n e f r o l o g i a. 2 0 1 5;35(5):421–447
RevistadelaSociedadEspañoladeNefrología www . r e v i s t a n e f r o l o g i a . c o m
Review
An
update
for
atypical
haemolytic
uraemic
syndrome:
Diagnosis
and
treatment.
A
consensus
document
夽
Josep
M.
Campistol
a,∗,
Manuel
Arias
b,
Gema
Ariceta
c,
Miguel
Blasco
a,
Laura
Espinosa
d,
Mario
Espinosa
e,
Josep
M.
Grinyó
f,
Manuel
Macía
g,
Santiago
Mendizábal
h,
Manuel
Praga
i,
Elena
Román
h,
Roser
Torra
j,
Francisco
Valdés
k,
Ramón
Vilalta
c,
Santiago
Rodríguez
de
Córdoba
l aServiciodeNefrología,HospitalClínic,Barcelona,SpainbServiciodeNefrología,HospitalUniversitarioMarquésdeValdecilla,Santander,Spain
cServiciodeNefrologíaPediátrica,HospitalUniversitariMaterno-InfantilValld’Hebrón,UniversidadAutónomadeBarcelona,Barcelona, Spain
dServiciodeNefrologíaPediátrica,HospitalLaPaz,Madrid,Spain eServiciodeNefrología,HospitalUniversitarioReinaSofía,Córdoba,Spain
fServiciodeNefrología,HospitalUniversitarideBellvitge,HospitaletdeLlobregat,Barcelona,Spain gServiciodeNefrología,HospitalVirgendelaCandelaria,SantaCruzdeTenerife,Spain
hServiciodeNefrologíaPediátrica,HospitalLaFe,Valencia,Spain
iServiciodeNefrología,HospitalUniversitario12deOctubre,Madrid,Spain jEnfermedadesRenalesHereditarias,FundacióPuigvert,Barcelona,Spain kServiciodeNefrología,ComplejoHospitalarioACoru ˜na,ACoru ˜na,Spain
lDepartamentodeMedicinaCelularyMolecular,CentrodeInvestigacionesBiológicas(CSIC),Madrid,Spain
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received9April2015 Accepted3July2015
Availableonline3December2015
Keywords:
Atypicalhaemolyticuraemic syndrome
Eculizumab Complement
Thromboticmicroangiopathy
a
b
s
t
r
a
c
t
Haemolyticuraemicsyndrome(HUS)isaclinicalentitydefinedasthetriadofnonimmune haemolyticanaemia,thrombocytopenia,andacuterenalfailure,inwhichtheunderlying lesionsaremediatedbysystemicthromboticmicroangiopathy(TMA).Differentcausescan induce theTMAprocessthatcharacterisesHUS.Inthisdocumentweconsideratypical HUS (aHUS)a sub-typeofHUSin whichtheTMA phenomenaare theconsequenceof theendotelialdamageinthemicrovasculatureofthekidneysandotherorgansduetoa disregulationoftheactivityofthecomplementsystem.Inrecentyears,avarietyof aHUs-related mutationshavebeenidentifiedingenesofthecomplementsystem,whichcan explainapproximately60%oftheaHUScases,andanumberofmutationsand polymor-phismshavebeenfunctionallycharacterised.ThesefindingshavestablishedthataHUSisa consequenceoftheinsufficientregulationoftheactivationofthecomplementoncell sur-faces,leadingtoendotelialdamagemediatedbyC5andthecomplementterminalpathway.
夽
Pleasecitethisarticleas:CampistolJM,AriasM,AricetaG,BlascoM,EspinosaL,EspinosaM,etal.Actualizaciónensíndromehemolítico urémicoatípico:diagnósticoytratamiento.Documentodeconsenso.Nefrologia.2015;35:421–447.
∗ Correspondingauthor.
E-mailaddress:[email protected](J.M.Campistol).
2013-2514/©2015SociedadEspa ˜noladeNefrología.PublishedbyElsevierEspaña,S.L.U.ThisisanopenaccessarticleundertheCC BY-NC-NDlicense(http://creativecommons.org/licenses/by-nc-nd/4.0/).
EculizumabisamonoclonalantibodythatinhibitstheactivationofC5andblocksthe gen-erationofthepro-inflammatorymoleculeC5aandtheformationofthecellmembrane attackcomplex.InprospectivestudiesinpatientswithaHUS,theuseofEculizumabhas shownafastandsustainedinterruptionoftheTMAprocessandithasbeenassociated withsignificativelong-termimprovementsinrenalfunction,theinterruptionofplasma therapyandimportantreductionsintheneedofdialysis.Accordingtotheexistingliterature andtheaccumulatedclinicalexperience,theSpanishaHUSGrouppublishedaconsensus documentwithrecommendationsforthetreatmentofaHUs(Nefrologia2013;33[1]:27–45). In the current online versionof this document, weupdate the aetiological classifica-tionofTMAs,thepathophysiologyofaHUS,itsdifferentialdiagnosisanditstherapeutic management.
©2015SociedadEspa ˜noladeNefrología.PublishedbyElsevierEspaña,S.L.U.Thisisan openaccessarticleundertheCCBY-NC-NDlicense (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Actualización
en
síndrome
hemolítico
urémico
atípico:
diagnóstico
y
tratamiento.
Documento
de
consenso
Palabrasclave:
Síndromehemolíticourémico atípico
Eculizumab Complemento
Microangiopatíatrombótica
r
e
s
u
m
e
n
Elsíndromehemolíticourémico(SHU)esunaentidadclínicadefinidaporlatríada ane-mia hemolíticano inmune,trombocitopenia einsuficienciarenal aguda, enlaquelas lesionessubyacentesestánmediadasporunprocesodemicroangiopatíatrombótica(MAT) sistémico. Distintascausaspueden desencadenarelprocesodeMATquecaracterizael SHU. Eneste documentoconsideramosSHUatípico(SHUa)como elsubtipode SHUen elquelosfenómenosdeMATsonfundamentalmenteconsecuenciadelda ˜noproducido enelendoteliodelamicrovasculaturarenalydeotrosórganospordesregulacióndela actividaddelsistemadelcomplemento.Enlosúltimosa ˜nossehanidentificadodiversas mutaciones engenesdelsistema delcomplementoasociadosa SHUa, queexplicarían aproximadamenteel60%deloscasosdeSHUa,ysehancaracterizadofuncionalmente numerosasmutacionesypolimorfismosasociadosaSHUaquehanpermitidodeterminar quelapatologíaseproducecomoconsecuenciadeladeficienteregulacióndelaactivación delcomplementosobrelassuperficiescelularesyquellevaalda ˜noendotelialmediadopor laactivacióndelC5ydelavíaterminaldelcomplemento.Eculizumabesunanticuerpo monoclonalhumanizadoqueinhibelaactivacióndelC5,bloqueandolageneracióndela moléculaproinflamatoriaC5aylaformacióndelcomplejodeataquedemembrana.En estudiosprospectivosenpacientesconSHUasuadministraciónhademostradola inter-rupciónrápidaysostenidadelprocesodeMAT,conunamejorasignificativadelafunción renala largoplazoyunareducción importantedelanecesidad dediálisisyelcesede laterapiaplasmática.Enfuncióndelasevidenciascientíficaspublicadasylaexperiencia clínicaacumulada,elGrupoEspa ˜noldeSHUapublicamosundocumentodeconsensocon recomendacionesparaeltratamientodelaenfermedad(Nefrología2013;33(1):27–45).Enla presenteversiónonlinedeldocumentoseactualizanloscontenidossobrelaclasificación etiológicadelasMAT,lafisiopatologíadelSHUa,sudiagnósticodiferencialysumanejo terapéutico.
©2015SociedadEspa ˜noladeNefrología.PublicadoporElsevierEspaña,S.L.U.Esteesun artículoOpenAccessbajolalicenciaCCBY-NC-ND (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Introduction
Haemolytic uraemic syndrome (HUS) is a clinical entity consisting of the triad of nonimmune microangiopathic haemolytic anaemia, thrombocytopenia, and acute renal failure.1 The histological lesions of HUS typically involve
systemicthromboticmicroangiopathy(TMA),largelyresulting inimpairedintrarenalvessels.AgreaternumberofHUScases arecausedbyaShigatoxin-producing(STEC)Escherichiacoli enteric infection or verotoxin-producing (VTEC) germs, resulting inthe so-calledtypicalHUSor STEC(VTEC)-HUS. Genetic or acquired (autoantibodies) dysregulation of the alternative complement pathway leading to endothelial
nefrologia.2015;35(5):421–447
423
damage and systemic TMA phenomena occur in nearly 10% of HUS reports.2 This kind of HUS related to the
complement dysregulation is known as atypical HUS (aHUS).
