concentrazione target del furfurale ... 104
7.3. Ottimizzazione delle condizioni della reazione di auto-idrolisi ed idrolisi acida a furfurale ... 105
7.4. Prove di concia con il furfurale effettuate presso PO.TE.CO. ... 107
6
7.6. Prove di concia con il furfurale da biomassa in presenza della biomassa residua ... 113
7.7. Prove di concia con il furfurale ad alta concentrazione ... 117
7.8. Introduzione alle prove con l’HMF ... 119
7.9. Ottimizzazione delle condizioni della reazione di idrolisi del fruttosio ad HMF con catalizzatore omogeneo ... 120
7.10. Prove di concia con l’HMF prodotto da fruttosio con catalizzatore H2SO4 ... 121
7.11. Ottimizzazione delle condizioni di idrolisi ad HMF in catalisi eterogenea ... 128
7.12. Produzione di HMF concentrati a partire da fruttosio ... 129
7.13. Prove di concia con HMF ad alta concentrazione ottenuto a partire da fruttosio alimentare ... 130
7.14. Produzione di HMF concentrato a partire da soluzioni di fruttosio alimentare al 10% ... 134
7.15. Prove di concia CHC-3 impiegando soluzione 92 g/l in HMF ... 135
7.16. Ottimizzazione delle condizioni di idrogenazione dell’HMF per produrre BHMF ... 139
7.17. Prove di concia con BHMF prodotto dall’HMF ... 141
7.18. Idrogenazione di soluzioni concentrate di HMF per produrre BHMF concentrato da impiegare come conciante... 146
7.19. Prove di concia con miscela 70 g/l in HMF e 30 g/l in BHMF ... 147
7.20. Riferimenti bibliografici... 153
8. Considerazioni conclusive ... 154
9. Parte sperimentale ... 155
9.1. Reattivi, solventi, precursori commerciali ... 155
9.2. Strumentazione ... 156
9.2.1. Cromatografia liquida ad alta pressione (HPLC) ... 156
9.2.2. Spettroscopia 1H-NMR ... 156
9.2.3. Spettroscopia FT – IR ... 157
7
9.2.5. Reazioni in microonde ... 157
9.2.6. Reazioni in autoclave ... 159
9.2.7. Reazioni di concia delle pelli ... 160
9.3 Determinazioni analitiche ... 160
9.3.1. Determinazione dell’emicellulosa, cellulosa, lignina e ceneri nelle materie prime ... 160
9.3.2. Determinazione del furfurale mediante tecnica HPLC ... 160
9.3.3. Determinazione dell’HMF ... 161
9.3.4. Determinazione dell’HMF mediante tecnica 1H-NMR ... 162
9.3.5. Determinazione del BHMF mediante 1H-NMR ... 164
9.3.6. Determinazione della temperatura di gelatinizzazione (Tg) ... 164
8
INTRODUZIONE E SCOPO DELLA TESI
I processi conciari stanno affrontando la sfida sempre più pressante di introdurre innovazione economicamente ed ambientalmente sostenibile.
In questo contesto, per la prima volta, è stata studiata la capacità conciante di due molecole furaniche quali la 5–idrossimetil–2–furaldeide (HMF) e il 2,5–bis–idrossimetil–furano (BHMF), facilmente ottenibili a partire da zuccheri e, più in generale, da biomasse lignocellulosiche, anche di scarto e a valore negativo. La prima molecola, 5–idrossimetil–2– furaldeide (HMF), può essere sintetizzata in acqua per disidratazione acido-catalizzata a partire da diversi tipi di carboidrati: zuccheri semplici (come fruttosio, glucosio, saccarosio) ma anche da oligo- e poli-saccaridi (come i fruttani, l’inulina, la cellulosa, l’amido ecc...) o direttamente da biomasse grezze o di scarto ricche di tali carboidrati (topinambur, biomassse erbacee, scarti di zuccherificio). Il BHMF è il prodotto di idrogenazione catalitica dell’HMF ed è quindi anch’essa un bioderivato.
Questa tesi si inquadra difatti nella ricerca di valorizzazione delle biomasse per diversi settori tecnologici abbandonando la vecchia logica delle alimentazioni da biomassa di prima generazione (come il mais o i semi da olio) che entravano in competizione con la produzione del cibo (food verso feed). Le biomasse di seconda generazione sono invece materiali lignocellulosici o oleaginosi non commestibili, impiegati in un concetto integrato di bioraffineria. In tale ambito rientrano le colture non commestibili a veloce crescita, come alcune erbacee o, ancora meglio, si può pensare alla valorizzazione della biomassa di scarto generata nella produzione di colture commestibili (la paglia del grano, la pula del riso, gli stocchi del mais, le bucce degli agrumi, dei pomodori..). Andando all’aspetto più estremo si può pensare agli scarti alimentari, sia industriali (industria dei cibi e delle bevande) che domestici (Food Supply Chain Waste, FSCW), così come all’enorme quantità di scarti prodotti nei processi di coltivazione e valorizzazione dei prodotti agricoli. La maggior parte di questi scarti viene usata per la concimazione del terreno e non ancora come materia prima per una economia innovativa, la “bio-based economy”, dove si persegue la valorizzazione integrale della biomassa.
Per renderci meglio conto della posta in gioco, si stima che la produzione globale annua di biomassa sia circa 1011 tonnellate, per il 60% circa si tratta di biomassa terrestre, il rimanente 40% di biomassa acquatica. Tale biomassa è composta per circa il 75% di
9 carboidrati, per circa il 20% di lignina ed il rimanente 5% è costituito di trigliceridi, terpeni, proteine ed altri componenti minoritari.
Per questo motivo abbiamo voluto valutare l’attività conciante di queste molecole furaniche bio-based mai prese in considerazione in ambito conciario e paragonarle con le prestazioni del furfurale (FA), unica aldeide furanica marginalmente studiata come conciante. In un’ottica green chemistry queste molecole verranno sintetizzate a partire da risorse rinnovabili, ottimizzando sia i processi di idrolisi/disidratazione acido-catalizzata per ottenere furfurale ed HMF, sia il processo catalitico di idrogenazione dell’HMF a BHMF per produrre le soluzioni acquose più idonee per il successivo processo di concia. In un’ottica sinergica tra diverse specializzazioni e competenze, le soluzioni di FA, HMF e BHMF verranno infatti impiegate in prove conciarie sperimentali e le pelli ottenute saranno quindi caratterizzate per valutare l’efficienza dei nuovi principi concianti. Se si riuscirà ad evidenziare delle prospettive in tal senso, si potrebbe aprire per i processi conciari, una prospettiva assolutamente nuova, in cui nuovi concianti “vegetali” non tossici e biodegradabili sono ottenibili da risorse rinnovabili, anche di scarto del territorio toscano.
10
1. Le biomasse: stato dell’arte
Per biomassa si intende "la frazione biodegradabile dei prodotti, rifiuti e residui di origine biologica provenienti dall’agricoltura (comprendente sostanze vegetali e animali), dalla silvicoltura e dalle industrie connesse, comprese la pesca e l’acquacoltura, nonché la parte biodegradabile dei rifiuti industriali e urbani". Questa è la formulazione prevista dalla Direttiva Europea 2009/28/CE, ripresa da tutta la legislazione ad essa riferente. E quindi, anche se sulla definizione stessa di biomassa vi sono e vi sono stati giudizi non univoci, essa è, al momento, quella universalmente più accettata.
In questa definizione di biomassa sono inclusi anche:
• materiali vegetali residui prodotti da trattamenti esclusivamente meccanici di coltivazioni agricole non dedicate;
• materiali vegetali prodotti da interventi selvicolturali, da manutenzione forestali e da potatura;
• materiali vegetali residui dalla lavorazione esclusivamente meccanica del legno vergine.
La biomassa vegetale viene prodotta mediante il processo di fotosintesi clorofilliana che, grazie all’apporto dell’energia del sole, consente di trasformare semplici elementi minerali in complesse molecole organiche. La biomassa vegetale assorbe biossido di carbonio dall’atmosfera durante la crescita e la restituisce durante la combustione. Il bilancio della CO2 di questi processi è pertanto nullo, e quindi non contribuisce all’effetto serra.
L’Italia e l’Europa si sono impegnate per ridurre le emissioni di CO2 e per soddisfare almeno in parte il proprio fabbisogno energetico con l’impiego di energie rinnovabili. In Europa vi è la potenzialità di soddisfare circa il 50% del fabbisogno energetico a partire da biomasse specificatamente coltivate (su terreni non più richiesti per le colture dedicate all’alimentazione), da residui agricoli e rifiuti urbani, assicurando autosufficienza e sicurezza delle risorse energetiche a lungo termine.
