ESTROGENI: GENERALITÀ E STRUTTURA CHIMICA
Estrogeni, androgeni e progestinici sono le tre classi di ormoni steroidei alle quali appartengono i più importanti ormoni sessuali che le gonadi maschili (testicoli) e femminili (ovaie) producono. Gli ormoni gonadici hanno varie funzioni, tra cui le principali sono:4
• Stimolazione dello sviluppo di ovociti e spermatozoi dalle rispettive cellule germinali, attraverso la loro azione locale (funzione paracrina e autocrina);
• Stimolazione dello sviluppo e delle funzioni degli organi sessuali secondari e regolazione della secrezione degli ormoni ipotalamo-ipofisari, agendo invece su cellule bersaglio lontane (funzione endocrina).
Nell’uomo, la modesta produzione di estrogeni avviene nelle cellule interstiziali del Leydig dei testicoli. Nelle donne, gli estrogeni sono prodotti nel follicolo ovarico dalle cellule della granulosa e della teca nella prima fase del ciclo mestruale, mentre, dopo l’ovulazione, vengono sintetizzati dalle cellule luteinizzate della granulosa e del corpo luteo, assieme al progesterone. Produzioni minime di estrogeni si hanno anche da parte dei muscoli scheletrici, del tessuto adiposo e, durante la gravidanza, da parte della placenta e del surrene fetale.1
Gli estrogeni sono coinvolti nel controllo del rilascio delle gonadotropine nell’asse ipotalamo-ipofisario.1, 4 Il fattore ipotalamico di liberazione delle gonadotropine (GnRH) è un decapeptide che stimola l’adenoipofisi (o ipofisi anteriore) a rilasciare l’ormone follicolo stimolante (FSH) e l’ormone luteinizzante (LH), che nella donna agiscono sull’ovaio mentre nell’uomo sul testicolo. La funzione di entrambi gli ormoni è quella, nella donna, di promuovere la gametogenesi e lo sviluppo di follicoli; nell’uomo, di promuovere la spermatogenesi. L’FSH è l’ormone principale che controlla la secrezione di estrogeni, infatti fa sì che nella donna gli androgeni della granulosa si convertano in estrogeni, mentre nell’uomo agisce sulle cellule del Sertoli e stimola la produzione di una proteina legante gli androgeni. L’LH invece, fa sì che nella donna si abbia l’ovulazione a metà ciclo ed è il principale ormone che controlla la successiva secrezione di progesterone da parte del corpo luteo; nell’uomo, agisce invece nelle cellule del Leydig, stimolando la produzione di testosterone. I prodotti dell’attività secretoria delle gonadi esercitano un
Estrogeni LH FSH GnRH OVAIO IPOFISI IPOTALAMO (e progesterone) Inibina
Figura 1. Controllo dell’asse ipotalamo-ipofisario.
Gli estrogeni producono anche effetti benefici sul cuore e sulla struttura ossea dell’organismo umano; infatti mantengono la densità ossea riducendo il rischio di fratture; Inoltre influenzano i centri cerebrali che sono deputati al mantenimento e alla regolazione della temperatura corporea.1
Con l’insorgenza della menopausa, le donne vanno in contro ad una diminuzione di estrogeni che innesca una serie di disagi, come ad esempio piccoli shock termici (vampate di calore), incremento del colesterolo (LDL) ematico con conseguente aumento della probabilità di insorgenza di patologie cardiache e diminuzione della densità minerale ossea che può portare a patologie come l’osteoporosi.1 Questa serie di eventi può essere tenuta sotto controllo con una terapia ormonale sostitutiva (HRT); spesso però, questo comporta l’insorgenza di patologie ormone-dipendenti, come neoplasie alla mammella e all’utero. Ci sono molteplici eventi che evidenziano come lo sviluppo e la progressione di neoplasie al seno (con il rischio di sviluppare metastasi) siano in stretta correlazione con l’esposizione ad estrogeni, endogeni ed esogeni. Infatti, il carcinoma mammario è stato il primo ad essere riconosciuto come patologia ormone-dipendente (fu dimostrato che l’asportazione totale delle ovaie induceva una regressione dei tumori mammari in un gruppo di pazienti nel periodo pre-menopausa).5
Gli estrogeni vengono utilizzati clinicamente, non solo per il trattamento ormonale nella donna in post-menopausa, ma anche per l’ipogonadismo primario, in cui si cerca di stimolare lo sviluppo dei caratteri sessuali secondari e la crescita ottimale.1
I principali estrogeni femminili sono l’estradiolo (17 β-estradiolo), l’estrone e l’estriolo.1 HO OH OH CH3 Estriolo HO O CH3 Estrone HO OH CH3 Estradiolo
L’estradiolo è il principale prodotto della secrezione ovarica, ma viene in parte prodotto anche dalla corteccia del surrene. Sebbene una certa quantità di estrone sia prodotta nell’ovaio, la maggior parte di questo ormone (ed anche di estriolo) è sintetizzata nel fegato a partire dall’estradiolo o, in tessuti periferici, a partire dall’androstenedione e da altri androgeni.