In 2011, Eculizumab (Soliris®; Alexion Pharmaceut-icals, Connecticut, USA) was approved by the American and European regulatory agencies for the treatment of aHUS.3 Eculizumab is a humanised monoclonal antibody
inhibitingC5activation and blockingthe productionofthe proinflammatoryC5aanaphylatoxin,aswellastheformation ofthe membrane-attack complex,leading to cell lysis.4 In
prospectivestudiesconductedinaHUSpatients,Eculizumab effectivelyhalted theTMAprocessandits effects,andwas associatedwiththerapid,significant,andlong-term improve-mentofhaematological and renal functionabnormalities,5
and with improved systemic involvement and high blood pressure.
In2012,theSpanishGroupforaHUSgatheredtodevelopa consensusdocumentincluding recommendationsfor treat-ing the disease.6 The group has been meeting every year
eversincetheninorder toupdateboththe understanding of the various aspects of interest related to the dis-ease (including the aetiological classificationof TMAs, the pathophysiology ofaHUS, andcompanion diagnostics) and treatment recommendations based on already published
scientificevidenceandclinicalexperience.Thecontents orig-inally published in Nephrology 2013;33(1):27–45 have been updated in the current online version of the consensus document.
Aetiological
classification
of
thrombotic
microangiopathies
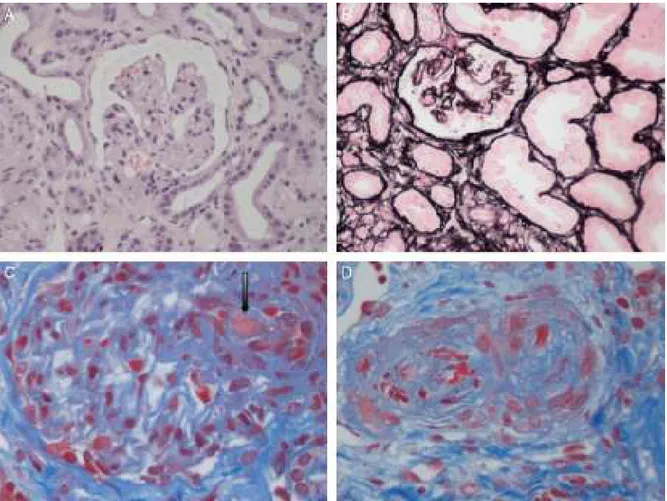
The term TMA describes a histological lesion ofthe arte-rioles and capillaries resulting in thickened and swollen vessel walls, detachment of endothelialcells, widening of thesubendothelialspacecausedbythebuild-upofproteins andcelllysismaterial,andthepresenceofplateletthrombi obstructingvascularlumen(Fig.1).1Twoclinicalentitieswith
differentaetiologyandpathophysiologyarecharacterisedby primaryTMAlesions:thromboticthrombocytopenicpurpura (TTP)andHUS.
IntravascularthrombosisinTTPresultsfromasevere defi-ciency in the metalloprotease activity of the A Desintegrin andMetalloproteinasewithThromboSpondintype1motif, mem-ber13(ADAMTS13),aplasmaenzymeresponsibleforsplitting theultra-largemultimersoftheVonWillebrandfactor.7This
deficiency may be genetic or acquired via IgG circulating
Fig.1–Renalhistopathologicallesionsfromhaemolyticuraemicsyndrome.(A)Ischaemicandretractedglomeruli.(B) Mesangiolysis(C)Thrombiintheglomerularcapillaries(arrow).(D)Arteryoccludedbyplateletthrombi.Photographs courtesyofDr.R.Ortega(HistopathologydepartmentoftheHospitalUniversitarioReinaSofía,Córdoba).
antibodies blocking ADAMTS13 (particularly in patients receivingplateletantiaggregants).8
NinetypercentofHUScasesarecausedbyaSTECenteric infectionresulting fromcontaminated food(typical HUSor STEC [VTEC]-HUS).2 TheShiga toxin causes a directinjury
on the vascular endothelium, triggering a number of cell andvasculareventswhichultimatelyleadtoTMA.2Clinical
presentationusuallyinvolvesabdominalpainanddiarrhoea, togetherwithacuterenalfailurewithin4–10days.Prognosis istypicallyfavourable,withamortalityratebelow5%and80% ofpatientsachievingcompleteclinicalrecovery,although pro-gressiontoseverechronicrenalfailureisobservedovertime inupto20–30%ofpatients.9,10
aHUS is essentially diagnosed by exclusion once ADAMTS13 (TTP) deficiency or STEC infection (STEC-HUS) are ruled out. In patients with aHUS, TMA phenomena are a consequence of the dysregulation of the alternative complement pathway on the cell surface. This abnormal-ity results in uncontrolled activity on own cells following complement activation (by severaltriggering factors), lead-ing to endothelial damage, inflammation, and secondary thrombosis, with an increasingnumber of casesinvolving geneticoracquired factors.Mutationshavebeen described inoneor morecomplement proteinsinnearly60% ofover 1000 patients with reports of aHUS in the literature,11–18
althoughageneticcomponent(involvingcomplementgenes or other, including coagulation genes) and/or unspecified autoimmunitymayalsobepresentintheremainingpatients. Ofnote, anti-FactorH(FH) autoantibodieshavebeen found in 5–10% of aHUS patients.19 Unlike STEC-HUS, which is
usually an isolated event, aHUS is a chronic and relaps-ing entity triggered by the uncontrolled activation of the complement system. Before Eculizumab became available, aHUShadbeenmostlyassociatedwithapoorprognosis:the mortalityratefollowingafirstepisodeofaHUSwas10–15%, and renal function remained unrecovered in up to 50% of patients.11,12,20
AtypeofaHUSresultingfromrecessivemutationsinthe DGKEgenecodingfortheDGK-(diacylglycerolkinase-) pro-teinhasrecentlybeendescribed.21 Thelossofthisenzyme
activityinendothelialcells,platelets,andpodocytesleadsto endothelialcellsapoptosisandimpairedangiogenicresponse, thereby resulting in a prothrombotic and inflammatory state.22PatientswithDGKEmutationsexhibitvarious
pheno-typesrangingfromaHUStomembranoproliferative glomeru-lonephritiswithhighproteinuriaandnephroticsyndrome.23
aHUS patients develop persistent high blood pressure and haematuria-proteinuria(includinginthenephroticrange)in theirfirstyearoflife.UnlikepaediatricaHUSassociatedwith complementgeneticalterations,progressiontochronicrenal diseaseamongthesepatientsisnotsudden,butdevelopsover years.21
In addition to STEC enteric infection (typical HUS), abnormalities in the regulation of complement activation, mutationsinDGKEorcoagulationgenes(aHUS),or(genetic or autoimmune)deficiency ofADAMTS13inTTP, thereare many other factors and clinical entities thatmay be asso-ciated with the development of TMA. This kind of TMA is included under the term secondary TMA. Some cases reported in children are associated with methylmalonic
aciduria24 or more commonly (5% of HUS reports in
chil-dren) with neuraminidase-producing invasive Streptococcus pneumoniaeinfections(resultingintheexposureofthe crypto-antigen T in the cell surface and unleashing the TMA phenomenon),25 or H1N1infection.26 TMAhasbeen
gener-ally associatedwithviral infections(CMV, HIV,parvovirus), neoplasticprocesses,drugs(antitumoragents,includingthe vascular endothelialgrowth factor inhibitors, immunosup-pressants such as calcineurin inhibitors [cyclosporine and tacrolimus]orthemammaliantargetofrapamycininhibitors [mTOR;sirolimus,everolimus],plateletantiaggregants, antivi-rals, or oral contraceptive drugs), malignant high blood pressure,bonemarroworsolidorgantransplantation, preg-nancy and postpartum, autoimmune systemic diseases, or glomerulonephritis.27
Importantly,theforegoingcausesofTMAmaynotalways be identified in all patients, whereas some may present morethan oneaetiology,resultinginaheterogeneous pre-sentation and achallenging diagnosis. Infact, overlapping entitiesare commonandup to25% ofpatientswith STEC-HUS and 86% of patients with pregnancy-associated HUS developcomplementsystemmutations,whereaHUSis actu-allytheunderlyingdisease.28,29Mutationsinthecomplement
systemhavealsobeenreportedinpost-transplantHUS asso-ciated with the use of calcineurin inhibitors and in HUS relatedtoautoimmunediseasesin27%and33%ofpatients, respectively.12Furthermore,severalcasesofsecondaryTMA
have been reportedtodate withsuccessfultreatment out-comeswithEculizumab(TMAassociatedwithdrugs,30solid
organs31orbonemarrow32transplantation,pregnancy33and
systemic erythematouslupus34).Thefactthat complement
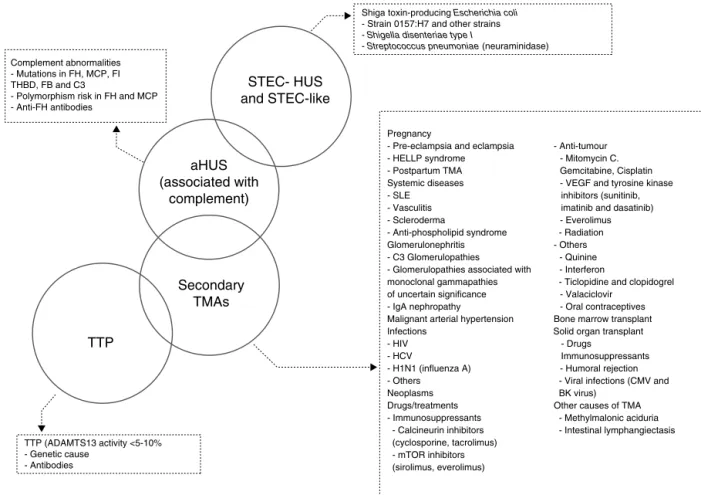
blockade (by Eculizumab) is associated with a favourable clinical responseand the reversibility ofTMAsuggests the potentialandimportantroleofnon-geneticcomplement dys-regulation in many cases of secondary TMA, predisposing patients to its development. On this basis, Fig. 2 sum-marisestheproposedaetiologicalclassificationofTMAsand illustrates the potentialoverlapping betweenthese clinical entities. The classification of TMAs should be understood as a current topic of interest and major debate is tak-ing place among the medical community as a result of the continuousprogressmade intheunderstanding ofthe pathophysiology of these entities.35 Given that the aHUS
mediatedbycomplementdysregulationisthe mainreason fordebate,onlythisentitywillbediscussedinthefollowing sections.