Nel corso dell’ultimo decennio, si è comunque sempre più consolidata una visione multifunzionale dell’agricoltura. I vantaggi associati all’impiego di queste colture sono molteplici:
• rappresentano una fonte di reddito aggiuntiva a quello tradizionalmente derivante dall’attività agroforestale;
11
• contengono i processi di abbandono delle aree meno competitive, in termini di qualità e quantità, per le produzioni convenzionali;
• rendono disponibile una fonte energetica rinnovabile e alternativa a beneficio dell’intera società, meno dipendente dai prodotti di origine fossile;
• si inseriscono in una più razionale gestione dello spazio rurale con potenziali effetti positivi sul piano paesaggistico e sulla salvaguardia della flora e della fauna selvatica;
• richiedono pratiche colturali poco intensive, tali da favorire il mantenimento di alti contenuti di sostanza organica nei suoli, evitando così processi di depauperamento che determinano un aumento del carbonio atmosferico;
• sono facilmente reperibili e stoccabili;
• possono essere sostituite senza onerosi interventi impiantistici;
• mantengono una buona accettabilità sociale, sia nella fase di approvvigionamento che in quella di utilizzo.
1.1. Tipologie di biomasse
Le colture dedicate possono essere raggruppate in tre tipologie principali [1]:
• Colture da biomassa lignocellulosica: sono specie a elevata produzione di sostanza secca che può essere sottoposta a differenti processi di conversione. Questa tipologia colturale può essere a sua volta distinta in due categorie:
• Short Rotation Forestry (SFR): queste tipologie di biomasse sono migliori per qualità della biomassa e per la capacità di ricrescita: si tratta o di una coltura fitta, a ciclo molto breve, che dopo il taglio delle piante viene reimpiantata ex novo, oppure di un impianto ultra fitto che viene ripetutamente tagliato a intervalli molto brevi (1 – 3 anni) nell’arco della vita utile della piantagione.
• Colture erbacee poliennali o annuali, tra cui sono identificabili come specie: il miscanto (Miscanthus sinesis Andrerss.), la canna comune (Arundo Donax L.) e il sorgo da fibra (Sorghum). Invece nelle aree dove la disponibilità idrica è limitata, presentano buone potenzialità anche il cardo (Cynara cardunculus L.) e il panico (Panicum virgatum L.).
• Colture oleaginose: Dall’esterificazione di oli vegetali di colza, girasole e soia si ottiene il biodiesel, con proprietà e prestazioni simili a quelle del gasolio minerale. Il
12 biodiesel si caratterizza per l’assenza di zolfo e di composti aromatici, il contenimento del particolato fine (PM10) e la capacità di contribuire alla riduzione dell’effetto serra.
• Colture da carboidrati: per fermentazione dei carboidrati, dalle colture zuccherine si produce il bioetanolo, che viene addizionato alle benzine. Tra le specie impiegabili a tal scopo, quelle più sperimentate e diffuse sono la canna da zucchero, il frumento, il sorgo e il mais.
1.2. Struttura e componenti della biomassa lignocellulosica
A un livello superiore, la biomassa può essere considerata come una matrice complessa di emicellulosa (23 – 32%), cellulosa (38 – 50%), lignina (15 – 25%) e altri componenti non strutturali, organici e inorganici (proteine, estratti e ceneri), in composizioni variabili. Analizziamo più nel dettaglio ogni singolo componente, in relazione alle diverse tipologie di biomasse.
1.2.1. Cellulosa
La cellulosa è il polimero naturale più abbondante al mondo: è un omopolisaccaride di formula generale (C6H10O5)n e un grado di polimerizzazione (DP) che può variare da 1000 per la cellulosa nella carta di giornale fino a 10000 per la cellulosa nel cotone. Il gradi di polimerizzazione per alcuni materiali lignocellulosici sono riportati qui di seguito, in Tabella 1.1:
Tabella 1.1: Grado di polimerizzazione (DP) della cellulosa per vari materiali lignocellulosici. Lino Juta Canapa Paglia di grano Faggio Abète Norvegese Salice
2990 3770 3270 4950 3630 3840 2580
Risultati ottenuti tramite misure SEC. Risultati del Riso National Laboratory (Plant Reseach Department, 2002).
La cellulosa forma la struttura di sostegno di tutte le biomasse presenti sulla terra e costituisceapprossimativamente il 50% del materiale della parete cellulare. Le molecole di cellulosa hanno come unità base un anello di β – D – glucopiranosio.
Ogni anello è ruotato di 180 °C rispetto al successivo; pertanto l'unità ripetitiva è formata da due anelli contigui, uniti a mezzo di legami 1,4 – β – glicosidici [2]. Tale conformazione assicura che i monomeri siano uniti gli uni agli altri in direzione equatoriale, minimizzando così gli ingombri sterici. L'elevata stabilità di tale conformazione porta ad una ridotta flessibilità del polimero, tanto che questo è di solito descritto come un vero e proprio nastro.
13 Le diverse unità costituenti la cellulosa manifestano una forte tendenza a formare legami a idrogeno intra - e inter - molecolari e interazioni più deboli di Van der Waals: i legami a idrogeno sono formati tra un atomo di H di un gruppo alcolico e un atomo di ossigeno della stessa catena (legame intra-molecolare) o di una catena parallela (legame inter – molecolare) (Figura 1.1).
Figura 1.1: Struttura della cellulosa e legami intra – e inter – molecolari.
Queste strutture parzialmente cristalline conferiscono alla cellulosa rigidità, resistenza meccanica e ad attacchi chimici e enzimatici: ad esempio, per attaccare e degradare chimicamente la cellulosa, è necessaria un’idrolisi a temperatura elevata (circa 200 – 250 °C), catalizzata generalmente da acidi forti. La cristallinità della cellulosa varia a seconda delle diverse forme. Sono note diverse forme della cellulosa, tra le quali le principali sono la cellulosa I e la II [3 , 4].
La regione amorfa della cellulosa può essere facilmente idrolizzata dagli acidi, mentre la cellulosa cristallina è più resistente [5]. Il risultato è che l'idrolisi con acido diluito tende a rimuovere prevalentemente le regioni amorfe dalla cellulosa cristallina, che necessita invece di condizioni di reazione più severe.
A livello supermolecolare, il raggruppamento di molecole di cellulosa mediante legami a idrogeno porta alla formazione di microfibrille (Figura 1.2), lunghi filamenti di cellulosa le cui dimensioni possono variare dalle “fibrille elementari”, con circa 36 catene, alle grandi microfibrille di alghe, con più di 1200 catene [6 , 7]:
14 Figura 1.2: rappresentazione dell’architettura molecolare della cellulosa, in relazione alle microfibrille e alla
parete cellulare [6].
Le microfibrille si organizzano in due regioni diverse: da una parte abbiamo strutture cristalline altamente ordinate, mentre dall’altra si formano zone amorfe (o para – cristalline). A causa delle forti interazioni delle catene di cellulosa nella regione cristallina, è difficile che i solventi e altri reattivi chimici possano penetrare attraverso tale struttura, mentre le regioni amorfe sono più accessibili [6].
Il legame a idrogeno della cellulosa contribuisce alla sua resistenza, al carattere fibroso e all'insolubilità nei solventi. La sua resistenza alla idrolisi è elevata rispetto alla maggior parte dei polisaccaridi, come ad esempio l’amido: mentre l’amido è una fonte importante di energia nella dieta umana, la cellulosa non può essere metabolizzata dall’organismo.
La cellulosa tende ad aggregarsi con altri componenti della pianta: interagisce con le emicellulose e con la lignina, incrementando ulteriormente la resistenza della pianta [8].
1.2.2. Emicellulosa
Le emicellulose sono polimeri eterogenei di pentosi (xilosio e arabinosio), esosi (principalmente glucosio, galattosio, mannosio), con piccole quantità di L – ramnosio, in aggiunta all’ acido 4 – O – metilglucuronico e a quello galacturonico: questi ultimi sono acidi glucuronici, ottenuti per ossidazione del gruppo alcolico primario ad acido carbossilico [5]. Si riportano in Figura 1.3 alcune tipologie di acidi uronici che si ritrovano nelle unità delle emicellulose.
15 Figura 1.3: Alcuni importanti acidi uronici delle emicellulose.
La percentuale dei diversi monomeri varia al variare delle materie prime, ma comunque la maggior parte delle emicellulose è costituita da pentosi, in particolare da xilosio [5].
Questi polisaccaridi sono simili alla cellulosa per la presenza di legami β – (1 → 4) nelle catene principali. Le differenze rispetto alla cellulosa sono dovute principalmente al fatto che le emicellulose non hanno una struttura cristallina altamente ordinata [8].
Il ruolo delle emicellulose all’interno delle cellule vegetali non è certo, anche se, essendo associate alla cellulosa e alla lignina, contribuiscono certamente a migliorare il sostegno strutturale della pianta [5].