Dal punto di vista chimico, gli estrogeni sono costituiti da un sistema a quattro anelli condensati, che richiamano la struttura del ciclopentanoperidrofenantrene.
17 16 15 14 13 12 11 10 9 7 8 6 5 4 3 2 1 A B C D
Nell’estradiolo l’anello A è idrossilato in posizione C-3 e l’ossidrile in posizione C-17 dell’anello a cinque termini conferisce l’attività estrogenica di tale ormone. L’assenza di quest’ultimo ossidrile nell’estrone e la presenza invece anche di un altro ossidrile a livello del C-16 nell’estriolo, determina una bassa attività ormonale di questi steroidi e quindi una minore affinità per il recettore estrogenino.
plasmatiche a bassa densità (LDL). Nelle donne non in menopausa, tale processo avviene principalmente nelle ovaie.
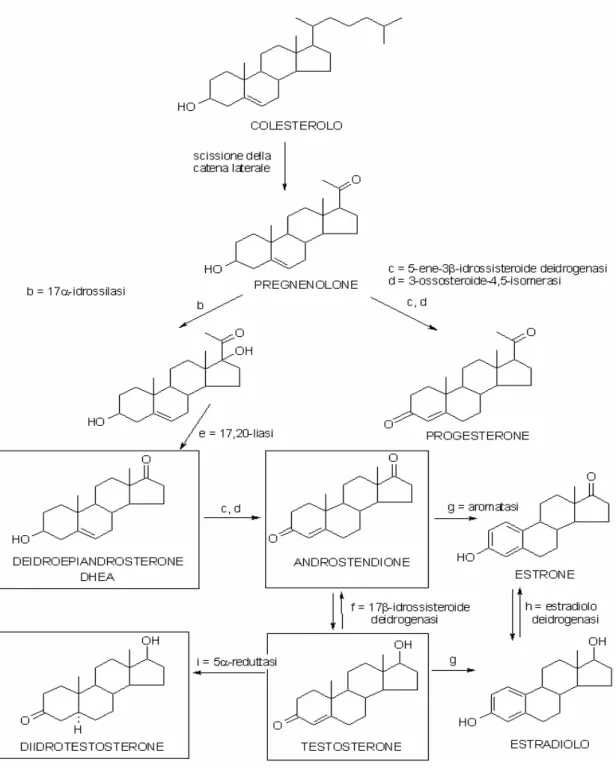
Figura 2. Biosintesi dell’estradiolo.
L’enzima P450scc (20,22-desmolasi) catalizza il distacco della catena laterale del colesterolo, trasformandolo in pregnolone. Il pregnenolone è convertito dalla 17α -idrossilasi (b) in 17α-idrossipregnenolone. Il passaggio successivo, catalizzato dalla 17,20 idrossilasi (e), porta alla rottura del legame fra C-17 e C-20. La perdita della catena
acetilica del 17α-idrossipregnenolone dà l’intermedio deidroepiandrosterone, che viene convertito in androstendione dall’enzima 5-ene-3β-idrossisteroide deidrogenasi (c). L’androstendione è interconvertibile con l’intermedio ridotto, il testosterone, per mezzo della 17β-idrossisteroide deidrogenasi (f).