Atypical
haemolytic
urinary
syndrome:
a
clinical
entity
Epidemiology
aHUSisconsideredanultra-raredisease.Dataavailableonits incidenceandprevalencearelimited,aswellasthe knowl-edgeoftheactualepidemiologyofthedisease.Theannual incidence of aHUS in the US has been estimated to beof ∼1–2cases/millioninhabitants.36Arecentmulticentrestudy
inEuropereportedanincidenceof0.11cases/million inhabi-tants.AccordingtotheEuropeanMedicinesAgency(EMA),the
nefrologia.2015;35(5):421–447
425
TTP (ADAMTS13 activity <5-10% - Genetic cause - AntibodiesTTP
Secondary
TMAs
aHUS
(associated with
complement)
Shiga toxin-producing Escherichia coli - Strain 0157:H7 and other strains - Shigella disenteriae type I
- Streptococcus pneumoniae (neuraminidase) Complement abnormalities
- Mutations in FH, MCP, FI THBD, FB and C3
- Polymorphism risk in FH and MCP - Anti-FH antibodies
STEC- HUS
and STEC-like
Pregnancy
- Pre-eclampsia and eclampsia - Anti-tumour - HELLP syndrome - Mitomycin C. - Postpartum TMA Gemcitabine, Cisplatin Systemic diseases - VEGF and tyrosine kinase - SLE inhibitors (sunitinib,
- Vasculitis imatinib and dasatinib) - Scleroderma - Everolimus - Anti-phospholipid syndrome - Radiation Glomerulonephritis - Others - C3 Glomerulopathies - Quinine - Glomerulopathies associated with - Interferon
monoclonal gammapathies - Ticlopidine and clopidogrel of uncertain significance - Valaciclovir
- IgA nephropathy - Oral contraceptives Malignant arterial hypertension Bone marrow transplant Infections Solid organ transplant - HIV - Drugs
- HCV Immunosuppressants - H1N1 (influenza A) - Humoral rejection - Others Neoplasms Drugs/treatments - Immunosuppressants - Calcineurin inhibitors (cyclosporine, tacrolimus) - mTOR inhibitors (sirolimus, everolimus)
- Viral infections (CMV and BK virus)
Other causes of TMA - Methylmalonic aciduria - Intestinal lymphangiectasis
Fig.2–Classificationoftheaetiologiesofthromboticmicroangiopathies.ADAMTS13:ADisintegrinAndMetalloproteinase withaThromboSpondintype1motif,member13;aHUS:atypicalhaemolyticuraemicsyndrome;CMV:cytomegalovirus;FB: complementfactorB;FH:complementfactorH;FI:complementfactorI;HCV:hepatitisCvirus;HELLP:Hemolysis,Elevated Liverenzymes,LowPlateletcount;HIV:humanimmunodeficiencyvirus;HUS:haemolyticuraemicsyndrome;MCP: membranecofactorprotein;mTOR:mammaliantargetofRapamycin;SEL:systemicerythematouslupus;STEC:Shiga toxin-producingEscherichiacoli;THBD:thrombomodulin;TMA:thromboticmicroangiopathy;TTP:thrombocytopenic thromboticpurpura;VEGF:vascularendothelialgrowthfactor.
prevalencemay beof∼3.3patients/millioninhabitants/year amongpatientsbelowtheageof18,withlowerratesamong adults.
ChildrenandadultsarepredominantlyaffectedbyaHUS, althoughitmaydevelopatanytimeinlife.11,12Theonsetofthe
diseaseusuallyoccursbeforetheageof18(60%vs.40%)and sexcharacteristics arewell-balanced (womenare primarily affectedwhenthediseaseisdevelopedinadulthood).11,13
Clinicalpresentation
Clinicalonsetisoftenabrupt,although20%ofpatientsmay developitprogressively(inweeksormonths),accompaniedby subclinicalanaemia,fluctuatingthrombocytopenia,and pre-servedrenal function.11 Theclinical picture comprises the
triadofnonimmunemicroangiopathichaemolyticanaemia, thrombocytopenia, and acute renal failure.1 High levels
of lactate dehydrogenase (LDH), undetectable haptoglobin and schistocytes confirm the presence of intravascular hemolysis20associatedwithhaematuria,proteinuria,and/or
acuterenalfailure(withorwithoutoligoanuria).Highblood pressureresultingfromvolumeoverloadorvascularlesionis common.1Insomepatients,thesinglemanifestationofTMA
maybeproteinuriawithhighbloodpressureandprogressive renalfailurewithouthaematologicalabnormalities.
Even though aHUS lesions are predominantly observed in renal vessels, the diffuse and systemic nature of the TMAphenomenonleadstotheinvolvementofthe microvas-culature of other organs (including, but not limited to the brain,heart,intestine, pancreas, and lungs),1 therefore
accounting for common extrarenal symptoms.11,12
Neuro-logical symptomsare the mostcommon (48%),37 including
irritability, somnolence, confusion,convulsions, encephalo-pathy,stroke,hemiparesis,visualabnormalities,hemiplegia, and coma.1,12,37,38 Myocardialinfarctionhasbeendescribed
inupto3%ofaHUSpatientsinrelationtosuddendeath.12,39
Myocardiopathy, heart failure, and peripheral ischaemic vasculopathy have also been reported,19,37,40,41 as well as
diarrhoea (30%) and other digestive symptoms (including, but not limited to colitis, nausea, vomit, abdominal pain,
hepatitis,cholestasis,andpancreatitis).12,19,38,42Skin
involve-mentincludingulcerlesionsinlowerlimbshasrecentlybeen reportedinaHUSpatients.43Heterogeneityofsymptomshas
posedachallengeforcompaniondiagnosticsofothercauses ofTMA.
Pathophysiology
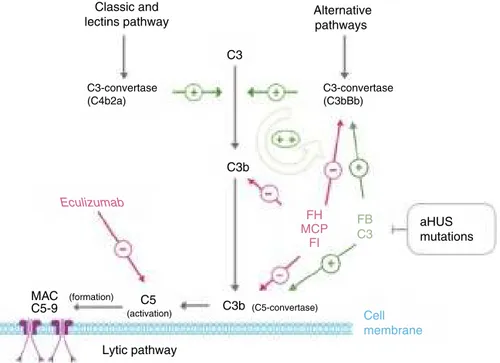
The complement system, consisting of several circulating plasmaandmembrane-associatedproteins,ispartofinnate immunity and is vital for fighting infections, processing immune complexes, antibody response, and the elimina-tionofapoptotic residues.Activationbyanyoftheexisting pathways(classical,lentin,andalternative)leadstothe for-mationofmultiproteincomplexeswithC3-convertaseactivity splitting the C3 protein and resulting in C3b (Fig. 3). The covalentbindingofthismoleculetothesurfacesactivating thecomplementfavoursphagocytosisbypolymorphonuclear leukocytesandmacrophages,thereforeresultinginactivated C5,directingtheattackcomplextothemembraneand caus-ingcelllysis.Inaddition,theresultingC3bleadstoarapidly enhancedcomplementactivationbypromotingtheformation of further C3-convertases, since it is one of the compo-nentsofC3-convertaseofthealternativepathway.44Inorder
toavoidtotaluptakebycomplement activation,aswell as damageto selftissues (C3bbinds indiscriminately to both pathogens and self cells), a number of process-regulating proteins,suchasFH,themembranecofactorprotein(MCP), andcomplement factorI(FI)dissociate C3-convertasesand resultinC3bdegradation.C3blevels,therefore,remainlow undernormalconditionsandtheybuildup following com-plement activation only in the structures related to this activity.