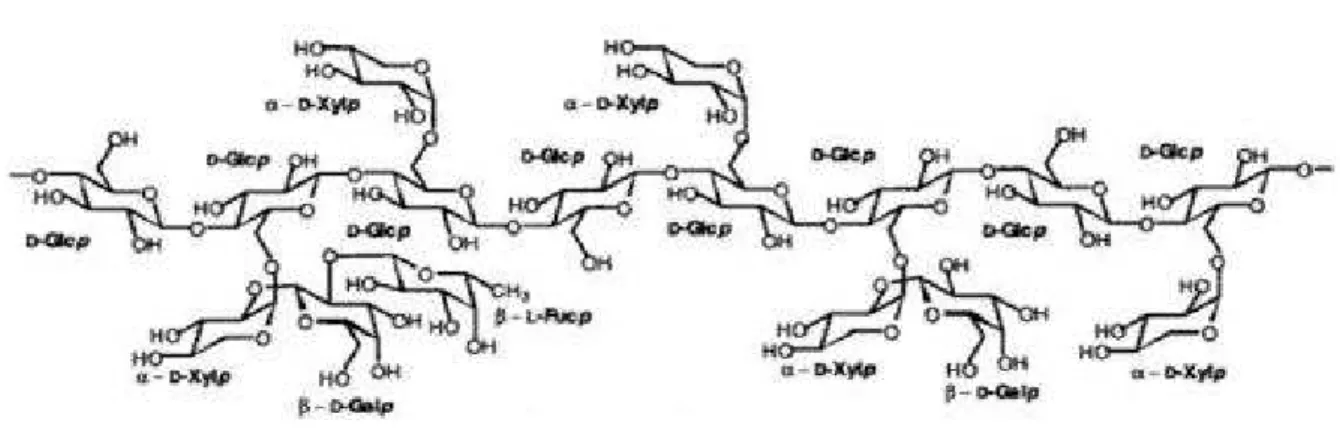
Il D – xiloglucano (Figura 1.4) è la tipologia di emicellulosa più studiata, in quanto rappresenta il costituente principale della parete cellulare delle piante [9]: come la cellulosa, contiene unità di glucosio connesse con legami β – (1 → 4) [10]. Diversamente dalla cellulosa, sono presenti catene laterali legate ai gruppi idrossili dei C-6 della catena principale.
Figura 1.4: Struttura del D – xiloglucano.
L’arabinoglucuronoxilano (Figura 1.5), detto più comunemente D – xilano, rappresenta dal
7 al 15% del legno di conifera [11]: è composto da unità di D – xilopiranosio legate mediante legame β – (1 → 4) a vari Tpi di unità ramificate [2 , 12]. Queste ramificazioni con legami α – (1 → 2) includono principalmente acidi uronici, ovvero zuccheri con il gruppo alcolico primario ossidato ad acido carbossilico [11].
16 Figura 1.5: Struttura del D – xilano.
Oltre a questi acidi uronici, sono presenti anche unità di L – arabinosio connesse mediante legami α – (1 → 3). Un’altra catena di ramificazione è rappresentata dalle unità di L – arabinofuranosile, unite mediante legame α – (1 → 3). Gli acidi uronici e le unità di L – arabinosio possono trovarsi sulla stessa catena dello D – xilano o su catene diverse. La catena principale del D – xilano è molto simile a quella della cellulosa, eccetto che per l’assenza di un gruppo alcolico primario per ogni residuo di zucchero. L’assenza del gruppo alcolico primario riduce la formazione dei legami a idrogeno intermolecolari. Inoltre, la sostituzione degli acidi uronici sulla catena dello xilano, rende questi componenti acidi e più solubili in acqua delle catene di cellulosa [11].
A causa della struttura furanosidica dell’arabinosio, è possibile rimuovere queste catene laterali in condizioni moderatamente acide, mantenendo intatto lo scheletro principale dello xilosio [11].
Nelle piante, lo xilano esiste sotto forma di complesso lignina – xilano [12]: sembra essere il principale componente di collegamento tra lignina e altri carboidrati nella parete cellulare delle piante. Gli arabinoxilani tendono a legarsi a polisaccaridi cellulosici e a residui di acido ferulico, formando legami esterei ed eterei molto stabili.
I D - mannani (Figura 1.6) sono le principali emicellulose presenti nelle conifere: le unità di mannosio sono connesse da legami β – (1 → 4) e formano microfibrille simili a quelle della cellulosa. La principale emicellulosa appartenente a questa famiglia è il
(galatto)glucomannano, che costituisce circa il 20% del peso secco [13]: le unità di D –
galattosio sono unite mediante legami α – (1 → 6) alla catena dell’ (1 → 4) – β – D – mannano [12, 14]. Il rapporto glucosio a mannosio è di circa 1 : 3, mentre il rapporto galattosio a glucosio varia tra 1 : 1 a 1 : 10 [15].
Figura 1.6: Struttura del D
L’emicellulosa più importante all’interno della famiglia del D
l’arabinogalattano (Figura 1.7), presente in bassa quantità sia
latifoglie. L’arabinogalattano è formato da catene principa legate con legami β – (1 → 3) con ramificazioni di galattopiranosio e, in misura minore, da legami α arabinofuranosil – (1→ 3) –
Figura 1.7
Infine, il ramnogalacturonano
galacturonico connesse con legame α legame α – (1 → 4) e α – (1
: Struttura del D – mannano (in alto) e del D – galattomannano (in basso)
L’emicellulosa più importante all’interno della famiglia del D
(Figura 1.7), presente in bassa quantità sia nelle conifere che nelle latifoglie. L’arabinogalattano è formato da catene principali di unità di D
→ 3) con ramificazioni di β – D – galattopiranosil
galattopiranosio e, in misura minore, da legami α – (1 → 6) con ramificazioni di – L – arabinofuranosio disaccaride [2 , 12 , 14]
Figura 1.7: Struttura dell’ L – arabino – D – galattano.
ramnogalacturonano (Figura 1.8) ha uno scheletro di unità di acido D
galacturonico connesse con legame α – (1 → 4) e unità di L – ramnosio unite me (1 → 2).
17
galattomannano (in basso).
L’emicellulosa più importante all’interno della famiglia del D – galattano è nelle conifere che nelle li di unità di D – galattopiranosio galattopiranosil – (1 → 6) – D- → 6) con ramificazioni di β – L –
[2 , 12 , 14].
(Figura 1.8) ha uno scheletro di unità di acido D - ramnosio unite mediante
18 Figura 1.8: Struttura del ramnogalacturonano.
In Tabella 1.2 sono riassunte le varie tipologie di emicellulose presenti nel legno [16]: Tabella 1.2: Classificazione delle differenti emicellulose nel legno [16].
L'associazione delle emicellulose con la cellulosa è una questione complessa e non ancora pienamente documentata e compresa; una piena comprensione di questo aspetto potrebbe permettere di ottenere rese superiori in zuccheri o in acido levulinico a partire da biomasse lignocellulosiche.
Si ritiene che non vi siano veri e propri legami chimici tra i polisaccaridi, mentre sono presenti interazioni basate prevalentemente su legami a idrogeno intermolecolari e su forze di Van der Waals [4]. Dato che lo scheletro di questo polisaccaride è molto simile a quello della cellulosa, tali interazioni possono verificarsi quando questo scheletro è non sostituito. L'associazione tra i polisaccaridi permette la formazione di un reticolo nella parete primaria che può rivestire un ruolo fondamentale nel mantenere l’integrità strutturale delle pareti [17, 18].
19
1.2.3. Pectine
È un insieme di polimeri altamente ramificati di zuccheri acidi, gli acidi glucuronici [5]. Tra questi abbiamo l’acido mannuronico, glucuronico e in particolare l’acido D – galacturonico, in cui le unità sono legate mediante legami glicosidici α – (1 → 4). Anche il ramnosio è una componente significativa [19].
Lo scheletro della struttura pectinica è formato da residui di acido α – (1 → 4) – D – galacturonico, parzialmente metilati (Figura 1.9). Comunque, sono presenti residui di α – (1→ 2) – L – ramnosil – α – (1 → 4) – D – galacturonosil disaccaridi [20].
Figura 1.9: Struttura base della pectina.
Anche se le pectine sono polisaccaridi acidi, non contengono acido glucuronico e acido 4 – metil – glucuronico, che sono presenti solo nelle emicellulose [21]. Tre dei più importanti tipi di polisaccaridi presenti nelle pectine sono il ramnogalatturonano I, il ramnogalatturonano II e l’omogalatturonano [22].
Per quanto riguarda il ramnogalatturonano I, circa la metà dei residui di ramnosio sono sostituiti in posizione C-4 da oligosaccaridi costituiti da L – arabinosio e da D – galattosio [23]. Invece, nel più complesso ramnogalatturonano II, lo scheletro principale è sostituito in posizione C-2 e C-3 con aldeidi – e cheto – zuccheri [20].