L’estradiolo e l’estrone vengono sintetizzati a partire dagli androgeni, rispettivamente testosterone e androstedione, per azione del complesso P-450 aromatasi (g) che catalizza, in sequenza, l’idrossilazione e l’ossidazione del gruppo metilico (C-19), la formazione di un doppio legame in pos. 1-2, la decarbossilazione in posizione 19 e la formazione del caratteristico anello benzenico degli estrogeni. L’estradiolo può essere anche prodotto a partire dall’estrone, mediante l’azione dell’enzima estradiolo deidrogenasi ( h).
Metabolismo
Una volta liberato in circolo, l’estradiolo si lega fortemente ad una α2-globulina legante gli ormoni sessuali, detta SHBG (sex-hormone binding globulin) e, con minore affinità, all’albumina. L’estrogeno legato è relativamente indisponibile per la diffusione all’interno delle cellule, pertanto è la frazione libera ad essere fisiologicamente attiva.
L’estradiolo è convertito dal fegato e da altri tessuti in:
1. Metaboliti coniugati. L’ossidrilazione delle posizioni 2 e 4 danno, rispettivamente, il 2-idrossiestradiolo e il 4-idrossiestradiolo. I metaboliti ossidativi vengono quindi coniugati con solfato o con acido glucuronico e, in queste forme, eliminati con le urine o mediante secrezione biliare. I coniugati possono essere tuttavia idrolizzati nell’intestino a dare composti attivi riassorbibili nel circolo portale. Il 2-idrossiestradiolo, essendo una specie catecolica, subisce una metilazione metabolica simile a quella delle catecolammine, ad opera della O-metiltrasferasi, producendo il 2-metossiestradiolo;
HO OH CH3 Estradiolo CH3 HO HO OH CH3 OH HO OH 2-idrossieatradiolo 4-idrossiestradiolo H3CO HO OH CH3 OH HO OCH3 CH3 2-metossiestradiolo 4-metossiestradiolo Figura 3a.
2. Estrone ed estradiolo. La trasformazione del gruppo alcolico secondario in posizione 17β in gruppo chetonico produce l’estrone; l’ossidrilazione della posizione 16 produce l’estriolo.
HO OH CH3 CH3 HO OH OH CH3 HO O CH3 HO O OH Estradiolo Estrone Estriolo 16 αααα idrossiestrone Figura 3b.
RECETTORI ESTROGENICI: RECETTORI DI MEMBRANA E
RECETTORI NUCLEARI
Come detto precedentemente, l’estradiolo, una volta entrato in circolo si lega alla α2 -globulina SHGB. Una volta raggiunto il recettore target, il complesso SHGB-estradiolo si dissocia e l’estrogeno va ad interagire con il proprio recettore.1 Recettori di membrana e recettori nucleari sono i due tipi di recettori che interagiscono con gli estrogeni e si trovano principalmente nella mammella, nell’utero, nel cervello, nel fegato e nelle ossa.
Recettori di membrana: struttura e meccanismo.
I recettori estrogenici di membrana sono stati scoperti di recente: attraverso lo studio dei vari tipi di risposta che si hanno quando l’estradiolo si lega al recettore, è emerso che alcune risposte sono rapide (da secondi a minuti) e non comportano nessun coinvolgimento genomico e quindi nessun tipo di trascrizione.7 I recettori membranari si trovano principalmente nel SNC. Sono recettori accoppiati a proteine G e sono costituiti da una singola catena polipeptidica che forma sette α-eliche transmebrana, avente come dominio extracellulare il residuo N-terminale e come dominio intracellulare il residuo C-terminale (figura 4). La proteina G si accocia al terzo loop citoplasmatico. Essa è formata da tre subunità (α, β, γ) e, quando l’agonista si lega al recettore, la subunità α si dissocia e va ad attivare un effettore, che in questo caso è la fosfolipasi C (PLC).3 Quest’ultimo porta alla formazione di IP3 (inositolo 1,4,5 trifosfato) che incrementa la concentrazione Ca++ intracellulare, stimolando il suo rilascio dal reticolo sarcoplasmatico, e di DAG (diacilglicerolo) che a sua volta va ad attivare una proteina chinasi di membrana, la PKC (protein chinasi C). Tale proteina favorisce l’entrata di ioni calcio all’interno della cellula. L’aumento della concentrazione di calcio intracellulare promuove l’attivazione delle NOS (NO-sintetasi) che rilasciano l’ossido d’azoto (NO), utilizzato nel SNC come neurotrasmettitore.7
Figura 4. Recettori di membrana accoppiati a proteine G.