Several studies have shown that around 60% of aHUS patientsarecarriersofmutationsincomplement-regulating genes(CFH,MCP,CFI,thrombomodulin[THBD],orinthe com-ponentsofC3-convertase,factorB[FB],andC3).45–54Allthese
mutationscausethedysregulationofthealternativepathway (Table 1). FH acts in plasma by controlling complement homeostasisandincellsurfacesbypreventingdamagetoself components. Mutationsin theC-terminal regionofFH are characteristicofaHUS.BecausethealteredFHregion medi-atescomplement activationincell surfaces,cell protection againstaccidentaldamageresultingfromcomplement acti-vationisdecreasedbythesemutations,withnoinvolvement ofcomplementregulationinplasma.55Thefunctionalassay
ofaHUS-associated mutationsfound in other complement genes,includingMCP,CFI,CFBorC3,hasalsoconfirmedthat all ofthemresult inadefective protectionofcell surfaces and thislossofcomplement regulationmay beduetothe decreasedactivityofregulatingproteinsortotheabnormally high activity ofC3-convertases. Thus,while theregulatory activityoftheseproteinsisimpairedbymutationsinFH,MCP, andFI,mutationsinFBorC3resultinfurtheractivationofthe C3-convertase.
Around5–10%ofaHUSpatientsdevelopanti-FHantibodies targetedtotheC-terminalregion,withsimilareffectstothose observedinFHmutations.56,57Theirroleinthepathogenesis
of aHUS has not been fully established, but seems to be
Table1–Riskfactorsinatypicaluraemichaemolytic
syndromea Mutations Lossoffunction CFH(∼13%) MCP(∼11%) CFI(∼10%) THBD(∼4%) Gainoffunction C3(∼4%) CFB(∼3%) Polymorphisms Increasingrisk CFH:c.-332C>T;c.2016A>G(p.Gln672Gln);c.2808G>T (p.Glu936Asp)
MCP:c.-652A>G;c.-366A>G;c.989-78G>A;*897T>C
Providingprotection CFH:c.184G>A(p.Val62Ile) Autoantibodies Anti-FH(∼5%) Environmentalfactors Infections Immunosuppressants Oralcontraceptives Anti-cancerdrugs
Anti-FH:anti-complementfactorHantibody;CFB:complement
fac-torBgene;CFH:complementfactorHgene;CFI:complementfactor
Igene;MCP:membranecofactorprotein.Gene;THBD:
thrombomo-dulingene.
a “Multiplehits”theory.aHUSisacomplexdiseasenormally
involv-ingvariousrisk,genetic,andenvironmentalfactors.Patientsare
commonlycarriersofmorethanonemutationincomplement
genesorcombinedmutationswithriskpolymorphisms.
Envi-ronmentalfactorsarealsonecessarytohelprevealthegenetic
disposition from mutations or polymorphisms. Concomitant
mutationswithriskpolymorphisms,autoantibodies,or
trigger-ingenvironmentalfactorsaccountfortheincompletepenetrance
ofaHUS,aswellasforthedifferencesinitspresentationand
progressionamongcarriersofcomplementgenemutations.
associatedwithdiseaseonsetorrecurrence.Giventhat anti-bodytitresmaydecreaseovertime,theyshouldbescreened earlyinthecourseofaHUS.Anti-FHantibodiesareassociated withcomplementfactorH-relatedprotein1deficiency(FHR1) inpatientswithaHUS.58
PenetranceofaHUSincarriersofmutationsinsomegenes isaround 50%,withonlyafewcarriersfrom familieswith identifiedmutationscommonlydevelopingaHUSand show-ing a variable clinical presentation. Clinical heterogeneity, whichresultsfromthe existenceofadditional(genetic and environmental)riskfactorsmediatingthedevelopmentand theoutcomeofthedisease,islargelyobservedamong unre-lated carriers of this mutation. Screening for complement mutationsinaHUSpatientsandconductingcase-control stud-ies basedon geneticpolymorphismsincandidate genesor geneticmarkersinthehumangenomehasallowedforthe identificationofsomevariants(polymorphisms)inCFHand MCPgenesmodulatingthepenetranceandseverityofthe dis-ease(Table1).49,59,60
Haplotypes CFH-H3 and MCPggaac are the most rele-vantpolymorphismsassociatedwiththeriskofaHUS.Both
nefrologia.2015;35(5):421–447
427
Classic and lectins pathway C3-convertase (C4b2a) C3-convertase (C3bBb) Alternative pathways aHUS mutations Cell membrane Eculizumab C3 C3b (formation) (activation) (C5-convertase) MAC C5-9 FH MCP FI C3b FB C3 C5 Lytic pathwayFig.3–Complementdysregulationinatypicalhaemolyticuraemicsyndrome.Complementactivationbyanyofthe3 pathways(detectionofforeignantigens,alternativepathway;ofantibodies,classical;ormannanpolysaccharides,lectin) leadstothebuild-upoflargequantitiesofC3bontheactivatorcellmembrane,causingopsonisationandC5activation (terminalorlyticpathway),resultingintheformationofthemembraneattackcomplexandcelllysis.Complement activationresultsininflammationandleucocyterecruitment.ThekeyprocessincomplementactivationisC3bformation, whichdependsonunstableenzymaticcomplexes–C3-convertases–catalysingtheruptureofC3tocreateC3b.Inturn,C3b hastheabilitytoformfurtherC3-convertaseofthealternativepathway(C3bBb),thusenhancingtheinitialactivation.The mediationofC3Bproductionistwo-fold:dissociationofC3-convertasesandproteolyticinactivationofC3bandC4b.Several regulatoryproteinsinplasmaandthecellmembranecarryoutthisregulatoryactivities,including,factorH,MCPandfactor I,whichplayanessentialroleinthedissociationofC3-convertaseofthealternativepathway(C3bBb)andtheproteolytic degradationofC3b.MutationsintheseproteinsfoundinpatientswithaHUSinterferewiththisregulatoryactivityofthe alternativepathwayactivation.SomepatientswithaHUSarecarriersofmutationsinproteinsC3andfactorBorganising C3-convertase.Thesemutationsareparticular,astheyincreasetheactivityofmutatedproteins(gain-of-function mutations),resultinginincreasedcomplementactivationandexceedingthecapacityofregulatoryproteins.
haplotypesincludesingle-nucleotidepolymorphisms(SNP)in theCFHandMCPgene-promoterregion,downregulatingFH andMCP.Thepresenceofbothpolymorphismsin homozygo-sismayprovidearationaleforaHUSdispositioninpatients withnomutationsinanyofthegenesassociatedwithaHUS. ArecentcollaborativestudybytheEuropeanWorkingParty on Complement Genetics in Renal Diseases including 795 patientswithaHUShasshownthat3%ofthesepatientswere carriersofcombinedmutationsinmorethanonegene. Addi-tionally,this largestudy hasproved that concomitant risk haplotypesCFH-H3 and MCPggaac also leadto significantly increaseddiseasepenetranceincarriersofcombined muta-tions,stressingtheideathatgenotypingoftheserisk polymor-phismshelpspredicttheriskofaHUSinmutationcarriers.61
Alongwiththepreviousgeneticalterations,anumberof triggeringenvironmental factorsare also implicatedinthe onsetofaHUS.Theabovemutationsarepredisposingfactors ofthedisease,preventingadequatecomplementregulationin cellsurfaceswhenthesystembecomesactivatedin microves-sels. aHUS is triggered by infectious events in 50–80% of
patients,11,12,40particularlythoseinvolving theupper
respi-ratorytract(influenzaH1N1virus).Diarrhoeacausedby gas-troenteritismayprecedeaHUSinupto30%ofcases(including diarrhoea by STEC11,12,19).12 Pregnancy, particularly during
post-partum,isacommonpredisposingfactorofaHUSamong women,12,29togetherwiththeuseoforalanovulatoryagents.
Mutationsinthegenecodingforthrombomodulin(THBD), ananticoagulantproteinactingasthrombincofactorandalso regulatingtheFI-mediatedC3binactivation,havebeen associ-atedwithaHUS.62Basedoncomplementdysregulationtypical
ofpatientswithaHUS,thefunctionalanalysisofTHBD muta-tionsassociatedwithaHUShasshownthatthrombomodulin mutationsimpairthecomplementregulatoryactivity.62
Nev-ertheless,theimpairmentoftheanticoagulationactivityby thrombodulinmutationsassociatedwithaHUSand the rel-evance ofthese abnormalities inaHUS remains unknown. Inthisregard,arecentstudyconductedin36patientswith aHUS hasassessedthepresenceofmutationsinthegenes of the complement system and coagulation through mas-siveDNAsequencing,detectingmutationsingenesfromboth
Table2–Clinicaloutcomeofpatientswithatypicalhaemolyticuraemicsyndromebasedoncomplementabnormalities (priortoEculizumab).