Le pectine si trovano in quantità significative in frutta e verdura; nelle piante, qualora siano presenti, si ritrovano sopratutto nelle pareti cellulari primarie dei semi e in particolare nella lamella media [24].
I gruppi carbossilici liberi di catene adiacenti sono legati mediante ioni Ca2+ e Mg2+ [25]. Questi ioni fanno da ponte tra due residui di catene adiacenti di ramnogalatturonani [19]. Il grado di estraibilità delle pectine dipende da queste associazioni molecolari [26]: quando tali ioni sono assenti, la pectina è solubile e quindi estraibile (generalmente con acqua calda) dalla matrice, mentre in caso contrario la pectina forma un gel amorfo.
Alcuni autori hanno avanzato l’ipotesi che le ramificazioni e la possibile associazione della pectina con l’emicellulosa e la cellulosa possano contribuire ad aumentare la porosità delle pareti cellulari, interferendo negativamente con l’idrolisi [17].
20
1.2.4. Lignina
La lignina è una macromolecola con una struttura complessa, estremamente ramificata. Svolge diverse funzioni fondamentali per la vita del vegetale: in particolare, il suo ruolo principale è quello di ridurre la permeabilità all’acqua intervenendo in tutti i processi di scambio di minerali, nutrienti e metabolici.
Mentre la cellulosa costituisce le fibre che conferiscono alle piante resistenza [6], la lignina opera da riempitivo e da collante tra queste fibre, fornendo loro resistenza alla compressione e agli urti. L’emicellulosa ha invece il ruolo fondamentale di tenere associate cellulosa e lignina. Tutto questo viene schematicamente riportato in Figura 1.10.
Figura 1.10: Interazione tra i principali costituenti della biomassa nella parete cellulare.
Nel complesso, è una macromolecola costituita da subunità di fenilpropano (Figura 1.11): Figura 1.11: Unità di fenilpropano.
Tali subunità sono l'alcool cumarilico (alcool 4–idrossicinnamilico), l'alcool coniferilico (alcool
4–idrossi–3–metossicinnamilico) e l'alcool sinapilico (alcool 4–idrossi–3,5–
21 Figura 1.12: Alcoli idrossicinnamilici costituenti la lignina.
Il rapporto di queste unità varia al variare delle piante [27]. Questi alcoli polimerizzano mediante un processo radicalico, che permette la formazione di legami carbonio - carbonio o di legami eterei e il polimero risultante ha una struttura tridimensionale, amorfa. Il legame può avvenire su differenti siti di ogni unità fenolica. I più comuni tipi di legami che si possono trovare nella lignina sono: β – O – 4, α – O – 4, β – 5, 5 – 5, 4 – O – 5, β – 1, e β – β [28 , 29]. Tali legami sono riportati in Figura 1.13.
Figura 1.13: Tipici legami intermonomerici in una molecola di lignina.
Osservando la struttura della lignina, è possibile notare la predominanza del legame β – O – 4 detto anche arilglicerolo – β – aril – etere.
22 Sebbene questi siano i legami principali, sono stati identificati almeno venti differenti tipi di legami: i legami eterei dominano nella lignina nativa, e si stima che siano fino a un terzo del totale dei legami. Questi legami sono molto resistenti da rimuovere e questo spiega le basse velocità con cui la lignina si degrada [5].
I monolignoli possono essere legati in maniera trifunzionale, formando punti di ramificazione lungo il polimero e permettendo di ottenere una struttura a rete. Data la varietà di legami che si possono avere, le molecole di lignina non possono essere considerate come una serie di unità regolari e definite, in quanto essa presenta una struttura estremamente irregolare che può variare anche dalla sua origine.
L’eterogeneità della lignina è ben riconoscibile per le diverse piante di diverse famiglie, ordini, generi e classi così come la struttura della lignina in una stessa specie può variare in base alla zona di provenienza ed alla età della pianta.
Sono stati proposti diversi modelli per definire la struttura della lignina ma, a causa della varietà di forme presenti e delle difficoltà inerenti alle tecniche analitiche disponibili, non è stata ancora identificata una struttura completa di tale macromolecola. I modelli che sono stati sviluppati sono solo rappresentazioni ottenute dalle analisi delle proporzioni relative di ogni unità e di ogni legame [28, 30, 31]. Tra queste strutture, riportiamo qui di seguito quella proposta da Freudenberg (Figura 1.14).
23 In base alla formula proposta da Freudenberg, la lignina naturale contiene principalmente i seguenti gruppi funzionali [32]:
• Gruppi metossilici: La quantità di questi gruppi dipende dalla tipologia della pianta considerata e dal metodo di isolamento. In generale, il contenuto dei gruppi metossili per i legnami di conifere (softwood) e per i legnami di latifoglie (hardwood) oscilla tra 0,92 e 0,94 per 1 unità di fenilpropano.
• Gruppi Idrossilici: Ci sono gruppi idrossili alifatici primari (legati all’atomo Cγ), gruppi idrossilici alifatici secondari (legati all’atomo Cα) e gruppi idrossilici fenolici (legati all’atomo C4 atomo dell’anello aromatico) nella lignina. Generalmente, la lignina contiene 0,2 gruppi idrossilici alifatici primari per 1 unità di fenilpropano, 0.84 gruppi idrossilici alifatici secondari per 1 unità di fenilpropano e 0,30 – 0,35 gruppi idrossilici fenolici per 1 unità di fenilpropano.
• Gruppi Carbossilici: la lignina naturale contiene una piccola quantità di gruppi – COOH, circa 0,05 per 1 unità di fenilpropano. L’incremento dei gruppi –COOH corrisponde a un incremento dell’idrofilicità della lignina. I gruppi carbossilici sono capaci di formare legame a idrogeno con altri gruppi funzionali e incrementare così la reticolazione della macromolecola.
• Gruppi carbonilici: La quantità totale dei gruppi carbonilici presenti nella lignina è di circa 0,21 per una unità di fenilpropano e comunque possono essere di 4 diversi tipi: ci sono gruppi aldeidici legati al γ – C – atomo (0.04 per 1 unità di fenilpropano). Il resto dei gruppi carbonilici (circa 0,17 per un’unità di fenilpropano) sono gruppi chetonici, 0,07 gruppi per un’unità di fenilpropano sono collocati all’α – C – atomo e 0,1 gruppi per un’unità di fenilpropano sono collocati al β – C – atomo. In aggiunta, la lignina contiene piccole quantità di gruppi chinonici.
La frazione di lignina rappresenta circa il 20 e il 35% della biomassa, a seconda delle specie arboree. Infatti, la lignina è in genere presente in quantità minore nelle latifoglie, dove è circa il 22%, mentre nelle conifere è il 27 – 29% [33]. Variazioni tra le diverse lignine softwood sono generalmente trascurabili [38], cosa che non avviene per le lignine hardwood [35].
La presenza di legami tra la lignina e le emicellulose è stata proposta per prima da Erdmann [36]. Recentemente, i legami lignina – emicellulosa sono stati riconsiderati e sono state individuate le seguenti principali tipologie di legami [37]:
• legami di tipo benzil
gruppo idrossile di un carboidrato;
• legami di tipo benzil
acido carbossilico di un carboidrato;
• legami glicosidici tra un gruppo idrossile alifatico o aromatico e un carboidrato;
• legami di tipo acetale tra due gruppi idrossili dei carboidrati e un gruppo carbonile della lignina.
Figura 1.15: Strutture proposte
La lignina può anche essere legata, mediante legami glicosidici, alle catene laterali degli arabinoxilani e a quelle dei
associazioni avvengano via acidi idrossicinnamici che agiscono da ponte
la lignina [39]: quando tali associazioni sono presenti, gli zuccheri possono manifestare una resistenza superiore all’idrolisi acida.
1.2.5. Componenti non-strutturali del legno
Sia le conifere che le latifoglie contengono generalmen unità di amilosio e di amilopectina (Figura 1.16):
legami di tipo benzil - etere tra un gruppo α – idrossile di un’unità di lignina e un gruppo idrossile di un carboidrato;
legami di tipo benzil - estereo tra un gruppo α – idrossile di un’unità di lignina è un acido carbossilico di un carboidrato;
licosidici tra un gruppo idrossile alifatico o aromatico e un carboidrato;
legami di tipo acetale tra due gruppi idrossili dei carboidrati e un gruppo carbonile
: Strutture proposte per i differenti legami lignina–emicellulosa [37
La lignina può anche essere legata, mediante legami glicosidici, alle catene laterali degli arabinoxilani e a quelle dei galattoglucomannani [38]. È probabile che molte di queste associazioni avvengano via acidi idrossicinnamici che agiscono da ponte
la lignina [39]: quando tali associazioni sono presenti, gli zuccheri possono manifestare una resistenza superiore all’idrolisi acida.
strutturali del legno
le latifoglie contengono generalmente amido [2]. L’ amilosio e di amilopectina (Figura 1.16):
24 idrossile di un’unità di lignina e un
idrossile di un’unità di lignina è un
licosidici tra un gruppo idrossile alifatico o aromatico e un carboidrato; legami di tipo acetale tra due gruppi idrossili dei carboidrati e un gruppo carbonile
emicellulosa [37].