Inoltre l’estradiolo è direttamente e indirettamente responsabile dell’inibizione del rilascio del GnRH a livello ipotalamico.3 Il meccanismo indiretto è il seguente: l’estradiolo va a bloccare l’entrata di ioni potassio attraverso i canali GIRK, inibendo i recettori µ degli oppiodi e i recettori GABAB a lui accoppiati, che si trovano sulla membrana plasmatica dei neuroni POMC e GABA, presinaptici rispetto ai neuroni GnRH. In questo modo i neuroni presinaptci non vengono iperpolarizzati e non si ha trasmissione sinaptica con i neuroni GnRH postsinaptici, con conseguente blocco del rilascio di GnRH.6
Recettori nucleari: struttura e meccanismo.8
I recettori nucleari sono formati da sei domini (A-F) (figura 5):
1. Un dominio N-terminale A/B altamente variabile, sia nella sequenza che nella lunghezza, contenente la regione ormone-indipendente AF1(Actvation Function-1) che è coinvolta nell’attivazione della trascrizione;
2. Un dominio centrale C altamente conservato che si lega con il DNA, chiamato DBD (DNA binding domain). È formato dallo “zinc-finger”, ossia da due anse unite da una catena con quattro residui di cisteina che circondano un atomo di zinco. Lo zinc-finger è responsabile del legame con la zona ERE del DNA;
3. Un dominio “cerniera” D, interposto tra il DBD e l’LBD (ligand-binding domain). La flessibilità della sua struttura secondaria è la responsabile del cambiamento conformazionale che si ha quando il ligando si lega al recettore; 4. Un dominio E, che lega l’ormone (LBD) e contenente anch’esso una zona che
attiva la trascrizione (AF-2), ma ormone-dipendente. È formato da 12 α-eliche disposte a “sandwich”, che costituiscono la tasca idrofobica responsabile del legame con il ligando. LBD è infatti formato da tre strati di α_eliche antiparallele: uno strato centrale a tre eliche (H5/6, H9 e H10), posto tra due strati addizionali di eliche (H1_4 e H7, H8, H11) ed infine possiede due tratti con struttura β-sheet (S1, S2) e un tratto α-elicoidale (H12);
5. Un dominio C-terminale F, che promuove la trascrizione da parte della RNA polimerasi.
Figura 5. Struttura recettori nucleari.
I recettori nucleari si trovano nel citoplasma sottoforma di complessi oligomerici con 1
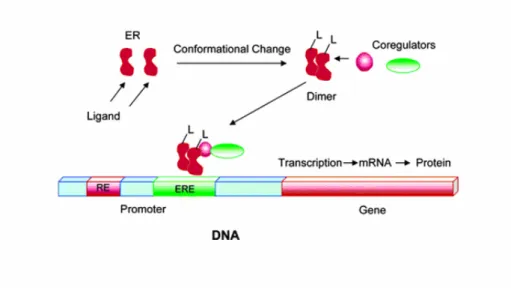
chiamate EREs (Estrogen Response Elements), localizzata all’estremità 5’della regione del promotore del gene. La successiva attivazione trascrizionale dei geni responsivi avviene mediante l’interazione diretta del complesso ligando-recettore con la zona ERE e con i coattivatori legati al sito AF-2 (figura 6).
Figura 6. Il ligando si lega al recettore per gli estrogeni inducendo un cambio
conformazionale che rende possibile la dimerizzazione e l’interazione con i coattivatori.
Sono stati caratterizzati due sottotipi recettoriali nucleari degli estrogeni: ERα e ERβ, quest’ultimo scoperto nel 1996.
Recettori nucleari: differenze tra ERαααα e ERββββ.
Confrontando la funzione che ha la sequenza AF-1 nell’attivare la trascrizione nei due sottotipi recettoriali α e β, è stato evidenziato che in ERα essa ha un ruolo importante, mentre in ERβ il suo ruolo non è così rilevante.8
I due sottotipi recettoriali α e β presentano un’omologia aminoacidica di circa il 96% per il dominio DBD (DNA Binding Domain) e un’omologia del 53% circa per l’LBD.8 Invece, per i domini A/B e D l’omologia è del 27% e del 26% rispettivamente. Infine, la regione F è quella che presenta la minore omologia (22%).