Gene RiskofdeathorESRFin
thefirstepisodeor
withinthenextyear
Riskofrelapse RiskofdeathorESRFat
3–5years
Riskofrelapsefollowing
renaltransplant CFH 50–70% 50% 75% 75–90% CFI 50% 10–30% 50–60% 45–80% MCP 0–6% 70–90% 6–38%a <20% C3 60% 50% 75% 40–70% CFB 50% 3/3withoutESRF 75% 100% THBD 50% 30% 54%a 1patient
Anti-FH 30–40% 40–60% 35–60%a Higherwithincreased
antibodytitres
Anti-FH:anti-complementfactorHantibodies;CFB:complementfactorBgene;CFH:complementfactorHgene;CFI:complementfactorIgene;
ESRF:end-stagerenalfailure;MCP:membranecofactorproteingene;THBD:thrombomodulingene.
a DataonESRF.
AdaptedfromLoiratandFremeaux-Bacchi.1
systems.63Thegeneinthecoagulationsystemwiththelargest
number ofmutations was plasminogen (PLG), a cymogene whichplaysanimportantroleinfibrinolysisfollowing con-versiontoplasmin.Eventhoughthesedatasuggestthat coag-ulationgenesmayaddtoaHUSdisposition(particularlyPLG), furtherstudiesarerequiredtoconfirmtheseobservations.
Thesearch ofnewgenesassociatedwithaHUShasalso beenaddressedbyLemaireetal.21throughexome
sequenc-ing.Theauthorshaveidentifiedhomozygoticmutationsinthe DGKEgenecodingfortheDGK-proteinin13patientswith aHUSfrom9families.Thesepatientshadaveryearlyonset ofaHUS,generallywithintheirfirstyearoflife,followedby multiplerecurrencesandcommonprogressiontoend-stage renalfailureintheseconddecadeoflife.21Deficiencyof
DGK-inendothelialcellshasbeenrecentlyshowntoinducethe expressionofICAM-1andtissuefactorbymeansofincreased p36-MAPK-mediatedsignalling,leadingtoapoptosis,altering theangiogenicresponse,anddeterminingaproinflammatory andprothromboticphenotype.Yet,theabsenceofDGK-is notdetrimentaltocomplementactivationincellsurfaces.21,22
The absence of DGK- in podocytes and endothelial cells mayprobablyimpairthediaphragmofglomerularfiltration, whichwould accountformassiveproteinuriaand the sus-ceptibilitytoglomerularconditionsamongthesepatients,21,23
althoughthereasonwhythesepatientstendtodevelop sev-eralglomerularconditionsremainsuncertain.Finally,despite theroleofthecomplementinthedevelopmentofrenal dis-ease amongcarriersofDGKE, mutationshad beeninitially ruledout,21patientswithDGKEmutationsadditionally
asso-ciatedwithothergenespreviouslyrelatedtoaHUS,including THBDandC3,64havebeenrecentlyidentified,thussuggesting
thatcomplementdysregulationmayplayaroleinthe modu-lationofdiseaseonsetandoutcomeatleastinsomecarriers ofDGKEmutations.
Prognosis
TheavailabilityofEculizumabhassignificantlyrevolutionised the prognosis of patients living with aHUS, a very severe
disease in mostcasesin spiteofintensive treatmentwith plasmatherapy(PT;Table2).FollowingafirstepisodeofaHUS, overall mortalitywas higherthan 10% and morethan half of the patients required dialysis and/or developed a more permanent renaldamageinthenext12months.11,12,20The
clinicaloutcomechangesrelatedlydependingonthepatient’s mutation.Inthisrespect,outcomeseemedtobeparticularly poorinpatients withFHandC3mutations,withmortality and end-stage chronicrenal failure (ESCRF)rates over 50% withinoneyearfromthefirstepisodeofaHUS.Furthermore, halfofthesepatientsrelapsed.MutationsinFI,FB,andTHBD werealsoassociatedwithhighratesofmortalityandESCRF atoneyear (50%),withrelapses occurringinnearlyonein 3 patients overcoming the first episode of aHUS. On the other side, less than 10% of patients withMCP mutations died or progressed to ESCRF, although the risk of relapse among these patients was higher and up to 90% of them developed new episodes of aHUS. Between 50 and 75% of patients withmutationsin FH,CI,C3, FB orTHBD died or developedESCRFwithin3–5 yearsfrom thefirstepisodeof aHUS.1
Recurrenceofatypicalhaemolyticuraemicsyndrome followingrenaltransplantation
The outcome ofrenal transplantation (RT) amongpatients withESCRFduetoaHUShasbeenhistoricallylimitedbythe increasedpercentageofrecurrencesofpost-transplant dis-ease(∼50%;graftlossrate:80–90%65,66),althoughitchanges
significantlybasedonthetypeofalteration.Inaseriesof57 patientswithaHUSreceivingRT,5-yearrecurrence-freegraft survivalwas significantlylower inpatientswithmutations in the genes coding for complement proteins compared to patients in whom only polymorphisms but no genetic abnormalities were found.67 Yet, itshould bestressed that
theriskofrecurrenceofaHUSfollowingRTinpatientswith nogeneticabnormalitiesisalsodeemedhigh.68Mutations
in FH are associated with a higher risk of recurrence or graft lossfollowingRT(75–90%;specificallythoserelatedto
n e f r o l o g i a. 2 0 1 5; 3 5(5) :421–447
429
Table3–Diagnostictestsandproceduresrecommendedforpatientswiththromboticmicroangiopathy.
Generaldiagnostictests
•Completemedicalrecords,includingdrugs,datafromsystemicdiseases,personalandfamilyhistory
•Completephysicalexamination,includingafundoscopicexam
•Generalroutinebloodandurinetests
•Haptoglobinlevels
•Serumcomplementlevels
•Peripheralbloodsmear
•Serologyforsystemicdiseases(ANA,anti-ADN,ANCA,antic-Scl-70,anticentromere)
•Anti-cardiolipinantibodiesandlupusanticoagulant
•SerologyforHIV,HCV,HBV,CMVandH1N1
•Completeclottingtest,withfibrinogen,fibrinogendegradationproductsanddimerD
•InvestigationsfortypicalHUS-causingbacterialinfectionsandShigatoxintest(ifclinicallysuspected)
Specificdiagnostictests
•STECinfection •Faecalsampleincaseofdiarrhoeaorrectalculture:STECcultures(MacConkeyforE.coliO157:H7);PCRforStxgenesO157:H7andother
serotypes,andotherviruscharacteristics;ELISAand/orVerocelltissuecultureassayforStxserum:anti-LPSantibodiesforprevalentserotypes
•Pneumococcalinfection •Bacterialculture(generally)ofsterilebodyfluids;DAT(Coombstest),viraltest(respiratory),chestx-ray(pleuraleffusionasacharacteristicin
mostcases),cytochemistry,andCSFcultureincasestopneumococcalmeningitis
•Alteredregulationofthecomplement •C3,C4(plasma/serum),AH50
•FH,FI,FB(plasma/serum)
•Anti-FHautoantibodies
•ExpressionofsuperficialMCPinleukocytes(poly-ormononuclearleukocytesusingaFACStest)
•MutationanalysisinFH,FI,MCP,C3,FB±THBD
•ADAMTS13deficiency(acquiredorhereditary) •PlasmaactivityofADAMTS13ordose(ELISA)±inhibitor
•Cobalaminmetabolism:methylmalonicaciduria •Aminoacidchromatographyinplasma/urine(hyperhomocysteinemia,hypomethioninemia;homocystinuria);organicacidchromatographyin
urine(methylmalonicaciduria)
•MutationanalysisforthegeneMMACHC
ADAMTS13:ADisintegrinAndMetalloproteinasewithaThromboSpondintype1motif,member13;ANA:antinuclearantibody;ANCA:Autoantibodiestoneutrophilcytoplasmicantigens;CMV:
cytomegalovirus;CSF:cerebrospinalfluid;DAT:directantiglobulintest;DNA:deoxyribonucleicacid;ELISA:enzyme-linkedimmunoabsorptionassay;FACS:fluorescenceactivatedcellsorting;FB:
complementfactorB;FH:complementfactorH;FI:complementfactorI;HIV:humanimmunodeficiencyvirus;HUS:haemolyticuraemicsyndrome;MCP:membranecofactorprotein;STEC:Shiga
toxin-producingEscherichiacoli;THBD:thrombomodulin;VHB:hepatitisBvirus;VHC:hepatitisCvirus.