La lignina può anche essere legata, mediante legami glicosidici, alle catene laterali degli È probabile che molte di queste associazioni avvengano via acidi idrossicinnamici che agiscono da ponte tra le emicellulose e la lignina [39]: quando tali associazioni sono presenti, gli zuccheri possono manifestare una
25 Figura 1.16: Struttura dell’amilosio (sinistra) e dell’amilopectina (destra).
L’amilosio ha una struttura lineare formata da unità di glucosio unite mediante legame α – (1 → 4) [2]. La natura assiale del legame glicosidico riduce la forza e l’abbondanza dei legami a idrogeno intermolecolari rispetto a quelli della cellulosa.
L’amilopectina ha la stessa struttura dell’amilosio, ma in ogni 24 – 30 unità di glucosio sono presenti ramificazioni di unità con legami α – (1 → 6). L’amilopecTna ha un DP più alto rispetto all’amilosio ma, a causa della presenza di queste ramificazioni, non ha una struttura ad elica estesa: questo significa che non è possibile alcun legame a idrogeno intermolecolare.
Seppur in minore quantità, nelle pareti cellulari delle piante sono presenti anche le proteine: possono essere legate covalentemente con la lignina e i polisaccaridi a formare delle strutture altamente reticolate [40].
Oltre alle proteine, nel legno sono presenti numerosi altri composti organici, localizzati in tutta la parete cellulare, i cosiddetti “estratti”, composti a basso peso molecolare che possono essere separati dai componenti insolubili della parete cellulare mediante estrazione con solventi organici o con acqua calda.
Tra questi componenti abbiamo principalmente monosaccaridi, polisaccaridi, tannini, terpeni, grassi, cere e composti aromatici.
Elevate concentrazioni di estratti sono presenti nelle biomasse solo in casi eccezionali, e comunque si tratta di specie tropicali. In alcuni alberi, i tannini possono rappresentare fino al 30% della massa, mentre il genere Larix può avere fino al 40% di arabinogalattano come estratto [2]. In generale, tuttavia, gli estratti costituiscono meno del 5 – 10% della biomassa. Come ulteriori componenti della biomassa lignocellulosica abbiamo le ceneri, ovvero i residui inorganici che rimangono a seguito dell’ incenerimento del legno [12]. Le ceneri non hanno quindi alcun valore energetico. Elevati contenuti di ceneri possono causare problemi nelle reazioni di pirolisi e nei sistemi di combustione di biomassa [40].
26 Ai fini di un’ eventuale idrolisi della biomassa, un elevato contenuto di ceneri può implicare un più elevato consumo di acido, a causa della natura alcalina di tali ceneri. Il contenuto di ceneri può variare notevolmente al variare delle specie vegetali; inoltre, tale contenuto dipenderà anche dalla fase di crescita delle piante, dal periodo di raccolta e dalle modalità di coltivazione.
I cationi più importanti presenti nelle ceneri di biomasse lignocellulosiche sono Si, Ca, K e
Mg. Altri elementi come Mn, Na e P sono presenti in quantità minori. Elementi come Al, Fe, Zn, Cu, Ti, Pb, Ni, V, Co, Ag e Mo si trovano in tracce nella maggior parte dei substrati [41 , 42]. Come anioni si ritrovano generalmente cloruri, carbonati, solfati, silicati, ossidi e idrossidi [43].
Gli elementi inorganici che costituiscono la cenere tendono ad essere presenti sia come componenti degli estratti che come cristalli. In generale le latifoglie contengono un po’ più di ceneri delle conifere, e le latifoglie delle zone tropicali contengono più ceneri delle latifoglie delle zone temperate, ma questo non è universalmente vero [44].
27
1.3. Riferimenti bibliografici
1) M. Galli, S. Pampana, Le fonti rinnovabili per la produzione di energia: il ruolo delle biomasse. Le colture dedicate ad uso energetico: il progetto Bioenergy Farm. Quaderno ARSIA 6/2004.
2) E. Sjöström, Wood Chemistry. Fundamentals and Applications, 2nd edition, Academic Press, San Diego, CA, USA, 1993, 293.
3) C. O’ Sullivan, Cellulose, 1997, 4, 173 – 207.
4) H. A. Krassig, in Cellulose: Structure Accessibility and Reactivity Polymer Monographs, Vol. 11, Gordon and Breach Science Publishers, Switzerland, 1992.
5) H. R. Bungay, Energy, the Biomass Options. John Wiley and Sons, New York, NY, 1981. 6) J. C. Roberts, The Chemistry of Paper. Royal Society of Chemistry, Cambridge, UK, 1996. 7) J. Sugiyama, Planta, 1985, 166, 8 – 161.
8) I. S. Goldstein, Overview of the chemical composition of wood. In. M. Lewin and I. S. Goldstein, Wood Structure and Composition. International Fiber Science and Technology, 11, 1991.
9) T. Doco, P. Williams, M. Pauly, M. A. O'Neill, P. Pellerin, Carbohydrate Polymers, 2003, 53, 61 – 253.
10)G. O. Aspinall, Ency. Plant Physiol. 1981, 13, 3 – 8.
11)J. P. Casey, Pulp and Paper: Chemistry and Chemical Technology. John Wiley and Sons, New York, NY, 1980
12)D. Fengel, G. Wegener, Wood: Chemistry Ultrastructure and Reactions. De Gruyter, Berlin, 1989, 613.
13)J. Lundqvist, A. Jacobs, M. Palm, G. Zacchi, O. Dahlman, H. Stålbrand, Carbohydrate Polymers, 2003, 51, 203 – 211.
14)K. Shimizu, Chemistry of hemicelluloses; in: Wood and Cellulosic Chemistry. 2nd edition, Eds. D. N. S. Hon, N. Shiraishi, Marcel Dekker Inc., New York, USA, 2001, 177 – 214.
15) P. Hakkila, Utilisation of Residual Forest Biomass. Springer-Verlag, Berlin, 1989. 16) T. E. Timell, Wood science and technology, 1967, 1, 45.
17)N. C. Carpita, D. M. Gibeaut, Plant Journal, 1983, 3, 1 – 30.
18)M. Pauly, P. Albersheim, A. G. Darvill, W. S. York, Plant Journal, 1999, 20, 39 – 629.
19)R. D. Hatfield, Cell Wall Polysaccharide Interactions and Degradability. In. H. G. Jung, D. R. Buxton, R. D. Hatfield and J. Ralph, Forage cell wall structure and digestibility. ASA-CSSA-SSSA, Madison, WI. 1993, 285 – 313.
20)P. Aman, Composition and structure of cell wall polysaccharides in forages. In. H. G. Jung, D. R. Buxton, R. D. Hatfield, J. Ralph, Forage cell wall structure and digestibility, ASA-CSSA-SSSA, Madision, WI., 1993, 99 – 183.
28
21)P. J. Van Soest, Cell Wall Matrix Interactions and Degradation - Session Synopsis. In. H. G. Jung, D. R. Buxton, R. D. Hatfield and J. Ralph, Forage cell wall structure and digestibility, ASA-CSSA-SSSA, Madison, WI., 1993, 96 –377.
22)P. M. Dey, Cabohydrates. Academic Press, London, 1990,1 , 42 – 415.
23)W. S. York, A. G. Darvill, M. McNeill, T. T. Stevenson, P. Albersheim, Methods Enzymol., 1986, 118, 3 – 40.
24)J. Priess, The biochemistry of plants. Academic Press, New York, NY, 1988, 14, 297 – 371. 25)S. K. Stombaugha, H. G. Jungb, J. H. Orfa, D. A. Somersa, Crop Science, 2000, 40, 12 – 408. 26)J. A. Monro, J. Food Comp. Anal., 1991, 4, 88 – 99.
27)H. G. Jung, D. R. Buxton, R. D. Hatfield, J. Ralph, Forage Cell Wall Structure and Digestibility. ASA-CSSA-SSSA, Madison, WI., 1993, 83 – 104.
28)E. Adler, Wood Sci. Technol., 1977, 11, 169 – 218.
29)E. Dorrestijn, L. J. Laarhoven, I. Arends, P. Mulder, J. Anal. Appl. Pyrolysis, 2000, 54 (1-2), 92 – 153.
30)K. Freudenberg, A. C. Neish, Constitution and Biosynthesis of Lignin, Springer-Verlag, Heidelberg, 1968, 47 – 122.