Il gene per il recettore α codifica per 595 amminoacidi, mentre quello per il recettore β codifica per 530 amminoacidi.
Figura 7b. Recettore β
Le due cavità di binding di ERα e ERβ differiscono per due residui amminoacidici: ERβ presenta infatti un residuo Met336 al posto di Leu384 dell’ERα e un residuo Ile373 al posto di Met421 dell’ERα. Inoltre, il volume della cavità di ERβ risulta più piccolo rispetto a quello di ERα.31
Figura 8. Sovrapposizione del recettore α (azzurro) con il recettore β (giallo). Il recettore α è maggiormente espresso a livello di mammella, utero, ossa, cervello, cellule della granulosa del follicolo9, fegato e sistema cardiovascolare.
Il recettore β si trova principalmente nel SNC, nel sistema immunitario, nell’apparato digerente, nei reni, nella prostata, nel tessuto polmonare, nelle ossa, nell’endotelio vascolare, nelle cellule della teca interna del follicolo9, nelle cellule luteiniche del corpo luteo9, nell’utero e in piccola parte anche nella mammella.
Questa differente distribuzione tissutale dei recettori α e β spiega, almeno in parte, le diverse e selettive azioni che gli estrogeni esercitano nei vari tessuti.8
AGONISTI ED ANTAGONISTI A CONFRONTO
Diverse conformazioni degli ERs in seguito ad interazione con agonisti ed antagonisti. 8
Attraverso lo studio cristallografico dei recettori estrogenici complessati con un agonista o un antagonista, è possibile evidenziare le diverse conformazioni che il recettore assume a seconda della natura del ligando. Consideriamo, a titolo di esempio, l’estradiolo (agonista) e il tamoxifene (antagonista).
Tamoxifene O N HO OH CH3 Estradiolo
Nel caso in cui sia l’estradiolo ad interagire con il recettore, l’α-elica H12 si chiude sull’α-elica H4, dando origine ad una conformazione “chiusa”, al cui interno rimane “intrappolato” il ligando. In questo modo si attiva il meccanismo che conduce, in ultima analisi, all’attivazione della trascrizione.
Se invece è un antagonista ad occupare il sito di binding del recettore, l’α-elica H12 non si chiude sull’α-elica H4; la conformazione rimane così “aperta” e i coattivatori non possono legarsi al complesso ligando-recettore; di conseguenza, non viene attivata la trascrizione. Tale conformazione aperta è dovuta all’ingombro sterico esercitato dalla catena laterale dell’antagonista (mancante nell’agonista) che, nel caso del tamoxifene, è di tipo N,N-dimetilamminoetilica. La conformazione è resa stabile da un legame a idrogeno che si forma tra tale catena ed il residuo Asp351 del sito di binding.
Interazioni di agonisti ed antagonisti con il sito di binding di ER. 8, 10
Vediamo ora le diverse interazioni a cui agonisti ed antagonisti danno origine all’interno del sito di binding del recettore estrogenico.
Agonista
Come si può vedere dalla Figura 9a, l’ossidrile dell’anello fenolico A dell’estradiolo stabilisce dei legami ad idrogeno con il Glu353, con l’Arg394 e con una molecola di acqua. Invece l’ossidrile dell’anello D instaura un legame a idrogeno con His524. Gli anelli facenti parte della molecola sono coinvolti in interazioni di tipo idrofobico all’interno del sito di legame.
Figura 9a. Interazione dell’estradiolo con il sito di binding di ER.
Antagonista
La Figura 9b rappresenta l’interazione del raloxifene con ER.
S HO OH O O N
legami ad idrogeno con Asp351. La restante parte della molecola è implicata in interazioni idrofobiche simili a quelle viste per l’estradiolo.
Figura 9b. Interazione del raloxifene con il sito di binding di ER.