abnormalities interminal 3′ and gene conversion between CFHandCFHR1,resultinginthehybridgeneCFH/CFHR1[both abnormalities impair the functionality of the C-terminal domain of FH]), posing a high risk with mutations in C3 andFIaswell(40–80%;Table2).12,42,48,65,67,69–71Todate,very
few transplants have been performed in patients with FB mutations,though recurrence ofaHUS and graft losswere reported inall cases.49,72 In general,plasma factors ofthe
complement involvedinaHUSare synthesisedinthe liver, and so patients with mutations in the complement genes codingforthesefactorsremainpronetoaHUSfollowingRT, asdysfunctional factors are stillbeing produced.MCP is a transmembraneproteinthatishighlyexpressedinthekidney and,asaresult,thisdefectcanbecorrectedbyRTby deliver-ingunchangedMCPintothegraft.Over80%ofpatientswith MCPmutationsdevelopnorecurrenceofaHUSfollowingRT, withalong-termsurvivalratecomparabletothatofpatients receivingtransplantsforotherreasons.40,65,66Theriskof
post-transplantrecurrenceinpatientswithTHBD62mutationsor
anti-FHantibodiesisnotwell-established,althoughitseems toberelatedtohighandpersistenttitresofantibodiesinthe latter.19,73
Diagnosis
of
atypical
haemolytic
uraemic
syndrome
In light ofthe rapid evolution and the severity ofTMA, a differential diagnosis should be immediately established fromthesyndromeperspective,allowingforsupportive mea-sures tobetaken within24–48hfrom patient’s admission. ConsiderationsforanaetiologicaldiagnosisofTMAwill sub-sequentlybemade.Table3summarisesthemajorprocedures anddiagnostictestsrecommendedforthediagnosisofTMA, including specific tests for companion diagnostics of the variousaetiologiesofTMA.
In patients with TMA, tests results include thrombo-cytopenia (platelet count <150,000/mm3 or decrease >25% frombaseline)20and microangiopathichaemolyticanaemia
(haemoglobin<10mg/dlwithanegativedirectCoombs test [thoughsomepatientswithpneumococcalorH1N1-related HUSmayshowpositivedirectCoombstest],25elevatedLDH,
decreasedhaptoglobin,reticulocytosis,andschistocytes).20,62
Inaretrospectiveseriesof50patientswithhistologicalTMA, 44% had a normal platelet count.74 Consequently,
diagno-sis of TMA should be considered in patients with renal failure and elevated LDH, but without thrombocytopenia. For schistocytes, even though they can be found in most patientswithrenaldisease,preeclampsiaormechanicvalves, TMA can be diagnosed with a schistocytecount >1% pro-vided thatthereare no other known causes.75 In contrast,
the absence of schistocytes does not rule out a diagnosis ofTMA.
Highlevelsofserumcreatinine,lowglomerularfiltration (GF)orthepresenceofproteinuriaand/orhaematuria11,20,69
areindicativeofrenalfailure.Arenalbiopsymayberequired foradultpatientsfollowingacuterenalfailuretodetermine the aetiology,rule out other processes, and assess progno-sis,although the indicationand timefor biopsiesmust be examinedindividuallyinpatientswithsuspectedTMAdue
Table4–Differentialdiagnosisbetweendisseminated
intravascularcoagulationandthrombotic
microangiopathy.
DIC TMA
Plateletcount ↓ ↓
Fibrinogen ↓ Normal
Fibrinogendegradationproducts ↑ Normal
DimerD ↑ Normal
Antithrombin ↓ Normal
Schistocytes Present Present
Haptoblogin Normal ↓
Coagulationtimes Long Normal
Bloodpressure ↓ ↑
DIC: disseminated intravascular coagulation; TMA: thrombotic
microangiopathy.
totheriskofbleeding.Inthissense,diagnostic renal biop-siesarenotrecommendedinpatientsconclusivelydiagnosed withaHUS(positivefamilyhistory,recurrence,etc.).Among paediatricpatients,thediagnosisisessentiallymadeonthe basisofclinicalpresentation,thoughrenalbiopsymay occa-sionallyberequired(especiallyincasesofsecondaryTMAor RT). Patients withclinical suspicion ofTMAshould always beexamined byanephrologistinlightofthe urgent treat-mentstrategyrequiredtoensureminimumirreversiblerenal damage.
Disseminated intravascular coagulation (DIC) is a syn-dromethatmaybeassociatedwithseveralmajorlaboratory and clinical findings related to TMA. DIC is characterised by a systemic activation ofcoagulation, secondary to sev-eral clinicalconditions(sepsis,traumaorcertain tumours), leading to thrombosis and bleeding, commonly involving renalfunction.76Keytestcriteriabasedoncoagulationtests
for differential diagnosis of DIC and TMA are listed in Table4.
Acompleteanddetailedclinicalhistoryshouldbemade forTMApatients,includingpersonalandfamilyhistory, trigg-ering factors (drugs, infections), and a thorough physical examination. As opposed to previous considerations made severalyearsago,signsandsymptomsofthedifferenttypes ofTMAarecurrentlythoughttobenonspecificandavoidthe companiondiagnosticsbetweenthesetwoentities.1The
dif-ferentiationbetweenHUSandTTPwasclassicallybasedon clinical criteria, with HUS and TTP being diagnosed when renal involvementand neurological involvementwere pre-dominant, respectively. However,50% ofpatients withTTP developrenalfailureand50%ofpatientswithaHUSdevelop neurologicalabnormalities.37,77
Clinicalfeaturesdonotallowforadifferentiationbetween STEC-HUS andaHUSaswell,giventhatupto30%ofaHUS cases are developed following gastroenteritis12 or patients
develop diarrhoea42 (a typicalsymptom ofSTEC-HUS). On
the other hand, platelet count and the severity of renal involvementcanactuallyguidecompaniondiagnostics. Over-all,TTPpresentswithseverethrombocytopenia(<20,000/mm3 in 73% of patients with acquired TTP)78 and moderate
nefrologia.2015;35(5):421–447
431
Thrombocytopenia
<150.000 or>25% decrease±
Microangiopathic haemolysis
Elevated LDH Haptoglobin decrease Schistocytes Haemoglobin decrease*Plus one or more than one of the following
Evaluate ADAMTS13 and Shiga toxin /STEC** test
TTP
<5-10% ADAMTS13 activity
>5-10% ADAMTS13 activity
Shiga toxin /STEC positive
aHUS
Secondary TMA
STEC-HUS***
Neurological symptoms
Confusion Seizures
Renal involvement
Elevated creatinine
Decrease in estimated glomerular filtration rate Urinary changes
Gastrointestinal involvement
Diarrhoea Nausea/vomiting Abdominal pain GastroenteritisFig.4–Algorithmforthedifferentialdiagnosisofprimarythromboticmicroangiopathy.ADAMTS13:ADisintegrinAnd MetalloproteinasewithaThromboSpondintype1motif,member13;aHUS:atypicalhaemolyticuraemicsyndrome;HUS: haemolyticuraemicsyndrome;LDH:lactatedehydrogenase;STEC:ShigatoxinproducingEscherichiacoli;TTP:thrombotic thrombocytopenicpurpura.*NegativedirectCoombstest.**TheShigatoxintest/STECisindicatedwhenthepatienthasa historyofdigestiveinvolvementorgastrointestinalsymptoms.***STECinfectioncanrarelytriggertheunderlyingdisease activityinsomepatientswithaHUS.
moderate thrombocytopenia (50–100,000/mm3) and severe renalinvolvement.Thisrulecanbedeemedaguide,but deter-minationofADAMTS13activityandtheShigatoxintestare essentialforanaccuratedifferentialdiagnosisbetweenTTP, STEC-HUS,andaHUS(Fig.4).ThediagnosisofSTEC-HUScan beconfirmedbythepresenceoftheShigatoxinorapositive cultureofSTECinpatientswithTMA,28whiletheactivityof
ADAMTS13inplasmashouldbe<5–10%inordertoconfirm thediagnosisofTTP.79,80Thediagnosisoftheremainingcases
shouldbedirectedtowardsaHUS,79andsoadditionaltestsare
requiredtoruleoutsecondaryTMAs.Testsamplesshouldbe collectedpriortoPT.