31)N.G. Lewis, S. Sarkanen, Lignin and Lignan Biosynthesis, American Chemical Society, ACS Symposium Series 697, Washington DC, USA, 131 – 147.
32)K. V. Sarkanen, Lignins: occurrence, formation, structure and reactions, University of Washington: Seattle, Washington, 1987.
33)P. Hakkila, Utilisation of Residual Forest Biomass. Springer-Verlag, Berlin, 1989. 34)W. G. Slater, H. Beevers, Plant Physiol., 1958, 33, 146.
35)J. B. Pridham, W. Z. Hassid, Biochem. J., 1966, 100, 21. 36)Erdmann, Ann. Chem. Pharm., 1866, 138, 1 – 19.
37)T. Watanabe, Analysis of native bonds between lignin and carbohydrate by specific chemical reactions (2003); in: Association Between Lignin and Carbohydrates in Wood and Other Plant Tissues, Eds. T. Koshijima, T. Watanabe, Springer-Verlag, Heidelberg, Germany, 91 – 130.
38)O. Eriksson, B. O. Lindgren, Svensk Papperstidn, 1977, 80, 59.
39)R. D. Hartley, C. W. Ford, Phenolic constituents of plant cell walls and wall biodegradability (1989); in: N. G. Lewis and M. G. Paice, ACS Symp. Ser. 339. Am. Chem. Soc., Washington, DC., 45 – 137.
40)Eds. J. Gullichsen, H. Paulapuro, P. Stenius, Forest Products Chemistry, Fapet, Finland, 11 – 57. 41)V. I. Naidenov, I. F. Koperin, V. R. Purin, Lesnoi Zhurnal, 1982, 2, 7 – 115.
42)K. Pohlandt, M. Strecker, R. Marutszky, Chemosphere, 1993, 26, 8 – 2121.
43)E. A. Osman, J. R. Goss, Am. Soc. Agric. Engineers Annual Meeting, 1983, 83, 1 – 16. 44)R. M. Rowell, The chemistry of solid wood. American Chemical Society, Washington, 1984.
29
2. Furfurale ottenuto da materie prime rinnovabili
2.1. L’idrolisi acida dei costituenti C5 della biomassa lignocellulosica a furfurale
Il furfurale o furaldeide, l’α – aldeide del furano, è stato isolato per la prima volta da J. W. Dobereiner nel 1832, che ne ottenne una minima quantità come coprodotto della sintesi dell’acido formico. Nel 1840, J. Stenhouse scoprì che il furfurale poteva essere prodotto distillando un’ampia varietà di cereali, avena e crusca. Nel 1901, C. Harries ne dedusse la struttura riportata in Figura 2.1.
Figura 2.1: Struttura del furfurale.
In Tabella 2.1, sono elencate alcune proprietà del furfurale:
Tabella 2.1: Alcune proprietà fisiche del furfurale.
Proprietà Valore Temperatura di fusione -36.5 °C Temperatura di ebollizione (760 mm Hg) 161.7 °C Densità 1.16 g/ml Indice di rifrazione nD20 1.52698 Viscosità a 25 °C 1.494 cP
Solubilità in acqua a 20 °C 8.3 g per 100 ml di acqua
Non esiste una via sintetica economicamente vantaggiosa per la produzione del furfurale nell’industria chimica, e quindi lo si produce esclusivamente per idrolisi di residui di piante ricche di xilani [1 - 5]. La produzione industriale di furfurale richiede un contenuto minimo di pentosani pari a circa 18 – 20% [6]. Solo circa un terzo dei pentosani presenti nelle materie prime utilizzate viene trasformato in furfurale con gli attuali processi industriali. Il contenuto di pentosani e i valori di rese in furfurale per alcune materie prime sono riportati in Tabella 2.2 [3]:
30 Tabella 2.2: Contenuto dei pentosani per varie materie prime e relativa resa ponderale % in furfurale [3].
Biomassa Pentosani totali
(in % in peso rispetto alla massa secca)
Resa ponderale % in furfurale nella produzione industriale rispetto alla biomassa di partenza
Pannocchia di mais 30-40 10 Bagassa 25-27 8-9 Cotone 27 8-9 Hardwoods 21-24 6-8 Corteccia di faggio 19-21 5-6 Guscio di riso 16-18 6 Guscio di girasole - 8-9
La formazione del furfurale avviene in due stadi. Nel primo, l’idrolisi acido–catalizzata fa sì che i pentosi polimerici siano idrolizzati a dare pentosi monomerici in soluzione acquosa. Poiché la struttura dei pentosani è quella di un poliacetale, l’idrolisi acida dei pentosani corrisponde all’idrolisi degli acetali [6]. Il meccanismo di idrolisi include I seguenti stadi [7, 8]:
a) diffusione dei protoni attraverso la matrice lignocellulosica umida;
b) protonazione del legame all’ossigeno, con formazione di un ossigeno trivalente; c) rottura del legame carbonio – ossigeno, con formazione di un carbocatione e di
un derivato idrossilato;
d) reazione del carbocatione con l’acqua e liberazione dello ione H+, lasciando il gruppo idrossile anche sull’altra parte della molecola;
e) diffusione dei prodotti dei reazione in fase liquida, se la loro struttura glielo consente.
Questa sequenza di reazioni (Figura 2.2) si ripete fino a quando la rottura dei legami con l’ossigeno è completa ed alla fine si ottengono i rispettivi monomeri [4, 9].
31 Nel secondo stadio, i pentosi sono convertiti in furfurale mediante liberazione di tre molecole di acqua [9] (Figura 2.3). Il pentoso è schematizzato nella sua forma ad anello, che rappresenta un emiacetale intramolecolare. La forma aperta aldeidica in equilibrio con quella chiusa non viene considerata in quanto presente in quantità inferiore all’1% rispetto ai pentosi totali presenti.
Figura 2.3: Meccanismo per la deidratazione dei pentosi a furfurale.
Gli stadi di trasformazione includono due eliminazioni - 1,2 e un’eliminazione - 1,4 di acqua. Le eliminazioni - 1,2 avvengono tra due atomi di carbonio adiacenti e portano alla formazione di un doppio legame, mentre l’eliminazione - 1,4 coinvolge due atomi di carbonio separati da altri due atomi di carbonio e porta alla formazione di un anello [10]. La formazione dell’anello è favorita dal fatto che gli atomi di carbonio che partecipano al doppio legame formano una struttura planare con angoli di legame di 120 °C.
Impiegare la velocità di scomparsa dello xilosio come mezzo per quantificare il tempo di residenza è una valida procedura in quanto la velocità di scomparsa dello xilosio non risente di reazioni di degradazione. Quindi, indipendentemente dal fatto che queste reazioni di degradazione avvengano o no (dipenderà dal disegno del reattore), la velocità di scomparsa dello xilosio è la stessa.
32 Uno studio cinetico dettagliato è stato pubblicato da Root, Saeman, Harris, e Neill [11]: impiegando fiale in vetro sigillate e riscaldate a varie temperature mediante bagno a olio, al fine di studiare la cinetica di reazione per l’idrolisi di soluzioni di xilosio in H2SO4 a diversa concentrazione. Gli autori hanno misurato la velocità di scomparsa dello xilosio e le rese in furfurale su un ampio intervallo di temperature (da 160 a 280 °C), di concentrazione di xilosio (da 3,125 a 200 g/l) e di acido solforico (0,0306 a 3,831% in peso). La velocità di scomparsa dello xilosio è espressa dalla seguente equazione:
[ ]
=
.( ∙ )
∙
∙
( !")
Dove [xilosio] è la concentrazione di xilosio (moli/litro), t è il tempo (minuti), CH è la concentrazione di acido iniziale (moli/litro) e T è la temperature assoluta (°K).
La precedente equazione può essere impiegata per calcolare il tempo di residenza per la completa scomparsa dello xilosio [11].
Questa è un’equazione molto importante per la progettazione di reattori batch per la produzione industriale del furfurale. Si riporta qui di seguito una rappresentazione grafica della precedente equazione, impiegando la KI ricavata precedentemente per varie concentrazioni in peso di H2SO4 e a diverse temperature.
Figura 2.4: Tempo di reazione nominale in funzione della concentrazione di H2SO4 e della temperatura.