SERMs: MODULATORI SELETTIVI DEI RECETTORI DEGLI
ESTROGENI
I SERMs sono molecole che esplicano un’attività agonista, cioè simile a quella degli estrogeni naturali, in alcuni tessuti, come ossa (mantenimento della densità minerale e riduzione di insorgenza di osteoporosi), sistema cardiovascolare (riduzione del colesterolo LDL e quindi dell’incidenza di cardiopatie) e SNC (riduzione delle vampate di calore); hanno invece proprietà antagoniste in altri, come cervello e mammella (ridotto effetto proliferativi e quindi ridotto rischio di insorgenza e sviluppo di tumori). Alcuni farmaci appartenenti a tale categoria (ad es. il tamoxifene), se usati in terapie a lungo termine, possono incrementare il rischio di insorgenza di neoplasie a livello dell’endometrio, a causa di un’azione agonista su tale tessuto.
I tre SERMs più conosciuti sono il tamoxifene (Nolvadex), il raloxifene (Evista) e il toremifene (Fareston).8
S HO OH O O N Raloxifene Tamoxifene O N Toremifene O N Cl Tamoxifene
È un derivato trifeniletilenico e può essere considerato il capostipite della categoria dei SERMs.8 È utilizzato per prevenire e curare i primi stadi del tumore estrogeno-dipendente della mammella, grazie alla sua attività antiestrogenica in questo tessuto. Inoltre è dotato di una debole attività agonista nelle ossa e nel sistema cardiovascolare.11 È stato usato anche come agente chemiopreventivo, approvato dell’FDA nel 1999, sempre per contrastare il rischio dell’insorgere del tumore alla mammella nelle donne in pre e post-menpausa.8 Tuttavia, come detto precedentemente, la terapia a lungo termine con questo farmaco può aumentare il rischio di insorgenza di neoplasie a livello dell’endometrio. 11
Raloxifene
Il Raloxifene è un derivato benzotiofenico che ha dimostrato un attività agonista sulle ossa e sul tessuto adiposo: preserva infatti la densità minerale ossea e ha un effetto ipolipemizzante; presenta inoltre un’attività antagonista a livello della mammella. Questo profilo farmacologico produce un effetto antiosteoporotico e cardioprotettore simile a quello degli estrogeni, in assenza di rischio di carcinoma della mammella. Si è osservato inoltre una riduzione delle probabilità di sviluppo di tumori all’utero. Non è invece efficace contro quei sintomi della menopausa, quali vampate di calore e sudorazione.8
Toremifene
Il toremifene è un derivato trifeniletilenico, come il tamoxifene. Esso presenta le stesse proprietà di quest’ultimo, anche se non sembra presentare il rischio di insorgenza di tumori
Meccanismo d’azione
Non è molto chiaro come i SERMs possano comportarsi da agonisti in alcuni tessuti e da antagonisti in altri. Questa loro proprietà farmacologica può essere spiegata con tre tipi di meccanismi, correlati tra loro:8
1. Diversa conformazione assunta dai recettori in seguito al legame con il SERM; 2. Diversi sottotipi recettoriali di ER (α e β) con cui il SERM interagisce a livello
dei vari tessuti e loro diversa modalità di attivazione della trascrizione; 3. Diverse proteine coregolatrici che si legano ai recettori.
Il punto n. 1 è già stato descritto nei paragrafi precedenti.
Per quanto riguarda la diversa modalità di attivazione della trascrizione (punto n. 2), è stato dimostrato che l’attività agonista possa sfruttare un meccanismo per cui il complesso ligando-ER interagisce con il DNA mediante particolari proteine (come jun e fos appartenenti alla proteina AP-1) e questo conduca all’attivazione della trascrizione genica.12
Per quanto riguarda il punto n. 3, una volta che si è formato il complesso SERM-ER, esso può interagire con un coattivatore (CoA), che promuove la trascrizione (come accade per gli estrogeni naturali), oppure con un corepressore (CoR) che inibisce il meccanismo di trascrizione, nonostante il complesso recettore-ligando sia unito al sito ERE del DNA (figura 10). La presenza di determinati coattivatori o corepressori e l’espressone della trascrizione dipende dal tessuto e dal sottotipo recettoriale di ER.
Figura 10. Meccanismo d’azione dei SERMs nell’attività antiagonistica legante un
CoR e come agonista ERE-non dipendente.