Treatment
options
for
atypical
haemolytic
uraemic
syndrome
Treatment for aHUS should involve two different strate-gies:on oneside,supportivetreatmentmeasuresaimedat managing the consequences of aHUS (acute renal failure,
highbloodpressure,anaemia,thrombocytopenia,etc.),anda targetedtherapytohaltandrevertTMA.Specificoptionsfor themanagementofaHUSwillbereviewedinthissection. Plasmatherapy
PT can be delivered as plasma infusion (PI) and plasma exchange (PE). In PI, patients are given virus-inactivated, non-nativefreshfrozenplasma(FFP),addingfunctional com-plement regulators.81 In PE, a patient’s plasma is replaced
with FFP, which not only results in the administration of high doses ofcomplement-regulating proteins, but also in theeliminationofdysfunctionalendogenoussoluble comple-mentinhibitors,minimisingtheriskofvolumeoverload.In addition,anti-FHantibodiesarealsoclearedinPE,together withpotential inflammatory/thrombogenicfactors involved in endothelial damage and platelet hyperaggregation. The treatmentofchoicerecommendedforepisodesofaHUS tra-ditionallyconsistedofearlyandintensivePEathighvolumes and ofvariable frequencybasedondisease activity.PIsare
Table5–Prognosisofpatientswithatypicalhaemolytic
uraemicsyndrometreatedwithplasmainfusionor
plasmaexchange. Remission Deathor end-stagerenal failure CFH 63%(complete:5%; partial:58%) 37% CFI 25%(complete: 12,5%;partial:12,5%) 75% C3 57%(complete:43%; partial:14%) 43% THBD 88%(complete:62%; partial:25%) 13%
Anti-FHantibodies 75%(complete:25%;
partial:50%) NA MCP 97%oftreated patients(complete: 90%;partial:7%)y 100%ofnon-treated patients NA
Anti-FH:anti-complementfactorHantibodies;CFH:complement
factorH gene; CFI:complement factorI gene; MCP:membrane
cofactor protein gene; NA: not available; complete remission:
haematologicalandrenal functionnormalisation;partial
remis-sion: haematological normalisation and renal sequels; THBD:
thrombomodulingene.
AdaptedfromNorisetal.12
usuallyineffective exceptin afew patients with complete deficiencyofFH82(circulatinglevelsofcomplementproteins
are normalinmostpatients). Overall,PTisnotconsidered effectiveinpatientswithisolatedMCPmutations,asthisis anon-circulatingproteinattachedtothecellmembrane,with virtuallyallpatientsrelapsingfollowinganepisodeofaHUS irrespectiveoftheuseofPT.12
Even though no prospective clinical trials are available, PT has empirically been the treatment of choice in aHUS foryears asmortalityin patientswith TTP-HUSdecreased over the last 3 decades. Table 5 summarises the results ofthe largest international registry ofPT in patients with aHUS(InternationalRegistryofRecurrentandFamilialHUS/TTP), including273patients diagnosedbetween1996and 2007.12
CompletehaematologicalandrenalrecoveryrateswithPTin thisregistryaregenerallybelow50%(exceptforpatientswith mutationsinTHBDandMCP),and particularlylowratesin patientswithmutationsinFHandFI(5y12.5%).12Mortality
and/oroutcomeofESRFaregenerallyhighin3outof4patients withFImutations.SomepapersprovethatearlyintensivePEis essentialtopreventpatientsfromdevelopingaHUS,and main-tenancecanpreventdiseaserecurrenceandESRF,11,81though
themosteffectivemanagementstrategyisstillnotknown, noristhelong-termimpactonrenalfunction.
Concomitant immunosuppressionand PT may improve outcomes in patients with anti-FH antibodies.19,83,84 High
antibodytitresarecorrelatedwithahigherriskofrelapseand renalsequelsinthesecases.19Eventhoughfurthertrialsare
requiredtoconclude on howanti-FH antibodies are devel-opedbypatientswithaHUS,thefactthatthevastmajority
of these patients have complete deficiency of FHR1 leads totheideathattheseantibodiesare reallytargetedagainst the FHR1 protein, and that the anti-FH activity is a cross reaction resulting from the remarkable homology existing betweenthese2proteins.Thispossibilityisawarningofthe potentialanti-FHR1/anti-FHsensitisationinhomozygotesfor CFHR3-CFHR1 deletionbythe exposuretoexogenousFHR1, discouragingtheuseofPIintheseindividuals.
ThepotentialcomplicationsofPIare anaphylactic reac-tionstoFFP,hypervolemia,highbloodpressure,heartfailure, or hyperproteinaemia. The main complications of PE are obstructedvenousaccess(6%),lowbloodpressure(5%),and allergy (4%)85, with a higher frequency among paediatric
patients.85Astudyconductedin71paediatricpatientswith
aHUS(59treatedwithPE)showedthat80%ofchildrenhad some renal sequels within one month of follow-up, 17% weredialysis-dependant,and31%developedcatheter-related complications.86
Eculizumab
Eculizumab isahumanised monoclonalIgG2/4kappa anti-body that binds to the C5 complement protein with high affinity, blocking the excision into C5a and C5b, and pre-ventingtheformationoftheC5b-9complexoftheterminal complement (membraneattackcomplex)(Fig.3).4 InaHUS,
the dysregulation of the alternative complement pathway leadstotheuncontrolledactivationofC5,causingadamage toselfstructuresviatheformationofthemembraneattack complex.Thisprocessisrapidlyandsustainablyreducedas aresultoftheblockadeoftheterminalcomplementpathway byEculizumab.AlargenumberofpatientswithaHUShave shownagoodclinicalresponsetothedrug(Table6).
TheefficacyandsafetyofEculizumabinaHUSwere ini-tiallyassessedintwophaseII,prospective,multicentretrials, including 37 patients older than 12 years of age and with primary or recurrent disease following RT, who received Eculizumab for 26 weeks,followed by long-termextension periods.5 Seventeen patients with aHUS (mean time from
diagnosis:9.7months)withevidenceofprogressiveTMA fol-lowing ≥4 sessions of PT the week before their inclusion (C08-002)wereenrolledinthefirststudy.Thesecond study recruited20patients(meantimefromdiagnosis:48.3months) receivingPT(1sessionevery2weeksand3sessionsaweek) wherenodecrease >25%wasreportedinplatelet countfor at least 8 weeks prior to first dosing of Eculizumab (C08-003). Geneticoranti-FH antibodymutationswere observed in 76% and 70% of patients from the first and the sec-ond study, respectively. Primary outcomes in both studies were:(a)inhibitionofcomplement-mediatedTMA(study1: increasedplateletcount;study2:TMA-freepatient≥12weeks [nodecreaseinplateletcount>25%,noPTandnodialysis]), and (b) haematologicalnormalisation (≥2 normal consecu-tivemeasurements ofplatelets and LDH,with aminimum interval of 4weeks).Secondary outcomes included change intherateofdailyinterventionsforTMA(PIorPEsessions, dialysis,orboth,perpatient/day),renalfunction,qualityof life,safetyandtolerability.Primaryoutcomesreportedat26 weeksandinextensionstudiesaredescribedinTable7.With regardtoprimaryoutcome,following26weeksoftreatment,
nefrologia.2015;35(5):421–447
433
Table6–PublishedcasesofpatientswithatypicalhaemolyticuraemicsyndromereceivingEculizumab(lastupdatedin April2014).