Quanto riportato permette di affermare che:
33
• una piccola variazione della temperatura causa enormi differenze nel tempo di reazione e quindi, ai fini cinetici, sono richieste, a livello di impianto, alte temperature, che però portano alla rapida degradazione del furfurale formato. La resa teorica di furfurale a partire dai pentosani è del 72,73% in peso. Generalmente le rese sono assai inferiori a questo valore perché, competitivamente alla disidratazione dei pentosi a furfurale, possono avvenire delle reazioni di degradazione [9], ed in particolare:
• Una reazione di polimerizzazione del furfurale, chiamata “resinificazione” del furfurale:
n furfurale → (furfurale)n
• Una reazione del furfurale con un intermedio della conversione pentoso – furfurale. Questa reazione è chiamata “condensazione” del furfurale:
furfurale + pentosio → furfural - pentosio
Queste due reazioni possono avvenire o no, a seconda che si mantenga in fase liquida o meno il furfurale prodotto. Quando il furfurale viene aggiunto a una soluzione di xilosio, e quando questa soluzione è sottoposta a catalisi acida per convertire lo xilosio a furfurale, si ottiene una resa in furfurale più bassa rispetto alla rispettiva soluzione senza furfurale. Comunque, una reazione del furfurale con lo xilosio può essere esclusa perché l’aggiunta del furfurale alla soluzione di xilosio non incrementa la velocità di scomparsa dello xilosio. Conseguentemente, il furfurale deve reagire con un intermedio della reazione di conversione xilosio – furfurale. Una possibilità è che una molecola di furfurale possa reagire con in primo intermedio a dare il furfural – xilosio. È anche possibile che due molecole di furfurale possano reagire con un intermedio, a dare il difurfural – xilosio (Figura 2.5).
34 Il contributo alla degradazione del furfurale dovuto alle reazioni di condensazione è significativamente superiore rispetto a quello dovuto alle reazioni di resinificazione.
Ci sono molti aspetti della reazione che non sono ancora completamente chiari, soprattutto se si considera la cinetica di formazione e di degradazione del furfurale. Le informazioni disponibili ricoprono un ampio intervallo di pH e di temperatura e mostrano incoerenze tra i differenti lavori, come proposto da Root [11], da Dunlop [12], da Garret e Dvorchik [13], da Antal [14] e da J. Qi [15].
Queste ricerche hanno comunque permesso di concludere in maniera chiara che, per incrementare la resa del furfurale, è necessaria una scelta accurata dei principali parametri: temperatura, concentrazione di acido e tempo di reazione.
Le più comuni condizioni impiegate per ottenere il furfurale da materiali lignocellulosici sono: temperature comprese tra 140 e 180 °C, alte pressioni (3,5 – 10 atm) e differenti tempi di reazione (1 min – 10 ore) [3, 16 – 21].
La reazione di idrolisi può essere condotta adottando generalmente due diversi approcci:
• nella prima metodologia, la depolimerizzazione dei pentosani a xilosio e la successiva deidratazione a furfurale avvengono contemporaneamente. La reazione è generalmente condotta in un’autoclave e le condizioni impiegate sono blande: temperature moderate, basse pressioni e brevi tempi di reazione [18, 22, 23]. Si riporta in Tabella 5.3 l’effetto della concentrazione di H2SO4 sulla resa in furfurale per un processo in un unico stadio [23], utilizzando come substrato una crusca di riso contenente il 28% di pentosani e adottando un rapporto liquido/solido pari a 25 ml/grammo.
Tabella 2.3: Effetto della concentrazione di H2SO4 sulla resa in furfurale in un processo a uno stadio. [H2SO4] (% peso) Furfurale (% peso)
3 1.1 5 1.3 7 1.9 10 2.3 15 3.2 20 3.3 25 3.0 30 2.2 35 0.7 T = 125 °C, P = 1.5 atm, t = 20 min, L/S = 25 ml/g
Dai valori riportati in Tabella 2.3, si nota come concentrazioni di acido superiori al 20% possano determinare la degradazione del furfurale a causa di reazioni collaterali:
35 un ulteriore contributo alla degradazione è dato anche dal fatto che il furfurale non è continuamente allontanato durante la sua formazione.
• La seconda metodologia impiega un I ° stadio di idrolisi di dissoluzione dei pentosani in condizioni più blande (preidrolisi); durante tale pretrattamento, non solo si ha l‘idrolisi dello xilano, ma anche di altri carboidrati contenuti nell’emicellulosa, come i galattoglucomannani e gli arabinogalattani. Essi sono idrolizzati e rimossi in maniera simile a quanto avviene per gli xilani [24].
La preidrolisi permette di recuperare in soluzione gli zuccheri e su quest’ultima frazione si effettua un II ° stadio di deidratazione dello xilosio a furfurale. Il vantaggio della tecnologia a due step sta nel fatto che il materiale lignocellulosico residuo subisce una minore degradazione e può essere usato per la conversione ad altri prodotti (glucosio, etanolo, fenoli, ecc..). L’effetto del tempo di preidrolisi sulla resa in furfurale in un processo a due stadi è riportato in Tabella 2.4 [23]:
Tabella 2.4: Effetto del tempo di preidrolisi sulla resa in furfurale in un processo a due stadi [26]. Tempo Furfurale (% in peso)
0 1.5
30 10.5
60 8.9
120 8.7
T = 110 °C, [H2SO4] = 15% (w/w), L/S = 25 ml/g
Anche in questo caso, la produzione di furfurale dopo 30 minuti diminuisce a seguito della successiva decomposizione del furfurale formato.
Kim e Lee [25] riportano una resa in xilosio maggiore all’80% nell’idrolisi dell’emicellulosa hardwood a 120 – 140 °C impiegando H2SO4 al 2,5%.
Pessora [26] ha realizzato l’idrolisi dell’emicellulosa della bagassa con H2SO4 in laboratorio (palloncino da 25 ml) e in un reattore semi - pilota (25 litri): in laboratorio, ottiene il 71 – 74% di xilosio, lavorando a temperatura di 140 – 150 °C, con un tempo di 10 – 20 minuti e concentrazione di acido di 70 – 100 mg acido/gbiomassa secca. Impiegando un reattore semi-pilota, ottiene invece una resa dell’83,3% in peso rispetto allo xilosio a 140 °C con 100 mgacido/gbiomassa secca, dopo 20 minuti.
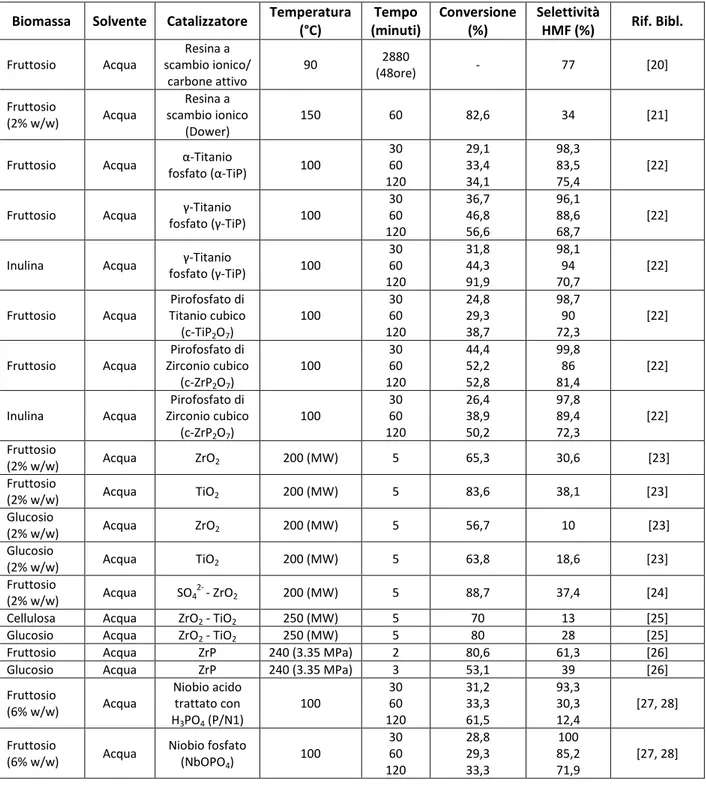
Vari ricercatori hanno studiato la deidratazione dello xilosio a furfurale impiegando catalizzatori acidi, includendo acidi minerali [23, 27, 28], zeoliti [29, 30] e eteropoliacidi [31]. Alte rese di furfurale, superiori al 75%, sono state ottenute con catalizzatori MCM-41
36 modificati con gruppi acidi solfonici in solventi quali DMSO e miscele acqua-toluene, a basse concentrazioni di xilosio (3% in peso) [30].
In letteratura, sono riportati numerosi esempi di catalizzatori che, impiegati a basse concentrazioni di acido, possono favorire ulteriormente l’idrolisi: tra questi, ritroviamo soprattutto TiO2, ZnCl2, AlCl3 [23]. Essi possono agire come acidi di Lewis, promuovendo la reazione di idrolisi, o possono stabilizzare gli intermedi della deidrociclizzazione dei pentosi, aumentando la resa a furfurale. Quando si impiegano ossidi di zirconio o di zinco, si osserva un effetto modesto mentre il TiO2 mostra una migliore attività catalitica sia nei processi a uno che in quelli a due step. Questo può essere spiegato con la formazione di specie solfato sulla superficie del TiO2 [32].