Studi fatti sui topi,8 dimostrerebbero che l’azione dei SERMs su ERα induca la trascrizione e che si abbia un’azione opposta su ERβ. Sembra addirittura che la
stimolazione di ERβ porti all’inibizione di ERα, andando a formare con esso un eterodimero. Sia il raloxifene che il tamoxifene legano entrambe le isoforme del recettore e agiscono come antagonisti puri quando il loro effetto è mediato dal recettore beta mentre funzionano da agonisti parziali quando l’effetto è mediato da ERα.
SERBAs: AGONISTI SELETTIVI PER ER
ββββ
I SERBAs (Selective Estrogen Receptor Beta Agonists) sono molecole non steroidee selettive per il sottotipo β dei recettori estrogenici. I SERBAs potrebbero presentare quelle caratteristiche “ideali” dei SERMs, ovvero la capacità di promuovere effetti benefici a livello del fegato, delle ossa, del SNC (tra cui migliorare le capacità mnemoniche) , 13degli adipociti (regolare il metabolismo degli acidi grassi andando a diminuire la concentrazione di colesterolo nel sangue)14 e del sistema cardiovascolare (prevenire l’ipertrofia del miocardio e danni post-ischemici)15 e, allo stesso tempo, essere privi dell’azione proliferativa su tessuti quali mammella ed utero (mediata da ERα). Gli agonisti selettivi per ERβ possono anche essere considerati anti-infiammatori specifici per alcune patologie (alcune forme di asma ed infiammazioni che coinvolgono i linfociti T-helper).16 Inoltre, è stata dimostrata l’utilità terapeutica di agonisti ERβ-selettivi nell’iperplasia/cancro della prostata, nella demineralizzazione ossea, nell’artrite e nell’infiammazione intestinale.17
È stato inoltre evidenziato che ERβ è in grado di esercitare un’azione modulatoria nei confronti di ERα;18 ciò è emerso da studi riguardanti la concentrazione di recettori progestinici (PR) a livello del nucleo ipotalamico preottico mediale (MPN) e del nucleo ventromediale (VPM), nel cervello neonatale di cavia femmina. Premesso che:
• nel MPN sono presenti entrambi i sottotipi ERα e ERβ;
• nel VPM è presente solo il sottotipo ERα;
• solo ERα promuove, una volta attivato, l’espressione dei recettori progestinici (PR); dopo somministrazione di estradiolo (che attiva entrambi i sottotipi recettoriali), solo nel VPM risulta aumentata la concentrazione dei PR, mentre nel MPN tale concentrazione rimane invariata. Si è giunti quindi alla conclusione che uno dei ruoli svolti dal recettore β
HSDs: ENZIMI COINVOLTI NELLA BIOSINTESI DEGLI ORMONI
STEROIDEI
Gli HDSs (Hydroxysteroid Dehydrogenases) sono degli enzimi che catalizzano reazioni di ossidoriduzione stereospecifche di alcool o carbonili, usando rispettivamente NAD(P)+ o NAD(P)H come cofattori. Nell’uomo sono presenti molti tipi di HSDs (3α-, 3β-, 11β-, 17β- e 20α-HSDs) che a loro volta si suddividono in altri sottotipi, ma soltanto il 17β-HSD sembra essere coinvolto negli ultimi passaggi della biosintesi degli ormoni steroidei.19
17ββββ-HSD e le sue isoforme
L’enzima 17β-HSD fu isolato nella placenta umana per la prima volta nel 1958, da Langer e Engel. Negli anni seguenti tale enzima fu purificato e furono caratterizzati alcuni suoi parametri, come cinetica, substrato e coenzimi.19
Il 17β-HSD appartiene alla superfamiglia delle SDR (short-chain dehydrogenases/reductases) che svolgono reazioni di ossido-riduzione NAD(H)-dipendenti. Sono enzimi oligomerici, formati da circa 250-350 aminoacidi. La struttura degli SDR è fatta a “sandwich”, dove al centro si trova una struttura β-sheet e ai lati ci sono numerose α-eliche. Il rilascio o l’acquisto dello ione idruro avviene grazie alla presenza del cofattore NAD(H) e i residui aminoacidici coinvolti in questo sistema di scambio (tra enzima/cofattore e substrato o viceversa), sono la Tyr che catalizza le reazioni acido-base (poiché la Tyr ha un gruppo OH nella catena laterale) e la Lys che, avendo un gruppo NH3+ nella catena laterale e essendo vicino alla Tyr, è in grado di “ricostruire” la catena laterale della Tyr. Le caratteristiche strutturali appena descritte si ritrovano in tutte le classi di enzimi appartenenti alle SDR.20
Figura 11. Modello di struttura del 17β-HSD. In rosso le α-eliche, in verde la struttura
β-sheet, in giallo il NAD(H) e in blu il ligando.