PatientswithaHUSinnativekidneys
Reference Mutation Responsetoplasma
therapy
Baselineserum
creatinine,mol/l
Patientoutcome Lastserum
creatininevalue,
mol/l
121,122 Unidentified Resistanttoplasma
exchange 265 Remissionafter3 years 35 123 CFH Partiallysensitiveto plasmaexchange 80 Remissionafter10 weeks 26
124 a Unidentified Resistanttoplasma
exchange
690 Recurrenceafter2
weeks
End-stagerenal
failure
125 a Unidentified Resistanttoplasma
exchange
∼310 Recurrenceafter2
weeks
End-stagerenal
failure
126 CFHS1191LV1197A Resistanttoplasma
infusion 108 Remissionafter15 months 44 127 CFI p.A258T Resistanttoplasma exchange 610 Remissionafter7 months 230
38,128 b Unidentified Noplasmatherapy 600 Remissionafter6
months 125 129 CFH C611Y Intoleranceto plasmaexchange ∼230 Remissionafter24 months ∼100
130 Unidentified Resistanttoplasma
exchange
∼325(dialysis) Remissionafter9
months
∼80
131 CFH Resistanttoplasma
exchange
∼310(dialysis) Remissionafter18
months ∼75 132 MCP c.286+1G>C Resistanttoplasma exchange Dialysis Haematological normalisation End-stagerenal failure
133 Unidentified Resistanttoplasma
infusion
Continuous hemodiafiltration
Remissionafterone
year 18 134 CFH 3355G>a; Asp1119Asn;SCR19 Resistanttoplasma exchange
Dialysis Remission>2.5years 26
43a Unidentified Sensitivetoplasma
exchange
Dialysis Resolutionof
thrombocytopenia
andskinlesions
Dialysis
135 CFH Partiallysensitiveto
plasmaexchange
723(dialysis) Remission Dialysis-free
136c Unidentified − ∼247(dialysis) Remissionafter6
months
Dialysis-free
137 C3 Resistanttoplasma
therapy
Dialysis Remissionafter2
years Dialysis 138 CFH 3514G>T Resistanttoplasma therapy
∼222(dialysis) Remissionafterone
year 117 139 C3 c3466G>A Resistanttoplasma exchange Dialysis Remission>11 months 115
140a C3 – Dialysis Remission Dialysis-free
141c CFB c.967A>C; p.Lys323Gln – 20 Favourableoutcome at6months,inspite ofthepersistenceof slightlyincreased plasmalevelsofLDH yC5b-9 Normallevels 142 CFH p.Arg53Cys;c.157C.T Resistanttoplasma exchange
Normallevels Remission Normallevels
143c CFH p.Lys1186Thr CFI p.Ile322Thr Partiallysensitiveto plasmaexchange Dialysis Remission 75 144 CFI c.786delA Resistanttoplasma exchange Dialysis Remission 88
Table6–(Continued)
Renaltransplantpatients
PreventiveuseofEculizumab
Reference Mutation Previoustransplants
(number)
Responsetoplasma
therapy
Baselineserum
creatinine,mol/l
Patientoutcome Lastserum
creatininelevel,
mol/l
107 CFHW1183C No Sensitivetoplasma
exchange
∼45 Norecurrence 44
108 CFHE1198stop No Noplasmatherapy Dialysis Norecurrence Normal
109 CFH/CFHR1 hybridgene No Sensitivetoplasma exchange Dialysis Norecurrence 80 110 CFH/CFHR1 hybridgene No Sensitivetoplasma exchange
Dialysis Norecurrence Normal
111 CFH/CFHR1
hybridgene
No Noplasmatherapy Dialysis Norecurrence 79
112 CFHc.3497C9T No Resistanttoplasma
exchange
Dialysis Norecurrence 76
UseofEculizumabforthetreatmentofpost-transplantaHUSrecurrence
145a CFHY475S Yes(1) Resistanttoplasma
exchange
132 Graftloss NS
146,147 C3R570Q Yes(1) Sensitivetoplasma
exchange 320 2recurrencesin cases ofdelayed Eculizumab 230
97 Unspecified No Resistanttoplasma
exchange
323 Remission 238
98 CFHS1191L Yes(2) Intoleranceto
plasmaexchange
131 Remission 130
148a Unidentified No Resistanttoplasma
exchange
415 Graftloss NS
42 CFH Yes(1) Resistanttoplasma
exchange
500 Remission 62
69 C3R570W Yes(2) Partiallysensitiveto
plasmaexchange 220 Remission 115 99 CFH E3514Stop No Partiallysensitiveto plasmaexchange 565(dialysis) Remission 229
149 Unidentified Yes(1) Resistanttoplasma
exchange 449(dialysis) Recurrence5 monthsfollowing withdrawalof Eculizumab.Graft loss NS 43 CFH No Partiallysensitiveto plasmainfusiond 220 Remission (disappearanceof skinlesions) 209
aHUS:atypicalhaemolyticuraemicsyndrome;CFB:complementfactorBgene;CFH/CFHR1:complementhybridgeneresultingfromCFH/CFHR1
conversion;CFH:complementfactorHgene;CFHR1:complementfactorH-relatedprotein1gene;CFI:complementfactorIgene;HUS:haemolytic
uraemicsyndrome;LDH:lactatedehydrogenase;TMA:thromboticmicroangiopathy;NE:notspecified.
a ReceivingonlyonedoseofEculizumab.
b ReduceddoseofEculizumab.
c EarlyuseofEculizumab(≤7daysfollowingdiagnosisofaHUS).
d SuspectedpersistenceofTMAactivityduetothepresenceofulcerativeskinlesionsinlowerlimbs.
instudy 1treatmentwithEculizumabwas associatedwith asignificantincreaseinthenumber ofplateletsfrom base-line(p<0.001)andarateofhaematologicalnormalisationof 76%.Instudy2,80%ofpatientswerefreeofTMAepisodes following26weeksoftreatment withEculizumaband 90% hadhaematologicalnormalisation.Intermsofsecondary out-comes,treatmentwithEculizumabat26weekswasassociated withasignificantreductionintherateofdailyinterventions
for TMA vs. baseline (p<0.001), as well with a continuous improvementofestimatedGFR(+32ml/min/1.73m2[p=0.001 vs. baseline] and +6ml/min/1.73m2 [p<0.001 vs. baseline] in studies 1 and 2, respectively), adecrease inproteinuria (p<0.05) and a reduced need fordialysis. Also, the earlier Eculizumab is introduced in trials (less time of evolution between the clinical manifestationofaHUS and the drug), themoresignificanttheimprovementoftheestimatedGFR
n e f r o l o g i a. 2 0 1 5; 3 5(5) :421–447
435
Table7–MainresultsfromprospectivestudieswithEculizumabinpatientswithaHUS.
C08-002(n=17) C08-003(n=20) C10-003(n=22) C10-004(N=41)
Week26 Week64 Week100 Week26 Week62 Week156 Week26 Week26
Changeinplateletcountfrombaseline(×109/l),mean
pvaluevs.baseline +73a <0.001 +91 0.001 +97 <0.0001 +5 NS NA NA +164 <0.0001 +135 <0.0001
Plateletcountnormalisation,bnumberofpatients(%) 14(82) 15(88) NA NA NA NA 21(95) 40(98)
NoTMAevents,cnumberofpatients(%) 15(88) 15(88) 15(88) 16(80)a 17(85) 19(95) 21(95) 37(90)
Haematologicalnormalisation(complete
haematologicalresponse)d,numberofpatients(%)
13(76) 15(88) 15(88) 18(90) 18(90) 18(90) 18(82) 36(88)
DailyinterventionrateforTMAe(numberof
events/patients/day)
BeforeEculizumab,mean
OnEculizumab,mean
pvaluevs.“beforeEculizumab”
0.88 0 <0.001 0.88 0 <0.001 NA 0.23 0 <0.001 0.23 0 <0.001 NA NA NA
CompleteresponseforTMA,fnumberofpatients(%) 11(65) 13(76) NA 5(25) 7(35) NA 14(64)a 30(73)a
OutcomeofestimatedGFR(ml/min/1.73m2)
pvaluevs.baseline +32 0.001 +32 <0.001 +38 ≤0.05 +6 <0.001 +9 0.003 +4 NS +64 <0.0001 +29 <0.0001
Reduction≥25%inserumcreatinine,numberof
patients(%)
11(65) 13(76) 13(76) 3(15) 7(35) 11(55) 16(73) NA
IncreasedestimatedGFR≥15ml/min/1.73m2,number
ofpatients(%)
8(47) 9(53) 10(59) 1(5) 3(15) 8(40) 19(86) 22(54)
Nodialysis,numberofpatients/numberofpatientsin
dialysisatthestartoftreatment(%)
4/5(80) 4/5(80) NA 0/2(0) 0/2(0) NA 9/11(82) 20/24(83)
ImprovementinCRDinatleastonestage,numberof
patients(%)
10(59) 11(65) 13(76) 7(35) 9(45) 12(60) 17(77) 26(63)
Reductionofproteinuriainatleastonegradein
patientswithbaselineproteinuriagrade≥1,number
ofpatients/totalnumberofpatients
12/15 9/11 NA 6/11 7/9 NA NA NA
Improvementinqualityoflife(changeinquestionnaire
scoring),meang pvaluevs.baseline +0.32 <0.001 +0.30 <0.001 +0.29 <0.0001 +0.10 <0.001 +0.13 <0.001 +0.16 ≤0.001 +19.7 <0.0001 +0.23 0.003
aHUS:haemolyticuraemicsyndrome;CRD:chronicrenaldisease;EQ-5D:EuroQoLGroup5-DimensionSelf-ReportQuestionnaire;FACIT-F:Functionalassessmentofchronicillnesstherapy-fatigue;
GFR:glomerularfiltrationrate;LDH:lactatedehydrogenase;NA:notavailable;NS:notsignificant;PE:plasmaexchange;PI:plasmainfusion;TMA:thromboticmicroangiopathy.
a Primaryoutcome.
b Platelets≥150×109/l.
c Noplateletdecrease>25%;noplasmatherapyandnostartofdialysisfor≥12weeks.
d ≥2normalconsecutivemeasurementsofplateletsandLDH,withatleast4weeksapart.
e PlasmaPIorPEsessions,dialysis,orboth,perpatient/day.
f Completehaematologicalresponseanddecrease≥25%inserumcreatininefrombaseline(twoconsecutivemeasurementswithatleast4weeksapart).
g TheEQ-5Dquestionnairewasusedinallstudies,exceptforC10-003study,wheretheFACIT-Fquestionnaireforpaediatricpatientswasused.