2.2. Potenziali applicazioni del furfurale e dei suoi derivati
A partire dai primi anni del 1990, la produzione mondiale del furfurale si è spostata dai paesi sviluppati a quelli in via di sviluppo: infatti, negli ultimi anni c’è stato un notevole incremento nella produzione di furfurale in Cina [33], dove si usano come materie prime gli scarti del riso mentre la produzione nell’Europa occidentale si è notevolmente ridotta. Grazie ai bassi costi di produzione associati alle tecnologie impiegate, i maggiori produttori mondiali di furfurale sono la Cina e la Repubblica Dominicana, come è riportato in Tabella 2.5 [34]:
Tabella 2.5: Produzione mondiale del furfurale (2005) [34].
Paese Materia prima Produzione
(tonnellate per anno)
Cina Pannocchia di riso 200,000 Tailandia Pannocchia di riso 8,500 Repubblica Dominicana Bagassa 32,000
Sud Africa Bagassa 20,000
Spagna Pannocchia 6,000
Altri (incluso India e Sud Africa) Pannocchia/Bagassa <15,000 Russia Pannocchia Dato non disponibile
TOTALE >280,000
Il costo di mercato del furfurale è approssimativamente 1,4 $/kg (2013), paragonato con il prezzo nel 1990 di 1,74 $/kg e di 1 $/kg nel 2005.
37 Figura 2.6: andamento del costo del furfurale nel tempo.
Il prezzo di mercato è dovuto principalmente alla richiesta del prodotto: ad esempio, il momentaneo calo del prezzo di mercato del furfurale e del furfuril alcol (il principale derivato del furfurale) nel 2001 è stato causato da un indebolimento del settore delle resine furaniche.
La richiesta del furfurale è vista in crescita, dagli attuali 300 Kton/anno alla previsione di 1 Mton/anno per il 2020, in quanto la domanda di prodotti verdi e bio-based è in costante aumento.
Il mercato del furfurale è assai ampio: può essere impiegato come tale o come composto di partenza per la sintesi di un’ampia varietà di composti chimici alifatici e eterociclici e nella produzione di resine, in particolare quelle fenoliche. Il 65% di tutto il furfurale prodotto è convertito in furfuril alcol. In aggiunta a ciò, i principali prodotti chimici preparati dal furfurale sono il tetraidrofurano (THF) e il diidropirano (DHP).
2.2.1. Impiego del furfurale come tale
Il furfurale può essere impiegato efficacemente come agente estraente [9]. Tale applicazione si basa sul fenomeno della coniugazione intermolecolare: quando molecole con doppi legami coniugati come il furfurale vengono in contatto con altre molecole che contengono doppi legami, si vengono a creare delle strutture complesse. Conseguentemente, il furfurale lega preferenzialmente molecole contenenti doppi legami. Per questa ragione, il furfurale è impiegato per:
38 1. rimuovere gli aromatico dagli oli lubrificanti per migliorare il rapporto
viscosità/temperatura;
2. rimuovere gli aromatici dai combustibili diesel per migliorarne le caratteristiche di accensione;
3. per ottenere composti poli insaturi dagli oli vegetali come l’olio di soia, utilizzabili ad esempio per vernici, in quanto la presenza di composti con doppi legami permette l’essicazione della vernice per ossidazione con aria e forma polimeri reticolati.
Nelle applicazioni 1 e 2, il prodotto desiderato (libero da aromatici) è il raffinato, mentre nell’applicazione 3 il prodotto desiderato (ricco di composti insaturi) è ottenuto dall’estratto. In perfetta analogia alle resine fenolo - formaldeide, il fenolo reagisce con il furfurale per formare delle resine fenolo – furfurale. I gruppi idrossilici del fenolo rendono reattive le posizioni orto e para. Conseguentemente, il fenolo reagisce con il furfurale, formando il furfurilol fenolo in composizione mista e variabile orto e para (Figura 2.7).
Figura 2.7: Formazione del furfurilol fenolo.
A temperature più alte di 100 °C o a temperature più basse in presenza di appropriati catalizzatori (generalmente basi o acidi), due molecole di furfurilol fenolo reagiscono tra loro formando un ponte etereo (Figura 2.8), con conversione elevata. Questi processi possono continuare a livello degli atomi di idrogeno reattivi in posizione orto e para, formando rispettivamente il difurfurirol fenolo e il trifurfurilol fenolo, con una struttura altamente reticolata.
39 In analogia alle resine formaldeide – urea, si possono ottenere anche resine furfurale – urea [35]. Anche in questo caso, il primo stadio consiste nella formazione della monofurfurilol urea (Figura 2.9). Anche in questo caso, lo studio successivo consiste nella formazione di un ponte etereo tra due di queste molecole, liberando acqua. In questo caso, però, essendoci ancora atomi di idrogeno reattivi sull’azoto, si riesce ad ottenere un sistema ancora più esteso e reticolato rispetto alle resine fenolo – furfurale.
Figura 2.9: Formazione della monofurfurilol urea.
Come ulteriori applicazioni del furfurale, ricordiamo il suo impiego per la formulazione di erbicidi e fungicidi [36]. Nel 1923 si trovò che persino quando vengono impiegate alte concentrazioni di formaldeide, non si riesce a evitare la crescita della muffa Penicillium, mentre l’impiego di una soluzione di furfurale allo 0,5% è invece sufficiente a evitare tale crescita.
2.2.2. Impiego dei derivati del furfurale
Il furfuril alcol è commercialmente il derivato più importante del furfurale. Attualmente, circa il 65% di tutto il furfurale prodotto è convertito a furfuril alcol, mediante idrogenazione catalizzata da cromito di rame.
L’applicazione più importante per il furfuril alcol riguarda il suo impiego per la sintesi di resine, analogamente a quanto accade per il furfurale [35]. In ambiente acido, il gruppo alcolico di una molecola di furfuril alcol reagisce con l’idrogeno attivo di un’altra molecola di furfuril alcol e si forma una struttura con ponte metilenico (Figura 2.10):
Figura 2.10: Dimero del furfuril alcol.
Nella stessa maniera, ulteriori molecole di furfuril alcol possono aggiungersi indefinitamente al polimero in crescita. La reazione può essere controllata mediante raffreddamento e può essere fermata a qualsiasi grado di viscosità mediante neutralizzazione. Grazie alla elevata
40 resistenza ottenuta da un processo termico di indurimento che permette di ottenere un materiale altamente reticolato, queste resine sono molto utilizzate ad esempio nelle fonderie e stanno progressivamente sostituendo le resine fenolo/formaldeide. Il vantaggio principale è che le resine a base di furfuril alcol sotto stress termico non rilasciano formaldeide, composto che nelle resine fenolo/formaldeide si libera a seguito di reazioni di degradazione termica e che costituisce un serio problema per i lavoratori della fonderia. Il tetraidrofurano THF è preparato da un processo in due stadi che comprende la decarbonilazione del furfurale, da cui si forma il furano, seguita dall’idrogenazione di questo composto, generalmente condotta a 100 °C e a 20 bar; il catalizzatore impiegato in entrambi gli stadi è palladio al 5% supportato su carbone microporoso.
Il tetraidrofurano può essere ulteriormente ridotto a n – butanolo, ossidato a butirro lattone o ad acido succinico, deidratato a butadiene, clorurato e fatto reagire con una serie di reagenti per dare gli 1,4 – derivati del n–butano [37] (Figura 2.11):
![Figura 1.2: rappresentazione dell’architettura molecolare della cellulosa, in relazione alle microfibrille e alla parete cellulare [6]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7979389.120836/14.892.320.621.126.428/figura-rappresentazione-architettura-molecolare-cellulosa-relazione-microfibrille-cellulare.webp)
![Tabella 2.2: Contenuto dei pentosani per varie materie prime e relativa resa ponderale % in furfurale [3]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7979389.120836/30.892.141.797.147.319/tabella-contenuto-pentosani-varie-materie-relativa-ponderale-furfurale.webp)
![Figura 3.3: meccanismo di formazione dell’HMF da fruttosio e da glucosio [3 , 4].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7979389.120836/46.892.162.768.691.1125/figura-meccanismo-formazione-hmf-fruttosio-glucosio.webp)

![Figura 3.4: Ruolo catalitico del fosfato di Niobio per la conversione di glucosio ad HMF [38]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7979389.120836/51.892.134.798.704.1029/figura-ruolo-catalitico-fosfato-niobio-conversione-glucosio-hmf.webp)


![Figura 6.1: rappresentazione schematica della interazione tra collagene e vari tipi di concianti [2]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7979389.120836/92.892.132.839.258.807/figura-rappresentazione-schematica-interazione-collagene-vari-tipi-concianti.webp)