L’enzima 17β-HSD fu inizialmente classificato come un enzima reversibile, ovvero in grado di catalizzare sia reazioni di riduzione che di ossidazione, in base ai cofattori ad esso legati.
Soltanto negli anni ’90 furono scoperte tutte le diverse isoforme del 17β-HSD, e si osservò che alcune di esse erano in grado di ridurre ed altre di ossidare a seconda dei cofattori a cui erano legate.
Attualmente si conoscono otto isoforme di 17β-HSD (1-8), nella specie umana. La prima ad essere stata scoperta e studiata è stata l’isofoma 1, ossia l’estradiolo 17β -deidrogenasi (17β-HSD1), isolata nella placenta.
La 17β-HSD1 è in grado di catalizzare la reazione di riduzione dell’estrone con conseguente formazione di estradiolo, sfruttando l’ossidazione del proprio cofattore che da NAD(P)H passa a NAD(P)+. 19
O
CH3 CH3OH
17ββββ-HSD1
Le strategie fino ad ora adottate per cercare di controllare le patologie ormone-dipendenti, come il tumore alla mammella, sono consistite in:
• Uso di SERMs con proprietà antiestrogeniche (es. tamoxifene), in modo tale da bloccare l’azione dell’estradiolo sugli ER;
• Uso di inibitori dell’enzima aromatasi, che catalizza la formazione di estradiolo a a partire da testosterone.
La 17β-HSD1 costituisce uno dei recenti target per controllare e bloccare la biosintesi dell’estradiolo. Inibitori di questo enzima potrebbero essere utili per il trattamento e la prevenzione di alcune malattie estrogeno-dipendenti come tumori al seno e all’utero.
Nell’organismo umano, la 17β- HSD1 si trova a livello della granulosa dei follicoli ovarici, della placenta, della prostata, del tessuto adiposo, della mammella e dell’endometrio; in particolare, in questi ultimi due tessuti risulta spesso sovraespresso. Tale enzima è un omodimero costituito da 327 aminoacidi ed avente una massa di 34.9 KDa.21
Il sito di binding della 17β-HSD1 è formato da una tasca idrofobica, al cui interno è presente una sorta di “tunnel”, delimitato da Val143, Leu149, Leu96, Phe159, Pro187, Phe226 e Val225, in cui si inseriscono gli steroidi. Al di fuori di tale tasca ci sono due aree polari, fondamentali per il legame degli estrogeni, una formata da His221 e Glu282 e l’altra, situata in posizione opposta, formata da Ser142 e Tyr155. Esiste anche una terza area polare, formata da Tyr218 e da Ser222, che sembra essere coinvolta nella stabilità della struttura tridimensionale dell’enzima.
Esaminando le interazioni che si instaurano tra il sito attivo della 17β-HSD1 e l’estradiolo (Figura 12), si nota che l’OH dell’anello fenolico A instaura legami ad idrogeno con una delle due aree polari menzionate prima, ossia quella formata da His221 e Glu282, mentre l’OH in posizione 17 stabilisce legami ad idrogeno con l’altra regione polare. La parte lipofila dell’estradiolo dà invece interazioni di tipo idrofobico con i residui aminoacidici che formano la tasca idrofobica. Di fronte al sito di binding, si trova la tasca che accoglie il cofattore (in questo caso NADP+); non sono stati ancora ben identificati tutti gli aminoacidi che delimitano tale tasca. 21
Figura 12. Sito di legame della 17β-HSD1 umana, con al suo interno l’estradiolo. I legami ad idrogeno sono segnati in giallo, gli aminoacidi idrofobici in arancione e quelli
polari in blu.